

Here, Cheng et al. present data from Tetrahymena that highlight a division of labor among ciliate piggyBac-derived genes, which carry out mutually exclusive categories of excision events mediated by either transposon-like features or RNA-directed heterochromatin.
Keywords: Tetrahymena, programmed genome rearrangement, domesticated transposon, piggyBac, vacuoles
Abstract
Ciliated protozoans perform extreme forms of programmed somatic DNA rearrangement during development. The model ciliate Tetrahymena thermophila removes 34% of its germline micronuclear genome from somatic macronuclei by excising thousands of internal eliminated sequences (IESs), a process that shares features with transposon excision. Indeed, piggyBac transposon-derived genes are necessary for genome-wide IES excision in both Tetrahymena (TPB2 [Tetrahymena piggyBac-like 2] and LIA5) and Paramecium tetraurelia (PiggyMac). T. thermophila has at least three other piggyBac-derived genes: TPB1, TPB6, and TPB7. Here, we show that TPB1 and TPB6 excise a small, distinct set of 12 unusual IESs that disrupt exons. TPB1-deficient cells complete mating, but their progeny exhibit slow growth, giant vacuoles, and osmotic shock sensitivity due to retention of an IES in the vacuolar gene DOP1 (Dopey domain-containing protein). Unlike most IESs, TPB1-dependent IESs have piggyBac-like terminal inverted motifs that are necessary for excision. Transposon-like excision mediated by TPB1 and TPB6 provides direct evidence for a transposon origin of not only IES excision machinery but also IESs themselves. Our study highlights a division of labor among ciliate piggyBac-derived genes, which carry out mutually exclusive categories of excision events mediated by either transposon-like features or RNA-directed heterochromatin.
Duality of germline and somatic functions can lead to dramatic differences in genome organization in germline versus somatic cells. In most organisms, germline genomes can be thought of as the “master copy,” stably transmitting genetic and epigenetic information across generations. In contrast, somatic genomes can be optimized for expression and specialized function (Sabour and Scholer 2012). A classic example of specialized somatic function driving genome rearrangements comes from vertebrate lymphocytes, which undergo V(D)J gene rearrangements, creating a remarkable diversity of T and B cells (Litman et al. 2010). In contrast to these locus-specific rearrangements, several vertebrate and invertebrate species exhibit genome-wide rearrangements. Genome-wide rearrangement is especially prevalent and well studied in ciliated protozoa, such as Tetrahymena thermophila, in which individual cells possess both a germline and a somatic nucleus. The germline “micronucleus” is transcriptionally silent except during mating, whereas the somatic “macronucleus” is transcriptionally active directing vegetative growth. Like in other ciliates, T. thermophila mating triggers degradation of the parental macronuclei and biogenesis of a new macronucleus via programmed genome rearrangement. During the sexual process of conjugation, the diploid micronucleus goes through meiosis, post-meiotic mitosis, and cross-fertilization to generate zygotic nuclei, which further divide and differentiate into new macronuclei and micronuclei of the progeny cell, while the old macronucleus degrades (Yao et al. 2014). Programmed genome rearrangement in Tetrahymena involves genome-wide deletion of thousands of segments (internal eliminated sequences [IESs]) that together make up ∼34% of the micronuclear genome. After IES elimination, the remaining macronuclear DNAs are fragmented at specific chromosome breakage sequences and endoduplicated to generate ∼67 copies of the somatic macronuclear genome (Yao et al. 2014). Some of the resulting minichromosomes are eliminated during subsequent vegetative growth (Lin et al. 2016).
Early studies in ciliates revealed that the DNA elimination process serves to remove transposons from the somatic genome. For example, the “telomere-bearing element” (TBE) in Oxytricha trifallax and the Tec element in Euplotes crassus resemble Tc1/mariner transposons and are present in thousands of copies in the micronucleus but completely eliminated from the macronucleus (Jahn et al. 1993; Doak et al. 1997). TBE transposases are required for this process (Nowacki et al. 2009). Transposon-like elements are also present in Paramecium and T. thermophila micronuclear genomes but eliminated from macronuclear genomes (Klobutcher and Herrick 1995; Patil et al. 1997). These findings led to a model in which IES deletion during programmed DNA elimination resembles transposon excision (Klobutcher and Herrick 1997).
Despite its attractiveness, this model lacked experimental evidence. For instance, Tetrahymena IESs lack conserved sequence motifs akin to transposon ends that could regulate deletion. In the few cases where sequence determinants are known, IES-flanking rather than IES internal sequences regulate deletion boundaries (Godiska and Yao 1990; Chalker et al. 1999). Furthermore, studies in Tetrahymena and Paramecium revealed that IES excision occurs through RNA-induced heterochromatin formation rather than via a transposition-like process (Madireddi et al. 1996; Mochizuki et al. 2002; Yao et al. 2003; Betermier and Duharcourt 2014). In Tetrahymena, dsRNA is transcribed from IESs during conjugation and processed into ∼27-nucleotide (nt) small RNAs called scnRNAs (scan RNAs) (Chalker and Yao 2001; Malone et al. 2005; Mochizuki and Gorovsky 2005; Woo et al. 2016). scnRNAs then interact with an Argonaute protein (Mochizuki et al. 2002) and target homologous IESs, leading to histone modifications, including H3K9me3 and H3K27me3 (Taverna et al. 2002; Liu et al. 2007), and recruitment of chromodomain proteins Pdd1p and Pdd3p (Madireddi et al. 1996; Coyne et al. 1999; Nikiforov et al. 2000) to form heterochromatin structures that are then deleted. The IES elimination process therefore appears to resemble genome surveillance systems that repress transposons through small RNAs and heterochromatin (Wheeler 2013) rather than a transposition reaction.
The model in which transposon and IES excision are functionally related was revived by the finding that “domesticated” piggyBac genes play a key role in programmed DNA elimination in Paramecium and Tetrahymena (Baudry et al. 2009; Cheng et al. 2010; Chalker and Yao 2011; Dubois et al. 2012; Vogt and Mochizuki 2013). In Tetrahymena, the TPB2 (Tetrahymena piggyBac-like 2) gene arose via ancient capture of a piggyBac-like element by the ciliate host genome. Although the transposase-encoding ORF and its catalytic residues have been preserved, other transposition-associated features, including terminal inverted repeats (TIRs), have been lost. Following domestication, introns and additional protein-coding sequence were acquired by TPB2. The encoded protein Tpb2p plays a key role in heterochromatin-dependent IES excision (Cheng et al. 2010), suggesting that TPB2 evolved to target and excise heterochromatin instead of TIRs, retaining ancestral excision activity. Using this altered targeting, Tpb2p mediates excision of IESs that do not resemble transposons. Following TPB2-mediated excision, flanking DNA is rejoined by nonhomologous end-joining mediated partly by Ku80 (Lin et al. 2012).
A subsequent study found that another domesticated transposase gene, LIA5, is also involved in RNA-mediated heterochromatin formation during programmed DNA elimination (Shieh and Chalker 2013). The discoveries of TPB2 and LIA5 in Tetrahymena and PiggyMac in Paramecium provided a link between transposon excision and IES elimination. However, they also challenged a direct connection between the two mechanisms because the excision machinery seems to recognize heterochromatin rather than transposon-like cues. We speculated that studying additional domesticated piggyBac genes might reveal the missing evolutionary link between transposons and IESs. Indeed, the T. thermophila genome contains three more domesticated piggyBac genes of unknown function: TPB1, TPB6, and TPB7 (Supplemental Table S1; Supplemental Figs. S1, S2). We focused on TPB1 and TPB6, which encode proteins with a piggyBac-derived transposase domain (with predicted intact catalytic activity) and a Ku homology domain, suggesting that Tpb1p and Tpb6p might have fused the excision and repair functions necessary for programmed DNA elimination.
In this study, we undertook a detailed functional analysis of TPB1. We found that TPB1 plays an active role in programmed DNA elimination but does not participate in the vast majority of excision events. Instead, TPB1 deficiency during mating results in failure to remove 20 regions from the macronuclear genome, 12 of which are clearly defined IESs that interrupt protein-coding exons. One of these retained IESs disrupts the DOP1 (Dopey domain-containing protein) gene, causing deleterious effects in TPB1-deficient progeny. Many of these defects can be rescued by artificially excising the DOP1 IES. Intriguingly, unlike most Tetrahymena IESs, TPB1-dependent IESs have highly conserved terminal sequences that resemble piggyBac TIRs. We show that these IESs are excised efficiently and precisely and that their TIRs are necessary to direct excision. We further show that a related gene, TPB6, also helps mediate excision of the same subset of IESs. Thus, although minor in fraction, TPB1- and TPB6-dependent IESs represent the most direct evidence for the evolution of programmed DNA elimination from ancestral transposon excision. Our study reveals that Tetrahymena uses dual means to eliminate IESs, whereby some are excised through TPB1, TPB6, and transposon-like TIRs, whereas others require TPB2 and heterochromatin-based cues.
Results
TPB1 is important for progeny fitness
To determine TPB1’s function, we first used quantitative RT–PCR (qRT–PCR) to examine whether its expression pattern was consistent with a role in programmed DNA elimination. We found that TPB1 is highly transcribed only at late stages of conjugation, coincident with DNA rearrangements in developing new macronuclei (Fig. 1A) and similar to the pattern described for TPB2 (Cheng et al. 2010).
Figure 1.
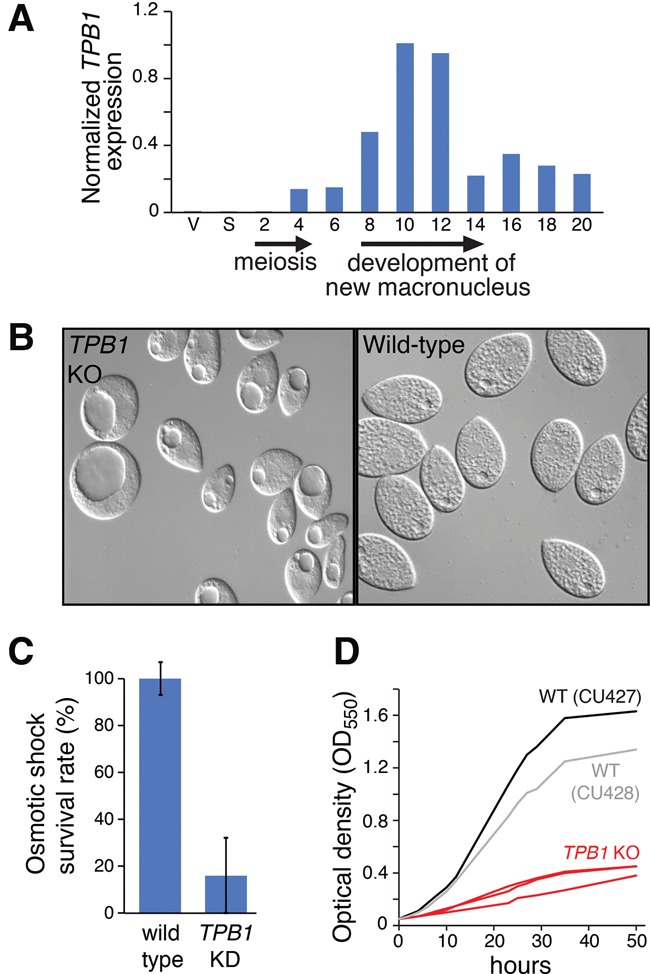
TPB1 deficiency leads to defects in progeny after mating. (A) TPB1 expression, as measured by qRT–PCR, is induced during meiosis and is highest 8–12 h after mating, coincident with programmed DNA rearrangement (Austerberry et al. 1984). RNA samples were extracted from vegetative (V), starved (S), and conjugating cells (2–20 h after mixing). (B) TPB1 knockout progeny (left panel) display a large vacuole defect. (C) TPB1 knockdown progeny have reduced survival after osmotic shock. Values represent the mean of three independent cell stocks. Error bars represent SD. (D) TPB1 knockout progeny (red) show dramatic growth defects compared with two wild-type strains (black and gray).
We next examined the consequences of depleting TPB1 using cadmium-inducible hairpin RNA (RNAi) knockdown, achieving >80% reduction of TPB1 transcripts (Supplemental Figs. S3A, S4). Reducing or eliminating TPB1 expression during vegetative growth did not affect cell morphology or growth rates, as expected given the lack of expression at this stage. Depleting TPB1 during mating also did not impair conjugation or progeny production. However, progeny of TPB1-deficient cells manifest severe defects, including a “giant vacuole” phenotype (Fig. 1B). TPB1-deficient progeny contain one very large vacuole that can occupy more than half of the cytoplasm and lack typical contractile vacuoles (CVs) (Patterson and Sleigh 1976). We speculated that the large vacuoles are enlarged CVs: By observing live cells, we observed fusion of multiple small vacuoles to form a central large vacuole that eventually contracted and emptied to complete a very slow contraction cycle (several minutes vs. <10 sec in wild-type cells) (Supplemental Movies S1–S3). Because one function of CVs is osmoregulation, we measured viability after osmotic shock. Whereas wild-type cells survived osmotic stress well, TPB1-deficient progeny had drastically reduced survival (Fig. 1C).
TPB1-deficient progeny grew slowly compared with wild-type cells, were unable to reach the same density (Fig. 1D), and showed impaired mating pair formation. Despite these severe defects, TPB1 depletion still yields a much milder phenotype than TPB2 depletion. Previously, we found that progeny of TPB2-depleted matings were completely inviable; cells arrested during late conjugation, failed to completely differentiate new macronuclei, and died (Cheng et al. 2010). This suggested that TPB1 either participates in a distinct process from TPB2 or has a weaker contribution to the same process.
We replicated our TPB1 knockdown findings using a knockout strategy in which TPB1 and flanking sequences were replaced with a neomycin resistance gene in the macronuclear and micronuclear genomes (Supplemental Fig. S3B). Subsequent examination of knockout breakpoints revealed that TPB1 was knocked out along with neighboring gene TTHERM_00309880A, which is expressed only weakly in vegetative stages, with slightly increased expression during conjugation. We found that knockdown and knockout strategies resulted in very similar phenotypes. We confirmed that expression of the neighboring gene was unaffected in our knockdown strains (Supplemental Fig. S4) and concluded that the defects that we saw in both strains were due solely to TPB1 deficiency and not the neighboring gene. We therefore used knockout as well as knockdown strategies for further studies of TPB1.
TPB1 performs a specific, not global, role in IES elimination
Our results showed that TPB1 is dispensable during vegetative growth but is critical for producing healthy progeny through conjugation. Since TPB1 is related to TPB2, we speculated that TPB1 controls IES excision during development of the new macronucleus, like TPB2. We therefore analyzed TPB1-deficient progeny for deletion of two well-studied IESs, the M and R elements (Fig. 2D; Supplemental Fig. S5). Although these IESs fail to delete upon TPB2 depletion, they deleted normally in TPB1-deficient progeny. This suggested that TPB1 depletion does not result in a gross defect in DNA deletion, which may explain its milder phenotype compared with TPB2. Nevertheless, we hypothesized that TPB1 is important for deletion of a subset of IESs; their retention could impact nearby genes, potentially explaining the TPB1-deficient phenotype (Fig. 1).
Figure 2.
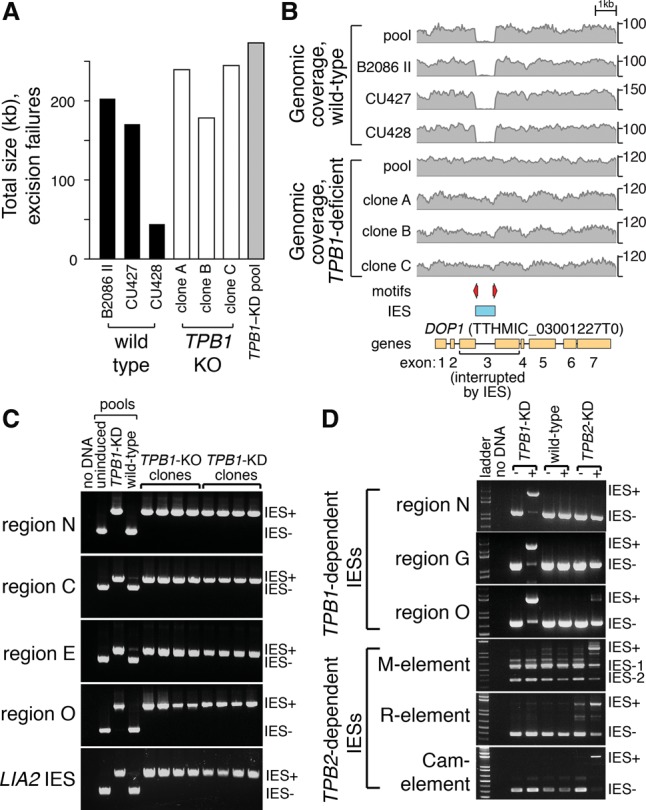
Specific genomic regions fail to excise in TPB1-deficient progeny. (A) TPB1-deficient progeny show no global IES excision defects. Using deep genomic sequencing, we identified “interesting” regions that were efficiently deleted in our reference sample (pooled wild-type progeny) but retained in each of the other samples (Materials and Methods). Surprisingly, individual wild-type strains (black bars) failed to delete many regions compared with the wild-type pool. TPB1-deficient clonal (white bars) and pooled (gray bar) progeny failed to delete a number and total size of regions comparable with wild-type cells. (B) Twenty regions fail to excise in TPB1-deficient cells. Genomic coverage in the DOP1 gene is depicted, showing that an IES in exon 3 is precisely excised in wild-type cells but fails to be excised in all TPB1-deficient samples. Also indicated (“motifs”) are TIRs that mark TPB1-dependent IESs (red arrows). (C) PCR assays using primers flanking five TPB1-dependent regions (N, C, E, and O and the LIA2 IES) reveal precise excision (“IES−”) in wild-type progeny and the progeny of cells in which TPB1 knockdown is not induced. However, in the progeny of TPB1 knockout cells and of cells in which TPB1 knockdown was induced by cadmium, IESs were retained (“IES+”). Expected product sizes are shown at the right ([IES+] micronuclear form; [IES−] macronuclear form). (D) TPB1 and TPB2 mediate excision of at least partially nonoverlapping sets of IESs. IESs in the N, G, and O regions fail to excise when TPB1 knockdown is induced by cadmium (IES+) but are properly excised in progeny of wild-type cells, TPB2 knockdown cells, and cells in which TPB1 knockdown is not induced (IES−). In contrast, excision of the M, R, and Cam elements is severely impaired upon TPB2 knockdown but unaffected in TPB1 knockdown cells. The M element undergoes variable excision in wild-type cells: Some cells delete 0.6 kb (IES-1), and others delete 0.9 kb (IES-2). Samples are labeled above gel images. (+) The presence of cadmium to induce knockdown; (−) the absence of cadmium to induce knockdown.
To address this possibility, we performed genome-wide analysis of TPB1-deficient progeny using deep genomic sequencing. We analyzed a pool of TPB1 knockdown progeny and a pool of wild-type cells after the completion of conjugation as well as three clonal wild-type strains and three clonal TPB1 knockout progeny lines. After mapping reads to the micronuclear reference assembly (Hamilton et al. 2016), we examined depth of coverage in each sample. Treating the wild-type pool as a “reference” sample, we looked for regions with low coverage in the wild-type pool and high coverage in other samples (Materials and Methods). Such regions represent those that successfully delete in most cells in the wild-type reference pool but fail to delete in another sample.
To our surprise, we found a large number of regions that apparently failed to delete even in one or more of the three clonal wild-type strains (Fig. 2A). The number and total extent of such regions vary widely among the three wild-type strains (293–953 regions; 44–203 kb in total). Upon closer examination (Supplemental Fig. S6), we found that some of these regions reflect variation in IES boundary usage, as seen for the well-studied M element, where some cells delete a 0.6-kb region and others delete 0.9 kb (Godiska and Yao 1990). However, in other regions, an IES appears to completely fail to delete in one or more strains. It is possible that some apparently variable regions may represent copy number polymorphisms among wild-type strains: An in-depth study of wild-type variation is needed to explore this possibility. No regions verifiably failed to delete in all three wild-type clones, but we note that because we compared coverage of each sample with a wild-type pool, we could not identify regions that might delete inefficiently in the wild-type pool. A recent study (Hamilton et al. 2016) also reported widespread heterogeneity in IES excision, with many IES boundary positions varying by 1–20 base pairs (bp); the variation that we were able to detect spans larger genomic extents (≥100 bp).
We found no evidence for a gross defect in IES removal in TPB1 mutant cells, which fail to excise a number and total size of regions similar to wild-type cells (Fig. 2A), suggesting that overall rearrangement efficiency is unaffected. We next searched for genomic regions that have an “all or nothing” dependence on TPB1, failing to be eliminated in all TPB1 mutant samples but in none of our wild-type samples or the wild-type line used for the macronuclear genome assembly (Eisen et al. 2006). Consistent with our hypothesis, we initially found 16 regions that may depend on TPB1 for removal (Fig. 2B; Supplemental Table S2; Supplemental Fig. S7).
We examined each of these 16 putative TPB1-dependent regions in detail (Supplemental Fig. S7; Supplemental Table S2). In eight cases, the region represents an IES that is reliably deleted in a stereotyped manner in wild-type cells but completely fails to delete in TPB1-deficient cells. A ninth case (region LM) (Supplemental Fig. S7C) represents an IES that completely deletes in a canonical fashion in wild-type cells, whereas TPB1-deficient samples delete smaller segments and retain most of this IES. We classify these nine cases as “high confidence” TPB1-dependent IESs. Although the other seven regions also show differences between wild-type and TPB1-deficient cells, their status is much less clear cut because we observed variation of neighboring sequences among wild-type cells (regions I and J) and/or because analysis of TPB1-deficient pool data suggests that a significant fraction of cells in the pool successfully deleted the region (regions J, Q, V, W, Y, and Z). Furthermore, in most of these less clear-cut cases, assembly gaps and/or errors in the micronuclear reference genome preclude understanding the structure of even wild-type excision events. We therefore focused our analysis on the nine high confidence TPB1-dependent IESs.
To our surprise, all nine high confidence IESs directly interrupt protein-coding exons (Supplemental Table S2; Supplemental Fig. S7). A recent genome-wide study found that only six of ∼12,000 Tetrahymena IESs interrupt exonic sequences (Hamilton et al. 2016), including one in the LIA2 gene (Yao et al. 2007; Fass et al. 2011; Motl and Chalker 2011). We wondered whether the LIA2 IES was TPB1-dependent. Although we could not initially analyze the LIA2 IES because it lies within a gap in the micronuclear reference genome assembly, we were successful using another approach. We generated an alternative de novo assembly using sequence reads from TPB1-deficient cells and repeated our search for TPB1-dependent regions (see the Materials and Methods). Using this method, we found three additional high confidence TPB1-dependent IESs (regions R, S, and T) (Supplemental Table S2; Supplemental Fig. S7), including the LIA2 IES, to yield a total of 12 high confidence TPB1-dependent IESs and one new ambiguous case (region AA). All three new high confidence IESs are within gaps in the micronuclear assembly. They reliably delete in a stereotyped manner in wild-type cells, completely fail to delete in TPB1-deficient cells, and interrupt genes. We further examined the six known exon-interrupting IESs identified in a recent genome-wide analysis (Hamilton et al. 2016): All six are among the TPB1-dependent IESs that we found. With a total of 12 exon-interrupting IESs, our study doubles the number of known examples of this IES class and suggests that TPB1 is necessary for removal of all exon-interrupting IESs.
The 12 high confidence TPB1-dependent IESs are diverse in size (136 to ∼4200 bp) (Supplemental Table S2), but are generally smaller than other IESs (median 419 bp, compared with 2947 bp; P < 10−5, Wilcoxon test). They interrupt 11 protein-coding genes; two IESs interrupt the same exon of a MIZ zinc finger gene (regions E and S). There is no obvious functional connection among all 11 genes, although two likely have vacuolar function (see below), and two (regions C and F) are paralogs of one another. Interestingly, the IESs in these paralogous genes are not in the same relative positions, suggesting that IESs inserted independently in these genes. Furthermore, four IESs contain internal transcripts, none of which has functional annotations. We note that the TPB1-dependent regions that we identified are the most obvious defects associated with TPB1 deficiency. If more subtle effects exist, such as reduced excision efficiency of some IESs, they would be difficult to detect above the surprising level of wild-type variation.
We validated five TPB1-dependent IESs using PCR in wild-type and mutant cells (Fig. 2C), confirming that their excision depends completely on TPB1. We further tested whether TPB2 is required for excision of five TPB1-dependent IESs. Surprisingly, excision of three of five TPB1-dependent IESs appears to be unaffected in the progeny of TPB2-depleted cells in a PCR assay (N, G, and O regions) (Fig. 2D). For the other two TPB1-dependent IESs, we observed partial retention in TPB2-depleted cells (Supplemental Fig. S8), suggesting that some TPB1-dependent IESs might also require TPB2. Alternatively, this partial retention may reflect the fact that TPB2-deficient cells arrest during conjugation, before TPB1-mediated rearrangement is complete. At the very least, our findings suggest that for some IESs, TPB1 and TPB2 activities are mutually exclusive.
Failure to excise the DOP1 IES results in large vacuoles
We investigated whether any of the TPB1-dependent IESs caused the “giant vacuole” phenotype and lower fitness of TPB1-deficient progeny (Fig. 1). All 12 high confidence TPB1-dependent IESs interrupt genes, and their retention would truncate or alter the interrupted proteins to varying extents (Supplemental Table S2). Two of the interrupted genes were excellent candidates to explain the large vacuole defect. The first candidate that we tested was VMA4 (vacuolar ATP synthase subunit E) (Ho et al. 1993), which is expressed during vegetative growth. A 1775-bp TPB1-dependent IES interrupts VMA4 13 nt after the start codon (Supplemental Fig. S9A). We predicted that IES retention would separate the body of the VMA4 gene from its promoter and start codon, entirely disrupting function. Unexpectedly, the expression level and length of the VMA4 mRNA are similar in TPB1-deficient and wild-type cells (Supplemental Fig. S9B,C), implying a new promoter and transcription start site (TSS) within the IES. Using 5′ RACE in TPB1-deficient cells, we indeed found that the IES encodes an alternative TSS, 5′ untranslated region (UTR), and in-frame start codon. Thus, failure to excise the IES results in replacement of the first five amino acids with an alternative seven-amino-acid sequence without affecting the rest of VMA4 (Supplemental Fig. S9D).
We investigated whether this subtle N-terminal change affects Vma4p function, possibly through a dominant-negative effect (Supplemental Figs. S3, S9). We tested this by introducing an IES-retaining copy of VMA4 into wild-type cells; this did not recapitulate the vacuole phenotype, ruling out a dominant-negative effect. We also attempted to rescue the vacuole phenotype by introducing a “prerearranged” gene (VMA4 with IES removed) into TPB1-deficient cells. Even after complete genomic replacement of all macronuclear copies (Supplemental Fig. S9D), this did not rescue the vacuole phenotype. Therefore, failure to remove the TPB1-dependent IES in VMA4 does not explain the phenotypes of the TPB1-deficient progeny.
We next examined another TPB1-dependent IES-containing candidate gene: DOP1. DOP1 is part of a small gene family that is widely conserved across eukaryotes (Rachidi et al. 2005). Fungal orthologs are essential for viability and proper cellular morphology (Pascon and Miller 2000; Barbosa et al. 2010), and mutation of a rat ortholog, Dopey1, causes abnormal vacuoles in neurons (Tanaka et al. 2014). In Tetrahymena, a TPB1-dependent IES interrupts the DOP1-coding region (Figs. 2B, 3A). IES retention would introduce premature stop codons, resulting in the absence of full-length Dop1p. We found that DOP1 mRNA expression is greatly reduced in TPB1-deficient progeny (Fig. 3B), suggesting that IES-containing DOP1 transcripts are degraded or that the transcript is severely 3′-truncated.
Figure 3.
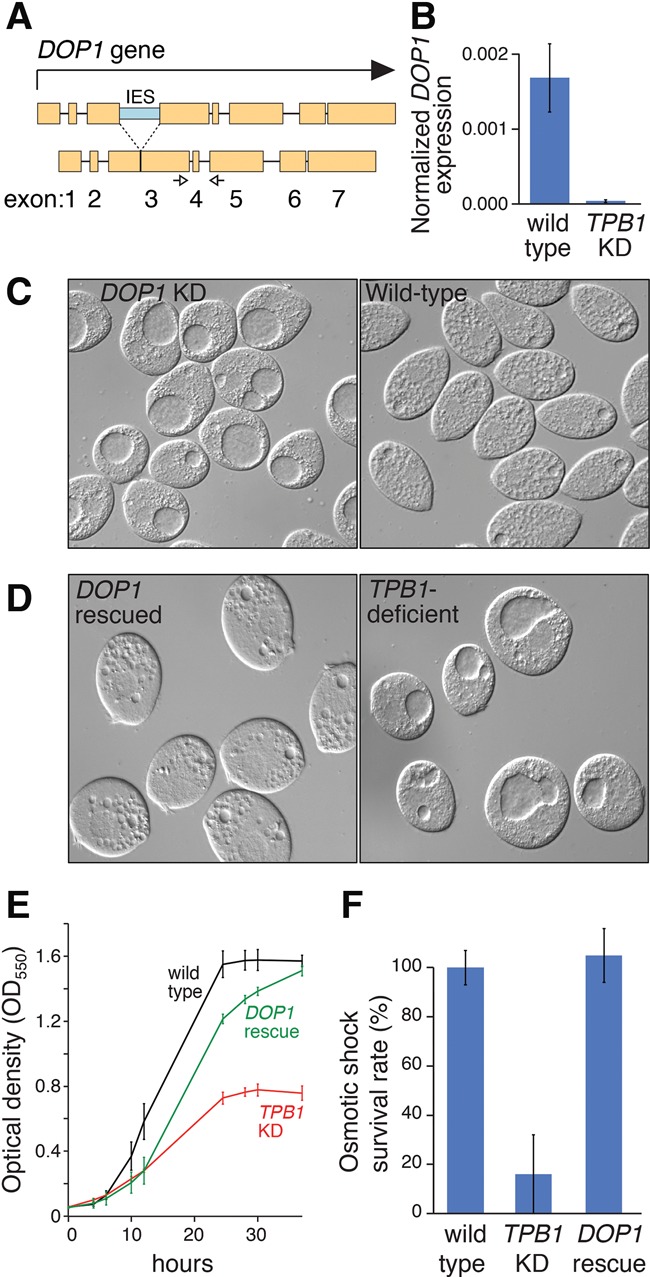
IES retention in DOP1 explains the vacuole phenotype of TPB1-deficient cells. (A) Schematic of DOP1 gene, showing coding exons (yellow) and the TPB1-dependent IES (blue) interrupting exon 3. White arrows show qRT–PCR primers (shown in B). (B) DOP1 expression in vegetatively growing cells is dramatically reduced in TPB1 knockdown progeny, as measured by qRT–PCR. (C) DOP1 knockdown (left panel) results in a large vacuole phenotype. (D–F) Compared with TPB1-deficient cells, DOP1-rescued (but still TPB1-deficient) cells no longer display large vacuoles (D, left), slow growth (E), or osmotic shock sensitivity (F). Values represent the mean of three independent cell stocks. Error bars represent SD.
To test DOP1’s role in the vacuole phenotype, we knocked down DOP1 in wild-type cells. Remarkably, knocking down DOP1 alone was sufficient to elicit a large vacuole phenotype (Fig. 3C) similar to that of TPB1-deficient progeny (Fig. 1B). We then tested the effects of rescuing DOP1 expression in TPB1-deficient progeny, replacing DOP1 with a “prerearranged” version (IES removed) (Supplemental Figs. S3, S10A). Upon DOP1 rescue, large vacuoles disappeared (Fig. 3D), and the contraction cycle was restored (Supplemental Movie S4). Even partial DOP1 rescue (the macronucleus has ∼67 copies of most genes) is sufficient for rescue (Supplemental Fig. S10B).
We next tested whether “fixing” DOP1 corrected all phenotypes of TPB1-deficient progeny. Indeed, slow growth rate and osmotic shock sensitivity were also largely corrected by DOP1 rescue (Fig. 3E,F). Although TPB1-deficient progeny cannot form mating pairs efficiently, restoring DOP1 also rescued this defect in mating pair formation (mating initiation). However, repairing DOP1 did not rescue the ability to complete mating and produce healthy progeny: Only ∼10% of progeny survive compared with 80%–90% in wild-type matings. We attribute this defect to retention of one or more of the other 11 gene-disrupting TPB1-dependent IESs. Together, our results demonstrate that retention of the DOP1 IES results in the large vacuoles associated with TPB1 deficiency but does not explain all TPB1 phenotypes. Thus, multiple TPB1-dependent IESs must be excised for full fitness.
TPB1-dependent IESs resemble piggyBac transposons
TPB1-dependent IESs are already different from most IESs because of their location in protein-coding exons. We wondered whether they have other unique features. We examined whether TPB1-dependent IESs form heterochromatin like other IESs, recruiting proteins such as Pdd1p (Kataoka and Mochizuki 2015). Upon analyzing H3K9me3, H3K27me3, and Pdd1p occupancy using published ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data (Kataoka and Mochizuki 2015; Kataoka et al. 2016), we found much lower occupancy of all three proteins in TPB1-dependent IESs than other IESs (Fig. 4B). We then examined three TPB1-dependent IESs in PDD1 knockout cells and in cells knocked out for the Argonaute-related gene TWI1. Most IES deletions are blocked in these mutants (Mochizuki et al. 2002), but the TPB1-dependent IESs that we tested delete normally (Fig. 4A). We also examined published data for scnRNAs (Schoeberl et al. 2012), the small RNAs that help recruit heterochromatin at most IESs; TPB1-dependent IESs were not enriched for scnRNAs (Fig. 4C). Together, these data suggest that TPB1-dependent excision does not depend on scnRNA-directed heterochromatin in the same way as TPB2-dependent excision does.
Figure 4.
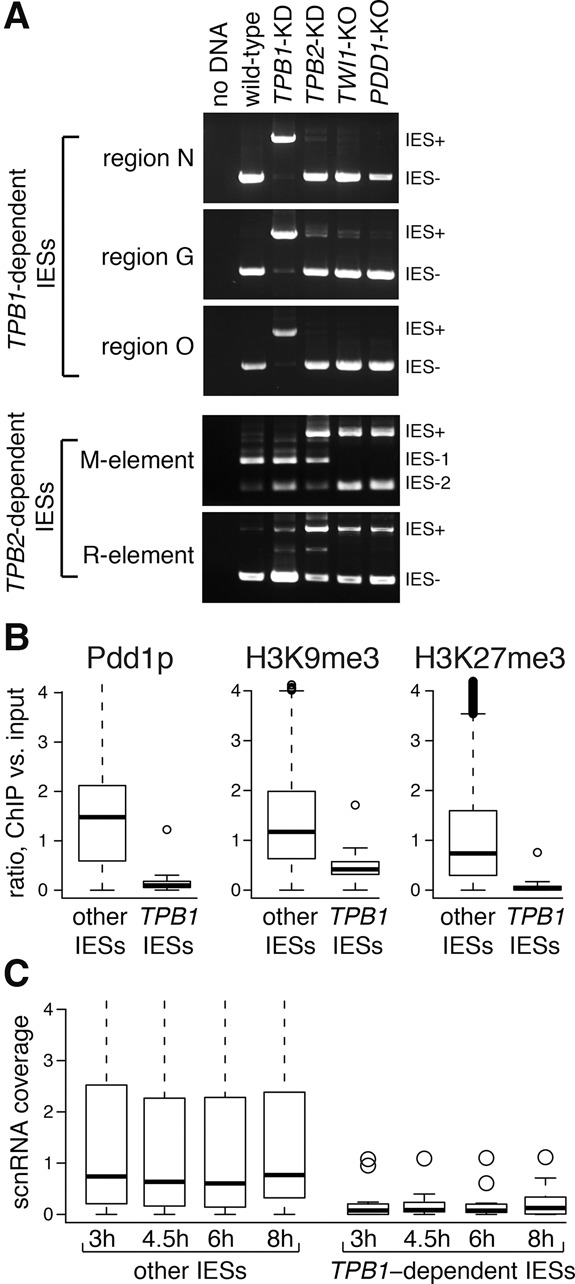
Differences between TPB1- and TPB2-dependent IESs. (A) TPB1-dependent IESs (regions N, G, and O) are excised efficiently in cells knocked out for TWI1, an Argonaute-related protein necessary for small RNA-directed heterochromatin formation at IESs, and in cells knocked out for PDD1, a protein that recognizes heterochromatin at IESs and is necessary for their removal. In contrast, excision of two TPB2-dependent IESs (M and R elements) is mostly blocked in TWI1 and PDD1 knockouts (the presence of IES− PCR products likely results from the continued presence of parental cells in the late mating pools used). TPB1 and TPB2 knockdown cells described in Figure 2D were also analyzed here for comparison. (B) Most IESs excise via an RNA-directed heterochromatin pathway, which depends on H3K9me3 and H3K27me3 accumulation at IESs and recruitment of heterochromatin proteins such as Pdd1p, but TPB1-dependent IESs appear to be excised independently of this pathway. Indeed, analysis of published ChIP-seq data shows that H3K9me3, H3K27me3, and Pdd1p are enriched on most IESs but not TPB1-dependent IESs. Box plots show the distribution of ChIP:input ratios for each IES. Thick horizontal lines show medians of each distribution, the open box covers the middle two quartiles, and circles represent outliers. (C) Similarly, scnRNAs are enriched at most IESs but not at most TPB1-dependent regions.
We next looked for conserved sequence features in TPB1-dependent IESs. We searched for sequence motifs at IES boundaries that could represent TPB1 recognition sites. Using the MEME algorithm, we found that all 12 high confidence TPB1-dependent IESs contain a characteristic 12-bp motif present at both IES boundaries in inverted orientations (Fig. 5A; Supplemental Fig. S7), much like transposon TIRs. We wondered whether a pair of motifs with suitable orientation and distance was sufficient to define TPB1-dependent IESs in the Tetrahymena genome. We searched the micronuclear reference assembly and found 127 motif pairs with appropriate relative orientation and distance (Materials and Methods). As expected, these 127 motif pairs included the nine TPB1-dependent IESs that do not have assembly gaps at their boundaries. The remaining 118 pairs did not delineate IESs even in wild-type cells, suggesting that additional determinants of TPB1-dependent excision exist outside the 12-bp TIRs.
Figure 5.
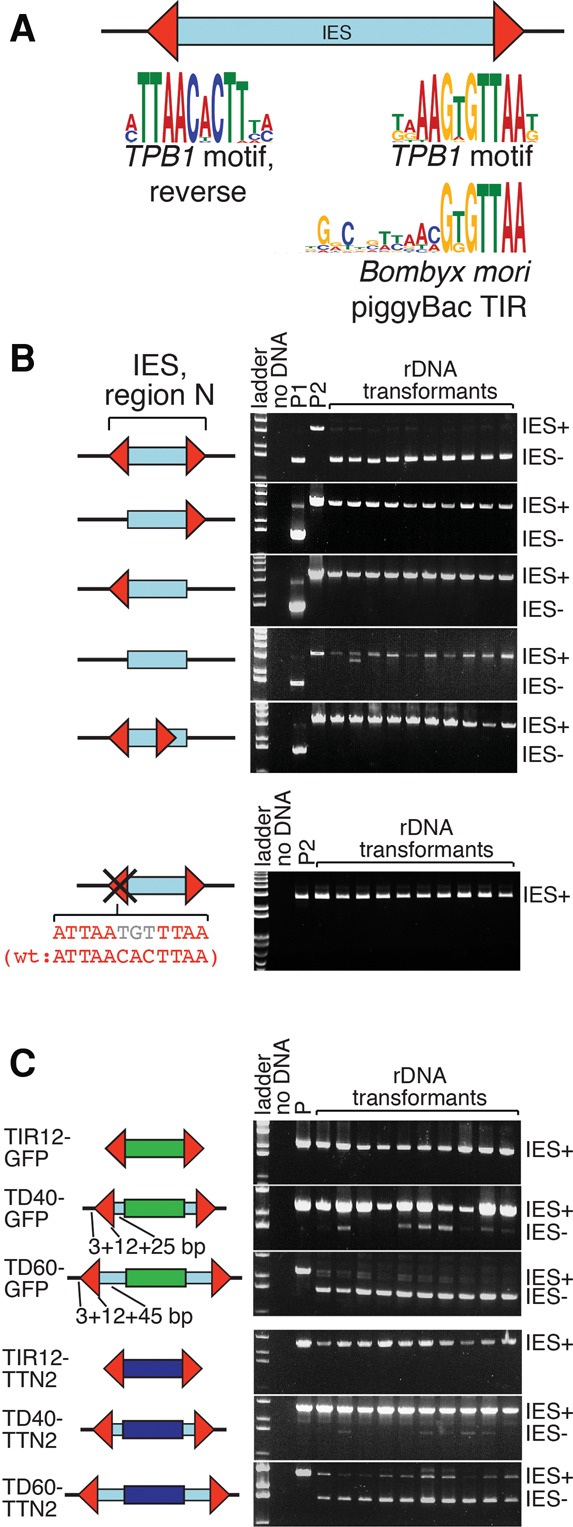
Transposon-like TIRs direct excision of TPB1-dependent IESs. (A) All 12 TPB1-dependent IESs have a characteristic boundary motif similar to TIRs of Bombyx mori piggyBac (P < 0.0002, TOMTOM motif comparison). (B) The TIRs are necessary to direct TPB1-dependent IES excision. We inserted the entire TPB1-dependent IES from the VMA4 gene—including TIRs (red arrows), 20-bp flanking sequences (black lines), and internal IES sequences (blue box)—into an rDNA vector. We introduced constructs into wild-type cells, allowed them to go through conjugation, and assayed deletion in 10 individual transformants by PCR. Successful excision required both TIRs in the correct relative positions. Even a small change to one TIR (bottom) abolishes correct excision. (Red letters) Wild-type nucleotides; (gray letters) altered nucleotides; (P1 and P2) control plasmids to show expected IES− and IES+ product sizes, respectively. (C) IES internal sequences are largely dispensable for TPB1-mediated excision. We modified our assay in B by introducing a heterologous “cargo” gene (GFP [green] or TTN2 [dark blue]) flanked by combinations of TIRs and adjacent IES internal and IES external sequences. The 12-bp TIRs alone did not support deletion (TIR12 constructs). However, larger terminal domains (TD) were sufficient to support deletion. The TD40 constructs containing the TIRs with 25 bp of adjacent IES internal sequence and 3 bp of adjacent IES external sequence did support excision, albeit inefficiently. More impressively, TD60 constructs that contain the TIRs with 45 bp of adjacent IES internal sequence and 3 bp of adjacent IES external sequence supported almost complete IES removal. (P) Control plasmid to show expected IES+ size.
We noticed that the motif that we identified is similar to the TIR motif of Bombyx mori piggyBac elements (P < 0.0002, TOMTOM motif comparison) (Fig. 5A), which is recognized by the piggyBac transposase for excision (Xu et al. 2006). This high similarity likely reflects the evolutionary origins of TPB1 as a domesticated piggyBac transposase. It further suggests that these 12 IESs are remnants of ancient transposon insertions.
To determine the significance of the TIRs in TPB1-dependent IESs, we used an rDNA vector (Chalker et al. 1999) to test cis-acting requirements for excision of the IES within VMA4. We inserted the entire IES (including TIRs, all internal sequences, and 20 bp of flanking sequences) in the rDNA vector, delivered it to conjugating cells before DNA deletion occurred, and analyzed IES excision in the progeny. Excision occurred precisely and nearly completely, indicating the robustness of this system (Fig. 5B). However, deleting either copy of the 12-bp TIR abolished deletion, indicating that the TIRs are necessary cis-acting sequences. Substituting only 3 bp of one TIR also abolished deletion, further establishing the importance of this sequence. Moving one copy of the TIR into the internal region of the IES was not sufficient to cause deletion from the new location or any other boundary, suggesting that additional sequences are necessary to direct deletion (Fig. 5B). This dependence on TIRs further distinguishes TPB1-dependent IESs from most other IESs, which are excised by TPB2 and have no essential shared boundary or internal sequences: Their excision is instead guided by scnRNAs and heterochromatin.
To further dissect the cis-acting requirements for TPB1-dependent excision, we assayed an artificial test IES using the same rDNA vector. This test IES comprised a segment of the GFP gene or the Tetrahymena macronuclear gene TTN2 as an internal “cargo” sequence bounded by the 12-bp TIRs from the VMA4 IES with various amounts of adjacent IES external and IES internal sequences (Fig. 5C). We found that a construct comprising only the 12-bp TIRs and cargo (TIR12) is insufficient to direct deletion, further supporting our model that appropriately arranged TIR motif pairs are insufficient to define TPB1-dependent IESs. Including 3 bp of flanking sequences and 25 bp of adjacent IES internal sequences (TD40) modestly improved excision frequency to 10%. However, increasing IES internal sequences to 45 bp (TD60) yielded near-complete deletion. These analyses showed that TPB1-dependent IESs are recognized through 45- to 60-bp terminal sequences, including the shared TIRs, but that additional IES internal sequences are dispensable for TPB1-dependent excision.
TPB6 cooperates with TPB1 to mediate transposon-like excision of some IESs
Our finding that TPB1 and TPB2 mediate excision of different subsets of IESs explains their evolutionary maintenance in Tetrahymena genomes. We wondered whether such an explanation might explain retention of other piggyBac-derived genes; i.e., they each excise different IES subsets. We next investigated TPB6, since it is most closely related to TPB1 and has similar domain architecture, with Ku-like and transposase-like domains (Supplemental Figs. S1, S2). Consistent with their important, nonredundant roles, TPB1, TPB2, and TPB6 are all conserved in other Tetrahymena species, with dN/dS ratios indicating strong purifying selection on the entire gene and on the transposase domain when analyzed alone (Supplemental Table S3).
We generated a germline TPB6 knockout strain (Supplemental Figs. S3F, S11) and found striking similarities between TPB6-deficient and TPB1-deficient phenotypes. Both knockouts have a milder phenotype than TPB2, successfully completing conjugation and progeny production. Furthermore, both TPB1 and TPB6 knockout progeny have large vacuoles, suggesting common IES targets (Fig. 6A). Indeed, all eight TPB1-dependent IESs that we tested are also retained in TPB6-deficient progeny (Fig. 6B). In contrast, TPB6 deficiency does not result in retention of a TPB2-dependent IES, the R element. Although we did not formally prove that TPB6 operates on all 12 TPB1-dependent IESs and no others, our results strongly suggest that both TPB6 and TPB1 might be required to excise a specialized subset of IESs that are distinct from TPB2-dependent IESs.
Figure 6.
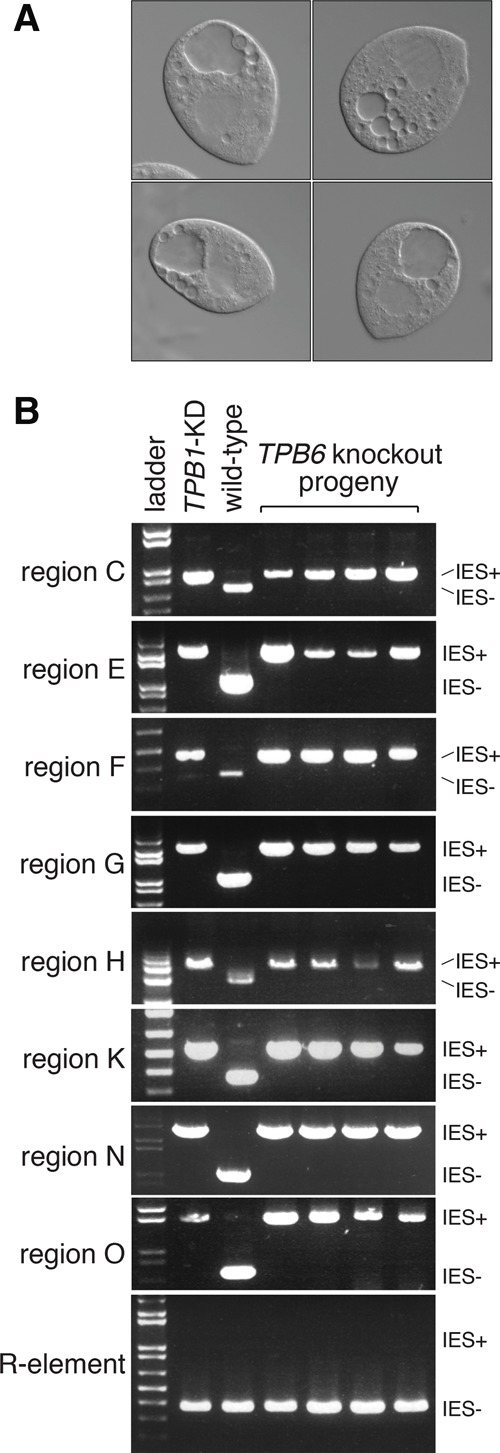
TPB6 knockout progeny share defects with TPB1 knockout progeny. (A) Representative TPB6 knockout progeny cells display the large vacuoles seen in TPB1-deficient progeny (Fig. 1B) and DOP1 knockdown cells (Fig. 3C). (B) TPB6-deficient progeny also fail to excise TPB1-dependent IESs. PCR assays of eight representative TPB1-dependent IESs (regions C, E, F, G, H, K, N, and O) show that excision fails in TPB6 knockout progeny. In contrast, excision of a TPB2-dependent IES, the R element, is unimpaired in TPB6-deficient progeny. Expected product sizes are shown at the right.
Discussion
There are fundamental similarities as well as surprising differences between different ciliate species in the process of programmed DNA rearrangement. In both Paramecium and Tetrahymena, piggyBac transposase-derived host genes as well as RNA-directed heterochromatin formation are required to mediate the excision of tens of thousands of IES elements (Baudry et al. 2009; Cheng et al. 2010; Shieh and Chalker 2013; Betermier and Duharcourt 2014; Yao et al. 2014). In this study, we found that the deletion of a distinct subset of IESs depends on additional domesticated transposase genes, TPB1 and TPB6, suggesting that there are two fundamentally different and mutually exclusive means to excise IESs in Tetrahymena. The first mechanism is used for the majority of IESs, which are found in intergenic or intronic regions. It is directed by scnRNA-guided heterochromatin at IESs followed by excision by the domesticated piggyBac-derived gene TPB2. Rather than recognizing terminal IES regions, TPB2 relies on heterochromatin formation within IESs, including recruitment of proteins such as Pdd1p (Kataoka and Mochizuki 2015). The second mechanism is the one that we describe here: transposon-like excision mediated by two other piggyBac-derived genes, TPB1 and TPB6, which together direct excision of all 12 IESs that are known to interrupt protein-coding exons. Retention of the TPB1-dependent IES in a vacuolar gene, DOP1, explains the large vacuole, osmotic shock sensitivity, and growth rate phenotypes of TPB1-deficient progeny but not their failure to complete mating. Thus, our findings point to a division of labor among the domesticated piggyBac-derived genes that direct programmed genome rearrangement in Tetrahymena. We anticipate that such division of labor might also exist in other ciliates, such as Paramecium and Oxytricha (Nowacki et al. 2009; Dubois et al. 2012).
We speculate that the form of IES elimination mediated by TPB1 and TPB6 is a direct descendant of transposon excision, preserving the transposon-like requirement for TIRs. Conventional transposition involves both integration and excision, but mutational analyses of piggyBac transposases show that the two functions are separable (Li et al. 2013). We propose that Tpb1p and Tpb6p acquired analogous mutations to lose the integration but not the excision function, retaining TIR dependence.
We propose that the invention of another form of IES excision via scnRNA-directed heterochromatin involved specialization of Tpb2p to recognize heterochromatin rather than TIRs. Available data do not reveal which excision mode evolved first, but it is clear that the TPB2/heterochromatin form of excision now dominates in T. thermophila. This would have the benefit of reducing the stringent requirement to maintain IES termini, reducing mutational load on the genome while widening the system's specificity to allow removal of other transposon types. Only TPB1-dependent IESs within protein-coding exons continue to exhibit transposon-like features, likely reflecting strong selective pressure to excise these IESs precisely and efficiently. Thus, this small subset of TPB1-dependent IESs is a traceable “fossil” record of the transposon origins of not only the excision machinery but also of IESs themselves. This insight, together with the domesticated piggyBac genes, suggests that IESs evolved from invasion of ciliate genomes by piggyBac transposons rather than another transposon type, Tc1/mariners, as surmised previously (Klobutcher and Herrick 1997). Moreover, unlike most Tc1/mariners, excision of piggyBac elements is seamless without overhangs, potentially rendering them more suitable for excision events in protein-coding regions.
Intriguingly, TPB1 and TPB6 have taken a further evolutionary step: Their common ancestor fused a Ku domain (important in nonhomologous end-joining) with the transposase domain, linking two of the activities needed for successful transposon/IES excision. In contrast, even though TPB2-dependent excision requires Ku for repair (Lin et al. 2012), Ku activity is encoded separately. We speculate that because TPB1/6-dependent IESs interrupt protein-coding exons, there is a much higher degree of stringency for successful excision that may have selected for fusion of the two domains.
Unexpectedly, this division of labor does not explain the simultaneous retention of TPB1 and TPB6, both of which are needed for excision of a subset of IESs. We propose that this is a result of subfunctionalization. Since both piggyBac transposases and Ku domains act as dimers (Walker et al. 2001), we speculate that Tpb1p and Tpb6p function as heterodimers. We propose that a common ancestor of TPB1 and TPB6 formed homodimeric proteins. However, following duplication, each gene acquired complementary debilitating changes (e.g., one in the transposase and one in the Ku domain), making retention of both genes essential. Thus, studying the function of domesticated transposon-like genes provides deep evolutionary insights into the origins of programmed DNA rearrangement in ciliates, one of the most dramatic examples of somatic genome rearrangements observed in any life form.
Materials and methods
Cell culture
Inbred T. thermophila strains B2086 II, CU428 (Mpr/Mpr [VII, mp-s]), and CU427 (Chx/Chx [VI, cy-s] were obtained from Peter Bruns (Cornell University, Ithaca, NY) and cultured as described previously (Gorovsky et al. 1975). Cells were grown in SPP or Neff medium at 30°C in a rotating incubator. Cells were prepared for mating by washing in 10 mM Tris-HCl (pH 7.4) and starvation in the same buffer for ≥10 h prior to mixing equal numbers of cells of complementary mating types. TPB1 heterozygous knockout strains were generated from parental mating of CU427 and CU428 strains using conventional gene transformation; these strains were mated to produce homozygous knockouts. Heterozygous TPB1 hairpin RNA-generating region-containing strains were also generated from parental mating of CU427 and CU428 and mated to produce knockdown progeny.
Knockout, knockdown, and knock-in plasmids (Supplemental Fig. S3)
Flanking regions (∼1 kb) of target genes were PCR-amplified and cloned into a plasmid with the neo cassette to allow drug selection (Gaertig et al. 1994; Mochizuki 2008). To generate knockdown constructs, a hairpin RNA-generating region (∼500 bp) within the ORF was PCR-amplified and cloned into the PCRII-I vector using two sets of restriction enzyme sites, generating two inverted copies, which were cloned into the pIBF rDNA vector. Hairpin expression was driven by the cadmium-inducible MTT1 promoter (Howard-Till and Yao 2006).
Cytological imaging
Living cells were loaded directly onto slides. For fixation, cells were treated with 2% paraformaldehyde. Images were captured using a Zeiss Axio Imager microscope.
Cell growth assay
Wild-type and mutant cultures were initiated using cells at OD550 ∼0.05 and grown in Neff medium at 30°C in a rotating incubator to log phase. Cell densities were measured by spectrophotometry to determine optical density (OD550). An OD550 value of 1 represents ∼1 × 106 cells per milliliter.
Osmotic shock assay
Cells were grown to log phase in hypertonic solution (Neff plus 200 mM sucrose), a sample was taken, and cells were fixed and counted under a microscope. The medium was then washed away from the remaining unfixed cells, which were moved to hypotonic solution (10 mM Tris-HCl at pH 7.4) and incubated overnight before fixing and recounting to determine survival rates.
IES elimination PCR assay in TWI1 and PDD1 complete knockout strains
PDD1 knockout strains ΔPDD1 39.1A and ΔPDD1 W3.3.4 were obtained from Dr. Douglas Chalker (Washington University, St Louis, MO), and TWI1 knockout strains cΔTWI1 2-1A and cΔTWI1 WG4 were obtained from the Tetrahymena Stock Center (Cornell University, Ithaca, NY). For each gene, we mated the two corresponding knockout strains and isolated genomic DNAs to use as PCR templates from late mating pools 30 h after mixing.
Pdd1p, H3K9me3, and H3K27me3 occupancy
We reanalyzed published ChIP-seq data for Pdd1p (Kataoka and Mochizuki 2015), H3K9me3, and H3K27me3 (Kataoka et al. 2016). First, we created an improved genome-wide IES data set because we noticed that a published data set (Noto et al. 2015) was missing a number of IESs (especially those bounded by micronuclear assembly gaps). We therefore used a less stringent BLAST-based method to identify additional candidate IESs, defined as regions of the micronuclear assembly missing from the macronuclear assembly. In detail, we compared the two assemblies using Blastn (Altschul et al. 1997), retained matches of ≥100 bp with ≥99% identity, and retained only reciprocal best matches. Regions of the micronuclear assembly without macronuclear matches formed a preliminary IES set, which we further filtered to remove regions of <150 bp (many smaller regions represent sequences close to CBS sites rather than IESs) and regions derived from minichromosomes that are eliminated from the somatic nucleus (Lin et al. 2016). We used the union of the published and BLAST-based sets for further analysis; the combined data set comprises 12,355 IESs.
Next, we examined Pdd1p, H3K9me3, and H3K27me3 occupancy. We downloaded data from NCBI (Pdd1p ChIP: SRR2071905; input for Pdd1p SRR2071903 and SRR2071904; H3K9me3 ChIP SRR3659032; H3K27me3 ChIP SRR3659033; input for H3K9me3/H3K27me3 SRR3659031), trimmed reads to retain the first 36 bp using fastx_trimmer (http://hannonlab.cshl.edu/fastx_toolkit), and used bwa (Li and Durbin 2009) to map reads to the micronuclear assembly. We counted reads mapping to each IES, divided counts by the number of mapped reads (in millions) in each sample, and divided counts by the size of each IES. We added a very small number (10−6) to avoid problems of division by zero and took ratios of counts in the ChIP-seq samples to counts in the corresponding input sample (for Pdd1p, we averaged the two input samples).
scnRNA analysis
We downloaded scnRNA sequence data from NCBI (3 h, SRR514963; 4.5 h, SRR514964; 6 h, SRR514965; 8 h, SRR514966) (Schoeberl et al. 2012). All possible 25-nt sequences were extracted using published lists (Gene Expression Omnibus database, GSM949307–GSM949310) and mapped to the micronuclear assembly using Bowtie2 (Langmead and Salzberg 2012). For each sample we calculated scnRNA coverage by counting reads mapping to each IES, dividing counts by the size of each IES in base pairs, and then dividing by the number of mapped reads (in millions).
Genome sequencing and analysis
Genomic DNA samples used for sequencing included a wild-type mating pool (strains CU427 and CU428, 24 h after conjugation), a pool of progeny from mated TPB1 knockdown cells, three individual wild-type strains (CU427, CU428, and B2086II), and three individual TPB1 knockout progeny clones (homozygous homokaryons). DNAs were isolated from unfractionated cells and contained mostly macronuclear DNA due to its high copy number. One-hundred-base-pair paired-end reads were generated for the individual wild-type strains and knockdown pool using an Illumina HiSeq 2000 platform at the Biotechnology Resource Center Next-Generation Sequencing Core Facility, Academia Sinica (Taiwan) to a depth of 49 million to 60 million pairs. Forty-nine-base-pair paired-end reads were generated for the knockout clones and wild-type pool using an Illumina HiSeq 2500 platform at Fred Hutchinson Cancer Research Center to a depth of 68 million to 74 million pairs. Sequence data are publically available (Sequence Read Archive accession nos. PRJNA326452 and PRJNA338999).
In order to ensure that mappability was comparable for all sequencing runs, we trimmed reads to 49 bp. We used bwa (Li and Durbin 2009) to map reads to the micronuclear reference assembly (Genbank no. GCA_000261185.1) and retained uniquely mapping reads (mapq ≥15). Coverage was calculated using genomeCoverageBed from the BedTools package (Quinlan and Hall 2010). Using R (Ihaka and Gentleman 1996) and Bioconductor (Huber et al. 2015), coverage was averaged in 100-bp windows (20-bp slide) and normalized for each sample by dividing by the number of mapped reads in millions.
We noticed that most samples showed strong GC-related coverage bias, so we used loess curve fitting to normalize for GC bias. We first identified a set of 100-bp windows that represent typical macronuclear regions: (1) They do not overlap assembly gaps. (2) They do not overlap 8721 known IESs (Schoeberl et al. 2012). (3) They have GC content ≤40% (initially, a few high-GC windows skewed our loess fits). We took a random subset of 60,000 windows and, for each sample, filtered out atypical windows with coverage <0.1 or >0.8 reads per million and performed a loess fit of the relationship between GC content and coverage. We then GC-corrected the observed coverage, dividing coverage in each window by predicted values based on the loess fit.
After normalization and GC correction, we calculated ratios of coverage in each sample to that of the wild-type pool and used stringent criteria to look for “interesting” windows with coverage ratios of ≥4 and coverage of ≥0.2 reads per million. We filtered out windows in the region affected in our TPB1 knockout strain and in the mating type locus, which we expected to vary. We combined overlapping interesting windows into “interesting regions” (Fig. 2A).
To define TPB1-dependent regions, we first looked for windows that were “interesting” in all four TPB1-depleted samples but no wild-type sample. However, we found that some regions passing this test had also failed to delete in one or more wild-type samples but did not meet the stringent “interesting” threshold. We therefore used looser criteria to define “suspicious” windows in wild-type clonal lines, requiring ratios of more than twofold (same coverage threshold), and excluded these windows from further analysis. Upon detailed sequence analysis, we found that six of the resulting 22 candidate TPB1-dependent regions were additional cases of wild-type variation. One of these six cases varies among the sequenced wild-type samples but with below-threshold coverage. Although the remaining five regions were efficiently deleted in all wild-type samples that we sequenced, they were retained in the macronuclear genome assembly, which was generated from an independent wild-type strain, SB210 (Eisen et al. 2006). After these additional filters, we retained 16 TPB1-dependent regions from our analysis of the micronuclear assembly.
Unfortunately, several of the 16 TPB1-dependent regions contained gaps in the micronuclear assembly, often near IES edges; such gaps precluded detailed IES boundary analyses. We therefore generated an alternative genome assembly using data from our TPB1 knockdown pool; because these cells contain only undeleted DNA at TPB1-dependent IESs, we hypothesized that assembling these regions might be easier than using wild-type DNA, where a mix of macronuclear and micronuclear forms could confuse assembly algorithms. We trimmed adapters and low-quality regions using Cutadapt (http://cutadapt.readthedocs.io) and ran the SOAPdenovo2 assembly pipeline (Luo et al. 2012) with k-mer size 85 and insert size 220 bp. As predicted, our assembly reconstructed most TPB1-dependent regions without gaps. We repeated our genome-wide analysis using this assembly, identifying most of the same TPB1-dependent regions as before as well as four new regions (regions R, S, T, and AA; Genbank accession nos. KX713989–KX713992 and KY064162–KY064164) (Supplemental Table S2), making a total of 20 regions.
Bioinformatic characterization of TPB1-dependent IES regions
We defined the exact boundaries of TPB1-affected regions (Supplemental Table S2) using our sequencing reads mapped back to the micronuclear assembly or our alternative assembly, viewed using IGV (Robinson et al. 2011), and by comparison of the macronuclear reference assembly with the micronuclear assembly or our assembly. We extracted sequences of TPB1-dependent regions with 200-bp flanking regions and ran the MEME motif search algorithm (Bailey et al. 2009) with the following parameters: -mod anr (any number of motif occurrences per sequence) and -revcomp (search both strands). We also supplied MEME with a fifth-order background model generated from the micronuclear assembly using MEME's Fasta-Get-Markov tool. We found one good motif that we refined using only the 24 “true” instances found at the edges of the 12 high confidence TPB1-dependent IESs. To compare the motif with B. mori piggyBac TIRs (Xu et al. 2006), we aligned the 42 published TIR sequences and generated a motif (Sites2meme, MEME suite) that we used as input to TOMTOM (MEME suite). We used MEME's fimo tool to locate motif instances in the micronuclear assembly, again with a fifth-order background model. We filtered motif instances for those with a score ≥50 (a threshold chosen to ensure inclusion of all 24 true instances and filter out many other weaker motifs). We searched for pairs of high-scoring motifs within 10 kb in the appropriate relative orientation (most IESs are <10 kb in size), finding 127 such pairs. We used IGV to examine sequence data from wild-type and TPB1-deficient progeny to determine whether these pairs delimited IESs or TPB1-dependent regions.
In order to determine gene content around TPB1-affected regions, we used several annotation sources: 30,079 transcripts predicted from the micronuclear assembly and downloaded from the Broad Institute Web site (September 2013), 26,996 CDSs predicted from the macronuclear assembly (accession no. GCA_000189635.1) and mapped to the micronuclear assembly with GMAP (Wu et al. 2016), and RNA sequencing (RNA-seq) generated by the Broad Institute (SRA SRR606330) or as published (Xiong et al. 2012) and mapped to the micronuclear assembly with TopHat2 (Kim et al. 2013).
TPB6 gene prediction
A Blastp search of predicted T. thermophila proteins revealed an annotated gene, TTHMIC_02656519T0, similar to TPB1 that we renamed TPB6. However, comparison of the annotation with RNA-seq data, SRR636700 (Xiong et al. 2012), showed gene prediction errors. We therefore used Assembly by Reduced Complexity (http://ibest.github.io/ARC) to perform targeted assembly of RNA-seq reads matching the TPB6 genomic region. Two of the resulting contigs covered most of TPB6; we manually completed the annotation, filling gaps using genomic sequence. The prediction is well supported by RNA-seq data; we extended 3′-ward to the first in-frame stop codon. Our TPB6 prediction has Genbank accession BK009981.
RT–PCR
Total RNA was prepared using a RNA isolation kit (Roche) and reverse-transcribed using an oligo-dT primer. qPCR analysis was performed in the LightCycler carousel-based PCR system using the LightCycler FastStart DNA Masterplus SYBR Green kit (Roche). Expression levels were normalized using α-tubulin mRNA levels. Primer sequences are in Supplemental Table S4.
Supplementary Material
Acknowledgments
We thank members of the Yao and Malik laboratories for helpful discussions, Rini Kasinathan and Lisa Kursel for comments on the manuscript, and an anonymous reviewer who suggested that we examine the LIA2 IES. We are grateful to Douglas Chalker for strains. We received funding from the Howard Hughes Medical Institute, the National Institutes of Health (grant R01-GM74108 to H.S.M.), the Ministry of Science and Technology of Taiwan (MOST 105-2311-B-001-057-MY2), and the Institute of Molecular Biology, Academia Sinica of Taiwan (M.-C.Y.). H.S.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.290460.116.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austerberry CF, Allis CD, Yao MC. 1984. Specific DNA rearrangements in synchronously developing nuclei of Tetrahymena. Proc Natl Acad Sci 81: 7383–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa S, Pratte D, Schwarz H, Pipkorn R, Singer-Kruger B. 2010. Oligomeric Dop1p is part of the endosomal Neo1p–Ysl2p–Arl1p membrane remodeling complex. Traffic 11: 1092–1106. [DOI] [PubMed] [Google Scholar]
- Baudry C, Malinsky S, Restituito M, Kapusta A, Rosa S, Meyer E, Betermier M. 2009. PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia. Genes Dev 23: 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betermier M, Duharcourt S. 2014. Programmed rearrangement in ciliates: Paramecium. Microbiol Spectr 10.1128/microbiolspec.MDNA3-0035-2014. [DOI] [PubMed] [Google Scholar]
- Chalker DL, Yao MC. 2001. Nongenic, bidirectional transcription precedes and may promote developmental DNA deletion in Tetrahymena thermophila. Genes Dev 15: 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Yao MC. 2011. DNA elimination in ciliates: transposon domestication and genome surveillance. Annu Rev Genet 45: 227–246. [DOI] [PubMed] [Google Scholar]
- Chalker DL, La Terza A, Wilson A, Kroenke CD, Yao MC. 1999. Flanking regulatory sequences of the Tetrahymena R deletion element determine the boundaries of DNA rearrangement. Mol Cell Biol 19: 5631–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Vogt A, Mochizuki K, Yao MC. 2010. A domesticated piggyBac transposase plays key roles in heterochromatin dynamics and DNA cleavage during programmed DNA deletion in Tetrahymena thermophila. Mol Biol Cell 21: 1753–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne RS, Nikiforov MA, Smothers JF, Allis CD, Yao MC. 1999. Parental expression of the chromodomain protein Pdd1p is required for completion of programmed DNA elimination and nuclear differentiation. Mol Cell 4: 865–872. [DOI] [PubMed] [Google Scholar]
- Doak TG, Witherspoon DJ, Doerder FP, Williams K, Herrick G. 1997. Conserved features of TBE1 transposons in ciliated protozoa. Genetica 101: 75–86. [DOI] [PubMed] [Google Scholar]
- Dubois E, Bischerour J, Marmignon A, Mathy N, Regnier V, Betermier M. 2012. Transposon invasion of the Paramecium germline genome countered by a domesticated PiggyBac transposase and the NHEJ pathway. Int J Evol Biol 2012: 436196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Coyne RS, Wu M, Wu D, Thiagarajan M, Wortman JR, Badger JH, Ren Q, Amedeo P, Jones KM, et al. 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol 4: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass JN, Joshi NA, Couvillion MT, Bowen J, Gorovsky MA, Hamilton EP, Orias E, Hong K, Coyne RS, Eisen JA, et al. 2011. Genome-scale analysis of programmed DNA elimination sites in Tetrahymena thermophila. G3 (Bethesda) 1: 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertig J, Gu L, Hai B, Gorovsky MA. 1994. High frequency vector-mediated transformation and gene replacement in Tetrahymena. Nucleic Acids Res 22: 5391–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godiska R, Yao MC. 1990. A programmed site-specific DNA rearrangement in Tetrahymena thermophila requires flanking polypurine tracts. Cell 61: 1237–1246. [DOI] [PubMed] [Google Scholar]
- Gorovsky MA, Yao MC, Keevert JB, Pleger GL. 1975. Isolation of micro- and macronuclei of Tetrahymena pyriformis. Methods Cell Biol 9: 311–327. [DOI] [PubMed] [Google Scholar]
- Hamilton EP, Kapusta A, Huvos PE, Bidwell SL, Zafar N, Tang H, Hadjithomas M, Krishnakumar V, Badger JH, Caler EV, et al. 2016. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. eLife 10.7554/eLife.19090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MN, Hill KJ, Lindorfer MA, Stevens TH. 1993. Isolation of vacuolar membrane H+-ATPase-deficient yeast mutants; the VMA5 and VMA4 genes are essential for assembly and activity of the vacuolar H+-ATPase. J Biol Chem 268: 221–227. [PubMed] [Google Scholar]
- Howard-Till RA, Yao MC. 2006. Induction of gene silencing by hairpin RNA expression in Tetrahymena thermophila reveals a second small RNA pathway. Mol Cell Biol 26: 8731–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. 2015. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. 1996. R: a language for data analysis and graphics. J Comput Graph Stat 5: 299–314. [Google Scholar]
- Jahn CL, Doktor SZ, Frels JS, Jaraczewski JW, Krikau MF. 1993. Structures of the Euplotes crassus Tec1 and Tec2 elements: identification of putative transposase coding regions. Gene 133: 71–78. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Mochizuki K. 2015. Phosphorylation of an HP1-like protein regulates heterochromatin body assembly for DNA elimination. Dev Cell 35: 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Noto T, Mochizuki K. 2016. Phosphorylation of an HP1-like protein is a prerequisite for heterochromatin body formation in Tetrahymena DNA elimination. Proc Natl Acad Sci 113: 9027–9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klobutcher LA, Herrick G. 1995. Consensus inverted terminal repeat sequence of Paramecium IESs: resemblance to termini of Tc1-related and Euplotes Tec transposons. Nucleic Acids Res 23: 2006–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klobutcher LA, Herrick G. 1997. Developmental genome reorganization in ciliated protozoa: the transposon link. Prog Nucleic Acid Res Mol Biol 56: 1–62. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Burnight ER, Cooney AL, Malani N, Brady T, Sander JD, Staber J, Wheelan SJ, Joung JK, McCray PB Jr., et al. 2013. piggyBac transposase tools for genome engineering. Proc Natl Acad Sci 110: E2279–E2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin IT, Chao JL, Yao MC. 2012. An essential role for the DNA breakage-repair protein Ku80 in programmed DNA rearrangements in Tetrahymena thermophila. Mol Biol Cell 23: 2213–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CG, Lin IT, Yao MC. 2016. Programmed minichromosome elimination as a mechanism for somatic genome reduction in Tetrahymena thermophila. PLoS Genet 12: e1006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman GW, Rast JP, Fugmann SD. 2010. The origins of vertebrate adaptive immunity. Nat Rev Immunol 10: 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD. 2007. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev 21: 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madireddi MT, Coyne RS, Smothers JF, Mickey KM, Yao MC, Allis CD. 1996. Pdd1p, a novel chromodomain-containing protein, links heterochromatin assembly and DNA elimination in Tetrahymena. Cell 87: 75–84. [DOI] [PubMed] [Google Scholar]
- Malone CD, Anderson AM, Motl JA, Rexer CH, Chalker DL. 2005. Germ line transcripts are processed by a Dicer-like protein that is essential for developmentally programmed genome rearrangements of Tetrahymena thermophila. Mol Cell Biol 25: 9151–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K. 2008. High efficiency transformation of Tetrahymena using a codon-optimized neomycin resistance gene. Gene 425: 79–83. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Gorovsky MA. 2005. A Dicer-like protein in Tetrahymena has distinct functions in genome rearrangement, chromosome segregation, and meiotic prophase. Genes Dev 19: 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. 2002. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in Tetrahymena. Cell 110: 689–699. [DOI] [PubMed] [Google Scholar]
- Motl JA, Chalker DL. 2011. Zygotic expression of the double-stranded RNA binding motif protein Drb2p is required for DNA elimination in the ciliate Tetrahymena thermophila. Eukaryot Cell 10: 1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov MA, Gorovsky MA, Allis CD. 2000. A novel chromodomain protein, pdd3p, associates with internal eliminated sequences during macronuclear development in Tetrahymena thermophila. Mol Cell Biol 20: 4128–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto T, Kataoka K, Suhren JH, Hayashi A, Woolcock KJ, Gorovsky MA, Mochizuki K. 2015. Small-RNA-mediated genome-wide trans-recognition network in Tetrahymena DNA elimination. Mol Cell 59: 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacki M, Higgins BP, Maquilan GM, Swart EC, Doak TG, Landweber LF. 2009. A functional role for transposases in a large eukaryotic genome. Science 324: 935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascon RC, Miller BL. 2000. Morphogenesis in Aspergillus nidulans requires Dopey (DopA), a member of a novel family of leucine zipper-like proteins conserved from yeast to humans. Mol Microbiol 36: 1250–1264. [DOI] [PubMed] [Google Scholar]
- Patil NS, Hempen PM, Udani RA, Karrer KM. 1997. Alternate junctions and microheterogeneity of Tlr1, a developmentally regulated DNA rearrangement in Tetrahymena thermophila. J Eukaryot Microbiol 44: 518–522. [DOI] [PubMed] [Google Scholar]
- Patterson DJ, Sleigh MA. 1976. Behavior of the contractile vacuole of Tetrahymena pyriformis W: a redescription with comments on the terminology. J Protozool 23: 410–417. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachidi M, Lopes C, Costantine M, Delabar JM. 2005. C21orf5, a new member of Dopey family involved in morphogenesis, could participate in neurological alterations and mental retardation in Down syndrome. DNA Res 12: 203–210. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabour D, Scholer HR. 2012. Reprogramming and the mammalian germline: the Weismann barrier revisited. Curr Opin Cell Biol 24: 716–723. [DOI] [PubMed] [Google Scholar]
- Schoeberl UE, Kurth HM, Noto T, Mochizuki K. 2012. Biased transcription and selective degradation of small RNAs shape the pattern of DNA elimination in Tetrahymena. Genes Dev 26: 1729–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh AW, Chalker DL. 2013. LIA5 is required for nuclear reorganization and programmed DNA rearrangements occurring during Tetrahymena macronuclear differentiation. PLoS One 8: e75337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Izawa T, Yamate J, Franklin RJ, Kuramoto T, Serikawa T, Kuwamura M. 2014. The VF rat with abnormal myelinogenesis has a mutation in Dopey1. Glia 62: 1530–1542. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Coyne RS, Allis CD. 2002. Methylation of histone h3 at lysine 9 targets programmed DNA elimination in Tetrahymena. Cell 110: 701–711. [DOI] [PubMed] [Google Scholar]
- Vogt A, Mochizuki K. 2013. A domesticated PiggyBac transposase interacts with heterochromatin and catalyzes reproducible DNA elimination in Tetrahymena. PLoS Genet 9: e1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J. 2001. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412: 607–614. [DOI] [PubMed] [Google Scholar]
- Wheeler BS. 2013. Small RNAs, big impact: small RNA pathways in transposon control and their effect on the host stress response. Chromosome Res 21: 587–600. [DOI] [PubMed] [Google Scholar]
- Woo TT, Chao JL, Yao MC. 2016. Dynamic distributions of long double-stranded RNA in Tetrahymena during nuclear development and genome rearrangements. J Cell Sci 129: 1046–1058. [DOI] [PubMed] [Google Scholar]
- Wu TD, Reeder J, Lawrence M, Becker G, Brauer MJ. 2016. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol 1418: 283–334. [DOI] [PubMed] [Google Scholar]
- Xiong J, Lu X, Zhou Z, Chang Y, Yuan D, Tian M, Zhou Z, Wang L, Fu C, Orias E, et al. 2012. Transcriptome analysis of the model protozoan, Tetrahymena thermophila, using deep RNA sequencing. PLoS One 7: e30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HF, Xia QY, Liu C, Cheng TC, Zhao P, Duan J, Zha XF, Liu SP. 2006. Identification and characterization of piggyBac-like elements in the genome of domesticated silkworm, Bombyx mori. Mol Genet Genomics 276: 31–40. [DOI] [PubMed] [Google Scholar]
- Yao MC, Fuller P, Xi X. 2003. Programmed DNA deletion as an RNA-guided system of genome defense. Science 300: 1581–1584. [DOI] [PubMed] [Google Scholar]
- Yao MC, Yao CH, Halasz LM, Fuller P, Rexer CH, Wang SH, Jain R, Coyne RS, Chalker DL. 2007. Identification of novel chromatin-associated proteins involved in programmed genome rearrangements in Tetrahymena. J Cell Sci 120: 1978–1989. [DOI] [PubMed] [Google Scholar]
- Yao MC, Chao JL, Cheng CY. 2014. Programmed genome rearrangements in Tetrahymena. Microbiol Spectr 10.1128/microbiolspec.MDNA3-0012-2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.