

Abstract
Melanin protects the skin against DNA damage induced by direct absorption of sunlight's UV radiation. Yet, irradiating melanin in vitro or in cultured cells also generates active oxygen species such as superoxide, which can indirectly induce oxidative base lesions and DNA strand breaks. This photosensitization is greater for pheomelanin (yellow and red melanin) than for eumelanin (brown and black). The in vivo photosensitizing ability of melanin is unknown. We used congenic mice of black, yellow, and albino coat colors to investigate the induction of DNA lesions and apoptosis after exposure to predominantly UVB (280–320 nm) or UVA (320–400 nm) radiation. Cyclobutane pyrimidine dimers induced by direct UVB absorption were equal in all three strains, as was apoptosis measured as sunburn cells or as keratinocytes containing active caspase-3. However, terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling (TUNEL)-positive cells were ≈3-fold more frequent in black and yellow mice after UVB or UVA irradiation than in albino. In epidermal sheets, TUNEL-positive cells lined the upper portion of the hair follicle, consistent with UV-induced photosensitization by melanin in the hair shaft. Because the concentration of eumelanin in black mice was three times that of pheomelanin in yellow mice, pheomelanin had 3-fold greater specific activity. We conclude that UV-irradiated melanin, particularly pheomelanin, photosensitizes adjacent cells to caspase-3 independent apoptosis, and this occurs at a frequency greater than the apoptosis induced by direct DNA absorption of UV. Melanin-induced apoptosis may contribute to the increased sensitivity of individuals with blonde and red hair to sunburn and skin cancer.
Constitutive skin pigmentation dramatically affects the incidence of skin cancer. Fair-skinned individuals are more susceptible to UV-induced skin damage than individuals with darker skin, resulting in a 10- to 100-fold higher frequency of nonmelanoma and melanoma skin cancer (1, 2). UV-induced cutaneous cancers are frequent in patients with albinism subtypes caused by the absence of melanin (3), and albino mice appear to be susceptible to skin cancers (4). Melanin is thought to filter out UV radiation and scavenge active oxygen species, thereby reducing UV damage in the cutaneous cells. In addition, a supranuclear melanin cap structure minimizes photodamage to the nucleus (5, 6).
In contrast to these effects, melanin also is known to act as a photosensitizer that generates active oxygen species upon UV irradiation (1, 7). The tyrosine-derived aromatic rings of the melanin chromophore are excited to the singlet state, decay to the triplet, and transfer an electron to oxygen to yield superoxide (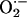 ) (8). Some evidence also indicates transfer of excitation from the chromophore to oxygen, giving singlet oxygen (1O2). Reaction of superoxide with iron (III) ions and hydrogen peroxide (created by dismutation of superoxide) can lead to the OH· radical, which is capable of causing DNA strand breaks. Melanins subsequently scavenge these active chemical species, but their scavenging capacity can be overwhelmed. In living skin exposed to UV, it is not known which of these opposing mechanisms predominates (7).
) (8). Some evidence also indicates transfer of excitation from the chromophore to oxygen, giving singlet oxygen (1O2). Reaction of superoxide with iron (III) ions and hydrogen peroxide (created by dismutation of superoxide) can lead to the OH· radical, which is capable of causing DNA strand breaks. Melanins subsequently scavenge these active chemical species, but their scavenging capacity can be overwhelmed. In living skin exposed to UV, it is not known which of these opposing mechanisms predominates (7).
To determine the photosensitizing function of eumelanin and pheomelanin after UV irradiation in vivo, we examined cell death after UV exposure in skin having different melanin contents. In human skin, it would be difficult to determine the site of a photosensitization reaction leading to the death of nearby cells, because melanin is distributed throughout the interfollicular epidermis, hair follicle, and hair shaft. In the dorsal skin of mice, however, melanin is limited to the hair shaft and keratinocytes of the hair bulb and dermal papilla (9). This limited spatial distribution makes it possible to identify the source of melanin-based photoreactions. It also makes it possible to measure the damaging effects of melanin without complications from its shielding function. We therefore used three congenic strains having different coat colors: black (C57BL/6J), yellow (B6.Cg-Ay; former name C57BL/6J-Ay), and albino (C57BL/6J-Tyrc-2J/Tyrc-2J). The yellow strain carries the Ay mutation in the A locus (agouti), resulting in production of pheomelanin instead of eumelanin in the murine hair (10). The albino strain carries a G-to-T base change at nucleotide 291 of the tyrosinase gene, changing the amino acid from arginine to leucine and destroying enzyme function. Three assays for apoptotic cells were used: sunburn cells (a morphological criterion), cells positive for active caspase-3 (an endstage effector of apoptosis), and terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) (cells containing DNA double-strand breaks). We found that after UV exposure, the hair follicles of both strains of pigmented mice contain TUNEL-positive keratinocytes not seen in albino mice, and that the frequency of these cells exceeds that of the sunburn cells and active caspase-3-positive cells induced in all three genotypes by direct DNA absorption of UV.
Methods
Mice, UV Irradiation, and Tissue Sections. Female C57BL/6J, B6.Cg-Ay, and C57BL/6J-Tyrc-2J/Tyrc-2J mice (The Jackson Laboratory) were used for experiments at 6–8 weeks of age. The coats of these congenic strains are black (C57BL/6J), yellow (B6.Cg-Ay), and albino (C57BL/6J-Tyrc-2J/Tyrc-2J). No tyrosinase activity is found in hair bulb extracts from the albino mouse (11). Mice in the resting phase of the hair follicle growth-regression cycle were shaved on the back with clippers and a shaver; the next day, they were exposed to predominantly UVB (280–320 nm) from three FS20T12-UVB lamps (National Biological, Twinsburg, OH) or to UVA (320–400 nm) from eight F20T12BL lamps (Spectra Mini, Daavlin, Bryan, OH) passed through an 11-mm glass plate filter to remove UVB and UVC (100–280 nm). The output of the UVB lamps was UVA, 27.1%; UVB, 72.8%; and UVC, 0.1%. The output of the filtered UVA lamps was UVA, 100%; UVB, 4 × 10–3%; and UVC, 8 × 10–4%. The UV output of the lamp was measured before each session by using a UVX meter (Ultraviolet Products). Mice were irradiated once with 1,250 J/m2 UVB or 100 kJ/m2 UVA. During irradiation, animals moved freely but were prevented from standing upright by a wire mesh with 1 × 1-cm openings. Animals were killed by isofluorane anesthesia and cervical dislocation immediately after irradiation for DNA lesion measurements, or 24 h later for examination of cell death in the epidermis. The animal protocol was reviewed and approved by the Yale Institutional Animal Care and Use Committee.
The central portion of the dorsal skin was removed and washed with PBS. A portion of the skin was embedded in OCT compound (Sakura Fine-Technical, Tokyo) on dry ice. Another portion was fresh-frozen, and sections were cut in a cryostat and fixed with 4% paraformaldehyde in PBS overnight at 4°C. Other skin samples were fixed with 4% paraformaldehyde in PBS overnight and embedded in paraffin. To generate epidermal sheets, whole skin was immersed into PBS/20 mM EDTA for 2 h at 37°C, and the epidermal sheet was separated from the dermis and fixed with acetone or 4% paraformaldehyde in PBS for 20 min as described (12).
Melanin Assays. Fontana-Masson stain for eumelanin. Five-micrometer paraffin sections of unirradiated skin were deparaffinized, hydrated, and stained for eumelanin by the Yale Dermatopathology service by using the Fontana-Masson procedure (13). Quantitation of eumelanin and pheomelanin. Skin samples were subjected to chemical degradation (KMnO4 oxidation and hydriodic acid reduction), and products were analyzed by HPLC as described (14, 15). Assays were performed in duplicate, and the values reported are the averages.
Localization of Cyclobutane Pyrimidine Dimers. Cyclobutane pyrimidine dimers were localized in UVB-irradiated epidermis by using horseradish peroxidase-labeled monoclonal antithymine dimer antibody (Kamiya Biomedical, Seattle). This antibody detects a TT-specific photoproduct that is not detected by antibody to (6-4) photoproducts (16) and so presumptively recognizes the TT cyclobutane dimer. The assay was performed as described (17) by using an antibody dilution of 1:50.
Apoptosis Assays. Sunburn cells. Five-micrometer paraffin sections were stained with hematoxylin/eosin, and sunburn cells were identified by light microscopy based on their characteristic morphology: condensed, pyknotic, darkly basophilic nuclei, eosinophilic cytoplasm, and intercellular gap (halo) formation (18). Sunburn cells were counted in the interfollicular and perifollicular epidermis and expressed as sunburn cells per linear cm of skin.
Active caspase-3. An affinity-purified rabbit polyclonal antibody that reacts with the cleaved mouse p20 subunit of caspase-3 (R & D Systems) was used for immunohistochemistry on paraffin sections as described (19). Active caspase-3-positive cells were counted in the interfollicular and perifollicular epidermis and expressed as active caspase-3-positive cells per linear cm of skin.
TUNEL. DNA double-strand breaks, whether introduced immediately via irradiated melanin or as a consequence of apoptosis, would be detectable by the TUNEL terminal transferase extension assay (20). Excision repair nicks or gaps do not appear to be substrates: (i) Terminal transferase is specific for DNA ends rather than nicks or gaps; (ii) in mouse skin, the frequency of interfollicular TUNEL-positive cells is ≈1% rather than the 100% undergoing excision repair; (iii) in double-labeling experiments, TUNEL-positive epidermal cells are the same cells undergoing morphological shrinkage (21); and (iv) mice defective for the first step of excision repair because of a knockout in the Xpa gene have more TUNEL-positive cells rather than fewer (22). Five-micrometer-thick paraffin sections were analyzed by using the ApopTag Fluorescein kit (Intergen, Norcross, GA) according to the manufacturer's instructions and counterstained with 1 μg/ml propidium iodide in PBS. For interfollicular regions, the frequency of positive cells was expressed as the number of TUNEL-positive cells per cm of length of each epidermal section. The frequency of positive cells in hair follicles was expressed as the number of TUNEL-positive cells per 1,000 hair follicle cells.
Localization of Cell Death in Epidermal Sheets. Epidermal sheets from the dorsal skin of UVB-irradiated mice fixed as described were subjected to TUNEL analysis. For cell permeabilization, epidermal sheets were placed in precooled 2:1 ethanol/acetic acid at –20°C for 10 min. The remaining procedures followed the ApopTag manufacturer's protocol for cell culture. Fluorescein-stained samples were observed by confocal microscopy (Bio-Rad MRC 1024), and a 3D picture was compiled by combining z-axis sections. Active caspase-3 detection in epidermal sheets used the APO ACTIVE 3 kit (Cell Technology, Minneapolis). Epidermal sheets were fixed with acetone for 30 min at room temperature and permeabilized in 0.2% Triton X-100 in PBS for 20 min. Subsequent steps were according to the manufacturer's protocol. Stained samples were observed by confocal microscopy as above by using FITC as the fluor and propidium iodide as nuclear counterstain.
Results
Eumelanin and Pheomelanin in the Dorsal Skin of Black, Yellow, and Albino Mice. Because most studies of murine melanin use tail skin, we first characterized the distribution and amount of eumelanin and pheomelanin in the three strains used here. As expected (9), Fontana-Masson staining showed no melanin granules in the interfollicular epidermis of murine dorsal skin (Fig. 1 A and B). In black mice, melanin granules were observed in the hair shaft (Fig. 1 A) as expected (10). In yellow mice, melanin granules were again distributed in the hair shaft but were less frequent (Fig. 1B). The skin and hair of albino mice contained no melanin granules, as expected (Fig. 1C). Next, the eumelanin and pheomelanin content of murine skin of each genotype was quantitated chemically (Table 1). The dorsal skin of black mice was predominantly eumelanin (100%), whereas yellow mouse skin was predominantly pheomelanin (100%). The total amount of skin melanin in black mice was ≈3-fold greater than that in yellow mice. As expected, albino mice contained no eumelanin or pheomelanin above the background of the chemical assay.
Fig. 1.

Melanin granules and UVB penetration in the dorsal skin of congenic black, yellow, and albino mouse strains. Fontana-Masson staining reveals melanin as black-brown granules in the black C57BL/6J strain (A) and yellow B6.Cg-Ay strain (B). Melanin granules were seen only in the hair shaft (A and B, arrow). No melanin was seen in the interfollicular or perifollicular epidermis. In the C57BL/6J-Tyrc-2J/Tyrc-2J albino mouse (C), no melanin granules were seen. Mice were unirradiated. (D–F) Equal UVB penetration among genotypes, as assessed by immunohistochemical detection of UV photoproducts. Skin harvested immediately after 1,250 J/m2 UVB irradiation was immunostained with an antibody specific for cyclobutane pyrimidine dimers. A similar depth of penetration of UVB is seen in all three strains (black, D; yellow, E; and albino, F). Cyclobutane pyrimidine dimer-positive cells were detected from the epidermal surface to the middle of the hair follicle. Arrows indicate the deepest dimer-positive cells. (Original magnification: A–C, ×400; D, ×350; E, ×250; F, ×240. Scale bars: 20 μm.)
Table 1. Melanin content of dorsal skin of black, yellow, and albino mice.
| Melanin type | Black | Yellow | Albino |
|---|---|---|---|
| PTCA | 28.7 | 0.9 | 0.9 |
| 4-AHP | 1.0 | 59.1 | 2.0 |
| Eumelanin* | 1,390 (100%)‡ | 0 | 0 |
| Pheomelanin† | 0 | 514 (100%)§ | 0 |
| Total melanin | 1,390 | 514 | 0 |
Values are ng/mg dry skin. Background values of eumelanin (43 ng/mg) and pheomelanin (18 ng/mg) were subtracted from each sample.
Eumelanin was obtained by multiplying the amount of PTCA (pyrrole-2,3,5-tricarboxylic acid) by a conversion factor of 50, after subtracting the level of PTCA in albino (14, 15).
Pheomelanin was obtained by multiplying the amount of 4-AHP (aminohydroxyphenylalanine) by a conversion factor of 9, after subtracting the level of 4-AHP in albino (14, 15).
Ratio of eumelanin to total melanin in black mouse dorsal skin.
Ratio of pheomelanin to total melanin in yellow mouse dorsal skin.
Skin Penetration of UVB and Induction of DNA Photoproducts. Melanins are well known to absorb UV, and their major biological role in humans is presumed to be the shielding of the nucleus from UV damage. Variations in UVB penetration between genotypes would confound assessments of melanin-sensitized DNA damage. To assess UVB penetration, we used immunofluorescence to examine the distribution of the major UVB photoproduct, the cyclobutane pyrimidine dimer, in the skin of the three pigmentation genotypes after irradiation. The epidermal surface was strongly immunopositive in all irradiated samples. At 1,250 J/m2, which corresponds to a minimal erythema dose (23), cyclobutane dimers were found in the upper hair follicle and interfollicular epidermis (Fig. 1 D–F). At a higher dose (4,000 J/m2), deeper penetration of UVB was seen (data not shown). There was no difference in penetration of UVB among the three pigmentation genotypes, as expected from the fact that melanin in mouse dorsal skin is located solely in the hair follicles and the mice were shaved before the experiment. The number of dimer-positive cells also appeared similar in the three genotypes. We also examined paraffin or frozen sections for 8-oxo-deoxyguanosine and single-strand breaks in the three genotypes; although signal was detectable from H2O2-treated positive controls, no signal above background was observed immediately after UVB or UVA irradiation (data not shown). These lesions may be present but obscured by introduction of DNA lesions while the tissue and DNA are being processed. These events are difficult to suppress in fixed tissue.
Apoptosis Induction: Sunburn Cells. The sunburn cell is a morphological hallmark of skin overexposed to sunlight; it also appears in certain skin diseases (18). These cells have been shown to be late-stage apoptotic keratinocytes (21, 24). To determine whether melanin photosensitizes cell death, we counted sunburn cells in the dorsal epidermis of UVB-irradiated mice for each pigmentation genotype. Skin samples were taken 24 h after a single UVB irradiation (1,250 J/m2). All three genotypes showed randomly distributed sunburn cells throughout both the interfollicular and perifollicular epidermis and the upper part of the hair follicle (Fig. 2 A, C, and E). The distribution of sunburn cells, penetrating almost to the middle level of the hair follicle, corresponded to the regions where cyclobutane dimers were present and is consistent with the involvement of UVB in sunburn cell induction. There was no apparent difference in location or distribution of sunburn cells among genotypes, nor were there differences after quantitation of sunburn cells located in the perifollicular plus interfollicular epidermis (Fig. 3E). After a single UVA exposure (100 kJ/m2), sunburn cells were not seen (data not shown), consistent with reports that sunburn cells are absent or rare after UVA (25, 26), although UVA can induce apoptosis in cultured cells (27). We conclude that apoptotic keratinocytes in skin are induced by direct DNA absorption of UVB radiation, independent of melanin.
Fig. 2.

Similar induction of sunburn cells (apoptotic epidermal keratinocytes) and active caspase-3-positive cells in black, yellow, and albino mice 24 h after UVB exposure. The location and frequency of sunburn cells (A, C, and E; arrows) or active caspase-3-positive cells (B, D, and F, arrows) are similar in the three strains of mice (black, A and B; yellow, C and D; albino, E and F). (Magnification: B–E, ×400.) Higher-magnification (×1,000) Insets in D–F show typical sunburn cells and active caspase-3-positive cells. In unirradiated skin, no active caspase-3-positive cells were seen below the keratinized layer (data not shown).
Fig. 3.

Excess of TUNEL-positive keratinocytes near hair follicles of black and yellow mice after UVB or UVA irradiation. (A–C) TUNEL-positive cells in skin sections 24 h after exposure to 1,250 J/m2 UVB (black, A; yellow, B; and albino, C). The red propidium iodide image (nuclei) is merged with the green FITC image (TUNEL). Pigmented mice show more TUNEL-positive cells than albino. In unirradiated skin, no TUNEL-positive cells were seen below the keratinized layer (data not shown). (Original magnification: ×320.) (D) Within the hair follicle, pigmented strains show ≈3-fold more TUNEL than albino mice (black vs. albino, P < 0.01; yellow vs. albino, P < 0.01 by Student's t test). Each value represents the mean of three mice, 1,250 J/m2. Approximately 2 cm of epidermis was examined per mouse. (E) Quantitative comparison of the three apoptosis assays in interfollicular plus perifollicular regions of the three strains. TUNEL showed a 2.5-fold difference between pigmented mice and the albino (black vs. albino, P < 0.02; yellow vs. albino, P < 0.01 by Student's t test). The contribution of pigmentation-dependent cell death revealed by TUNEL exceeds the pigmentation-independent apoptosis present in the albino strain or measured by the sunburn cell or as active caspase-3 assays. (F) UVA-irradiated mouse skin (100 kJ/m r2) also demonstrates a 3- to 4-fold increase in TUNEL-positive cells in hair follicles of black and yellow mice (black vs. albino, P < 0.01; yellow vs. albino, P < 0.02 by Student's t test).
Apoptosis Induction: Caspase-3. As a molecular assay for early-stage apoptosis, we immunostained paraffin sections with an anticaspase-3 antibody that recognizes the active cleaved form. The distribution and frequency of the caspase-3-positive cells after UVB were similar to that of the sunburn cells (Fig. 2 B, D, and F). Typical caspase-3-positive cells showed the same condensed and pycnotic nuclei and extracellular gaps as sunburn cells. Quantitation in the perifollicular plus interfollicular epidermis revealed no difference in frequency between the three pigmentation genotypes (Fig. 3E), and this frequency was similar to that of sunburn cells. We conclude that there is substantial overlap between caspase-3-positive cells and sunburn cells and that the two assays detect the same events in UVB-irradiated murine skin.
Induction of TUNEL in Follicles of Pigmented Mice. A molecular marker for late-stage apoptosis is the presence of DNA double-strand breaks (TUNEL) (24). The same UVB-irradiated samples studied above were therefore examined for epidermal cell death by using TUNEL. In all samples, most TUNEL-positive cells were seen in the interfollicular and perifollicular epidermis and in the upper end of the hair follicle (Fig. 3 A–C). Unexpectedly, TUNEL-positive cells were much more frequent in the black and yellow mice than in albino mice. Moreover, in the albino mice, TUNEL-positive cells were randomly distributed throughout the epidermis, whereas the positive cells in pigmented mice tended to concentrate around the hair follicle. Quantitation revealed that the frequency of TUNEL-positive cells in the follicle was 3-fold greater in black and yellow mice than in the albino (Fig. 3D); in perifollicular plus interfollicular epidermis, the difference was 2.5-fold (Fig. 3E). TUNEL-positive cells were almost exclusively keratinocytes, as indicated by keratin staining (data not shown).
It seemed possible that the excess TUNEL positivity in pigmented strains might represent DNA double-strand breaks induced nonenzymatically by melanin-sensitized photoreactions, rather than reflecting apoptosis. We therefore examined skin from black and yellow strains for TUNEL positivity at 0 h after UVB irradiation; no TUNEL-positive keratinocytes were seen (data not shown).
Numerical comparison of the three genotypes for each of the apoptosis assays is instructive (Fig. 3E). A UVB dose of 1,250 J/m2 induced ≈45 apoptotic cells per cm in albino mice and in pigmented mice assayed as sunburn cells or by active caspase-3. Superimposed on this basal level of “classic” apoptotic cells, pigmented strains sustained an additional ≈75 cells per cm of TUNEL-positive cells that did not evince the other indications of apoptosis. These cells apparently represent apoptosis independent of active caspase-3 and not showing the classical sunburn cell morphology. Because the number of excess TUNEL-positive cells was approximately the same in black and yellow mice, whereas the concentration of eumelanin in black mice was 3-fold that of pheomelanin in yellow mice (Table 1), the specific activity of TUNEL induction per ng of melanin was 3-fold greater for pheomelanin.
We also performed TUNEL analysis after 100 kJ/m2 UVA irradiation, less than a minimal erythema dose for UVA (23). Few TUNEL-positive cells (three or four per cm) were detected in the perifollicular plus interfollicular epidermis (data not shown). Within the hair follicle, more were seen and the number was greater in pigmented mice (Fig. 3F).
3D Distribution of TUNEL and Caspase-3-Positive Cells in Epidermal Sheets. In the paraffin cross sections, it appeared that the TUNEL-positive cells were more frequent along the hair shaft. Because the proximity to the hair shaft is critical to interpreting the role of melanin in generating the excess TUNEL-positive cells, we sought to confirm this result by performing the TUNEL assay on epidermal sheets. Strikingly, the TUNEL-positive cells in the pigmented mouse strains were indeed concentrated adjacent to the acellular hair shafts in the hair follicles (Fig. 4 A and B). TUNEL-positive cells were not located along the entire length of the follicle, presumably because UVB penetrated only to the midpoint of the follicle (Fig. 1). In contrast, in the albino mouse, TUNEL-positive cells were less frequent and randomly distributed throughout the interfollicular epidermis and hair follicle (Fig. 4C). These results indicate that keratinocytes located near hair shafts containing melanin are susceptible to cell death after UVB irradiation. We also examined caspase-3-positive cells in the epidermal sheets from UVB-irradiated mice. Consistent with the results in paraffin cross sections, caspase-3-positive cells were randomly distributed in the epidermal sheet of both pigmented and albino mice (Fig. 4 G–I).
Fig. 4.

3D distribution of TUNEL-positive cells and active caspase-3-positive cells along hair shafts in epidermal sheets from pigmented mice irradiated with UVB. Mice were irradiated with 1,250 J/m2 UVB, and 24 h later epidermal sheets were prepared. Images were constructed by assembling confocal z axis sections. Shown are black (A, D, and G), yellow (B, E, and H), and albino (C, F, and I) strains. (A–C) TUNEL (green, fluorescein). (D–F) TUNEL merged with propidium iodide (red) and overlap in yellow. In pigmented mice exposed to UVB, TUNEL-positive cells were concentrated in the hair follicle and along the hair shafts. In the albino mouse (C and F), TUNEL-positive cells were distributed randomly and unrelated to the location of hair follicle. G–I show active caspase-3 only (green). Active caspase-3-positive cells were distributed randomly with respect to hair follicles in all irradiated genotypes. (Original magnification: A–F, ×100; G–I, ×200.)
Discussion
Melanin Photosensitizes Cell Death. The occurrence of UV-induced TUNEL-positive cells adjacent to melanin-containing hair follicles reveals that, in vivo, melanin is a photosensitizer as well as a photoprotector. This experiment was made possible by the absence of extrafollicular melanin in mouse skin. The frequency of perifollicular plus interfollicular TUNEL-positive cells in albino mice equaled the frequency of sunburn cells or caspase-3-positive cells in each of the three genotypes (Fig. 3E). These results suggest that UVB (and, to a lesser extent, UVA) has two actions as diagrammed in Fig. 5: (i) Directly inducing classical apoptotic cells, which are identifiable by sunburn cell morphology, active caspase-3, or TUNEL. These are independent of melanin and evenly distributed throughout the region of epidermis penetrated by UVB. Sunburn cells are initiated by cyclobutane pyrimidine dimers (28) in actively transcribed genes (22) and require the p53 gene (24). Because typical caspase-3-positive cells resembled sunburn cells, they apparently also require a DNA damage signal. Extranuclear signals may be required in addition (18). (ii) Inducing a melanin-dependent mode of cell death detectable only by TUNEL. These sites are determined by UV penetration plus the distribution of melanin. In the mouse, this combination limits this mode of cell death to the upper half of the hair follicle. Melanin also is present in the hair bulb (data not shown), but UVB did not reach this site. Strikingly, the magnitude of the melanin-dependent cell death was approximately twice that of classic apoptosis.
Fig. 5.

Schematic diagram of the location of apoptotic cells in black, yellow, and albino mouse skin after UVB exposure. (Upper) Sunburn cells (SBC) and active caspase-3-positive cells were distributed randomly with respect to the hair follicles; there was no difference in location or frequency between pigmented and albino strains. (Lower) TUNEL-positive cells, in contrast, were more frequent in the black and yellow strains and were concentrated in the hair follicle along the hair shaft. In the albino strain, TUNEL-positive cells were less frequent and were randomly distributed.
Although black and yellow mice showed equal levels of TUNEL positivity, the level of eumelanin in black mice was 3-fold the level of pheomelanin in yellow mice, indicating a 3-fold greater specific activity of pheomelanin photosensitization. This activity is consistent with a report that pheomelanin produces almost five times as much superoxide as eumelanin after UV exposure (29). UV-irradiated melanin may thus also generate other consequences of superoxide, such as point mutations.
Caspase-Independent Cell Death from Melanin Photosensitization. Although apoptosis and necrosis are well known forms of cell death, several caspase-independent cell death modes have recently emerged. In these modes, cells undergo nuclear fragmentation without chromosome condensation or cell shrinkage. In one class of pathways, nuclear fragmentation results from nucleases which, unlike DFF45, do not require activation by caspases. These include AIF (apoptosis inducing factor) and endonuclease G (30, 31). Other modes of cell death achieve proteolysis without caspases, instead using lysozomal cathepsins [autophagy, resulting in double-membrane vesicles (32)], proteosomal degradation, or calpain proteases from the endoplasmic reticulum (30). The TUNEL-positive cells seen in the present experiments clearly exhibit nuclear fragmentation without chromosome condensation, otherwise they would have been scored as sunburn cells. We do not yet know whether the cells contain double-membrane vesicles or whether the process is inhibited by in vivo treatment with wide-spectrum caspase inhibitors or inhibitors of the autophagy pathway (32). TUNEL would also detect the DNA fragmentation in necrotic cells (33).
Potential Role of Melanin Photosensitization in Human Skin. These photosensitization processes may contribute to clinical manifestations in humans. Individuals with blonde or red hair are susceptible to sunburn and have a risk for skin cancer that is elevated ≈1.5-fold (blonde) to ≈3- to 6-fold (red), compared with Caucasian controls (34, 35). Skin cancer risk is correlated epidemiologically with the propensity to burn rather than tan. This “Celtic phenotype” is associated with red and blonde hair but also with light skin, so the greater transmission of UV through the skin has been thought responsible. The present studies indicate that melanin photosensitization may be an important contributor. If so, photosensitization would lead to a broader spatial distribution of TUNEL in human than mouse, because human epidermal melanocytes are distributed throughout the basal layer. The resulting sunlight sensitivity of individuals with any particular melanin type would reflect the balance between protection and damage, with pheomelanin contributing 3-fold greater damage than eumelanin and perhaps less efficient protection. In this scenario, when a pheomelanin-containing individual visits the beach, his melanosomes act as microscopic x-ray sources to generate superoxide and OH· radicals. The resulting TUNEL-positive response may be protective, with melanin acting as a screen at low UV doses and as a “poison pill” that removes damaged cells at higher doses (24, 36, 37). Yet when death-resistant mutant cells are already present, this cell death may instead be detrimental and favor clonal expansion of the mutants (12, 24).
Acknowledgments
We thank W. D. Vieira and G. E. Costin for technical advice and assistance and J. DiGiovanna and Y. Yamaguchi for helpful discussions. This work was supported by National Institutes of Health Grants CA78735 and CA55737 (to D.E.B.) and the National Institutes of Health intramural program (K.H.K. and V.J.H.).
Author contributions: V.J.H., K.H.K., and D.E.B. designed research; S.T., W.Z., K.W., and S.I. performed research; S.T., V.J.H., K.H.K., and D.E.B. analyzed data; and S.T., K.H.K., and D.E.B. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling.
References
- 1.Kollias, N., Sayre, R. M., Zeise, L. & Chedekel, M. R. (1991) J. Photochem. Photobiol. B 9, 135–160. [DOI] [PubMed] [Google Scholar]
- 2.Halder, R. M. & Bridgeman-Shah, S. (1995) Cancer 75, Suppl. 2, 667–673. [DOI] [PubMed] [Google Scholar]
- 3.Perry, P. K. & Silverberg, N. B. (2001) Cutis 67, 427–430. [PubMed] [Google Scholar]
- 4.Kripke, M. L. (1977) Cancer Res. 37, 1395–1400. [PubMed] [Google Scholar]
- 5.Montagna, W. & Carlisle, K. (1991) J. Am. Acad. Dermatol. 24, 929–937. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi, N., Nakagawa, A., Muramatsu, T., Yamashina, Y., Shirai, T., Hashimoto, M. W., Ishigaki, Y., Ohnishi, T. & Mori, T. (1998) J. Invest. Dermatol. 110, 806–810. [DOI] [PubMed] [Google Scholar]
- 7.Hill, H. Z. (1992) BioEssays 14, 49–56. [DOI] [PubMed] [Google Scholar]
- 8.Chedekel, M. R., Smith, S. K., Post, P. W., Pokora, A. & Vessell, D. L. (1978) Proc. Natl. Acad. Sci. USA 75, 5395–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silvers, W. K. (1979) The Coat Colors of Mice (Springer, New York).
- 10.Ozeki, H., Ito, S., Wakamatsu, K. & Hirobe, T. (1995) J. Invest. Dermatol. 105, 361–366. [DOI] [PubMed] [Google Scholar]
- 11.Townsend, D., Witkop, C. J., Jr., & Mattson, J. (1981) J. Exp. Zool. 216, 113–119. [DOI] [PubMed] [Google Scholar]
- 12.Zhang, W., Remenyik, E., Zelterman, D., Brash, D. E. & Wikonkal, N. M. (2001) Proc. Natl. Acad. Sci. USA 98, 13948–13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luna, L. G., ed. (1968) in Manual of the Histologic Staining Methods of the AFIP (McGraw–Hill, New York), pp. 104–105.
- 14.Ito, S. & Wakamatsu, K. (1994) Pigm. Cell. Res. 7, 141–144. [DOI] [PubMed] [Google Scholar]
- 15.Wakamatsu, K., Ito, S. & Rees, J. L. (2002) Pigm. Cell. Res. 15, 225–232. [DOI] [PubMed] [Google Scholar]
- 16.Eller, M. S., Maeda, T., Magnoni, C., Atwal, D. & Gilchrest, B. A. (1997) Proc. Natl. Acad. Sci. USA 94, 12627–12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu, Y. P., Lou, Y. R., Yen, P., Mitchell, D., Huang, M. T. & Conney, A. H. (1999) Cancer Res. 59, 4591–4602. [PubMed] [Google Scholar]
- 18.Sheehan, J. M. & Young, A. R. (2002) Photchem. Photobiol. Sci. 1, 365–377. [DOI] [PubMed] [Google Scholar]
- 19.Lu, Y. P., Lou, Y. R., Xie, J. G., Peng, Q. Y., Liao, J., Yang, C. S., Huang, M. T. & Conney, A. H. (2002) Proc. Natl. Acad. Sci. USA 99, 12455–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stadelmann, C. & Lassmann, H. (2000) Cell Tissue Res. 301, 19–31. [DOI] [PubMed] [Google Scholar]
- 21.Brash, D. E., Ziegler, A., Jonason, A., Simon, J. A., Kunala, S. & Leffell, D. J. (1996) J. Invest. Dermatol. Symp. Proc. 1, 136–142. [PubMed] [Google Scholar]
- 22.Brash, D. E., Wikonkal, N. M., Remenyik, E., van der Horst, G. T. J., Friedberg, E. C., Cheo, D. L., van Steeg, H., Westerman, A. & van Kranen, H. J. (2001) J. Invest. Dermatol. 117, 1234–1240. [DOI] [PubMed] [Google Scholar]
- 23.de Laat, A., Kroon, E. D. & de Gruijl, F. R. (1997) Photochem. Photobiol. 65, 730–735. [DOI] [PubMed] [Google Scholar]
- 24.Ziegler, A., Jonason, A. S., Leffell, D. J., Simon, J. A., Sharma, H. W., Kimmelman, J., Remington, L., Jacks, T. & Brash, D. E. (1994) Nature 372, 773–776. [DOI] [PubMed] [Google Scholar]
- 25.Willis, I. & Cylus, L. (1977) J. Invest. Dermatol. 68, 128–129. [DOI] [PubMed] [Google Scholar]
- 26.Lavker, R. & Kaidbey, K. (1997) J. Invest. Dermatol. 108, 17–21. [DOI] [PubMed] [Google Scholar]
- 27.Godar, D. E. (1999) J. Invest. Dermatol. 112, 3–12. [DOI] [PubMed] [Google Scholar]
- 28.Ley, R. D. & Applegate, L. A. (1985) J. Invest. Dermatol. 85, 365–367. [DOI] [PubMed] [Google Scholar]
- 29.Persad, S., Menon, I. A. & Haberman, H. F. (1983) Photochem. Photobiol. 37, 63–68. [DOI] [PubMed] [Google Scholar]
- 30.Jaattela, M. & Tschopp, J. (2003) Nat. Immunol. 4, 416–423. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzo, H. K. & Susin, S. A. (2004) FEBS Lett. 557, 14–20. [DOI] [PubMed] [Google Scholar]
- 32.Ohsumi, Y. (2001) Nat. Rev. Mol. Cell Biol. 2, 211–216. [DOI] [PubMed] [Google Scholar]
- 33.Dong, Z., Saikumar, P., Weinberg, J. M. & Venkatachalam, M. A. (1997) Am. J. Pathol. 151, 1205–1213. [PMC free article] [PubMed] [Google Scholar]
- 34.Urbach, F., Rose, D. B. & Bonnem, M. (1971) Environment and Cancer (Williams and Wilkins, Baltimore), pp. 355–371.
- 35.Long, C. C. & Marks, R. (1995) J. Am. Acad. Dermatol. 33, 658–661. [DOI] [PubMed] [Google Scholar]
- 36.Riley, P. A. (1974) in The Physiology and Pathophysiology of the Skin, ed. Jarrett, A. (Academic, New York), Vol. 3, pp. 1104–1127. [Google Scholar]
- 37.Quevedo, W. C., Holstein, T., Dyckman, J. & Isaacson, E. L. (1995) in Melanin: Its Role in Human Photoprotection, eds. Zeise, L., Chedekel, M. R. & Fitzpatrick, T. B. (Valdenmar, Overland Park, KS), pp. 221–226.