

Abstract
Histone deacetylases (HDACs) are a family of enzymes involved in the regulation of gene expression, DNA repair, and stress response. These processes often are altered in tumors, and HDAC inhibitors have had pronounced antitumor activity with promising results in clinical trials. Here, we report the crystal structure of human HDAC8 in complex with a hydroxamic acid inhibitor. Such a structure of a eukaryotic zinc-dependent HDAC has not be described previously. Similar to bacterial HDAC-like protein, HDAC8 folds in a single α/β domain. The inhibitor and the zinc-binding sites are similar in both proteins. However, significant differences are observed in the length and structure of the loops surrounding the active site, including the presence of two potassium ions in HDAC8 structure, one of which interacts with key catalytic residues. CD data suggest a direct role of potassium in the fold stabilization of HDAC8. Knockdown of HDAC8 by RNA interference inhibits growth of human lung, colon, and cervical cancer cell lines, highlighting the importance of this HDAC subtype for tumor cell proliferation. Our findings open the way for the design and development of selective inhibitors of HDAC8 as possible antitumor agents.
The epigenetic control of gene expression is operated through a series of posttranslational modifications of chromatin that influence the electrostatics of DNA–protein interactions and generate docking sites for a large number of chromatin-interacting proteins (1, 2). The acetylation status of lysine residues found in the accessible N termini of core histones is one of the posttranslational chromatin modifications that impinge on gene expression. Acetylation and deacetylation of histones are controlled by the enzymatic activity of histone acetyltransferases and histone deacetylases (HDACs) (3, 4). Alterations of gene expression are a hallmark of cancer, and mounting evidence suggests that at least a part of these alterations is mediated by epigenetic mechanisms (5, 6). Importantly, the aberrant recruitment of HDACs has been mechanistically linked to malignancy in leukemias and lymphomas (7, 8), and small-molecule HDAC inhibitors show antitumor activity in preclinical models and in clinical trials and have the promise to become effective, new antineoplastic therapeutics (9).
At least 18 HDAC subtypes exist, and they are subdivided into three classes (10): class I (HDACs 1–3 and 8), homologous to the yeast Rpd3 deacetylase; class II (HDACs 4–7, 9, and 10), related to the yeast Hda1 deacetylase; and class III proteins (Sirtuins 1–7), which are yeast Sir2 homologs. HDAC11 has homology to both class I and II enzymes but cannot unambiguously be assigned to either class. Class I and II HDACs, as well as HDAC11, are all zinc-dependent hydrolases. The therapeutically relevant HDAC inhibitors are thought to be nonselective or poorly selective inhibitors of all or most of class I and II enzymes but do not inhibit class III HDACs (9). It is not clear whether the antitumor properties of HDAC inhibitors are due to their lack of specificity or are the consequence of targeting one or few “crucial” subtypes. This question is of considerable interest because it may open the way for the development of novel, more selective compounds, possibly with enhanced efficacy and/or tolerability.
Our understanding of the catalytic mechanism of HDACs and inhibition by small molecules relies on the crystal structure of an archeobacterial homolog of eukaryotic deacetylases (HDLP) that is widely used as a model (11). The availability of structural information on the molecular architecture of human HDACs could have a significant impact on the design of subtype selective inhibitors. Here, we report a previously undescribed structure of a eukaryotic zinc-dependent HDAC, human HDAC8, complexed with the hydroxamic acid inhibitor, N-hydroxy-4-{methyl[(5-pyridin-2-yl-2-thienyl)sulfonyl]amino}benzamide (Compound 1 in Table 1). We also show that HDAC8 is important for the growth of human tumor cell lines and has a distinct inhibition pattern that differs from that of HDAC1 and -3, which both share 43% sequence identity with HDAC8. Our findings open the way to the development of selective inhibitors of this subtype as potential novel anticancer therapeutics.
Table 1. HDAC enzymatic inhibition.
| Compound | HDAC8 IC50, nM | HDAC1 IC50, nM | HDAC3 IC50, nM |
|---|---|---|---|
| TSA | |||
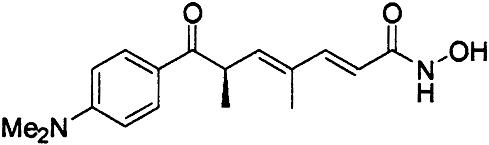 |
490 | 1.5 | 0.6 |
| SAHA | |||
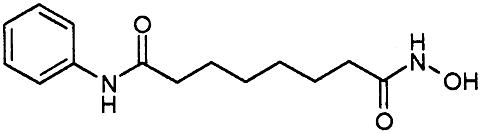 |
4,000 | 119 | 106 |
| Compound 1 | |||
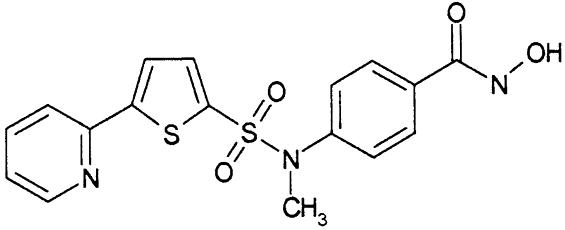 |
175.5 | 86.0 | 44 |
| MS27-275 | |||
 |
>10,000 | 185 | 201 |
| Apicidin | |||
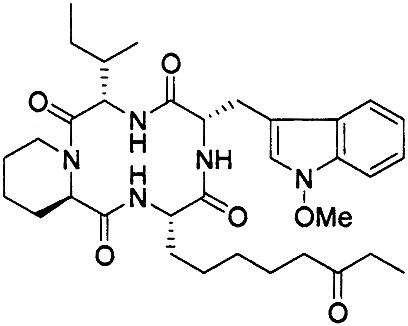 |
>1,000 | 2 | 0.7 |
Materials and Methods
Expression and Purification. The human HDAC8 gene was expressed and purified essentially as described in ref. 12, with minor modifications (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). For details on human HDAC1 and -3, also see Supporting Materials and Methods.
CD Spectroscopy. Spectra were collected by using a Jasco (Easton, MD) J-710 spectropolarimeter (see Supporting Materials and Methods).
Crystallization and Data Collection. For crystallization experiments, 130 μM HDAC8 was incubated for 1 h at room temperature with 6.7 molar excesses of inhibitor. Crystals were grown after 1 week at room temperature by the sitting-drop vapor-diffusion technique. Briefly, 4 μl of protein solution in gel filtration buffer was mixed with an equal volume of reservoir solution (0.1 M Mes, pH 5.3/1–5% polyethylene glycol 6000/2 mM Tris-2-carboxyethyl-phosphine) and 0.4 μl of 0.3 M glycyl-glycyl-glycine, and the mixture was allowed to equilibrate against 600 μl of reservoir solution. Data were collected at 100 K by using synchrotron radiation (see Supporting Materials and Methods).
Structure Determination and Refinement. The structure was solved by molecular replacement, using a truncated homology model, residues 63–320 of HDAC8, based on the crystal structure of HDLP in complex with trichostatin A (TSA) (11) (Protein Data Bank ID code 1C3R). Model building and refinement were performed with the programs quanta 2000 and cnx (both from Accelrys, San Diego). The structure was refined to 2.5 Å with final R and Rfree of 0.21 and 0.25, respectively. See Supporting Materials and Methods, Table 2, and Fig. 7, which are published as supporting information on the PNAS web site.
siRNA Transfection, Cell Growth, and Activity Assays. See Supporting Materials and Methods.
Results and Discussion
RNA Interference. To examine the role of HDAC8 in cell proliferation of tumor cells, we used RNA interference to disrupt HDAC8 expression in three human tumor cell lines of different origin (A549, HeLa, and HCT116). Two different short interfering RNAs (siRNAs), both targeting HDAC8, inhibited cell proliferation in all three cell lines. Data obtained with A549 cells are shown in Fig. 1. A siRNA targeting Eg5, a mitotic kinesin whose interference is known to cause mitotic arrest (Fig. 1B and ref. 13), gave comparable growth inhibition to HDAC8 siRNAs, whereas an unrelated siRNA (Luc) had no effect (Fig. 1 B and C). HDAC8 recently was reported to associate specifically with the inv(16) fusion protein found in acute myeloid leukemia, and TSA has been shown to impair inv(16)-mediated repression (14). From these data and our findings, we conclude that HDAC8 could have an important role in the physiology of solid or hematologic tumors.
Fig. 1.

Role of HDAC8 in cell proliferation of tumor cells. (A) Quantitative RT-PCR analysis of HDAC8 mRNA knockdown in A549 cells by an unrelated siRNA (Luc) or two HDAC8-specific siRNAs (8.6 and 8.7). (B) Growth inhibition evaluated by [14C]thymidine incorporation. Luc siRNA was used as a negative control, and Eg5 siRNA was used as a positive control. Each data set is the mean of triplicates. (C) The percentage of growth inhibition was normalized to the mock-transfected negative control. The data reported are mean values from three independent experiments. Error bars indicate SD.
HDAC Inhibition. We next tested a panel of HDAC inhibitors on class I HDACs 1, 3, and 8 (Table 1). Whereas HDACs 1 and 3 were inhibited by all of the inhibitors, HDAC8 was inhibited not appreciably by apicidin or MS27-275 and to a much lower extent than HDAC1 and -3 by suberoyl anilide hydroxamic acid (SAHA) and TSA. Compound 1, an aryl hydroxamic acid identified by screening a collection of small molecules, instead showed relatively similar potencies (<4-fold differences) on all tested proteins. Thus, HDAC8 shows a more restricted inhibition pattern than other class I enzymes, possibly indicating that it is feasible to develop selective inhibitors of this subtype, whereas development of inhibitors selective between more closely related HDACs, such as HDAC1 and -3, may be more complicated. This finding prompted us to attempt the cocrystallization of HDAC8 in the presence of compound 1 and to solve its three-dimensional structure to a resolution of 2.5 Å.
Overall Structure. The overall structure of HDAC8 consists of two molecules packed as a head-to-head dimer (Fig. 2A). Each molecule binds one Zn2+ ion and two K+ ions. The two molecules are essentially identical (rms deviation 0.34 Å for the Cα trace), forming a dimer interface that buries ≈1,300 Å2 of molecular surface area. The dimeric arrangement in the crystal is mediated by the inhibitor with the two capping groups (i.e., pyridine and thiophene moieties) of each inhibitor molecule stacking against each other. This finding could well explain the failure of our previous attempts to crystallize the apoprotein. Furthermore, each capping group interacts with protein residues Pro-273 and Tyr-306 of the opposite molecule, forming an extensive hydrophobic sandwich and giving rise to the twofold axis relating the two molecules in the asymmetric unit (Fig. 2 A). The oligomerization state observed in the crystal is unlikely to occur in solution, because the results of size-exclusion chromatography and light-scattering experiments are compatible with a monomeric state for both the apoprotein and the protein–inhibitor complex (data not shown). The structure of the HDAC8 monomer, similarly to the structure of HDLP (11) (30% sequence identity, Cα rms deviation of 2.4 Å), forms a single compact α/β domain composed of a central eight-stranded parallel β-sheet and 11 α-helices (Fig. 2B). The twisted β-sheet (topology β2, -1, -3, -8, -7, -4, -5, and -6) is flanked by five helices on one side (topology α2, -10, -9, -8, and -11) and three helices (topology α5, -6, and -7) on the other. Striking differences between the two proteins are observed for most of the loops emerging from the core of the protein, for the distal helices (α1, -3, and -4) and for the long extended loop (residues 325–356), which connects helices α10 and -11. This region in HDAC8 is completely different from that in HDLP, resulting in a large change of the loop between residues 332 and 345, including the loss of two helices (Fig. 2C). Overall, these variations give rise to a more accessible active site in HDAC8. In particular, the loop connecting α1 and -2 (residues 31–36) is shorter in HDAC8. In addition, the loops connecting β5 and -6 (residues 202–212) and β7 and α9 (residues 265–279) are more open in HDAC8, giving rise to a slightly deeper and larger pocket (Fig. 2C). Most strikingly, the loop 332–345, part of the long extended linker between α10 and -11, is 10 residues shorter than that observed in HDLP, leaving the active site less shielded from one side, which results in the lack of interactions between the protein and the inhibitor capping group. It is conceivable that structural variability in the loop regions emerging from the conserved core will be found across the entire family of HDACs, possibly reflecting the critical role of these regions in protein–protein interactions or in conferring different substrate specificities to individual members of the family.
Fig. 2.

Overall HDAC8 structure. (A) Ribbon diagram of the asymmetric unit of human HDAC8 crystals. The two monomers, A and B, are shown in indigo and yellow, respectively. The two inhibitor molecules and residues involved in the head-to-head packing are drawn in stick representation. Red, oxygen; blue, nitrogen; orange, sulfur. Carbon is shown in indigo (molecule A) and yellow (molecule B). Violet spheres, Zn2+ ions; red spheres, K+ ions. The chemical structure of the hydroxamic acid inhibitor is drawn in the upper left. (B) Ribbon diagram of human HDAC8 monomer with α-helices and β-strands labeled and colored yellow and indigo, respectively. The inhibitor is drawn in stick representation. Red, oxygen; blue, nitrogen; orange, sulfur; green, carbon. (C) Superposition of human HDAC8 (yellow) and HDLP (gray) backbone. Compound 1 and TSA are drawn in stick representation. Oxygen, nitrogen, and sulfur are colored as in A. Carbon is shown in yellow (compound 1) and gray (TSA). Dashed circles highlight structural variations of loops emerging from the core of the two structures.
Zinc-Binding Site. In the active site, the van der Waals surface of the enzyme reveals an ≈12-Å-deep, narrow pocket likely to accommodate the acetylated lysine during the catalytic reaction. At the bottom of this tunnel, a Zn2+ ion is found (Fig. 3). The architecture of the active site and its immediate surroundings are conserved between HDLP and HDAC8 and, based on sequence homology, are likely to be common to all zinc-dependent HDACs (Fig. 4). Similar to HDLP, the active site Zn2+ ion of HDAC8 is pentacoordinated, having Asp-178 (Oδ1), His-180 (Nδ1), and Asp-267 (Oδ1) as ligands in addition to the hydroxamic acid group of the inhibitor (Fig. 3). Immediately below the active site, a tube-like internal cavity filled by several water molecules could act as shuttle for the reaction product acetate (Fig. 5). In HDLP, this pocket has a different shape, by the effect of numerous substitutions of buried residues that line out the cavity (Figs. 4 and 5). Arg-37 and Cys-153, which are conserved strictly in class I and II HDACs, could be hydrogen-bond partners for the acetate byproduct of the hydrolysis reaction. In addition, Arg-37 creates a positive electrostatic potential that would be in accordance with binding the negatively charged acetate ion. The release of the acetate and its exchange with the bulk water could be accomplished by movements of the side chains of Tyr-18, Tyr-20, and His-42, located in close proximity to the external surface (Fig. 5). Ser-39, the target residue of PKA-catalyzed phosphorylation of HDAC8, also is facing the external surface of this cavity (Fig. 5). The phosphorylation of this residue was shown to inhibit HDAC8 enzymatic activity (15). A negatively charged phosphate group in this position possibly could impair the release of acetate, resulting in the decrease of enzyme activity. Therefore, HDAC8 phosphorylation has consequences opposite from those of the phosphorylation of other class I HDACs (16).
Fig. 3.

Molecular surface representation and architecture of HDAC8 active site. Residues relevant for catalysis and involved in inhibitor binding as well as the inhibitor and the modeled acetyllysine are drawn in stick representation. Red, oxygen; blue, nitrogen; orange, sulfur; gray, carbon. Carbon is colored green for the inhibitor and wheat for the docked acetyllysine. Polar interactions are shown as dashed yellow lines. Numbers in parentheses represent the zinc-ligand distances.
Fig. 4.

Multiple sequence alignment of selected deacetylases. The alignment encompasses the entire HDAC8 protein and residues 1–375 of HDAC1 (out of 482), 1–373 of HDLP (out of 375), and 643–1056 of HDAC4 (out of 1080). HDAC8 secondary structure and residue numbering are shown above the alignment. Red vertical bar, identical residues; blue vertical bar, conserved residues; green vertical bar, residues coordinating Zn2+ ion; filled violet circles, residues coordinating K+ ions at site 1; open violet circles, residues coordinating K+ ions at site 2; black stars, residues contacting the inhibitor; black triangles, residues lining the internal cavity.
Fig. 5.

Surface representation of the internal cavities of HDAC8 (yellow) and HDLP (light blue). The ribbon diagram of the HDAC8 backbone is shown in gray, and the water molecules found in the buried cavity of HDAC8 are shown as cyan spheres. The inhibitor, ions, and several residues lining the cavity are drawn in stick representation and colored as in Fig. 3.
Inhibitor-Binding Site. The structural characteristics of the hydroxamic acid inhibitor cocrystallized with HDAC8 are summarized in Fig. 2 A. It consists of a metal-binding domain, a linker domain, and a hydrophobic capping group. The inhibitor coordinates the zinc in a bidentate fashion and simultaneously contacts residues likely to be involved in catalysis (His-142, His-143, and Tyr-306) by its hydroxamate moiety (Fig. 3). In this regard, the situation is very similar to that observed for the inhibited structures of HDLP (11) and to zinc–metalloprotease–hydroxamate complexes (17, 18). The cyclic linker region of the inhibitor fits in the hydrophobic channel, with its aromatic aryl moiety stacked between side chains of Phe-152 and -208. Additional van der Waals interactions occur between the linker region and the side chain of Met-274 and the main chain of Gly-151. In the HDLP–TSA and HDLP–SAHA complexes (11), the linker region of the two inhibitors is formed by a five- and a six-carbon-long aliphatic chain, respectively. These chains act as optimal linkers for the hydroxamic acid and the capping group, allowing contacts both at the bottom and the entrance of the pocket. Instead, in HDAC8–compound 1 complex, the linker region is more involved in direct interactions with the hydrophobic pocket, and the pyridine moiety capping group does not show any relevant interaction with the protein (Fig. 2 B and C). Therefore, this group does not seem to contribute to the binding affinity of the compound, given that the dimeric arrangement seen in the crystal has not been found in solution. This observation should be true also for TSA and SAHA, both poor HDAC8 inhibitors (Table 1), because the loop region featuring the equivalent of Pro-22 from HDLP (Ala-32 in HDAC8), a residue that interacts with the capping group of TSA inhibitor (11), is shorter in HDAC8, and therefore this interaction is not possible (Fig. 2C). A Pro is present in this position in all class I and II HDACs, with the exception of HDAC10, which has an Ile, suggesting that in most HDACs this loop region should be structurally more similar to HDLP than to HDAC8. This finding is in line with the increased potency of TSA and SAHA observed on HDAC1 and -3 with respect to HDAC8. In HDAC8, the increase in potency going from SAHA to TSA and compound 1 (Table 1) is correlated with a progressively bulkier linker region, which maximizes hydrophobic interactions. All residues contacting the linker are conserved except for Met-274, which is a Leu in all other class I and II HDACs with the exception of HDAC10, which has a Glu in this position. The lack of measurable inhibition of HDAC8 by MS27-275, a compound that inhibits both HDAC1 and -3 (Table 1), can be rationalized by the presence of Trp-141 at the bottom of the tunnel (Fig. 6A), which should impede an effective zinc chelation by the bulky metal coordinating group, a N-(2-aminophenyl) formamide moiety, of MS27-275. Trp-141 is replaced by Leu in HDAC1 and -3, which should leave more space to accommodate this inhibitor and is in line with the increased potency toward HDAC1 and -3.
Fig. 6.

Inhibitor and potassium binding sites. (A) Closeup view of the molecular surface of HDAC8 at the active site entrance. The inhibitor is drawn in stick representation and colored as in Fig. 3. Red, Ala-32; yellow, Asp-101; violet, Trp-141; orange, Met-274. Regions that could be explored for the design of more potent HDAC8 inhibitors are labeled as a, b, c, and d. (B) Architecture of the two potassium-binding sites, K1 and K2. Residues coordinating K+ ions are drawn in stick representation. Red, oxygen; gray, carbon; red spheres, K+ ions; cyan spheres, water molecules W1 and W2. Coordination bonds are drawn as dashed yellow lines, and corresponding distances are reported in parentheses. The Fo – Fc map (contoured at 10 σ) is displayed around the K+ ions, which were omitted for map calculation.
Fig. 6A shows the molecular surface near the exit of the channel. Three shallow pockets could be explored for the design of more potent HDAC8 inhibitors. The capping group of the inhibitor is oriented in the direction of pocket d, even if this moiety does not interact with the protein but points toward the solvent. An inhibitor with additional meta substituents in the aryl moiety, with respect to the hydroxamic acid, could possibly interact with pockets b and d, providing selective interactions in addition to those in the channel. Further selectivity can be gained by maximizing interactions of the inhibitor with pocket c and with the loop region designated as “a” (Fig. 6A).
Potassium-Binding Site. Unexpectedly, the structure of HDAC8 reveals K+ ions bound at two sites: Site 1 is in close proximity (7.0 Å) to the zinc-binding site, whereas site 2 is located more toward the periphery of the molecule, 15 Å away from site 1 (Fig. 6B). The two sites were identified by strong positive peaks (19 and 13 σ, respectively) in the Fo – Fc maps and had typical distances and hexacoordination geometry for potassium binding (19). The final concentration of K+ ions in the crystallization drop is 75 mM, a value lower than its intracellular physiological value of ≈150 mM, suggesting that the potassium-binding sites could be biologically relevant. At site 1, the K+ ion is hexacoordinated by six oxygen ligands from Asp-176 (main-chain carbonyl oxygen and Oδ1), Asp-178 (main-chain carbonyl oxygen), His-180 (main-chain carbonyl oxygen), Ser-199 (Oγ), and Leu-200 (main-chain carbonyl oxygen), arranged in a distorted octahedral geometry (Fig. 6B). Potassium-binding site 1 is directly connected with the zinc-binding site, because the K+ ion is coordinated by the main-chain carbonyl oxygens of Asp-178 and His-180, whose side chains are involved in zinc chelation (Fig. 4A). The presence of K+ in the active site can be expected to influence the catalytic mechanism of the deacetylation reaction in several aspects. First, its presence increases the positive electrostatic potential in the active site and therefore could contribute to stabilize the oxyanion formed in the transition state of the deacetylation reaction and/or to stabilize the negatively charged acetate product. Furthermore, the K+ ion is coordinated by Asp-176 by means of both backbone carbonyl and side-chain oxygens (Figs. 4 and 6B). This coordination is expected to influence the correct orientation and chemical properties of Asp-176, a residue proposed to form a buried chargerelay system with His-142 (11). By analogy to Ca2+-binding proteins (20), upon chelation with the cation, the two CO bonds of the Asp-176 side chain no longer should be equivalent, with the bond of the chelating oxygen having more single-bond character and that of the nonchelating oxygen, hydrogen-bonded to His-142, more double-bond character. Therefore, the presence of potassium at site 1 should have a direct effect on the pK value of His-142, possibly facilitating its deprotonation after product release. Residues at potassium-binding site 1 are conserved in all zinc-dependent HDACs, with the exception of Leu-200, which can be substituted with Phe (HDAC1, -2, and -3) or Trp (HDAC10). Potassium-binding site 2 (Fig. 6B) is formed by the main-chain carbonyl oxygen of residues Phe-189, Thr-192, Val-195, and Tyr-225 and two water molecules. The relevance of this site for the catalytic activity of the enzyme is not obvious from the structure. It could help to maintain a specific protein conformation. The sequence conservation at this site is somewhat lower than at site 1: Thr-192 is Asp in class II HDACs; Val-195 is different only in HDAC9 (Ile); and Tyr-225 is different only in HDAC10 (Phe). The two potassium-binding sites are placed on the same face of the β-sheet and contribute equally to anchoring helix α7 and its loops to the central β-sheet (Fig. 6B). Residues forming the two potassium-binding sites come from loops contiguous to helix α7 (Asp-178 and His-180 at site 1; Phe-189 and Thr-192 at site 2) and from strands β4, -5, and -6 (Asp-176, Ser-199, and Leu-200 at site 1; Val-195 and Tyr-225 at site 2).
CD Spectroscopy. To further support the importance of the K+ ion to the structure, we have investigated the thermal unfolding of HDAC8 in solutions containing KCl or NaCl by CD spectroscopy. The melting midpoint of the secondary structure in a buffer containing NaCl was lower than that measured in the presence of KCl (Tm ≈ 35.5°C vs. Tm ≈ 41°C) (Fig. 8, which is published as supporting information on the PNAS web site). This finding suggests, in accordance with the structure, that K+ ions are required for the structural stability of HDAC8. The exchange with Na+ ions, which share with K+ the same external electronic configuration but a remarkably smaller ionic radius, could alter the overall folding of the enzyme, in agreement with the decreased protein stability observed when the protein was purified in the presence of NaCl (data not shown). The finding of a direct involvement of K+ ions in the architecture of the active site and in the stabilization of structural elements of HDAC8 could rationalize previously observed potassium dependence of the activity of this enzyme (12) and of other subtypes (P.G., unpublished results). Furthermore, the conservation of potassium-binding residues in zinc-dependent HDACs suggests that the presence of these metal ions could be relevant for the whole family of enzymes.
Supplementary Material
Acknowledgments
We thank Dominique Bourgeois for assistance in the data collection at beamline ID14-H3, European Synchrotron Radiation Facility, Grenoble, France; Michael Goetz (Merck, Rahway, NJ) for providing Apicidin; and Philip Jones (Istituto di Ricerche di Biologia Molecolare P. Angeletti) and Sylvie Bourrain (Merck, Harlow, U.K.) for providing the other HDAC inhibitors. This work was supported in part by a grant from the Ministero dell'Istruzione, dell'Università, e della Ricerca.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HDAC, histone deacetylase; HDLP, archeobacterial homolog of eukaryotic deacetylases; SAHA, suberoyl anilide hydroxamic acid; siRNA, short interfering RNA; TSA, trichostatin A.
Data deposition: The coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1W22).
Note Added in Proof. While this work was under review, a paper by Somoza et al. (21) was published. The authors describe the structure of human HDAC8 with four inhibitors. Their structures are comparable to ours.
References
- 1.Wolffe, A. P. & Guschin, D. (2000) J. Struct. Biol. 129, 102–122. [DOI] [PubMed] [Google Scholar]
- 2.Jenuwein, T. & Allis, C. D. (2001) Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 3.Kurdistani, S. K. & Grunstein, M. (2003) Nat. Rev. Mol. Cell Biol. 4, 276–284. [DOI] [PubMed] [Google Scholar]
- 4.Hassig, C. A. & Schreiber, S. L. (1997) Curr. Opin. Chem. Biol. 1, 300–308. [DOI] [PubMed] [Google Scholar]
- 5.Egger, G., Liang, G., Aparicio, A. & Jones, P. A. (2004) Nature 429, 457–463. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki, H., Gabrielson, E., Chen, W., Anbazhagan, R., van Engeland, M., Weijenberg, M. P., Herman, J. G. & Baylin, S. B. (2002) Nat. Genet. 31, 141–149. [DOI] [PubMed] [Google Scholar]
- 7.Kramer, O. H., Gottlicher, M. & Heinzel, T. (2001) Trends Endocrinol. Metab. 12, 294–300. [DOI] [PubMed] [Google Scholar]
- 8.Lee, S. C., Bottaro, A. & Insel, R. A. (2003) Mol. Immunol. 39, 923–932. [DOI] [PubMed] [Google Scholar]
- 9.Johnstone, R. W. (2002) Nat. Rev. Drug Discovery 1, 287–298. [DOI] [PubMed] [Google Scholar]
- 10.Gregoretti, I. V., Lee, Y. M. & Goodson, H. V. (2004) J. Mol. Biol. 338, 17–31. [DOI] [PubMed] [Google Scholar]
- 11.Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., Breslow, R. & Pavletich, N. P. (1999) Nature 401, 188–193. [DOI] [PubMed] [Google Scholar]
- 12.Hu, E., Chen, Z., Fredrickson, T., Zhu, Y., Kirkpatrick, R., Zhang, G. F., Johanson, K., Sung, C. M., Liu, R. & Winkler, J. (2000) J. Biol. Chem. 275, 15254–15264. [DOI] [PubMed] [Google Scholar]
- 13.Weil, D., Garcon, L., Harper, M., Dumenil, D., Dautry, F. & Kress, M. (2002) BioTechniques 33, 1244–1248. [DOI] [PubMed] [Google Scholar]
- 14.Durst, K. L., Lutterbach, B., Kummalue, T., Friedman, A. D. & Hiebert, S. W. (2003) Mol. Cell. Biol. 23, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, H., Rezai-Zadeh, N. & Seto, E. (2004) Mol. Cell. Biol. 24, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galasinski, S. C., Resing, K. A., Goodrich, J. A. & Ahn, N. G. (2002) J. Biol. Chem. 277, 19618–19626. [DOI] [PubMed] [Google Scholar]
- 17.Nishino, N. & Powers, J. C. (1978) Biochemistry 17, 2846–2850. [DOI] [PubMed] [Google Scholar]
- 18.Kam, C. M., Nishino, N. & Powers, J. C. (1979) Biochemistry 18, 3032–3038. [DOI] [PubMed] [Google Scholar]
- 19.Harding, M. M. (2002) Acta Crystallogr. D 58, 872–874. [DOI] [PubMed] [Google Scholar]
- 20.Mizuguchi, M., Nara, M., Kawano, K. & Nitta, K. (1997) FEBS Lett. 417, 153–156. [DOI] [PubMed] [Google Scholar]
- 21.Somoza, J. R., Skene, R. J., Katz, B. A., Mol, C., Ho, J. D., Jennings, A. J., Luong, C., Arvai, A., Buggy, J. J., Chi, E., et al. (2004) Structure 7, 1325–1334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}