

ABSTRACT
Biosimilars are biological products that are highly similar to existing products approved by health authorities. Demonstration of similarity starts with the comprehensive analysis of the reference product and its proposed biosimilar at the physicochemical and functional levels. Here, we report the results of a comparative analysis of a proposed biosimilar adalimumab MSB11022 and its reference product, Humira®. Three batches of MSB11022 and up to 23 batches of Humira® were analyzed by a set of state-of-the-art orthogonal methods. Primary and higher order structure analysis included N/C-terminal modifications, molecular weight of heavy and light chains, C-terminal lysine truncation, disulfide bridges, secondary and tertiary structures, and thermal stability. Purity ranged from 98.4%–98.8% for MSB11022 batches (N = 3) and from 98.4%–99.6% for Humira® batches (N = 19). Isoform analysis showed 5 isoform clusters within the pI range of 7.94–9.14 and 100% glycan site occupancy for both MSB11022 and Humira®. Functional analysis included Fab-dependent inhibition of tumor necrosis factor (TNF)-induced cytotoxicity in L929-A9 cell line and affinity to soluble and transmembrane forms of TNF, as well as Fc-dependent binding to Fcγ and neonatal Fc receptors and C1q complement proteins. All tested physicochemical and functional parameters demonstrated high similarity of MSB11022 and Humira®, with lower variability between MSB11022 and Humira® batches compared with variability within individual batches of Humira®. Based on these results, MSB11022 is anticipated to have safety and efficacy comparable to those of Humira®.
KEYWORDS: adalimumab, analytical similarity, biosimilars, FcR, Humira®, MSB11022, TNF
Abbreviations
- 4PL
4-parameter logistic
- Asn
asparagine
- AUC
analytical ultracentrifugation
- CD
circular dichroism
- Cmax
maximum observed concentration
- DTT
dithiothreitol
- EC50
half-maximal effective concentration
- ELISA
enzyme-linked immunosorbent assay
- EMA
European Medicines Agency
- Fab
fragment antigen-binding
- Fc
fragment crystallizable
- FDA
US Food and Drug Administration
- Gln
glutamine
- HC
heavy chain
- HMW
high-molecular weight
- icIEF
imaged capillary isoelectric focusing
- LC
light chain
- Lys
lysine
- mAbs
monoclonal antibodies
- Met
methionine
- MS
mass spectrometry
- MW
molecular weight
- pI
isoelectric point
- PK
pharmacokinetic
- Pro
proline
- Q-TOF
quadrupole time of flight
- SDS
sodium dodecyl sulfate
- SEC
size-exclusion chromatography
- SPR
surface plasmon resonance
- Tm
thermal transitions
- tm-TNF
transmembrane TNF
- TNF
tumor necrosis factor
- Tris
tris(hydroxymethyl)aminomethane
- UPLC-MS/MS
ultra-performance liquid chromatography-tandem mass spectrometry
- UV
ultraviolet
- VH
variable domain of the heavy chain
- VL
variable domain of the light chain
Introduction
Biological products, or biologics, such as growth factors and antibodies are medical products derived from natural sources or produced recombinantly in cells.1 Biologics are widely used for the treatment, prophylaxis and diagnosis of diseases. The expiration of patents for biological products enables the development of so-called biosimilars, which are biologics approved by regulatory agencies (based on very stringent guidelines), and are highly similar to their reference products.2,3
Biosimilar development aims to provide high-quality biological treatment to a broader patient population by increasing consumer access to drugs that are more affordable than the original product.2,4 The increased use of monoclonal antibody (mAb) therapies represents a major cost burden for healthcare providers. The high cost of treatment with originator biologics limits access, leading to an unmet medical need in the US and other parts of the world.5 The development of biosimilars can have a critical effect on the affordability and access to biologics in all markets, allowing more patients to be treated. For example, the cost savings upon the introduction of biosimilar infliximab for the treatment of autoimmune diseases in 5 EU countries would enable up to an additional 7561 patients to be treated (with 10–30% discount scenarios projecting €26–77 million in savings).6
The development of biosimilars is guided by scientifically rigorous principles mandated by regulatory agencies in the EU, the US, and other highly regulated regions of the world.3 Much greater analytical scrutiny and in-depth functional characterization are required for the approval of a biosimilar compared with the characterization performed for the approval of the original drug,3 as these are the foundations for comparable safety and efficacy performance in the clinic. The use of state-of-the-art orthogonal analytical technologies for the characterization of biosimilars, which are typically more extensive and diverse than those used during the development of the original product, are important in establishing confidence in biosimilars.5 Owing to their quality-driven development, currently marketed biosimilars that have been available in the EU for several years demonstrate similar efficacy and safety profiles compared with their reference products.7
Adalimumab (Humira®, AbbVie Inc.) is a recombinant human monoclonal immunoglobulin G1 (IgG1) that binds specifically to tumor necrosis factor (TNF) and blocks its general cytokine effects by preventing the interaction with p55 and p75 cell surface TNF receptors.8,9 As a consequence, adalimumab modulates TNF-mediated cellular functions.
The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved adalimumab for the treatment of the following immune system-mediated diseases: rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, plaque psoriasis, adult and pediatric Crohn's disease, ulcerative colitis, hidradenitis suppurativa, and uveitis.8-10
We report here the physicochemical and functional comparability of a proposed adalimumab biosimilar (MSB11022, Merck) and Humira®. We compared 3 MSB11022 batches (AL1D001–AL1D003) generated from independent drug substance produced at large scale with 13–23 batches of Humira® sourced from 2 regions (US and EU). A large panel of methods using orthogonal approaches was applied, as shown in Table 1. This allowed in-depth characterization at the physicochemical and functional level of MSB11022 in comparison to Humira®. The data presented here demonstrate that the manufacturing process of MSB11022 generates a high-quality product that is well aligned with the variability observed in Humira®.
Table 1.
Physicochemical and Functional Quality Attributes.
| Attribute | Analytical Technique | Aim of the test |
|---|---|---|
| Primary structure and posttranslational modifications | Peptide mapping by UPLC-MS/MS | Primary structure confirmation and coverage |
| Deamidation/oxidation | ||
| N/C-terminal modifications | ||
| Whole-molecule analysis by LC-MS | Intact LC and HC | |
| Glycation level | ||
| Misincorporation | ||
| High-order structure | Circular dichroism | Secondary and tertiary structures |
| Nano DSC | Thermal stability (identification of thermal transitions) | |
| Peptide mapping by UPLC-MS/MS | Assessment of disulfide bridges | |
| Purity and impurities | Purity by Bioanalyzer | Evaluation of purity |
| Aggregation by SEC* | Aggregation, purity levels | |
| HMW by AUC | Monomer purity and HMW | |
| Product variants | Charge variants by icIEF* | Isoform distribution Isoform pI values |
| Peptide mapping by UPLC-MS/MS | Glycan site occupancy | |
| Fab binding and potency | In vitro bioassay (rel. pot. [%EC50])* | Inhibition of TNF-induced L929-A9 cytotoxicity |
| SPR (affinity [KD]) | Affinity to TNF | |
| FACS (relative binding [%EC50]) | Binding to tm-TNF cell line | |
| Fc binding | SPR (affinity [KD]) | Affinity to Fcγ RI |
| Affinity to Fcγ RIIa R131 & H131 | ||
| Affinity to Fcγ RIIb | ||
| Affinity to Fcγ RIIIa V158 & F158 | ||
| Affinity to Fcγ RIIIb | ||
| Affinity to neonatal FcR | ||
| ELISA (rel. binding [%EC50]) | Binding to C1q |
Method fully validated in line with ICH Q2(R1) guidelines (ICH Guidelines).28
AUC, analytical ultracentrifugation; ELISA, enzyme-linked immunosorbent assay; HC, heavy chain; HMW, high-molecular weight; icIEF, imaged capillary isoelectric focusing; LC, light chain; LC-MS, liquid chromatography-mass spectrometry; SEC, size-exclusion chromatography; SPR, surface plasmon resonance; tm-TNF, transmembrane TNF; TNF, tumor necrosis factor; UPLC-MS/MS, ultra-performance liquid chromatography-tandem mass spectrometry.
Results
Primary structure and posttranslational modifications
Amino acid sequences for MSB11022 and Humira® were determined by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS and UV [UV] absorbance). The tryptic peptide maps were identical among 3 batches of MSB11022 and 21 batches of Humira®, without missing peaks or shifts in retention time (representative data shown in Fig. 1). The peptide mapping analysis confirmed that the primary structure was identical with 100% of sequence coverage (data not shown).
Figure 1.
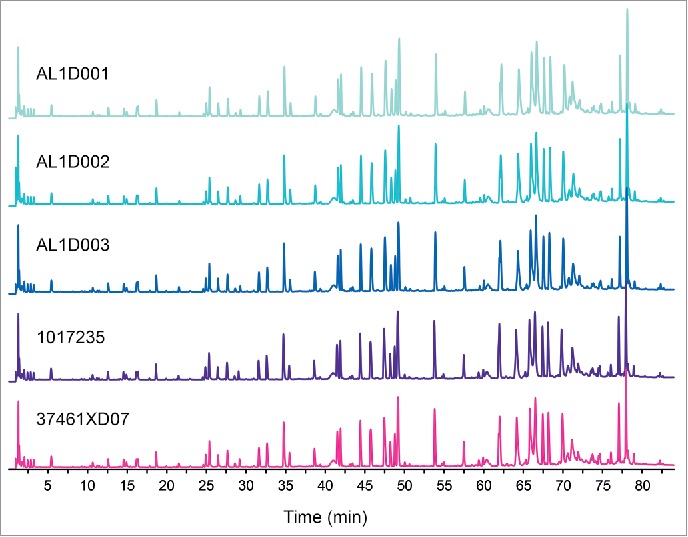
Comparison of total UPLC peptide mapping profiles between MSB11022 and Humira®. Representative UPLC peptide mapping profiles (total ion chromatograms) are presented for 3 batches of MSB11022 (AL1D001, AL1D002, and AL1D003) and 2 Humira® batches (1017235 and 37461XD07).
Peptide mapping by UPLC-MS/MS also enabled the assessment of N- and C-terminal heterogeneity. The N-/C-terminal sequence of the light chain (LC) was confirmed to be identical between MSB11022 and Humira®, without any missing amino acids in the primary structure. For the heavy chain (HC), minor heterogeneities between MSB11022 and Humira® were found. The N-/C-terminal modifications (pyro-glutamic acid (Glu), lysine (Lys) truncation, C-terminal proline (Pro) amidation) of MSB11022 were less variable among the batches and fell within the respective ranges observed for Humira® (Table 2). Overall, N-/C-terminal modifications were shown to be similar among the 3 batches of MSB11022 and 21 batches of Humira® (Table 2).
Table 2.
Primary Structure, High-Order Structure, and Purity Quality Attribute Ranges of MSB11022 and Humira®.
 |
Abbreviations: AUC = analytical ultracentrifugation; HC = heavy chain; HMW = high molecular weight; LCMS = liquid chromatography-tandem mass spectrometry; LMW = low molecular weight; <LOD = below limit of detection; MS = mass spectrometry; N = number; NR = non-reducing; R = reducing; SEC = size exclusion chromatography; SD = standard deviation; Tm = thermal transition; VH = variable domain of the heavy chain; VL = variable domain of the light chain. The limit of detection (LOD) of the oxidation level determination is 0.05%. 1US and EU batches of Humira®. 2Bar diagrams represent MSB11022 max range normalized to Humira® min/max range for each quality attribute 3Labelled according to ImmunoGenetics (IMGT) numbering. 4Labelled according to EU numbering.
Oxidation/deamidation sites and levels were also determined by peptide mapping by UPLC-MS/MS. The levels of oxidized methionine (Met) for all 5 major oxidation sites were less variable among the batches of MSB11022 (N = 3) and fell within the respective ranges observed for Humira® (N = 21; Table 2). Met252 was the most oxidized HC Met in all tested samples. The highest levels of Met252 oxidation were 10-fold higher in Humira® batches compared with those in MSB11022 batches. Overall, lower oxidation levels were seen in MSB11022 compared with Humira®. Similarly, deamidation and succinimidation of asparagine (Asn) and glutamine (Gln) residues were less variable among the batches of MSB11022 (N = 3) and fell within the respective ranges observed for Humira® (N = 21, data not shown).
The expected molecular weight (MW) of the LC and HC was in agreement with the theoretical MWs and similar for all batches Humira® and MSB11022 by whole-domain liquid chromatography-mass spectrometry (LC-MS; data not shown). Potential micro-heterogeneity was also evaluated. The possibility of incorporation of serine into Asn positions in Chinese hamster ovary cells was reported;11 such misincorporated species were not observed in any of the tested MSB11022 and Humira® batches (data not shown).
The relative distribution of the intact and glycated forms of both LC and HC, as well as C-terminal Lys truncation of HC level of MBS11022, fell within the respective ranges of Humira® batches (Table 2). All batches showed as main species the intact LC and the HC processed at its C-terminus (Lys removal).
High-order structure
When we assessed disulfide bridges by peptide mapping UPLC-MS/MS, all 9 expected disulfide bridges were identified in non-reducing condition and the expected configuration was confirmed in all tested samples (3 batches of MSB11022 and 21 batches of Humira®; representative data shown in Fig. 2).
Figure 2.
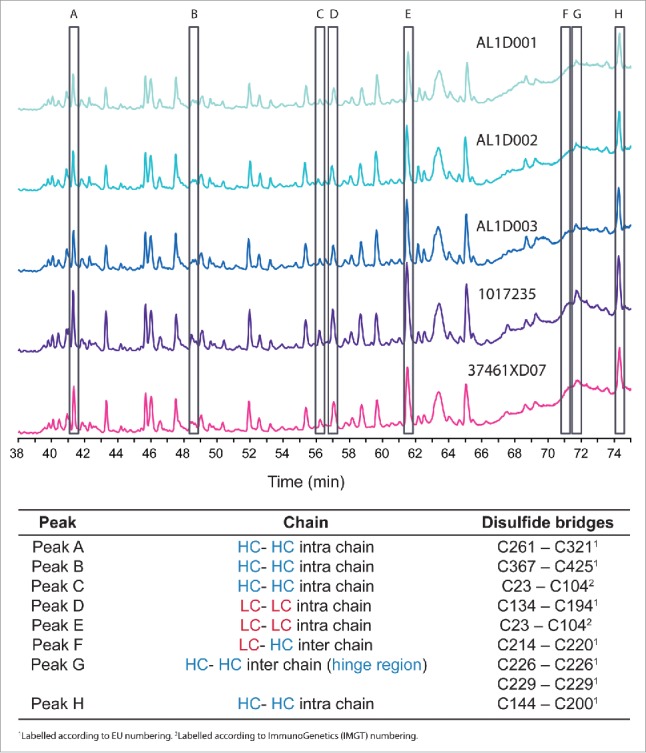
UV overlay of MSB11022 and Humira® disulfide bridges. Representative UPLC peptide mapping profiles without a reduction step of 3 batches of MSB11022 (AL1D001, AL1D002, and AL1D003) and 2 batches of Humira® (1017235 and 37461XD07). Peaks corresponding to disulfide bridges A through H are highlighted (open black rectangles) and the position of each bridge is specified below the chromatograms.
Thermal stability was determined by Nano DSC. All the tested batches presented 3 thermal transitions (Tm) with similar transition temperatures (Table 2). These findings were consistent with the 3 Tms reported for humanized monoclonal IgG1 in the literature.12 Tm1 and Tm2 are attributable to the denaturation of the constant CH2 and the antigen-binding fragment (Fab) domains, respectively, and Tm3 to the denaturation of the constant CH3 domain, as described by Garber et al.13
No differences in the secondary (α-helix, ß-sheet, turn) and tertiary structures (folding properties) were observed between Humira® and MSB11022 batches by circular dichroism (CD) measuring both far-UV and near-UV CD spectra (Fig. 3).
Figure 3.
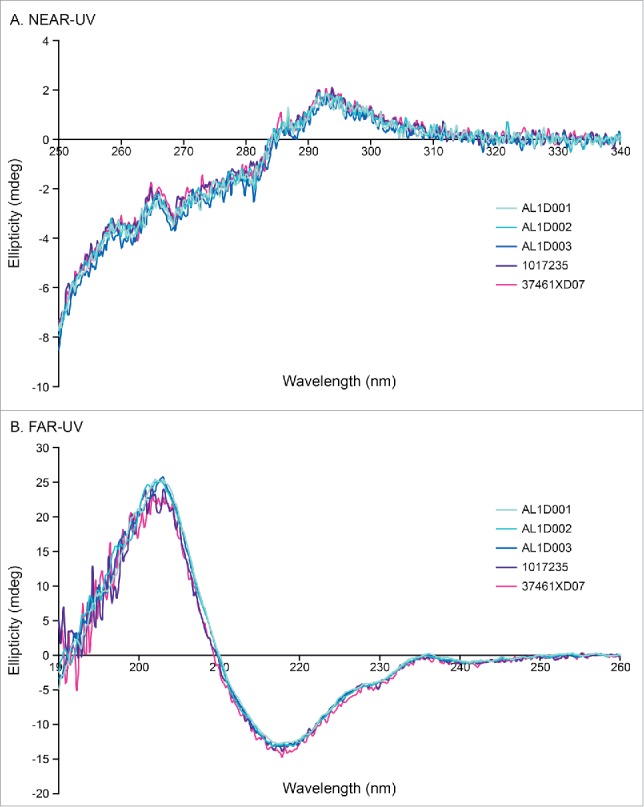
High-order structure analysis by CD. Representative near-UV CD spectrum (A) and far-UV CD spectrum (B) of MSB11022 and Humira® suggesting similar of tertiary and secondary structures, respectively.
Purity and impurities
The purity of MSB11022 and Humira® was evaluated by Bioanalyzer. Similar purity levels and low–molecular-weight (LMW) species were found for MSB11022 and Humira®. In both reducing and non-reducing conditions, purity ranges were similar between MSB11022 and Humira® batches. Minor impurity peaks were detectable between lower and upper MW markers, and the sum of these peaks accounted for less than 1.7% of the integrated areas in the non-reducing conditions (Fig. 4).
Figure 4.
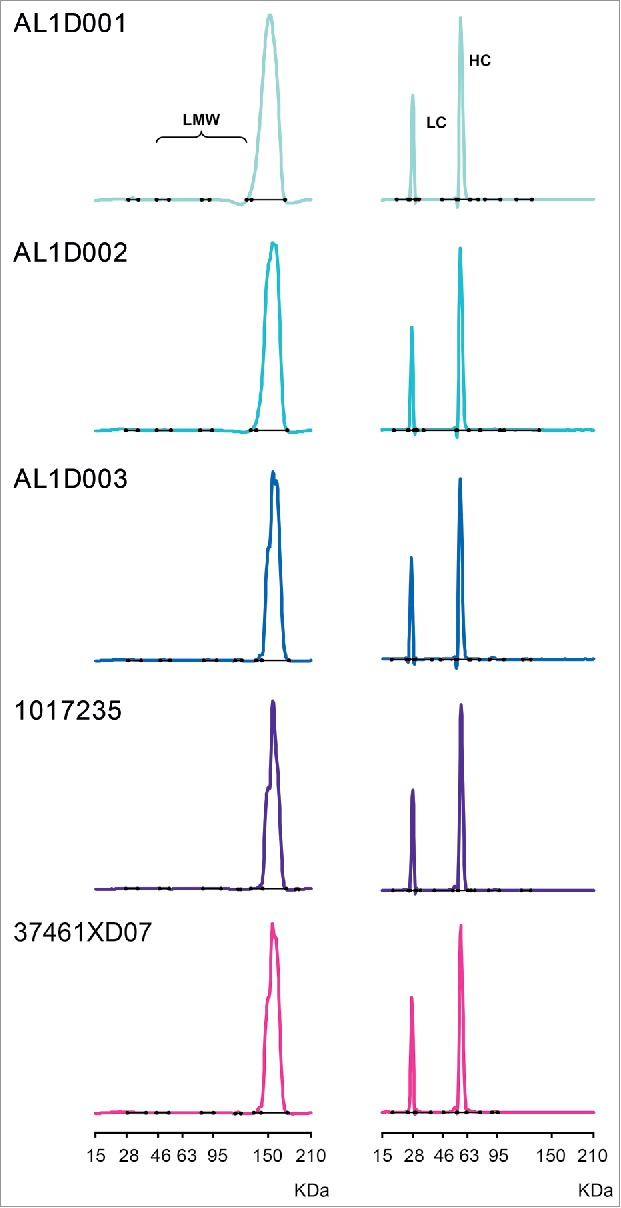
Purity of MSB11022 and Humira® by Bioanalyzer. Representative Bioanalyzer spectra of MSB11022 and Humira® in presence of SDS in non-reducing (left panel) and reducing conditions (right panel). Peaks representing the light chain (LC), heavy chain (HC), and low-molecular weight (LMW) aggregates are labeled.
Similar aggregates/high–molecular-weight (HMW) species levels and monomer purity were observed in MSB11022 and Humira® batches by size-exclusion chromatography (SEC; Table 2).
These results were confirmed by an orthogonal approach, analytical ultracentrifugation (AUC), showing comparable proportions of monomer, aggregates/HMW, dimers and fragments between MSB11022 and Humira® (Table 2). With three different methods, comparable purity and impurity levels were demonstrated for MSB11022 and Humira®.
Product variants
As measured by imaged capillary isoelectric focusing (icIEF) for the analysis of charge variants, 5 isoform clusters have been identified containing a total of 10 adalimumab charge variant isoforms (2 isoforms per cluster) migrating in the isoelectric point (pI) range of 7.94–9.14 (Table 3). For all isoform clusters, isoform areas of MSB11022 batches fell into the ranges observed for Humira® batches, suggesting a comparable charge variants profile between the 2 products (Table 3).
Table 3.
Charge Variants by icIEF of MSB11022 and Humira®.
| Min/max ranges (% area) |
||||||
|---|---|---|---|---|---|---|
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | ||
| N | pl 7.94–8.42 | pl 8.49–8.67 | pl 8.65–8.80 | pl 8.80–8.95 | pl 8.94–9.14 | |
| MSB11022 | 3 | 6.9–8.8 | 12.1–12.6 | 55.8–56.9 | 18.8–19.4 | 4.1–4.6 |
| Humira® EU | 10 | 6.3–10.5 | 11.6–13.5 | 54.7–61.4 | 15.6–20.1 | 3.4–5.4 |
| Humira® US | 11 | 6.5–8.9 | 10.9–13.2 | 54.9–59.7 | 16.6–20.0 | 4.3–5.5 |
icIEF, imaged capillary isoelectric focusing.
A 100% occupancy of the glycosylation site Asn297 (HC) was confirmed for Humira® batches and MSB11022 by peptide mapping/LC-MS/MS (data not shown), demonstrating similar glycan occupancy.
Fab binding and potency
Measurements of inhibition of TNF-induced L929-A9 cytotoxicity showed highly similar potency among 19 batches of Humira® and 3 batches of MSB11022 (Table 4). This result was confirmed by a ‘head-to-head’ comparative study of the Fab-dependent inhibition of TNF cytotoxic effect (representative data shown in Fig. 5).
Table 4.
Fab Binding and Fc Binding Quality Attribute Ranges of MSB11022 and Humira®.
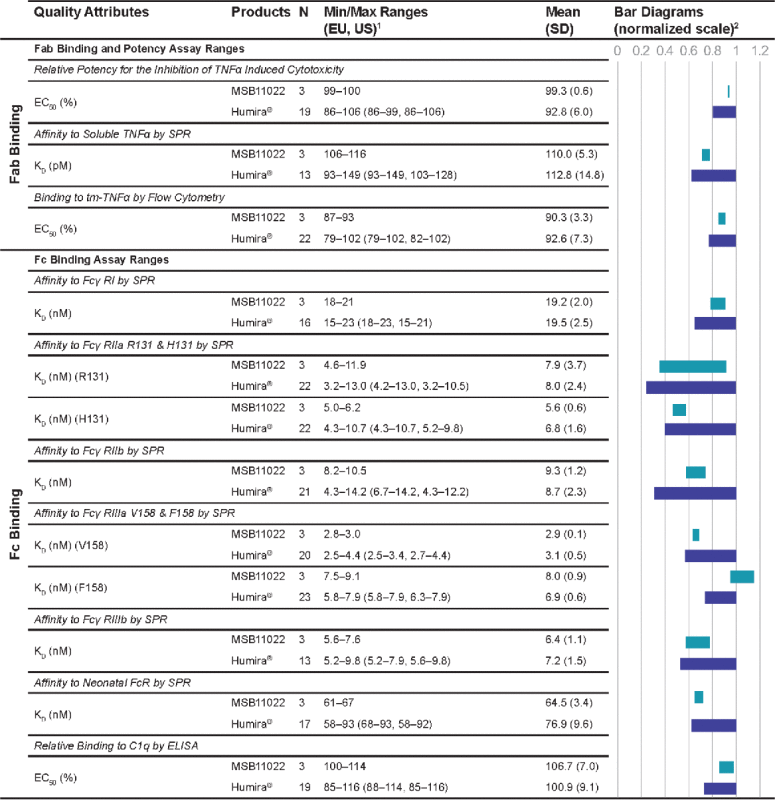 |
Abbreviations: EC50 = half-maximal effective concentration; ELISA = enzymelinked immunosorbent assay; Fab = fragment antigen-binding; Fc = fragment crystalizable; FcR = Fc receptor; KD = dissociation constant, N = number; SD = standard deviation; SPR = surface plasmon reasonance; tm-TNF = transmembrane TNF; TNF = tumor necrosis factor. 1US and EU batches of Humira®. 2Bar diagrams represent MSB11022 max range normalized to Humira® min/max range for each quality attribute. 3Labelled according to immunoGenetics (IMGT) numbering. 4Labelled according to EU numbering.
Figure 5.
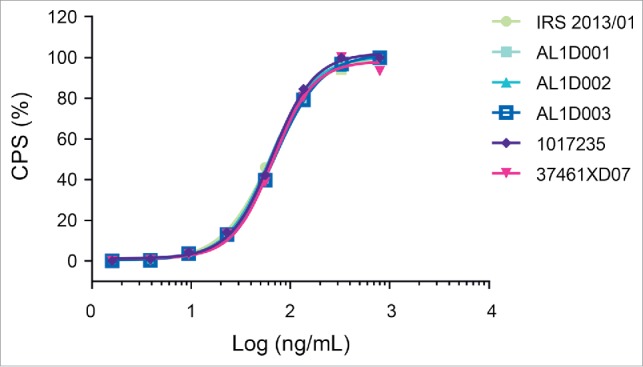
Inhibition of TNF-induced cytotoxicity. Representative graphs with an overlay of full-dose response curves (CPS: count-per-second) from one MSB11022 internal reference standard (IRS 2013/01), 3 batches of MSB11022 (AL1D001, AL1D002, and AL1D003) and 2 batches of Humira® (1017235 and 37461XD07).
Using binding kinetic analysis by surface plasmon resonance (SPR), highly similar affinity to soluble TNF, reflected by the dissociation constant (KD), was found among 13 Humira® batches and 3 MSB11022 batches (Table 4).
Binding to transmembrane TNF (tm-TNF) by flow cytometry was shown to be highly similar, as expressed by the half-maximal effective concentration (EC50), among the 22 Humira® batches and the 3 MSB11022 batches (Table 4).
Fragment crystallizable (Fc) binding
Highly similar Fc binding properties were found among both Humira® and MSB11022 batches, as measured by affinity to various Fcγ extracellular domains (RI, RIIa R131 & H131, RIIb, RIIIa V158 & F158, RIIIb), neonatal FcR, and by relative binding to C1q (Table 4). Kinetic binding sensorgrams were not distinguishable between MSB11022 and RP/RMP, as illustrated for the binding to the high-affinity allotype, FcγRIIIa V158 and to the low-affinity allotype, FcγRIIIa F158. While the affinity range of MSB11022 for binding to FcγRIIIa V158 is within the range of Humira®, overlapping binding patterns for binding to FcγRIIIa F158 were observed for MSB11022 and Humira®. Considering method variability using SPR, MSB11022 (3 batches with a mean affinity of 8 nM and a standard deviation of 0.9 nM) and Humira® (23 batches with a mean affinity of 6.9 nM and a standard deviation of 0.6 nM) are therefore also considered highly similar for binding to FcγRIIIa F158 (Table 4, Fig. 6).
Figure 6.
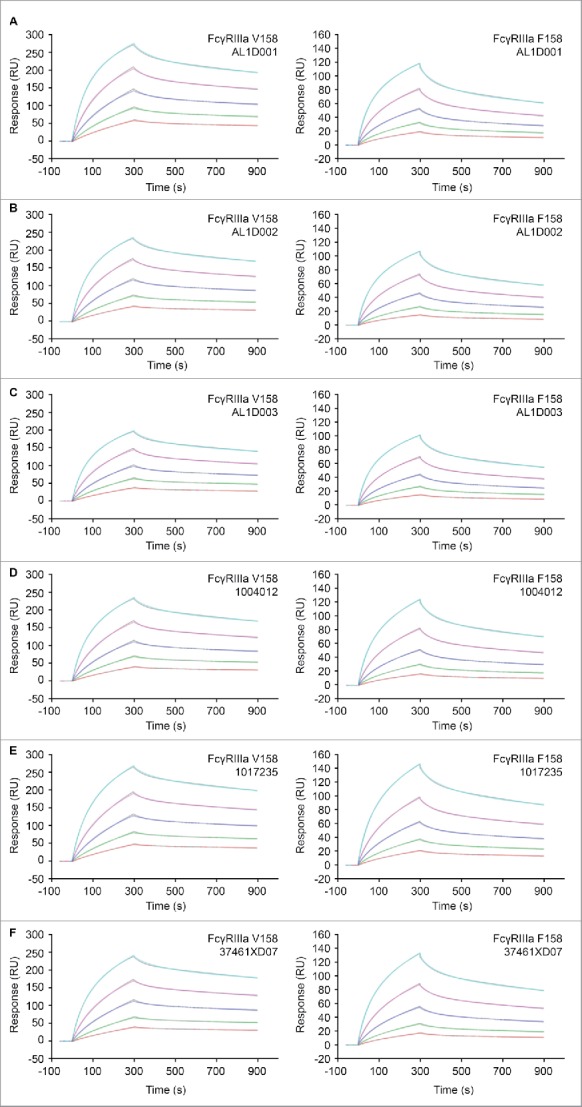
Comparison of FcγRIIIa binding by SPR between MSB11022 and Humira®. Representative surface plasmon resonance (SPR) sensorgrams of MSB11022 (batches AL1D001 [A], AL1D002 [B], AL1D003 [C]), Humira® US (batches 1004012 [D], 1017235 [E]), and EU (batch 37461XD07) binding to FcγRIIIa V158 and F158. The calculated curves are colored gray. The initial concentration of 50 nM (orange curve) was followed by 4 2-fold serial dilutions (other colored curves).
Degradation under stress conditions
Accelerated and stress stability studies are expected to be part of the demonstration of similarity to establish degradation profiles and provide direct comparison of the proposed biosimilar product with the reference product.14 Therefore, the comparative analysis of MSB1022 and Humira® was complemented by a pilot forced degradation study. A range of stress conditions were applied to MSB11022 and Humira® in a head-to-head setting. These conditions, comprising thermal stress, oxidation, deamidation, acidic pH stress, and mechanical stress, were chosen to generate different degradation pathways, and to provide an orthogonal assessment of degradation trends. No substantial differences were noted between MSB11022 and Humira® behavior upon various stress treatments (data not shown).
Discussion
Structural elements of a protein are critical determinants for its function; therefore, biosimilar development depends on extensive structural and functional characterization.
Requirements for the level of comparability between biosimilar mAbs and their reference products are as strict as, yet more comprehensive, than those for small molecules and less-complex biologics.5 State-of-the-art analytical methodologies measuring physicochemical and biological properties are applied to compare biosimilars with their reference products with a high level of scrutiny. Analytical programs should have at least 3 levels of testing: 1) release testing (and routine in-process testing) providing information on product attributes; 2) extended physico-chemical and biological characterization that comprehensively characterizes the biochemistry of a product; and 3) stability studies analyzing any changes over time.15 Variability of product characteristics is intrinsic to complex living cell production processes, and acceptable changes in quality attributes have been described.16 The quality range of an originator drug becomes more broad with changes in the production process, and this whole range could be used for claiming biosimilarity; however, biosimilars' sponsors, in collaboration with health authorities, typically select tighter quality ranges for their products.5
In this study, we investigated the physicochemical and functional comparability of a proposed adalimumab biosimilar (MSB11022) and Humira®. The manufacturing process was designed to achieve a quality that is similar to Humira®. In the early phase of experiments (data not shown), MSB11022 was selected based on rigorous testing for analytical similarity to Humira®. Cell line selection and the process development activities were performed in a stepwise manner using the principles of quality by design, and were guided by the target ranges of the quality attributes of multiple batches of Humira®. Multiple cell lines and hundreds of upstream and downstream process iterations were tested on a small scale with varying process parameters. The resulting materials from these multiple process versions were tested for analytical similarity to Humira® to support the selection of the final cell line and upstream/downstream process. The selected process was subsequently scaled up to 5000 L for the production of MSB11022. In the current work, MSB11022 was submitted to a comprehensive comparison to Humira® with a panel of methods.
The primary structure and high-order structure of MSB11022 and Humira® were thoroughly compared using orthogonal methods. The peptide mapping mass spectrometry (MS) and whole-molecule MS results demonstrated across a large number of batches that the primary structure of MSB11022 and Humira® was identical (data not shown), N-/C-terminal heterogeneity and posttranslational modification patterns were similar, and disulfide bridge pattern was identical. The oxidation level in MSB11022 batches fell within the range observed for Humira® (Table 2).
The Nano DSC results demonstrated MSB11022 and Humira® have similar thermal unfolding patterns, and these combined with the CD results suggest that MSB11022 and Humira® have similar secondary and tertiary structures (Fig. 3).
Three bioanalytical methods (Bioanalyzer, SEC, and AUC) demonstrated similar monomer purity levels, LMW species, and aggregation states for MSB11022 and Humira® (Table 2, Fig. 4). A comparable charge variants profile between the 2 products was observed by icIEF (Table 3). Taken together, the set of orthogonal approaches demonstrated that the primary, secondary, and tertiary structures, purity, aggregation states, and charge profile of MSB11022 and Humira® were similar. Functional comparability assays (cell-based bioassay, SPR, flow cytometry, and enzyme-linked immunosorbent assay [ELISA]) demonstrated highly similar relative potency for Fab-dependent inhibition of the TNF cytotoxic effect between MSB11022 and Humira®, as well as highly similar affinity for binding to soluble TNF, cellular tm-TNF, various Fcγ receptor extracellular domains, and C1q (Table 4).
Moreover, highly similar affinity of binding was also observed for MSB11022 and Humira® toward the neonatal FcR (Table 4). Binding to FcRn by SPR has been shown to be sensitive to oxidation levels in the Fc portion of IgG1,17,18 and high-affinity binding to this receptor is known to increase the serum elimination half-life of mAbs.19,20 This comparable binding affinity is expected to be indicative of similar pharmacokinetic (PK) profiles of MSB11022 and Humira®, and recently published data confirm this.21 Data from a phase 1 study comparing the PK profiles of MSB11022 and Humira® show PK equivalence for MSB11022 for all primary endpoints (area under the curve, maximum observed concentration [Cmax], and area under curve from time 0 to the last quantifiable concentration.21 Therefore, these results confirmed that both drugs have highly similar structural determinants and functional properties.
Analytical similarity assessment is ongoing, including the characterization of structure-function relationships, investigation of other quality attributes and orthogonal assessment of the current analytical panel via additional analytic methods, side-by-side analysis of the degradation profile/kinetics of Humira® and MSB11022 in stress conditions, characterization of product impurities/variants, and nonclinical pharmacodynamic studies aimed at probing mechanisms of action. It is anticipated that the data generated during these investigations will support the conclusion that MS11022 is analytically similar to Humira®.
In-depth assessment of MSB11022 and Humira® with state-of-the-art methods demonstrated high physicochemical and functional comparability between these 2 products. Our data also showed a similar or potentially greater variability among the different batches of Humira® than between Humira® and MSB11022 batches. This high variability among Humira® batches may be surprising, but could be attributable to changes in the manufacturing process. This level of variability has also been observed for other originator biologics. In a study examining the comparability of rituximab (MabThera®/Rituxan®; a mAb targeting the protein CD20) and a biosimilar rituximab, certain quality attributes shifted among batches of the originator.22 Product quality shifts have also been noted for Humira®. Up until 2013, 21 changes in the manufacturing process of the drug had been made.23 Nevertheless, it remains unclear what acceptable variations in batches for marketed biologics are.
In certain cases reported in Table 2, for example with regard to C-terminal amidated Pro, a minor population representing <5% of the mAb species in Humira® and 0.2% in MSB11022 was observed. Therefore, variability normalized to Humira® min–max ranges might be exacerbated by low levels of variants. The same might be true for truncated HC Delta (C-term GK), LMW species by SEC, aggregates/HMW species by AUC, and oxidation levels. Humira® also shows a larger range than MSB11022, with a similar mean, for glycated LC and HC and aggregates by SEC. It is worth highlighting that potential effects of drug age at the time of testing are unlikely to affect the relative comparison of MSB11022 and Humira®, as all MSB11022 samples were tested at mid-shelf life. It should be noted that this Humira® intra-batch variability could also be attributed to the greater number of Humira® batches in this analysis.
Based on these findings, MSB11022 is anticipated to show pharmacokinetics, potency, safety, and efficacy comparable to those of Humira®.
Materials and methods
Materials
MSB11022 batches were manufactured on a large scale at the Merck Aubonne-Vevey site (Switzerland). In total, up to 23 batches of Humira® from various origins (EU, US) were analyzed to build a target quality range for MSB11022. The analytical similarity study presented here included 3 MSB11022 drug product batches, at least 3 Humira® drug product batches sourced from the EU, and at least 3 Humira® drug product batches sourced from the US. An MSB11022 internal reference standard (IRS 2013/01) obtained from a representative engineering run of MSB11022 drug substance was used as reference standard throughout the analytical similarity study.
The tested quality attributes obtained with either qualified or fully ICH Q2 (R1) validated methods are summarized in Table 1. Three US-licensed and 3 EU-approved Humira® batches were analyzed head-to-head against 3 MSB11022 batches, and up to 16 additional batches of Humira® were analyzed in different analytical sessions. Some analytical methods such as CD, Nano DSC, and AUC did not allow a head-to-head analysis. In such cases, batches of Humira® were tested in the same analytical session with MSB11022 batches in the presence of an internal reference standard, if needed.
Peptide mapping
The complete primary structure and posttranslational modifications including deamidation and oxidation and occupancy of the glycosylation site; Asn297 (HC) of MSB11022 and Humira® were assessed by peptide mapping by UPLC-MS/MS (numbering as per IMGT [the international ImMunoGeneTics information system®] nomenclature for variable domain regions and as per EU nomenclature for constant domain regions). First, disulfide bridges were reduced in the presence of 12.5-mM dithiothreitol (DTT) and 4 M guanidine-HCl in a tris(hydroxymethyl)aminomethane (Tris) buffer at pH 7.6 at 37°C for 1 hour. Then, alkylation of the tested drugs was performed in the dark in the presence of 29 mM iodoacetamide for 1 hour. The buffer was exchanged for 6 M urea and Tris buffer at pH 8.0. Prior to hydrolysis, the buffer was diluted 3-fold with Tris buffer pH 8.0. The digestion of MSB11022 and Humira® (0.5 mg) by trypsin (24 µg) provided specific peptides that were subsequently separated by UPLC using a C18 column (Acquity UPLC BEH, 1.7 µm, 2.1 µm × 100 mm; Waters) and acetonitrile gradient containing formic acid at a flow rate of 0.2 mL/min at 60°C. Absorbance was monitored at 214 nm. The separated peptides were further identified by a coupled online quadrupole time of flight (Q-TOF) mass spectrometer.
The primary structure of the tested proteins was compared with a list of theoretical tryptic digest peptides using BiopharmaLynx 1.3 software.
Identification of disulfide bridges (cysteine pairing) was performed in similar conditions, except that MSB11022 and Humira® were alkylated by maleimide and iodoacetamide, and digested by trypsin without a reduction step.
Whole-domain LC-MS
The whole-molecule analyses were performed by LC-MS by using a Q-TOF mass spectrometer. Samples were analyzed after N-glycanase treatment. Disulfide bridge reduction was performed prior to LC-MS analysis to reduce the complexity of the molecule and to separate the HC from the LC.
Circular dichroism
CD spectra were measured in the far-UV (190–260 nm) and the near-UV (250–340 nm) spectral regions with 0.3 mg/mL and 5 mg/mL of the tested drugs (diluted in deionized water; Millipore), respectively. Spectra were recorded with a JASCO spectropolarimeter (Japan Spectroscopic Co., model J-810) in a quartz cuvette (1-mm cell length; Hellma) at 20°C. Four scans were accumulated for each spectral region with a scan rate of 20 nm/min and 4-s response time. The data pitch and the bandwidth were 0.2 nm and 1 nm, respectively. For each measurement, the sample concentration was normalized by the absorbance measured at 280 nm for the near-UV and 215 nm in the far-UV regions. Spectra with deionized water were used for baseline correction. When baseline drifts occurred, spectra were corrected using a narrow interval in which the signal was practically zero compared with the main spectral features (far UV, 250–260 nm; near UV, 330–340 nm)
Nano DSC
MSB11022 and Humira® samples (0.5 mg/mL in deionized water; Millipore) were analyzed by nanometer-range differential scanning nanocalorimetry using Nano DSC Autosampler (TA Instruments model 602001). After 10 minutes of equilibration at 10°C, the thermograms were obtained with a scan rate of 1°C/min and for the range 40°C–100°C. After baseline correction, thermograms normalized to protein concentration were analyzed with NanoAnalyze Data Analysis (version 3.1.2 by TA Instruments) by mathematical deconvolution using a polynomial baseline model that defined the 3 Tms of the tested drugs.
Bioanalyzer
The purity of MSB11022 and Humira® samples (0.1 mg/mL) was analyzed using an automated ‘gel-on-a-chip’ electrophoretic method (Agilent 2100 Bioanalyzer and Agilent Protein 230 Kit [catalog number 5067-1518]). This method separates analytes based on differences in MW, allowing the identification of degradation products and impurities. Samples are loaded after 5 min incubation at 70°C in the presence of sodium dodecyl sulfate (SDS) and maleimide under either non-reducing (without DTT) or reducing conditions (with DTT). The detection was performed by fluorescence (excitation at 458–482 nm [max. 470 nm], detection at 510–540 nm [max. 525 nm]). Markers of 4.5 kDa and 240 kDa were used to calibrate the electropherograms.
SEC-HPLC
SEC-HPLC was performed under non-denaturing conditions with Alliance 2695 (Waters) HPLC system using a TSKgel SuperSW3000 column (internal diameter [ID] 4.6 mm, Tosoh) at a flow rate of 0.35 mL/min in sodium phosphate/perchlorate buffer at pH 6.3. The isocratic elution profile was monitored using UV absorbance at 214 nm.
AUC
Monomeric purity and the level of purity of HMW and LMW species were evaluated by AUC in sedimentation velocity experiments using Optima XL-I ultracentrifuge (Beckman Coulter) and MSB11022 and Humira® at 0.5 mg/mL. Sedimentation was performed at 40,000 rpm with 70 scans taken at 8-min intervals at 20°C using An-50Ti analytical rotor. The path length was 12 mm and analytes were detected at 280 nm. Data were assessed using the SEDFIT program to obtain the c(s) profile of sedimentation coefficient (s) values. A diffusion coefficient was assigned to each value based on the assumption that all species have the same overall hydrodynamic shape and using a frictional coefficient ratio relative to that of a sphere (f/f0) fitted to the data. The partial specific volume used for adalimumab was 0.73 mL/g.
Charge variants by icIEF
Isoform profile and abundance were characterized in MSB11022 and Humira® samples by icIEF. Samples included MSB11022 or Humira® (1.0 mg/mL), carrier ampholytes (Pharmalyte 5–8 and 8–10.5), methylcellulose, and internal pI markers (pI 7.05 and 9.50; Master mix solution, ProteinSimple). Electrophoresis was performed on icIEF capillary cartridge FC-coated (ID 100 μm × 50 mm, ProteinSimple) in a pH gradient created by the carrier ampholytes under the influence of an electric field (pre-focusing 1 min at 1500 V, focusing 6 min at 3000 V). Analytes were detected by absorbance at 280 nm.
In vitro bioassay
Biological activity of MSB11022 and Humira® was measured in a murine fibroblast L929-A9 cell line (A9 cell line derived from the L929 cell line) by evaluating the inhibition of cytotoxicity induced by a fixed concentration of TNF in the presence of cycloheximide as described previously.24 L929-A9 cells were loaded into a 96-well plate. MSB11022 or Humira® was then added together with cycloheximide (Sigma) and TNF (R&D). After an incubation of 24 h at 37°C, 5% CO2, viable cells were measured by “ATPlite 1 step” luminescence assay (Perkin Elmer); an ATP monitoring system based on Firefly luciferase. The count-per-second values (y-axis) were plotted versus each own Log10 transformed MSB11022 or Humira® concentration (x-axis) and fit by using a 4-parameter logistic (4PL) algorithm (GraphPad Prism). For each data set, the concentration of MSB11022 or Humira® able to inhibit TNF cytotoxicity at EC50 was automatically calculated. The relative potency was expressed as percentage of activity of drug test preparation with respect to IRS 2013/01. The relative potency of MSB11022 and Humira® for the neutralization of TNF cytotoxic activity was calculated as the mean of 3 independent experiments. The variability of this validated method was evaluated for both repeatability (intra-session) and intermediate precision, which showed 17% and 9% coefficients of variation, respectively.
Surface plasmon resonance
Binding of soluble TNF to MSB11022 or Humira® was monitored by real-time biomolecular interaction analysis based on the SPR approach. The kinetics and affinity were measured using Fc capture Biacore method and a T200 Biacore (GE Healthcare) instrument as described previously.25 Interactions of the analyte in solution (TNF) with the ligand (MSB11022 or Humira®) captured beforehand by the Fc portion on Protein A covalently linked on the CM5 sensor chip were monitored. Kinetics rate constants and affinity were determined using the 1:1 fitting model.
Affinity to FcγR proteins was measured after covalent binding of FcγR extracellular domain proteins (FcγRI, R&D; FcγRIIa, Novus and Merck Serono; FcγRIIb, Novus; FcγRIIIa, R&D and Merck Serono; FcγRIIIb, R&D) to a CM5 sensor chip surface by amine coupling. Based on literature findings showing quantifiable detection of anti-TNF antibody binding to low-affinity Fcγ receptors only in the presence of soluble TNF,26 MSB11022 or Humira® was complexed with TNF homotrimer (2:1 molar ratio) for interaction studies with FcγRIIa/b, and IIIa/b. Serial dilutions of TNF-bound MSB11022/Humira® or MSB11022/Humira® alone (FcγRI) were injected sequentially. For FcRn (Merck Serono), the protein is covalently bound to a CM4 sensor chip surface by amine coupling. Serial dilutions of MSB11022 or Humira® alone at pH 6 were injected sequentially. Kinetics and affinity sensorgrams were analyzed with a 2-state reaction-fitting model using Biacore Evaluation software. The Fab and Fc affinities of MSB11022 and Humira® were expressed as average of results measured from 2 to 3 independent experiments.
FACS
Binding of MSB11022 and Humira® to tm-TNF was measured. A dose-response curve of adalimumab was applied on tm-TNF expressing cells using flow cytometry. HEK293 recombinant cells (Merck Serono) overexpressing non-cleavable tm-TNF variant27 at their surface were incubated with serial dilutions of adalimumab and then stained with 5 µg/mL secondary PE-labeled goat anti-human IgG1 Fc-specific polyclonal antibody (Jackson). The specificity of tm-TNF binding was verified with human IgG1 isotype control (Sigma). After washing, the cells were analyzed on a flow cytometer (BD FACSCalibur). The background autofluorescence was subtracted from the mean fluorescence intensity of each peak determined with the CellQuest Pro Software (BD). The intensity of the fluorescent signal is directly proportional to the amount of adalimumab bound to tm-TNF on cells. Dose-response curves were evaluated using 4PL algorithm. The relative potency was expressed as EC50 of drug test preparation with respect to IRS 2013/01. The tm-TNF binding activity was calculated as the mean of 3 independent experiments.
ELISA
MSB11022 and or Humira® binding capability to the human C1q was determined by the ELISA. Different concentrations of adalimumab were coated onto a 96-well plate. Unbound sites were blocked with 1% bovine serum albumin, then a fixed concentration of C1q (2 µg/mL, Sigma) was added and incubated for 2 hours at room temperature. The reaction was completed by the addition of anti-C1q-HRP antibody conjugate (1:2000 Abcam) and a substrate (TMB Sigma) that generated a colorimetric reaction. The intensity of the colorimetric signal read at 450 nm is directly proportional to the C1q protein bound to the coated antibody. The experimental data were interpolated with the 4PL algorithm. The relative potency was expressed as mean relative EC50 of drug test preparation with respect to IRS 2013/01. The C1q binding activity was calculated as the mean of 3 independent assays.
Disclosure of potential conflicts of interest
This study was supported by Merck and Merck Biosimilars. The authors are employees of Merck and Merck Biosimilars. No other potential conflicts of interest were reported.
Acknowledgments
We thank colleagues at Merck and Merck Biosimilars who supported the work. We would like to thank Fabio Camerini, Sabrina Fiumi, Fabio Valensisi, Cristian Ferrao, Luisa Iozzino, Anna Izzo, Gabriella Leo, Erika Birolo, Abhijeet Satwekar, Ombretta Caruso, Alessia Pellegrini, Elisabetta Zannoni, Gabriella Angiuoni, and Francesca Cutillo for supporting physicochemical analysis; Francesca Cavaliere, Angela Pelliccia, Giordana Bruni, Anna Rita Pezzotti, Michele Mascia, Laure Bonnet, and Aurore Glez for supporting functional analysis; and Robert Schnepf from Merck Global Manufacturing & Supply and Fabrice Romanet, Michael Muenzberg, Georg Schmies, and Georg Feger for critical reading of the manuscript. Editorial support during the preparation of the manuscript was provided by Guillaume Schoch of Fishawack Archimed AG, part of the Fishawack Group of Companies, supported by Merck.
References
- 1.U.S. Food and Drug Administration FDA is a biological product? [accessed 2016 Dec 21] http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194516.html [Google Scholar]
- 2.Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim DI, Lee SY, Chang SJ. Physicochemical characterization of Remsima. mAbs 2014;61163-1173; PMID:25517302; http://dx.doi.org/ 10.4161/mabs.32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, Gravanis I, Heim HK, Heinonem E, Ho K, et al.. Biosimilars: what clinicians should know. Blood 2012;120:5111-5117; PMID:23093622; http://dx.doi.org/ 10.1182/blood-2012-04-425744 [DOI] [PubMed] [Google Scholar]
- 4.Cornes P. The economic pressures for biosimilar drug use in cancer medicine. Target Oncol 2012; 7:S57-S67; PMID: 22249658; http://dx.doi.org/21441787 10.1007/s11523-011-0196-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs 2011;3:209-217; PMID:21441787; http://dx.doi.org/ 10.4161/mabs.3.2.15005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jha A, Upton A, Dunlop WC, Akehurst R. The budget impact of biosimilar infliximab (Remsima®) for the treatment of autoimmune diseases in five European countries. Adv Ther 2015;32:742-756; PMID:26343027; http://dx.doi.org/ 10.1007/s12325-015-0233-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood 2014;124:3191-3196; PMID:25298038; http://dx.doi.org/ 10.1182/blood-2014-06-583617 [DOI] [PubMed] [Google Scholar]
- 8.AbbVie Inc HUMIRA (adalimumab) injection, for subcutaneous use. 2016 Oct. [accessed 2016December21] http://www.rxabbvie.com/pdf/humira.pdf
- 9.AbbVie Inc SPC Humira 40 mg/0.4 ml Pre-filled Syringe and Pre-filled Pen. 2016 Sep 23. [accessed 2016December21] https://www.medicines.org.uk/emc/medicine/21201
- 10.EMA. Summary of opinion (post authorisation): Humira (adalimumab). 2016 May 26. [accessed 2016December21] http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/000481/WC500207152.pdf
- 11.Wen D, Vecchi MM, Gu S, Su L, Dolnikova J, Huang YM, Foley SF, Garber E, Pederson N, Meier W. Discovery and investigation of misincorporation of serine at asparagine positions in recombinant proteins expressed in Chinese hamster ovary cells. J Biol Chem 2009;284:32686-32694; PMID:19783658; http://dx.doi.org/ 10.1074/jbc.M109.059360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci 2008;97:1414-1426; PMID:17721938; http://dx.doi.org/ 10.1002/jps.21104 [DOI] [PubMed] [Google Scholar]
- 13.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun 2007;355:751-757; PMID:17321501; http://dx.doi.org/ 10.1016/j.bbrc.2007.02.042 [DOI] [PubMed] [Google Scholar]
- 14.U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. Guidance for Industry. [accessed 2016 Dec 21] http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291134.pdf [Google Scholar]
- 15.Brorson K, Kendrick B. Perspectives on well-characterized biological proteins. In: Schiel JE, Davis Dl, Borisov OV, editors. State-of-the-art and emerging technologies for therapeutic monoclonal antibody characterization. Vol. 1. Monoclonal anti-body therapeutics: structure, function, and regulatory space. Washington, DC: ACS; 2014. p. 99-116; doi: 10.1021/bk-2014-1176.ch004 [DOI] [Google Scholar]
- 16.Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol 2011; 29:310-312; PMID:21478841; http://dx.doi.org/ 10.1038/nbt.1839 [DOI] [PubMed] [Google Scholar]
- 17.Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Preksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol 2011;48:860-866; PMID:21256596; http://dx.doi.org/ 10.1016/j.molimm.2010.12.009 [DOI] [PubMed] [Google Scholar]
- 18.Bertolotti-Ciarlet A, Wang W, Lownesa R, Pristatskya P, Fang Y, McKelvey T, Li Y, Li Y, Drummond J, Prueksarita T, et al.. Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fcγ receptors. Mol Immunol 2009;46:1878-1882; PMID:19269032; http://dx.doi.org/ 10.1016/j.molimm.2009.02.002 [DOI] [PubMed] [Google Scholar]
- 19.Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281(33):23514-23524. Epub 2006 Jun 21; PMID:16793771; http://dx.doi.org/ 10.1074/jbc.M604292200 [DOI] [PubMed] [Google Scholar]
- 20.Robbie GJ, Criste R, Dall'acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother 2013; 57(12):6147-6153; PMID:24080653; http://dx.doi.org/ 10.1128/AAC.01285-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyland E, Mant T, Vlachos P, Attkins N, Ullmann M, Roy S, Wagner V. Comparison of the pharmacokinetics, safety, and immunogenicity of MSB11022, a biosimilar of adalimumab, with Humira® in healthy subjects. Br J Clin Pharmacol 2016;82:983-993; PMID:27285856; http://dx.doi.org/ 10.1111/bcp.13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013; 27(5):495-507; PMID:23649935; http://dx.doi.org/ 10.1007/s40259-013-0036-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tebbey PW, Varga A, Naill M, Clewell J, Venema J. Consistency of quality attributes for the glycosylated monoclonal antibody Humira® (adalimumab). mAbs 2015;7(5):805-811; PMID:26230301; http://dx.doi.org/ 10.1080/19420862.2015.1073429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terlizzese M, Simoni P, Antonetti F. In vitro comparison of inhibiting ability of soluble TNF receptor p75 (TBP II) vs. soluble TNF receptor p55 (TBP I) against TNF-alpha and TNF-beta. J Interferon Cytokine Res 1996; 16:1047-1053; PMID: 8974008; https://dx.doi.org/ 10.1089/jir.1996.16.1047 [DOI] [PubMed] [Google Scholar]
- 25.Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, Salfield J, Sasso EH. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol 2009; 131:308-316; PMID:19188093; http://dx.doi.org/ 10.1016/j.clim.2009.01.002 [DOI] [PubMed] [Google Scholar]
- 26.Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR, Louie JS, Kohno T. Differences in binding and effector functions between classes of TNF antagonists. Cytokine 2009; 45:124-31; PMID:19128982; http://dx.doi.org/ 10.1016/j.cyto.2008.11.008 [DOI] [PubMed] [Google Scholar]
- 27.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, et al.. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995; 83:793-802; PMID:8521496; http://dx.doi.org/ 10.1016/0092-8674(95)90192-2 [DOI] [PubMed] [Google Scholar]
- 28.ICH harmonised tripartite guideline Validation of analytical procedures: text and methodology Q2(R1). Current Step 4 version. 2005 Nov. [accessed 2016 Dec 21] http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf [Google Scholar]