

Abstract
Obesity as a multifactorial disorder involves low-grade inflammation, increased reactive oxygen species incidence, gut microbiota aberrations, and epigenetic consequences. Thus, prevention and therapies with epigenetic active antioxidants, (−)-Epigallocatechin-3-gallate (EGCG), are of increasing interest. DNA damage, DNA methylation and gene expression of DNA methyltransferase 1, interleukin 6, and MutL homologue 1 were analyzed in C57BL/6J male mice fed a high-fat diet (HFD) or a control diet (CD) with and without EGCG supplementation. Gut microbiota was analyzed with quantitative real-time polymerase chain reaction. An induction of DNA damage was observed, as a consequence of HFD-feeding, whereas EGCG supplementation decreased DNA damage. HFD-feeding induced a higher inflammatory status. Supplementation reversed these effects, resulting in tissue specific gene expression and methylation patterns of DNA methyltransferase 1 and MutL homologue 1. HFD feeding caused a significant lower bacterial abundance. The Firmicutes/Bacteroidetes ratio is significantly lower in HFD + EGCG but higher in CD + EGCG compared to control groups. The results demonstrate the impact of EGCG on the one hand on gut microbiota which together with dietary components affects host health. On the other hand effects may derive from antioxidative activities as well as epigenetic modifications observed on CpG methylation but also likely to include other epigenetic elements.
1. Introduction
Metabolic syndrome, a multifactorial disorder, results from a long-term imbalance of diet and physical activity, genetic predisposition, and an imbalanced gut microbiota influencing several metabolic pathways including epigenetic regulation. In 2015, the prevalence of metabolic syndrome was determined at 1.9 billion adults being overweight (BMI ≥ 25 kg/m2 (body mass index)) and more than 600 billion obese (BMI ≥ 30 kg/m2) [1]. Thus, this high incidence of obesity and associated diseases like type 2 diabetes (hyperglycemia on basis of an insulin resistance) are a challenge and financial burden for national health care systems.
Increased adipose tissue is an important energy storage but also a key endocrine organ with active metabolism relevant in energy homeostasis, lipid and glucose metabolism, fibrinolysis, coagulation, blood pressure, and inflammation (like interleukin-6 (IL-6), tumor necrosis factor α (TNF-α)) [2, 3], but also in increased reactive oxygen species (ROS) and free fatty acids (FFAs) production, and increased oxidative stress [4, 5]. Oxidative stress in turn is associated with both genome-wide hypomethylation and promoter hypermethylation of the DNA [6], resulting in transcriptional silencing of key antioxidant enzymes as well as tumor suppressor genes [7, 8]. Diets rich in bioactive anti-inflammatory compounds, such as polyphenols, have been recommended to reduce oxidative stress and to decrease inflammation [9].
EGCG ((−)-Epigallocatechin-3-gallate), the main catechin of green tea (50–75%), has been shown to support many potential health effects, including antioxidant, anticarcinogenic, hypocholesterolemic, and cardioprotective epigenetic activities [10]. It was found to increase energy expenditure and weight loss, reduce fat mass, and facilitate weight maintenance after weight loss [11]. Antiobesity effects might be mediated through antioxidative and singlet oxygen quencher properties. Inhibition of destructive effects of ROS might act through selective inhibition of specific enzyme activities such as (Dnmts) DNA methyltransferases, repair of chromosomal aberrations [12], and suppressing inflammation in the development of obesity [9]. EGCG supplementation (0.1%) in obese and diabetic C57BL/KsJ-db/db mice decreased the levels of insulin, IGF-1 (insulin-like growth factors), IGF-2, free fatty acid, and expression of TNF-α, interleukin- (IL-) 6, IL-1β, and IL-18 mRNAs in hepatic tissue [13]. EGCG inhibits Dnmt activity resulting in a decreased 5-methylcytosine concentration; 20 μmol/L of EGCG already inhibited Dnmt activity in oesophageal (KYSE-150), colon (HT-29), prostate (PC-3), and breast (MCF7 and MDA-MB-231) cancer cells, although no effects of EGCG on Dnmt activity (2–50 μmol/L) are also shown in cancer cells. Another Dnmt inhibitory pathway of flavan-3-ols results from an increase of S-adenosyl-l-homocysteine (SAH) [14]. A reduced Dnmt activity reactivates methylation-silenced genes in a dose (5–50 μmM of EGCG) and time dependent (12–144 h) manner [25].
According to Fang et al. (2003) MutL homologue 1 (MLH1) and O6-methylguanine-deoxyribonucleic acid methyltransferase (MGMT), both part of the DNA mismatch repair (MMR) system, are hypomethylated in the CpG rich promoter region [25] accompanied by a higher expression of mRNA [25]. In contrast, ROS-induced oxidative stress contributes to hypermethylation of normally unmethylated promoter regions, resulting in transcriptional silencing of key antioxidant enzymes as well as tumor suppressor genes [7, 8]. EGCG also alters histone acetyl transferase (HAT) activity, resulting in aberrant chromatin structures, as well as miRNA expression in hepatocellular carcinoma cells [14].
However, above small-intestinal absorption, tea catechins could reach the large intestine and be processed by gut microbiota into gallic acid and epigallocatechin (EGC) [26, 27]. Alterations in gut microbiota composition and differences in gut microbial metabolite profile due to different dietary feeding offer insights that may be relevant for several chronic conditions, including obesity. The obesity related gut microbiota is composed by a less bacterial diversity and altered abundance, gene-representation, and metabolic pathways [28]. These differences include a higher abundance of Firmicutes, whereas Bacteroidetes are reduced resulting in a higher Firmicutes/Bacteroidetes ratio in obese individuals. An increased abundance of Lactobacilli is mentioned as a growth promoter and is associated with weight gain and inflammatory processes during obesity [29]. Genes encoding for carbohydrate metabolism enzymes are increased in the gut microbiome of obese mice, provoking a greater capacity to extract energy from the diet and to generate short-chain fatty acids (SCFAs) [30, 31]. SCFAs are essential for the microbial community and play a role in regulation of energy balance, inflammatory processes, health, and obesity [32].
Meanwhile interventions with EGCG resulted in a reduction of Clostridium spp. abundance, increased Bacteroides, but also influenced Bifidobacterium and Prevotella, to a lesser extent resulting in lower levels of acetic and butyric acids and little influence on propionic acid levels in caecum. Thus, effects on weight reduction and weight maintenance due to dietary intervention with EGCG could be responsible for the regulation of energy metabolism in the body [26].
In the present study, we investigated the effects of a physiologically applicable dose of EGCG on gut microbiota, DNA damage, DNA methylation, and gene expression of inflammatory mediators: IL-6; Dnmt1, and DNA repair: MLH1 in liver and colon due to metabolic syndrome induced by a high fat diet (HFD) and due to control diet (CD) with a focus on associations between alteration of DNA repair processes, immune functions, and genomic instability. We investigated the colon as direct nutrient contact is given, but also hepatic tissue, the main organ in glucose and insulin metabolism. In addition, gut microbiota composition and diversity were analyzed on the basis of stool samples. Effects of dietary EGCG in alleviating conditions associated with obesity and metabolic syndromes are reported.
2. Materials and Methods
The Ethical Committee of the Medical University of Vienna approved the animal experiment (BMWFW-66.009/0329-WF/V/3b/2014) implemented with 6-week-old C57BL/6J male mice (n = 60) (Janvier Labs, France). Three animals were kept per cage (Macrolon type III, Techniplast GmbH, Germany) under standard conditions (24 ± 1°C, humidity 50 ± 5%, 12 hrs light/dark cycle). The animals were divided into four groups after an acclimatization time of two weeks with control diet (EF R/M control, 12% fat ssniff Spezialdiäten GmbH, Soest, Germany): (1) CD group (EF R/M control, 11% kJ fat, ssniff Spezialdiäten GmbH, Soest, Germany), (2) CD plus EGCG group (CD + EGCG) (EGCG: 25 mg/kg body weight per day), (3) HFD group (54% kJ fat ssniff EF acc.D12492 (I) mod., ssniff Spezialdiäten GmbH, Soest, Germany), and (4) a HFD plus EGCG group (HFD + EGCG). Food and water were provided ad libitum and supplemented with EGCG in the intervention groups (25 mg/kg b.w.). EGCG was provided as pure EGCG (EGCG-Uji-XP, System Biologie, Wollerau). Water/EGCG uptake was measured daily. Once per week the mice were weighted and food intake was determined (Table 1). After 4 months, the animals were killed by cervical dislocation.
Table 1.
Body weight and food and water intake of C57BL/6J male mice over a period of 4 months.
| Chow intake [g per day] | Water intake [ml per day] | Weight [g] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Month | ||||||||||||
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| Intervention | ||||||||||||
| CD | 2.64 ± 0.17 | 2.11 ± 0.62 | 2.08 ± 0.61 | 1.99 ± 0.64 | 5.60 ± 0.99 | 5.36 ± 0.99 | 5.40 ± 0.91 | 5.05 ± 1.27 | 23.99 ± 1.50 | 25.92 ± 1.09 | 26.94 ± 1.42 | 27.88 ± 1.49 |
| CD + EGCG | 2.67 ± 0.12 | 2.61 ± 0.13 | 2.63 ± 0.12 | 2.62 ± 0.28 | 5.19 ± 0.61 | 5.13 ± 0.54 | 4.83 ± 0.65 | 4.60 ± 0.39 | 24.16 ± 1.52 | 25.81 ± 1.32 | 26.98 ± 1.53 | 28.43 ± 1.76 |
| HFD | 2.56 ± 0.10 | 2.59 ± 0.15 | 2.60 ± 0.13 | 2.56 ± 0.13 | 5.30 ± 0.43 | 5.13 ± 0.47 | 4.97 ± 0.54 | 5.07 ± 0.39 | 30.54 ± 3.43 | 37.62 ± 4.11 | 42.88 ± 4.58 | 45.99 ± 4.46 |
| HFD + EGCG | 2.57 ± 0.10 | 2.50 ± 0.10 | 2.58 ± 0.13 | 2.55 ± 0.16 | 5.07 ± 0.61 | 4.56 ± 0.40 | 4.43 ± 0.41 | 4.55 ± 0.31 | 30.74 ± 3.43 | 38.79 ± 3.07 | 44.72 ± 3.68 | 47.69 ± 3.45 |
2.1. Single-Cell Gel Electrophoresis (SCGE) Assay
DNA migration in an electric field was analyzed in hepatocytes and colonocytes from mice in SCGE assays (comet assay) [33, 34]. This approach has the advantage that measurement can be conducted with variety of organs [35] and is increasingly used in genetic toxicology Collins, (2015) [36]. A typical picture from a “comet” as marker for DNA-instability is shown in (Figure 1).
Figure 1.

Photographic images of “comets” in an intact nucleus (a) and in a nucleus with damaged DNA (b). Stain: ethidium bromide.
Nuclei from livers and cells from colons were collected according to the method developed by Sasaki et al. (2002) [37]. The procedure for the measurement of DNA migration in liver cells and colonocytes was employed by us in a number of earlier studies [38–42]. 1.0 g liver tissue was homogenized by use of a Potter Elvehjem-type (B. Braun, Melsungen, Germany) at 400 rpm in 4.0 mL chilled homogenization buffer (pH 7.5). Subsequently, the homogenates were centrifuged (800g, 10 min, 4°C). Colon cells were isolated by scratching mucosa from the colons and were kept on ice in 2.0 mL homogenization buffer. The nuclei were suspended in LMPA (0.5%, Gibco, Paisley, UK) and transferred to slides which were precoated with NMPA (1.0%, Gibco, Paisley, UK).
The experiments were carried out according to Burlinson et al. (2007) [43]. After lysis (pH 10.0) and electrophoresis (20 min, 300 mA, 25 V, at 4°C, and pH > 13), the gels were stained with ethidium bromide (20 μg/mL, Sigma-Aldrich, Germany).
With nuclei from each organ, three slides were prepared per experimental point and 50 cells were evaluated from each slide. Slides were examined under a fluorescence microscope (Nikon EFD-3, Japan) using 25-fold magnification. DNA migration was determined with a computer aided comet assay image analysis system (Comet Assay IV, Perceptive Instruments, UK).
2.2. Gene Expression Analysis
Liver and colon samples were stored at −80°C until RNA and DNA isolation using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen, Germany) according to the manufacturer's protocol. Concentration, respectively, purity was controlled with a Picodrop100 (Picodrop, UK). Complementary DNA (cDNA) was synthesized by reverse transcription using RT2 First Strand Kit (Qiagen, Germany). cDNA was analyzed with real-time polymerase chain reaction (PCR) using qPCR Primer Assays (Qiagen, Germany) and RT2 SYBR Green Master Mix (Qiagen, Germany) according to manufacturer's protocol. PCR conditions were as follows: initial step of 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, ending with melting curve analysis (gradient melting of the products was performed at 0.5°C/10 s from 65°C to 95°C). Each sample was analyzed in duplicate with normalization to the housekeeping gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH).
2.3. Methylation Analysis
Genomic DNA was bisulfite converted with EpiTect® Fast Bisulfite Conversion kit (Qiagen, Germany) and amplified by PCR using the PyroMark PCR Kit (Qiagen, Germany) according to manufacturer's instructions with primers for Dnmt1 and MLH1 designed by PyroMark Assay Design SW 2.0 Software (Table 2).
Table 2.
Sequence to analyze primers for CpG methylation analysis.
| Gene | Primer | Sequence (5′-3′) | Size (bp) | GC% |
|---|---|---|---|---|
| Dnmt1 | FW | Biotin - GTA GGT TGT AGA AGA TAG AAT AGT TTT GA | 29 | 31 |
| RW | CCC ACT CTC TTA CCC TAT ATA ATA CAT | 27 | 37 | |
| Seq | CCC CTC CCA ATT AAT TTC | 18 | 44.4 | |
|
| ||||
| Sequence ID: gb|AH009208.2| | ||||
| Dnmt1: at reverse strand of chromosome 9: 20907205–20959888 (52684 bp). | ||||
|
| ||||
| Sequence to analyze | 7104 – CGCGCGCGCGAAAAAGCCGGGGTCTCGT - 7131 | 27 | 7 CpGs | |
|
| ||||
| MLH1 | FW | AGG GTA TTT TAG TTT TTA TTG GTT GGA GA | 29 | 31 |
| RW | TTA CAC CTC AAT TCC TAA AAT CTC TAT CCC – Biotin | 30 | 37 | |
| Seq | TTT AGT TTT TAG AAA TGA GTT AAT A | 25 | 16 | |
|
| ||||
| Sequence ID: ref|XR_379849.3| | ||||
| MLH1: at reverse strand of chromosome 9: 111228228–111271786 (43559 bp) | ||||
|
| ||||
| Sequence to analyze | 19 - GAAGAGCGGACCGTGAACTTTGACGCGCAAGCGCG | 64 | 8 CpGs | |
| TTGCCTTCTA-GCCTGGTGTCGGGCCGCTG - 82 | ||||
The PCR was carried out in a total reaction volume of 25.0 μL, containing 12.5 μL PyroMark 2x PCR master mix, 10 pmol (Dnmt1) or 7 pmol (MLH1) of each primer, 2.5 μL CoralLoad Concentrate 10x (Qiagen, Germany), and 10.0 ng (Dnmt1) or 15.0 ng (MLH1) bisulfite converted DNA. Thermocycling started with initial denaturation at 95°C for 15 min, followed by 45 cycles at 94°C for 30 s, 55.5°C for 45 s, 72°C for 45 s, and a final extension at 72°C for 10 min. PCR product quality was investigated with agarose gel electrophoresis. CpG methylation analysis was performed in a PyroMark Q24 MDx (Qiagen, Germany).
2.4. Gut Microbiota Analysis
Stool was collected before intervention and continuously after 1 month until the end of the study period and stored at −20°C until microbial DNA extraction by using the QIAamp® Fast DNA Mini Kit (Qiagen, Germany) following the manufacturer's protocol including two steps of 45 sec beadbeating at 4000 rpm with a 60 sec break in between to increase the DNA yield. DNA concentration and purification were measured using Pico100 (Picodrop Ltd., Cambridge, UK).
The abundance of gut microbial subgroups was determined by 16s rDNA using quantitative real-time PCR with SYBR green or TaqMan-Probe Master Mix with specific primer pairs (Tables 3 and 4) in a Rotor Gene 3000 (Corbett Life Science, Australia). The PCR reaction mixtures and serial DNA dilution of typical strains were prepared according to Remely et al. (2013) [44].
Table 3.
Primers and TaqMan®-Probes targeting 16rRNA coding regions of bacteria.
| Target organism | Primer/probe | Sequence (5′-3′) | Size (bp) | Conc. [pmol/µL] | Reference |
|---|---|---|---|---|---|
| All bacteria | Fwd primer | ACT CCT ACG GGA GGC AG | 468 | 10 | [15] |
| Rev primer | GAC TAC CAG GGT ATC TAA TCC | 10 | |||
| Probe | (6-FAM)-TGC CAG CAG CCG CGG TAA TAC-(BHQ-1) | 2 | |||
|
| |||||
| Clostridium cluster IV (Ruminococcaceae) | Fwd primer | GCA CAA GCA GTG GAG T | 239 | 4 | [16] |
| Rev primer | CTT CCT CCG TTT TGT CAA | 4 | |||
| Probe | (6-FAM)-AGG GTT GCG CTC GTT-(BHQ-1) | 2 | |||
|
| |||||
| Cluster XIVa (Lachnospiraceae) | Fwd primer | GCA GTG GGG AAT ATT GCA | 477 | 5 | [16] |
| Rev primer | CTT TGA GTT TCA TTC TTG CGA A | 5 | |||
| Probe | (6-FAM)-AAA TGA CGG TAC CTG ACT AA-(BHQ-1) | 1,5 | |||
|
| |||||
| Bacteroidetes | Fwd primer | GAG AGG AAG GTC CCC CAC | 106 | 3 | [17] |
| Rev primer | CGC TAC TTG GCT GGT TCA G | 3 | |||
| Probe | (6-FAM)-CCA TTG ACC AAT ATT CCT CAC TGC TGC CT-(BHQ-1) | 1 | |||
|
| |||||
| Bifidobacterium spp. | Fwd primer | GCG TGC TTA ACA CAT GCA AGT C | 125 | 3 | [18] |
| Rev primer | CAC CCG TTT CCA GGA GCT ATT | 3 | |||
| Probe | (6-FAM)-TCA CGC ATT ACT CAC CCG TTC GCC-(BHQ-1) | 1.5 | |||
Table 4.
Primers (SYBR® Green) targeting 16rRNA coding regions of bacteria, butyryl-coenzyme A (CoA) CoA transferase genes, and butyrate kinase gene.
| Target organism | Primer | Sequence (5′-3′) | Size (bp) | Conc. [pmol/µL] | Reference |
|---|---|---|---|---|---|
| Lactobacilli | Fwd primer | AGC AGT SGG GAA TCT TCC A | 352–700 | 4 | [19] |
| Rev primer | ATT YCA CCG CTA CAC ATG | 4 | |||
|
| |||||
| Enterobacteria | Fwd primer | AGC ACC GGC TAA CTC CGT | 492–509 | 3 | [20] |
| Rev primer | GAA GCC ACG CCT CAA GGG CAC AA | 834–856 | 3 | ||
|
| |||||
| Prevotella | Fwd primer | CACCAAGGCGACGATCA | 1458 | 2,5 | [21] |
| Rev primer | GGATAACGCCYGGACCT | 2,5 | |||
|
| |||||
| Akkermansia | Fwd primer | CAGCACGTGAAGGTGGGGAC | 1505 | 2,5 | [22] |
| Rev primer | CCTTGCGGTTGGCTTCAGAT | 2,5 | |||
|
| |||||
| BCoAT gene | Fwd primer | GCIGAICATTTCACITGGAAYWSITGGCAYATG | ~540 | 27 | [23] |
| Rev primer | CCTGCCTTTGCA ATRTCIACRAANGC | 27 | |||
|
| |||||
| Butyrate kinase | Fwd primer | TGCTGTWGTTGGWAGAGGYGGA | 273 | 18 | [24] |
| Rev Primer | GCAACIGCYTTTTGATTTAATGCATGG | 18 | |||
The diversity of gut microbiota was analyzed using denaturing gradient gel electrophoresis (DGGE) [44].
2.5. Statistical Analyses
For statistical analyses of SCGE assay results GraphPad Prism 5.02 (GraphPad Software, USA) was used. The means and standard deviation (SD) of % DNA in the comet tails of the nuclei from the different treatment groups were calculated. Comparisons of groups were done with Student's t-test based on the means of three slides/animal.
All statistical analyses of gene expression and methylation analysis were performed using IBM SPSS Advanced Statistics 20.0 (SPSS, USA). All data are shown means ± SD. ΔCT values were calculated by normalization to GAPDH (ΔCT = CT-Target − CT-GAPDH). The ΔΔCT value shows the difference between the two groups (ΔΔCT = ΔCT + EGCG − ΔCT-Control). Relative changes in gene expression between the intervention and control group are determined by the 2−ΔΔCT equation (fold change = 2−ΔΔCT). Kolmogorov-Smirnov-Test was used to test the normalization of the data. The Mann-Whitney U-Test was used to examine significant relationships. For all comparisons p values ≤ 0.05 were considered as statistically significant.
3. Results
3.1. Body Weight, Food Intake, and EGCG Uptake
Body weight and food intake were measured weekly, water/EGCG uptake daily. According to Table 1 food intake and total water consumption did not differ between the groups (Table 1). Mean EGCG uptake was about 0.64 ± 0.07 mg in the CD group and about 0.97 ± 0.09 mg in the HFD group of each mouse per day. Mice fed a HFD (T1: 30.54 ± 3.43 g; T4: 45.99 ± 4.46 g) and HFD + EGCG (T1: 30.74 ± 3.43 g; T4: 47.69 ± 3.45 g) increased significantly more weight in comparison to CD fed mice (T1: 23.99 ± 1.50 g; T4: 27.88 ± 1.49 g) and CD + EGCG (T1: 24.16 ± 1.52 g; T4: 28.43 ± 1.76 g) (p ≤ 0.01, Figure 2). The body weight increase over study period was significant in all groups (p ≤ 0.01).
Figure 2.
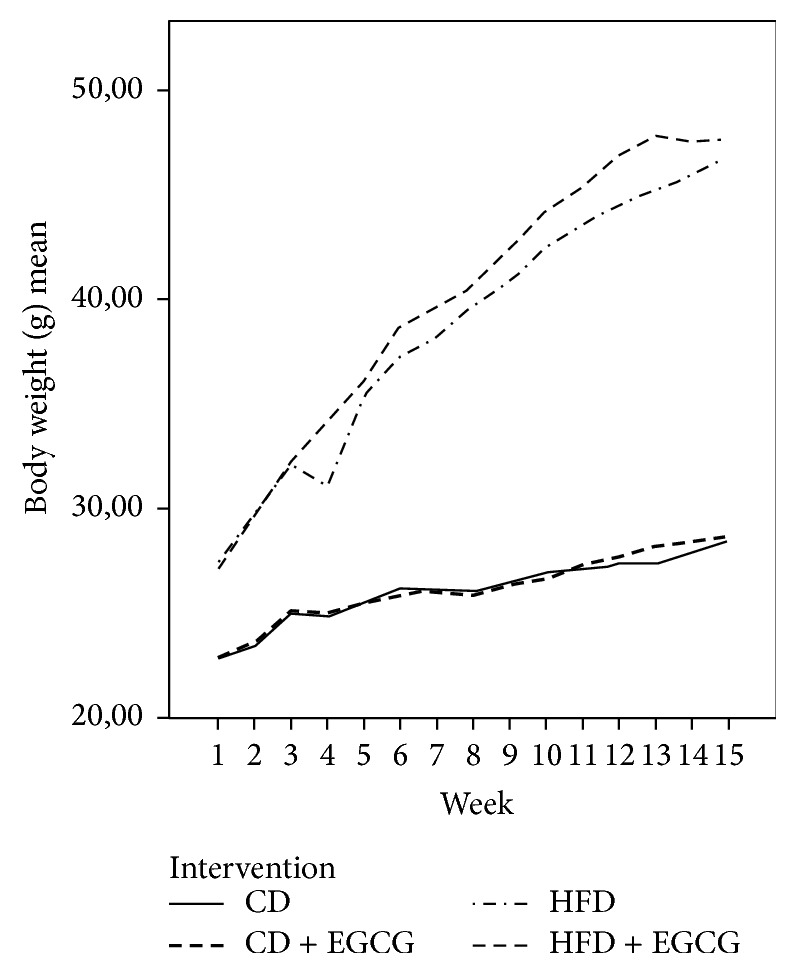
Body weight gain of C57BL/6J male mice over 4 months (CD = control diet, HFD = high fat diet, CD + EGCG = control diet plus EGCG, and HFD + EGCG = high fat diet plus EGCG).
3.2. SCGE Experiments with Nuclei from Colon and Liver Cells
HFD induced significant DNA damage in liver and colon compared with CD. In liver of animals fed with HFD and supplemented with EGCG in drinking water the extent of DNA migration was significantly decreased by 31.5% (p ≤ 0.05) after supplementation (Figure 3(a)). No effect was detected in colon of HFD group after EGCG supplementation Figure 3(b).
Figure 3.

Impact of EGCG supplementation on DNA damage in liver (a) and colon (b) of C57BL/6J male mice. Bars indicate means ± SD of results obtained with 15 animals per group. From each sample, three slides were made and 50 cells were evaluated per slide (CD = control diet, HFD = high fat diet, and CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG; stars indicate significance: ∗ p value ≤ 0.05).
Supplementation with EGCG in CD group in colon of animals caused slight DNA damage, while no effect was detected between CD and CD + EGCG in liver.
3.3. Relative Gene Expression and CpG Methylation of the Promoter Region of MLH1 in Liver and Colon Cells
In liver cells the relative gene expression of MLH1 decreased significantly in HFD fed mice compared to the CD (49%, p ≤ 0.001). EGCG supplementation showed significant reduction in CD + EGCG compared to CD (56%; p ≤ 0.001) and in HFD + EGCG compared to HFD (44%; p ≤ 0.01). Moreover the protective effect by EGCG was significantly higher in HFD + EGCG than in CD + EGCG (38%; p ≤ 0.05) (Figure 4(b)).
Figure 4.

Relative gene expression of MLH1 in colon (a) and liver (b) of C57BL/6J male mice. All gene expression data are relative to CD and were normalized to the house keeping gene GAPDH. Error bars represent 95% confidence intervals (CD = control diet, HFD = high fat diet, and CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG; stars indicate significance: ∗ p value ≤ 0.05, ∗∗ p value ≤ 0.01, and ∗∗∗ p value ≤ 0.001).
The relative gene expression of MLH1 in colon did not result in any significant difference between the interventions (Figure 4(a)).
In MLH1 promoter region the relative methylation of six CpGs was investigated in liver and in colon (Figures 5 and 6, Table 5). In liver cells CD + EGCG the mean methylation was higher compared to CD (CD: 2.83%; CD + EGCG: 3.23%) but in HFD + EGCG a decreased mean methylation status compared to HFD was shown (HFD: 3.36%; HFD + EGCG: 3.18%). In particular, at CpG1 HFD (57%) either CD + EGCG (44%) showed a significant decrease in methylation status compared to CD (p ≤ 0.01). Furthermore significant hypomethylation in the HFD + EGCG group was seen compared to CD + EGCG (24%; p ≤ 0.01; Figure 5(b)). At CpG4 (73%; p ≤ 0.01) and at CpG6 (60%; p ≤ 0.05) the methylation status significantly increased with HFD compared to CD. Hypermethylation was seen in both supplementation groups at CpG4 CD + EGCG (172%) and HFD + EGCG at CpG2 with an increase of 80% normalized to, respectively, CD or HFD (p ≤ 0.01). By comparison of CD + EGCG with HFD + EGCG the CD group showed significant hypomethylation at CpG3 (22%; p ≤ 0.01) and CpG5 (20%; p ≤ 0.01; Figure 5(a)).
Figure 5.

Relative CpG methylation status in promotor region of MLH1 in liver of C57BL/6J male mice. Mean methylation data are shown for MLH1 in each intervention group. All methylation data are relative to CD. Error bars represent 95% confidence intervals. In Figure 5(a) significance is shown for CpG 4, 5, and 6. Figure 5(b) shows significant differences in the methylation status of CpG1 (CD = control diet; HFD = high fat diet; CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG; stars indicate significance: ∗ p value ≤ 0.05 and ∗∗ p value ≤ 0.01).
Figure 6.

Relative CpG methylation status in promotor region of MLH1 in colon. Mean methylation data are shown for MLH1 as an overview (Figure 6(a)) (significant for CpG 1, 2, and 4) and CpG5 (Figure 6(b)). All methylation data are relative to CD. Error bars represent 95% confidence intervals (CD = control diet; HFD = high fat diet; CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG; stars indicate significance: ∗ p value ≤ 0.05 and ∗∗ p value ≤ 0.01).
Table 5.
DNA methylation results, presented as relative methylation (mean ± SD) compared to CD or in the HFD groups, respectively, for every CpG (stars indicate significance: ∗ p value ≤ 0.05, ∗∗ p value ≤ 0.01).
| Mean ± SD in % | CD + EGCG compared to CD | HFD compared to CD | HFD + EGCG compared to HFD |
|---|---|---|---|
| Dnmt1 liver | |||
| CpG1 | 1.71 ± 0.32∗ | 0.93 ± 0.25 | 1.62 ± 0.33∗ |
| CpG2 | 1.14 ± 0.21 | 0.81 ± 0.12 | 1.10 ± 0.39 |
| CpG3 | 1.37 ± 0.14∗∗ | 1.48 ± 0.73 | 1.18 ± 0.19 |
| CpG4 | 1.19 ± 0.40 | 0.88 ± 0.48 | 1.39 ± 0.21 |
| Dnmt1 colon | |||
| CpG1 | 1.07 ± 0.16 | 1.17 ± 0.24 | 0.89 ± 0.06 |
| CpG2 | 1.15 ± 0.30 | 1.12 ± 0.14 | 1.19 ± 0.14∗ |
| CpG3 | 0.98 ± 0.27 | 1.01 ± 0.19 | 1.23 ± 0.42 |
| CpG4 | 1.28 ± 0.09∗ | 1.17 ± 0.12 | 0.98 ± 0.13 |
| MLH1 colon | |||
| CpG1 | 0.38 ± 0.20∗ | 1.08 ± 0.46 | 0.98 ± 0.48 |
| CpG2 | 0.66 ± 0.39 | 0.40 ± 0.03∗ | 0.65 ± 0.13∗ |
| CpG3 | 1.04 ± 0.27 | 1.36 ± 0.44 | 0.87 ± 0.31 |
| CpG4 | 0.62 ± 0.11∗ | 2.13 ± 0.12∗ | 0.76 ± 0.42 |
| CpG5 | 1.75 ± 0.10∗ | 1.24 ± 0.09∗ | 0.55 ± 0.11∗ |
| CpG6 | 0.37 ± 0.14∗ | 1.53 ± 0.37 | 0.68 ± 0.70∗ |
| MLH1 liver | |||
| CpG1 | 0.56 ± 0.02∗∗ | 0.43 ± 0.03 | 0.98 ± 0.21 |
| CpG2 | 1.65 ± 0.61 | 1.27 ± 0.37∗∗ | 1.80 ± 0.42∗∗ |
| CpG3 | 1.11 ± 0.09 | 0.75 ± 0.13 | 1.16 ± 0.10 |
| CpG4 | 2.73 ± 1.06∗∗ | 1.73 ± 0.08∗∗ | 1.13 ± 0.15 |
| CpG5 | 1.20 ± 0.08∗∗ | 1.12 ± 0.26∗∗ | 0.86 ± 0.05∗∗ |
| CpG6 | 1.20 ± 0.10∗ | 1.60 ± 0.47 | 0.68 ± 0.16∗∗ |
The methylation of MLH1 in colon cells increased with EGCG in the CD group (CD: 2.79%; CD + EGCG: 2.97%) but decreased in the HFD groups (HFD: 3.41%; HFD + EGCG: 3.01%; Figure 6). In colon cells the methylation status of MLH1 showed a significant reduction at CpG2 in HFD compared to CD (60%; p ≤ 0.01). Furthermore, EGCG supplementation significantly reduced the methylation status of CpG2 in HFD + EGCG group compared to HFD (35%; p ≤ 0.01). A significant decrease at CpG1 was seen in CD with EGCG supplementation compared to CD (62%, p ≤ 0.01) whereas HFD + EGCG was significantly higher methylated compared to CD + EGCG (82% p ≤ 0.05). At CpG4 HFD resulted in a hypermethylation of 113% compared to CD. Furthermore, supplementation with EGCG in the CD decreased the methylation status significantly by 38% when normalized to CD (p ≤ 0.01). CpG5 of MLH1 was significantly higher methylated in HFD (24%; p ≤ 0.01) and in CD + EGCG (75%; p ≤ 0.01) compared to CD whereas in the HFD + EGCG significant hypomethylation was shown compared to CD (45%; p ≤ 0.01). A significant reduction was observed between CD + EGCG and HFD + EGCG animals (61%; p ≤ 0.01). CD + EGCG resulted in a significant hypomethylation at CpG6 of MLH1 in colon cells compared to CD (63%; p ≤ 0.05) and, respectively, in HFD with (34%; p ≤ 0.01).
3.4. Relative Gene Expression of IL-6 in Colon
A significant lower expression of IL-6 has been measured between CD and HFD animals (p ≤ 0.01) and also in CD and HFD + EGCG mice (p ≤ 0.01) in the colon (Figure 7). In liver the gene expression of IL-6 was under detection limit in the study group.
Figure 7.
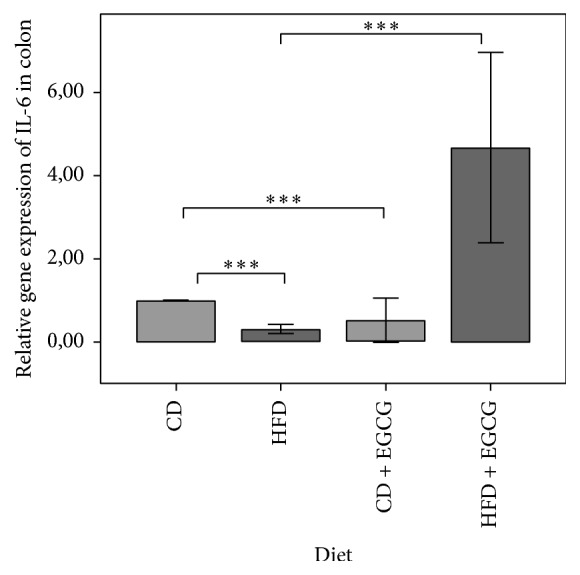
Relative gene expression of IL-6 in colon. All gene expression data are relative to CD and normalized to the house keeping gene GAPDH. Error bars represent 95% confidence intervals (CD = control diet; HFD = high fat diet; CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG; stars indicate significance: ∗∗∗ p value ≤ 0.001).
3.5. Relative Gene Expression and CpG Methylation of the Promoter Region of Dnmt1 in Liver and Colon Cells
Relative gene expression of Dnmt1 in liver cells was lower in HFD compared to CD (61%; p ≤ 0.01). Supplementation with EGCG resulted in a significantly reduced gene expression compared to, respectively, CD (75%; p ≤ 0.01) and HFD (51%; p ≤ 0.01) (Figure 8(b)).
Figure 8.

Relative gene expression of Dnmt1 in colon (a) and liver (b) of C57BL/6J male mice. All gene expression data are relative to CD and normalized to the house keeping gene GAPDH. Error bars represent a 95% confidence intervals (CD = control diet; HFD = high fat diet; CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG; stars indicate significance: ∗∗ p value ≤ 0.01).
In colon the relative gene expression of Dnmt1 decreased significantly in HFD compared to CD (69%, p ≤ 0.01) although the decrease of HFD compared to CD is compensated by EGCG in HFD with a more than three times higher gene expression of Dnmt1 compared to HFD (p ≤ 0.01, Figure 8(a)).
In the promoter region of Dnmt1 in liver and colon four CpGs were analyzed (Table 5). Mean methylation of Dnmt1 in liver cells was higher in CD + EGCG (3.21%) and HFD + EGCG (3.09%) compared to each diet CD (2.28%) and HFD (2.37%). Significant differences in methylation status of Dnmt1 were determined in CpG1 and CpG3. In both diets EGCG significantly increased the methylation status of CpG1 (CD + EGCG: CD 71%; p ≤ 0.05; HFD + EGCG: HFD 62%; p ≤ 0.05). The same effect of EGCG was seen at CpG3 in the CD group with a significant increase (37%; p ≤ 0.01). Furthermore the supplementation with EGCG resulted in a significantly higher methylation of CpG3 in HFD + EGCG than in CD + EGCG (28%, p ≤ 0.05). CpG2 and CpG4 did not show any significant changes in methylation status (Figure 9(b)).
Figure 9.

Relative CpG methylation status in promoter region of Dnmt1 in colon (a) and liver (b) of C57BL/6J male mice. Mean methylation data are shown relative to control diet. Error bars represent a 95% confidence interval (CD = control diet; HFD = high fat diet; CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG. Stars indicate significance: ∗ p value ≤ 0.05 and ∗∗ p value ≤ 0.01).
In colon cells CpG2 and CpG4 showed significant changes in the methylation status of Dnmt1. A significant higher methylation status in CpG2 was observed due to EGCG in the HFD group (19%; p ≤ 0.05). In CD + EGCG supplementation resulted in a significant hypermethylation at CpG4 (28%; p ≤ 0.05). No significant changes were observed at methylation status of Dnmt1 at CpG1 and CpG3 (Figure 9(a)).
3.6. Gut Microbiota Composition and Diversity
Differences in total bacterial abundance were shown between CD and HFD (p < 0.0001) and between CD + EGCG and HFD + EGCG but also between HFD and HFD + EGCG (p = 0.039; Figure 10(a)). HFD feeding caused a lower bacterial abundance in both HFD groups resulting in a lower microbial diversity compared to CD (HFD bands = 21.4 ± 5.08, HFD + EGCG bands = 19.6 ± 3.84, CD bands = 20.4 ± 4.62, and CD + EGCG bands = 17.02 ± 5.07).
Figure 10.

16S rDNA qPCR quantification of total bacterial abundance (a), Firmicutes/Bacteroidetes ratio (b), and F. prausnitzii (c). Error bars represent 95% confidence intervals (CD = control diet; HFD = high fat diet; CD + EGCG = control diet plus EGCG; HFD + EGCG = high fat diet plus EGCG. Stars indicate significance: ∗ p value ≤ 0.05, ∗∗ p value ≤ 0.01, and ∗∗∗ p value ≤ 0.001).
The Firmicutes/Bacteroidetes ratio is significantly higher in both HFD groups compared to CD groups (p < 0.0001). EGCG treatment induced a significantly lower ratio in HFD + EGCG compared to HFD (p < 0.0001) but a significantly higher ratio in CD + EGCG compared to CD (p < 0.0001; Figure 10(b)). Lactobacilli decreased with EGCG intervention. Both clostridial clusters (Clostridium cluster IV, Clostridium cluster XIVa) were significantly lower in HFD groups compared to CD groups (p < 0.0001). Clostridium cluster IV significantly increased in HFD + EGCG compared to HFD (p = 0.005). Clostridium cluster XIVa increased in CD + EGCG compared to CD (p = 0.189) and was significantly more abundant in comparison to HFD + EGCG (p < 0.0001). In turn, F. prausnitzii was less abundant in HFD groups compared to CD groups (p < 0.0001) and was more abundant in the CD groups compared to all other groups (CD + EGCG : HFD + EGCG : p < 0.0001; CD : CD + EGCG: p = 0.001; Figure 10(c)).
In HFD mice, butyryl CoA: acetate CoA-transferase gene was significantly lower in comparison to CD mice (p < 0.0001). Intervention with EGCG in the CD group resulted in a significant decrease (p < 0.0001) whereas with HFD no significant effect was shown. Results of the butyrate kinase gene showed similar results whereas HFD + EGCG group showed a significant increase (CD : HFD p < 0.0001; CD : CD + EGCG p = 0,001; HFD : HFD + EGCG p = 0.005).
Bacteroidetes were significantly lower abundant in HFD compared to CD (p < 0.0001) and significantly increased with EGCG intervention in HFD mice (p = 0,001). An increase was shown in CD mice due to EGCG treatment.
Akkermansia showed a lower abundance in HFD fed mice compared to CD (p = 0.092). EGCG treatment resulted in a lower abundance in CD + EGCG (p = 0,001) but no significant change in abundance of HFD + EGCG was observable (p = 0.574).
4. Discussion
We showed that HFD induces significant DNA damage in liver and colon compared with CD. These results were also reflected by a lower gene expression of Dnmt1 in liver and colon of HFD fed mice. In turn, DNA methylation status was higher in this group. MLH1 methylation status was also higher compared to CD, but gene expression lower due to HFD feeding in both organs. In particular CpG1 showed a decreased methylation status in contrast to CpG4 with an increased methylation in liver. In colon CpG2, CpG4, CpG5, and CpG6 of MLH1 promoter region were affected due to different feeding. IL-6 gene expression was significantly higher in HFD compared to CD.
The gut microbial composition differs between CD and HFD: lower total bacterial abundance due to HFD, lower microbial diversity, higher Firmicutes/Bacteroidetes ratio, lower abundance of F. prausnitzii, and Akkermansia, and reduced incidence of butyryl CoA: acetate CoA-transferase gene and butyrate kinase gene.
On the basis of microbial analysis we are able to support the results of our previous publications on human fecal analysis [44]. Mice fed a HFD differ in microbial subpopulations especially the Firmicutes/Bacteroidetes ratio which is already handled as a marker in obesity epidemic. Although other researches also show converse or not diverging results [45–49]. However, additionally we can support results of gut microbial metabolites/cell wall components influencing the host via epigenetic mechanisms too. An increased Firmicutes/Bacteroidetes ratio occurs or even causes low-grade inflammation, increased IL-6 gene expression, but also increased DNA damage and by increased MLH1 methylation status and reduced expression of the gene. However, the aspect if gut microbial dysbiosis first induces immunological disequilibrium or genomic instability remains. Pateras et al. (2015) extensively investigated the “cross-talk” between genomic instabilities and alteration of immune functions in regard to health implications in humans [50]. Ray and Kidane (2016) indicated a microbial dysbiosis as conducive for the release of bacterial metabolites triggering chronic inflammation followed by DNA damage [51]. Karakasilioti et al. (2013) support another theory that persistent DNA damage triggers chronic inflammation in adipose tissue [52]. Inhibition of cyclooxygenase 2, implicated in the generation of prooxidant eicosanoids, resulted in a decrease of 8-isoprostane, plasma thiobarbituric acid reactive substance, and an increased GSH/GSSG (glutathione/glutathione-disulfide) ratio in rats [53]. Thus, microbial dysbiosis may induce four different mechanisms of DNA damage: (1) mitogen released by a dysbiotic microbiota possesses the capacity to enter the cell and enhance reactive oxygen and nitrogen species (RONs) through enzyme expression, (2) the gut microbial dysbiosis provokes chronic inflammation through pattern recognition receptors to initiate DNA damage and cellular transformation, (3) the dysbiotic microbiota directly generates RONs to induce DNA damage and base excision repair (BER) intermediates, and (4) the release of bacterial genotoxins and metabolites causes chronic inflammation, which in turn promotes DNA damage [51].
Interventions with EGCG as an epigenetic active antioxidant may provide valuable impact in the therapy of metabolic syndrome. Bose et al. (2008) showed an effect of EGCG on HFD fed mice: the percentage of body fat and the visceral fat weight were reduced significantly (p ≤ 0.05) due to the supplementation of EGCG (3.2 g/kg diet) for 16 weeks when compared to control mice [11]. However, the bioavailability is low; intragastric administration of EGCG (75 mg/kg) resulted in 20.6 ng/g in the small intestine and 3.6 ng/g in colon. Oral administration of EGCG (equivalent with two or three cups of green tea) induced plasma levels of 0.2–0.3 μM. It is regulated by the active efflux through the multidrug resistance-associate protein 2 on the apical surface of the intestine and the liver. The uptake predominantly takes place from enterocytes into the intestinal lumen and from the liver to the bile with excretion in the feces and little to none in the urine. EGCG and other tea catechins undergo extensive biotransformation: methylated by catechol-O-methyltransferase (COMT), glucuronidated by UDP-glucuronosyltransferases, and sulfated by sulfotransferases [54]. However, depending on polymerization degree, EGCG also influences gut microbiota composition: higher polymerization results in a higher bioavailability in the gut as the absorption in the small intestine is negligible in contrast to low degree of polymerization [55]. Methylated catechins, ring fission products (like valerolactone), and phenolic products further degraded to phenylacetic and phenylpropionic acids are indicative of gut microbial transformations. However, the biological activities of these metabolites are lower than the activities of their parent compounds [54]. On the other hand EGCG has valuable direct impact as antioxidant, but also as epigenetic active substance influencing histone modification and/or DNA methylation patterns [27].
4.1. EGCG Protected DNA Damage Caused by HFD in the Liver
EGCG supplementation caused a decrease of DNA migration in liver while no effects were found in the colon in the HFD group. In the CD + EGCG even increased DNA damage was found in the colon. The divergent effects in the two organs may be due to the effect that catechin is present in the colon in higher concentration as in the liver. It was shown earlier that high doses of EGCG cause formation of ROS, as a consequence DNA damage in human cells [56].
As mentioned above, comets reflect single and double strand breaks as well as apurinic sites; our findings are indicative for a protective effect of the green tea catechin toward these types of lesions. Our findings are in partial agreement with results published by Kager et al. (2010) who gave EGCG orally to normal weight mice (as in the present experiment). The authors showed no evidence for a protective effect in standard SCGE experiments in colons and livers. However, a clear protective effect was seen in hepatocytes but not in colonocytes in this experiment in regard to formation of oxidized DNA bases [41]. The molecular mechanisms which account for the DNA-protective properties of EGCG involve scavenging of radicals that are typical for obesity [57] which has been found in vivo in rodents and also in vitro [58, 59] alternatively; indirect effects caused by activation of antioxidant enzyme via the transcription factors nuclear factor erythroid 2-related factor 2 (Nrf2) may play a role [60]
Oršolić et al. (2013) published results which were obtained with diabetic mice; they found even an increase of DNA migration in the liver after injection of EGCG for 7 days [61]. Notably, the authors used a relatively high dose of the catechin in this experiment (i.e., 50 mg/kg body weight) and it is known from in vitro studies that high concentrations of EGCG and other phenolics cause DNA damage as a consequence of radical formation [62, 63].
The patterns of gene expression of Dnmt1 and MLH1 which were observed in the liver are in partial agreement with results from the SCGE experiments; a decrease was observed in hepatocytes in obese animals compared to controls. However, EGCG did not compensate this effect but caused a further decline of the expression of both genes.
On the contrary a clear increase of Dnmt1 was seen with the catechin in the colons of HFD animals while obesity itself had no impact on the transcriptional activity of both genes in colonocytes. It is well documented that the MLH1 and Dnmt1 play a key role in DNA repair processes, in particular mismatch repair [64, 65]. However, distinct differences which we found between the induction of DNA migration in SCGE experiments and decreased gene expression levels distract from the assumption of a direct relation between comet formation and repair processes which are controlled by MLH1 and Dnmt1.
4.2. EGCG Decreased Inflammatory IL-6 and MLH1 Gene Expression Reflected by Higher MLH1 Promoter Methylation
In colon a significantly lower IL-6 gene expression was induced by EGCG but did not affect significantly MLH1 gene expression. In liver EGCG reduced MLH1 gene expression in both diet groups. The mean methylation was higher in CD + EGCG compared to CD whereas in HFD + EGCG a decrease was shown in comparison to HFD. However, methylation status varies CpG specific and additionally diet specific.
EGCG also reduced the expression levels of TNF-α, IL-6, IL-18, and IL-1β mRNAs, the serum levels of TNF-α, and the activation of Stat3 and JNK proteins in diethylnitrosamine- (DEN-) induced liver tumor genesis treated C57BL/KsJ-db/db (db/db) obese mice [13], but also IL-6 synthesis in rat adjuvant-induced arthritis by administration of 100 mg/kg EGCG, intraperitoneally daily [66]. Ahmed et al. (2008) showed an increase in the synthesis of soluble gp130 protein, an endogenous inhibitor of IL-6 signaling and transsignaling [66]. Additionally EGCG induced a concentration and time dependent reversal hypermethylation of tumor suppressor genes such as p16, RAR, MGMT, and MLH1 genes in human esophageal cancer cells [12].
4.3. EGCG Increased Dnmt1 DNA Methylation Resulting in Tissue Specific Variances in Gene Expression
EGCG supplementation resulted in a significantly reduced gene expression of Dnmt1 compared to, respectively, CD and HFD in the liver. In the colon EGCG compensates the decrease in gene expression due to HFD and results equalized to CD and with three times higher gene expression of Dnmt1 compared to HFD. The methylation status in the promoter region of Dnmt1 was higher in supplemented groups compared to both control groups (CD, HFD). A significant increase was shown in CpG1 and CpG3 in the liver. In colon CpG2 and CpG4 were affected, respectively, in HFD + EGCG or in CD + EGCG.
EGCG was already shown to be the most efficacious inhibitor of enzymatic DNA methylation in vitro in comparison to other tea polyphenols (catechin, epicatechin) and bioflavonoids (quercetin, fisetin, and myricetin). Inhibitory effects for SssI nmt- and Dnmt1-mediated DNA methylation were shown at a half maximal inhibitory concentration (IC50) of 0.21 and 0.47 μM, respectively. Inhibitory mechanisms are mentioned on one hand via direct pathways and on the other hand indirect via Dnmt-mediated DNA methylation through increased formation of SAH, a potent inhibitor of S-adenosylmethionine- (SAM-) mediated reactions [67] and via altering the availability of methyl groups which are used to methylate catechol groups on polyphenols by catechol-O-methyltransferase [27]. Although Lee et al. (2005) mentioned rather an important influence of the presence of a physiologically relevant concentration of Mg2+ (such as 2 mM) on inhibitory potency of EGCG compared to a dependence on its own methylation [67]. EGCG is also suggested to induce Foxp3 promoter demethylation inducing differentiation and expansion of Treg via Dnmt inhibitory activity and to reduce T cell proliferation and cytokine production [68]. Inhibition of Dnmts together with an inhibition of histone deacetylase is suggested to prevent the hypermethylation and the silencing of key genes [67]. Either gut microbial derived metabolites of EGCG, gallic acid (GA), and epigallocatechin (EGC) influence epigenetic gene expression via HAT inhibitors to a lesser extent [27].
4.4. EGCG Changed Obese Gut Microbial Profile to Lean Phenotype
HFD feeding caused a significant lower bacterial abundance in both HFD groups (HFD, HFD + EGCG) resulting in a lower microbial diversity compared to CD. The Firmicutes/Bacteroidetes ratio is significantly lower in HFD + EGCG but higher in CD + EGCG compared to corresponding control group. Main changes due to EGCG intervention are decreased Lactobacilli abundance and also a lower abundance of F. prausnitzii in HFD groups with highest abundance in CD. Butyryl CoA: acetate CoA-transferase gene significantly increased in CD + EGCG whereas in HFD + EGCG the butyrate kinase significantly increased. Akkermansia are reduced to be abundant with EGCG supplementation.
It is already known that HFD, Western lifestyle, impacts gut microbiota composition. Diet quality and quantity are important influencing factors on bacterial community composition and metabolic/immunological activity of the host gut microbiota. Microbiota-mediated genomic instability but also immunological disequilibrium may be reduced due to phytochemicals. However, they are generally poorly absorbed in the small intestine; thus the impact of the close contact with the gut microbiota affects health benefits attributed to natural compounds. Results from green tea polyphenols, EGCG, give evidence to have a positive influence on gut microbiota composition. Unno et al. (2014) show not only changes in body and stool composition due to EGCG treatment but also changes in gut microbiota composition. However, these changes were dependent on dosage: 0.3% EGCG supplementation induced Bifidobacterium, Lactobacillales, and Bacteroides but reduced the abundance of Clostridium clusters. A concentration of 0.6% EGCG supplementation increased the abundance of Lactobacillales and Bacteroides but nearly depleted the abundance of Clostridium clusters and Bifidobacterium [26]. However, Unno et al. (2014) used Wistar rats and fed a commercial chow; thus comparison of both projects in gut microbiota composition, especially Lactobacilli, may be impaired on induced metabolic syndrome due to HFD feeding. However, we showed also an increase in Bacteroidetes and a decrease in Clostridium clusters impairing butyrate metabolism. Unno et al. (2014) showed lower levels of acetic and butyric acid but little influence on propionic acid due to 0.6% EGCG supplementation [26]. We even showed differences in butyrate formation pathways. The butyrate kinase pathway is more related to a Western diet, reflected in HFD fed mice (main fat resource: lard). Meanwhile the butyryl CoA: acetate CoA-transferase gene is associated with vegetarian feeding and to a lesser content available in omnivores [69].
In addition to EGCG as an epigenetically active antioxidant we also supplemented C57BL/6J mice with vitamin E (4.5 mg/kg b.w.) as a positive control which showed similar results compared to EGCG but with profound effects which will be published elsewhere. DNA damage showed a significant reduction in both organs in the HFD group but not in the CD group (tail intensity in CD-animals + vitamin E: 6.03 ± 0.95 in liver and 6.00 ± 1.13 in colon). The relative gene expression of Dnmt1 in the colon increased with vitamin E supplementation in CD but significantly decreased in HFD. In all intervention groups a decreased gene expression was noted in the liver. The methylation status of Dnmt1 in colon was lower. Furthermore, a positive correlation of Dnmt1 mean methylation and DNA damage has been observed in liver whereas in CD a correlation has been found in the colon. Vitamin E supplementation affects specific CpG sites of MLH1 inducing a lower gene expression of MLH1 with HFD in liver and colon.
5. Conclusions
According to our results, EGCG might be suggested for the potential use for the prevention or in the therapy of obesity related and oxidative stress-induced health risks. One effect may derive from changes in GI microbiota and their anti-inflammatory effects by metabolites. Another effect may derive from antioxidative activities as well as epigenetic modifications observed on CpG methylation but also likely to include other elements of the epigenetic machinery. Interactions between antioxidative and epigenetic effects, for example, via ROS mediated breaks of Dnmt pathways, need to be explored.
Acknowledgments
The work was funded by the Austrian Science Fund (FWF; AP2658721).
Abbreviations
- CD:
Control diet
- CD + EGCG:
Control diet plus EGCG
- cDNA:
Complementary DNA
- COMT:
Catechol-O-methyltransferase
- Dnmt1:
DNA methyltransferase 1
- DGGE:
Denaturing gradient gel electrophoresis
- EGC:
Epigallocatechin
- EGCG:
(−)-Epigallocatechin-3-gallate
- FFAs:
Free fatty acids
- GAPDH:
Glyceraldehyde-3-phosphate-dehydrogenase
- HAT:
Histone acetyl transferase
- HFD:
High fat diet
- HFD + EGCG:
High fat diet plus EGCG
- IC:
Inhibitory concentration
- IGF:
Insulin-like growth factors
- IL-6:
Interleukin-6
- MGMT:
O6-Methylguanine-deoxyribonucleic acid methyltransferase
- MLH1:
MutL homologue 1
- MMR:
DNA mismatch repair
- Nrf2:
Nuclear factor erythroid 2–related factor 2
- ROS:
Reactive oxygen species
- SAH:
S-Adenosyl-l-homocysteine
- SAM:
S-Adenosylmethionine
- SCFA:
Short-chain fatty acids
- SCGE:
Single-cell gel electrophoresis
- SD:
Standard deviation
- TNF-α:
Tumor necrosis factor α.
Ethical Approval
The animal experiment was approved by the Ethical Committee of the Medical University of Vienna (BMWFW-66.009/0329-WF/V/3b/2014).
Competing Interests
The authors declare no actual or potential competing interests that might be perceived as influencing the results or interpretation of a reported study.
References
- 1. World Health Statistics 2015, 2015.
- 2.Fernández-Sánchez A., Madrigal-Santillán E., Bautista M., et al. Inflammation, oxidative stress, and obesity. International Journal of Molecular Sciences. 2011;12(5):3117–3132. doi: 10.3390/ijms12053117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kershaw E. E., Flier J. S. Adipose tissue as an endocrine organ. Journal of Clinical Endocrinology and Metabolism. 2004;89(6):2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 4.Savini I., Catani M. V., Evangelista D., Gasperi V., Avigliano L. Obesity-associated oxidative stress: strategies finalized to improve redox state. International Journal of Molecular Sciences. 2013;14(5):10497–10538. doi: 10.3390/ijms140510497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Lay S., Simard G., Martinez M. C., Andriantsitohaina R. Oxidative stress and metabolic pathologies: from an adipocentric point of view. Oxidative Medicine and Cellular Longevity. 2014;2014:18. doi: 10.1155/2014/908539.908539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thanan R., Oikawa S., Hiraku Y., et al. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. International Journal of Molecular Sciences. 2014;16(1):193–217. doi: 10.3390/ijms16010193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziech D., Franco R., Pappa A., Panayiotidis M. I. Reactive Oxygen Species (ROS)—induced genetic and epigenetic alterations in human carcinogenesis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2011;711(1-2):167–173. doi: 10.1016/j.mrfmmm.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Franco R., Schoneveld O., Georgakilas A. G., Panayiotidis M. I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Letters. 2008;266(1):6–11. doi: 10.1016/j.canlet.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 9.Wang S., Moustaid-Moussa N., Chen L., et al. Novel insights of dietary polyphenols and obesity. The Journal of Nutritional Biochemistry. 2014;25(1):1–18. doi: 10.1016/j.jnutbio.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y., Cheung C., Reuhl K. R., et al. Effects of green tea polyphenol (-)-epigallocatechin-3-gallate on newly developed high-fat/Western-style diet-induced obesity and metabolic syndrome in mice. Journal of Agricultural and Food Chemistry. 2011;59(21):11862–11871. doi: 10.1021/jf2029016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bose M., Lambert J. D., Ju J., Reuhl K. R., Shapses S. A., Yang C. S. The major green tea polyphenol, (-)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice. The Journal of Nutrition. 2008;138(9):1677–1683. doi: 10.1093/jn/138.9.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh B. N., Shankar S., Srivastava R. K. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochemical Pharmacology. 2011;82(12):1807–1821. doi: 10.1016/j.bcp.2011.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu M., Sakai H., Shirakami Y., et al. Preventive effects of (−)-epigallocatechin gallate on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db mice. Cancer Prevention Research. 2011;4(3):396–403. doi: 10.1158/1940-6207.capr-10-0331. [DOI] [PubMed] [Google Scholar]
- 14.Busch C., Burkard M., Leischner C., Lauer U. M., Frank J., Venturelli S. Epigenetic activities of flavonoids in the prevention and treatment of cancer. Clinical Epigenetics. 2015;7(1, article 64) doi: 10.1186/s13148-015-0095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y., Lee C., Kim J., Hwang S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnology and Bioengineering. 2005;89(6):670–679. doi: 10.1002/bit.20347. [DOI] [PubMed] [Google Scholar]
- 16.Matsuki T., Watanabe K., Fujimoto J., Takada T., Tanaka R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Applied and Environmental Microbiology. 2004;70(12):7220–7228. doi: 10.1128/AEM.70.12.7220-7228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Layton A., McKay L., Williams D., Garrett V., Gentry R., Sayler G. Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Applied and Environmental Microbiology. 2006;72(6):4214–4224. doi: 10.1128/aem.01036-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Penders J., Vink C., Driessen C., London N., Thijs C., Stobberingh E. E. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiology Letters. 2005;243(1):141–147. doi: 10.1016/j.femsle.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 19.Walter J., Hertel C., Tannock G. W., Lis C. M., Munro K., Hammes W. P. Detection of Lactobacillus, Pediococcus, Leuconostoc, and Weissellaspecies in human feces by using group-specific pcr primers and denaturing gradient gel electrophoresis. Applied and Environmental Microbiology. 2001;67(6):2578–2585. doi: 10.1128/aem.67.6.2578-2585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ootsubo M., Shimizu T., Tanaka R., et al. Oligonucleotide probe for detecting Enterobacteriaceae by in situ hybridization. Journal of Applied Microbiology. 2002;93(1):60–68. doi: 10.1046/j.1365-2672.2002.01668.x. [DOI] [PubMed] [Google Scholar]
- 21.Larsen E. C., Kondo Y., Fahrenholtz C. D., Duncan I. D. Current Protocols in Stem Cell Biology. chapter 2, unit 2D.1. Hoboken, NJ, USA: John Wiley & Sons; 2008. Generation of cultured oligodendrocyte progenitor cells from rat neonatal brains; pp. 1–13. [DOI] [PubMed] [Google Scholar]
- 22.Collado M. C., Cernada M., Baüerl C., Vento M., Pérez-Martínez G. Microbial ecology and host-microbiota interactions during early life stages. Gut Microbes. 2012;3(4):352–365. doi: 10.4161/gmic.21215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis P., Flint H. J. Development of a semiquantitative degenerate real-time PCR-based assay for estimation of numbers of Butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Applied and Environmental Microbiology. 2007;73(6):2009–2012. doi: 10.1128/aem.02561-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vital M., Penton C. R., Wang Q., et al. A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome. 2013;1(1):p. 8. doi: 10.1186/2049-2618-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang M. Z., Wang Y., Ai N., et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Research. 2003;63(22):7563–7570. [PubMed] [Google Scholar]
- 26.Unno T., Sakuma M., Mitsuhashi S. Effect of dietary supplementation of (−)-epigallocatechin gallate on gut microbiota and biomarkers of colonic fermentation in rats. Journal of Nutritional Science and Vitaminology. 2014;60(3):213–219. doi: 10.3177/jnsv.60.213. [DOI] [PubMed] [Google Scholar]
- 27.Hullar M. A. J., Fu B. C. Diet, the gut microbiome, and epigenetics. The Cancer Journal. 2014;20(3):170–175. doi: 10.1097/ppo.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tremaroli V., Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 29.Armougom F., Henry M., Vialettes B., Raccah D., Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS ONE. 2009;4(9) doi: 10.1371/journal.pone.0007125.e7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turnbaugh P. J., Ley R. E., Mahowald M. A., Magrini V., Mardis E. R., Gordon J. I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 31.Turnbaugh P. J., Hamady M., Yatsunenko T., et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delzenne N. M., Cani P. D. Interaction between obesity and the gut microbiota: relevance in nutrition. Annual Review of Nutrition. 2011;31(1):15–31. doi: 10.1146/annurev-nutr-072610-145146. [DOI] [PubMed] [Google Scholar]
- 33.Tice R. R., Agurell E., Anderson D., et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environmental and Molecular Mutagenesis. 2000;35(3):206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 34.Collins A. R. The comet assay for DNA damage and repair: principles, applications, and limitations. Molecular Biotechnology. 2004;26(3):249–261. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki Y. F., Kawaguchi S., Kamaya A., et al. The comet assay with 8 mouse organs: results with 39 currently used food additives. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2002;519(1-2):103–119. doi: 10.1016/s1383-5718(02)00128-6. [DOI] [PubMed] [Google Scholar]
- 36.Collins A. R. The comet assay: a heavenly method! Mutagenesis. 2015;30(1):1–4. doi: 10.1093/mutage/geu079. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki Y. F., Kawaguchi S., Kamaya A., et al. The comet assay with 8 mouse organs: results with 39 currently used food additives. Mutation Research. 2002;519(1-2):103–119. doi: 10.1016/s1383-5718(02)00128-6. [DOI] [PubMed] [Google Scholar]
- 38.Ferk F., Chakraborty A., Jäger W., et al. Potent protection of gallic acid against DNA oxidation: results of human and animal experiments. Mutation Research. 2011;715(1-2):61–71. doi: 10.1016/j.mrfmmm.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Wallner M., Antl N., Rittmannsberger B., et al. Anti-genotoxic potential of bilirubin in vivo: damage to DNA in hyperbilirubinemic human and animal models. Cancer Prevention Research. 2013;6(10):1056–1063. doi: 10.1158/1940-6207.capr-13-0125. [DOI] [PubMed] [Google Scholar]
- 40.Ferk F., Huber W. W., Filipič M., et al. Xanthohumol, a prenylated flavonoid contained in beer, prevents the induction of preneoplastic lesions and DNA damage in liver and colon induced by the heterocyclic aromatic amine amino-3-methyl-imidazo[4,5-f]quinoline (IQ) Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2010;691(1-2):17–22. doi: 10.1016/j.mrfmmm.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Kager N., Ferk F., Kundi M., Wagner K., Mišík M., Knasmüller S. Prevention of oxidative DNA damage in inner organs and lymphocytes of rats by green tea extract. European Journal of Nutrition. 2010;49(4):227–234. doi: 10.1007/s00394-009-0068-0. [DOI] [PubMed] [Google Scholar]
- 42.Chakraborty A., Ferk F., Simić T., et al. DNA-protective effects of sumach (Rhus coriaria L.), a common spice: results of human and animal studies. Mutation Research. 2009;661(1-2):10–17. doi: 10.1016/j.mrfmmm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Burlinson B., Tice R. R., Speit G., et al. Fourth International Workgroup on Genotoxicity testing: results of the in vivo Comet assay workgroup. Mutation Research. 2007;627(1):31–35. doi: 10.1016/j.mrgentox.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 44.Remely M., Dworzak S., Hippe B., et al. Abundance and diversity of microbiota in Type 2 diabetes and obesity. Journal of Diabetes and Metabolism. 2013;4, article 253 [Google Scholar]
- 45.Duncan S. H., Lobley G. E., Holtrop G., Ince J., Johnstone A. M., Louis P. Human colonic microbiota associated with diet, obesity and weight loss. International Journal of Obesity. 2008;32(11):1720–1724. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 46.Schwiertz A., Taras D., Schäfer K., et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18(1):190–195. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 47.Cani P. D., Delzenne N. M. Interplay between obesity and associated metabolic disorders: new insights into the gut microbiota. Current Opinion in Pharmacology. 2009;9(6):737–743. doi: 10.1016/j.coph.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Arumugam M., Raes J., Pelletier E., et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abdolrasulnia M., Menachemi N., Shewchuk R. M., Ginter P. M., Duncan W. J., Brooks R. G. Market effects on electronic health record adoption by physicians. Health Care Management Review. 2008;33(3):243–252. doi: 10.1097/01.hmr.0000324904.19272.c2. [DOI] [PubMed] [Google Scholar]
- 50.Pateras I. S., Havaki S., Nikitopoulou X., et al. The DNA damage response and immune signaling alliance: is it good or bad? Nature decides when and where. Pharmacology and Therapeutics. 2015;154:36–56. doi: 10.1016/j.pharmthera.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 51.Ray D., Kidane D. Gut microbiota imbalance and base excision repair dynamics in colon cancer. Journal of Cancer. 2016;7(11):1421–1430. doi: 10.7150/jca.15480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karakasilioti I., Kamileri I., Chatzinikolaou G., et al. DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metabolism. 2013;18(3):403–415. doi: 10.1016/j.cmet.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yara S., Lavoie J.-C., Levy E. Oxidative stress and DNA methylation regulation in the metabolic syndrome. Epigenomics. 2015;7(2):283–300. doi: 10.2217/epi.14.84. [DOI] [PubMed] [Google Scholar]
- 54.Yang C. S., Zhang J., Zhang L., Huang J., Wang Y. Mechanisms of body weight reduction and metabolic syndrome alleviation by tea. Molecular Nutrition and Food Research. 2016;60(1):160–174. doi: 10.1002/mnfr.201500428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marín L., Miguélez E. M., Villar C. J., Lombó F. Bioavailability of dietary polyphenols and gut microbiota metabolism: antimicrobial properties. BioMed Research International. 2015;2015:18. doi: 10.1155/2015/905215.905215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elbling L., Herbacek I., Weiss R.-M., Jantschitsch C., Micksche M., Gerner C. Hydrogen peroxide mediates EGCG-induced antioxidant protection in human keratinocytes. Free Radical Biology and Medicine. 2010;49(9):1444–1452. doi: 10.1016/j.freeradbiomed.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 57.Khoo N. K. H., Cantu-Medellin N., Devlin J. E., et al. Obesity-induced tissue free radical generation: an in vivo immuno-spin trapping study. Free Radical Biology and Medicine. 2012;52(11-12):2312–2319. doi: 10.1016/j.freeradbiomed.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson M. K., Loo G. Effects of epigallocatechin gallate and quercetin on oxidative damage to cellular DNA. Mutation Research. 2000;459(3):211–218. doi: 10.1016/s0921-8777(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 59.Hu C., Kitts D. D. Evaluation of antioxidant activity of epigallocatechin gallate in biphasic model systems in vitro. Molecular and Cellular Biochemistry. 2001;218(1):147–155. doi: 10.1023/A:1007220928446. [DOI] [PubMed] [Google Scholar]
- 60.Romeo L., Intrieri M., D'Agata V., et al. The major green tea polyphenol, (-)-epigallocatechin-3-gallate, induces heme oxygenase in rat neurons and acts as an effective neuroprotective agent against oxidative stress. Journal of the American College of Nutrition. 2009;28(4):492S–499S. doi: 10.1080/07315724.2009.10718116. [DOI] [PubMed] [Google Scholar]
- 61.Oršolić N., Sirovina D., Gajski G., Garaj-Vrhovac V., Jazvinšćak Jembrek M., Kosalec I. Assessment of DNA damage and lipid peroxidation in diabetic mice: effects of propolis and epigallocatechin gallate (EGCG) Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2013;757(1):36–44. doi: 10.1016/j.mrgentox.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 62.Tobi S. E., Gilbert M., Paul N., McMillan T. J. The green tea polyphenol, epigallocatechin-3-gallate, protects against the oxidative cellular and genotoxic damage of UVA radiation. International Journal of Cancer. 2002;102(5):439–444. doi: 10.1002/ijc.10730. [DOI] [PubMed] [Google Scholar]
- 63.Lu L. Y., Ou N., Lu Q.-B. Antioxidant induces DNA damage, cell death and mutagenicity in human lung and skin normal cells. Scientific Reports. 2013;3, article 3169 doi: 10.1038/srep03169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loughery J. E. P., Dunne P. D., O'Neill K. M., Meehan R. R., McDaid J. R., Walsh C. P. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Human Molecular Genetics. 2011;20(16):3241–3255. doi: 10.1093/hmg/ddr236.ddr236 [DOI] [PubMed] [Google Scholar]
- 65.Jin B., Robertson K. D. DNA methyltransferases, DNA damage repair, and cancer. Advances in Experimental Medicine and Biology. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahmed S., Marotte H., Kwan K., et al. Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proceedings of the National Academy of Sciences. 2008;105(38):14692–14697. doi: 10.1073/pnas.0802675105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee W. J., Shim J.-Y., Zhu B. T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Molecular Pharmacology. 2005;68(4):1018–1030. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 68.Wong C. P., Nguyen L. P., Noh S. K., Bray T. M., Bruno R. S., Ho E. Induction of regulatory T cells by green tea polyphenol EGCG. Immunology Letters. 2011;139(1):7–13. doi: 10.1016/j.imlet.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hippe B., Zwielehner J., Liszt K., Lassl C., Unger F., Haslberger A. G. Quantification of butyryl CoA:acetate CoA-transferase genes reveals different butyrate production capacity in individuals according to diet and age. FEMS Microbiology Letters. 2011;316(2):130–135. doi: 10.1111/j.1574-6968.2010.02197.x. [DOI] [PubMed] [Google Scholar]