

Abstract
The epidermal growth factor receptor (EGFR)/ErbB family of receptor tyrosine kinases includes oncogenes important in the progression of breast and other cancers, and they are targets for many drug development strategies. Each member of the ErbB family possesses a unique, structurally uncharacterized C-terminal tail that plays an important role in autophosphorylation and signal propagation. To determine whether these C-terminal tails are intrinsically disordered regions, we conducted a battery of biophysical experiments on the EGFR and HER3 tails. Using hydrogen/deuterium exchange mass spectrometry, we measured the conformational dynamics of intracellular half constructs and compared the tails with the ordered kinase domains. The C-terminal tails demonstrate more rapid deuterium exchange behavior when compared with the kinase domains. Next, we expressed and purified EGFR and HER3 tail-only constructs. Results from circular dichroism spectroscopy, size exclusion chromatography with multiangle light scattering, dynamic light scattering, analytical ultracentrifugation, and small angle X-ray scattering each provide evidence that the EGFR and HER3 C-terminal tails are intrinsically disordered with extended, non-globular structure in solution. The intrinsic disorder and extended conformation of these tails may be important for their function by increasing the capture radius and reducing the thermodynamic barriers for binding of downstream signaling proteins.
Keywords: analytical ultracentrifugation; C-terminal domain (carboxyl tail domain, CTD); circular dichroism (CD); epidermal growth factor receptor (EGFR); hydrogen exchange mass spectrometry; intrinsically disordered protein; phosphotyrosine signaling; small angle X-ray scattering (SAXS); dynamic light scattering (DLS); size exclusion chromatography
Introduction
The epidermal growth factor receptor (EGFR)3/ErbB family of receptor tyrosine kinases (RTKs) contains four member proteins: EGFR/ErbB1, HER2/ErbB2/neu, HER3/ErbB3, and HER4/ErbB4. These RTKs carry out important signaling functions via the sequential process of ligand binding by the extracellular domain, homo- or heterodimerization, activation of their intracellular kinase domain, and recruitment of downstream signaling proteins. These RTKs are also important oncogenic drivers in many breast, lung, and other human cancers (1). Several structural biology studies on the ErbB family have been published, and this has helped advance drug development for HER2-positive breast cancer (2). Protein crystallography studies published in 2004 showed the structure of pertuzumab bound to the extracellular domain of HER2 and lapatinib bound to the kinase domain of EGFR (3, 4). Since that time, growing structural biology-based understanding of how EGFR, HER2, and HER3 function at the atomic level has dramatically reshaped our understanding of RTKs (1, 2). Despite these advances, there is a domain in each EGFR/ErbB family protein for which little structural biology information is available; this domain is the C-terminal tail (CTT) domain. The CTTs contain numerous autophosphorylation sites that are essential for recruiting downstream signaling proteins and initiating intracellular signaling (5, 6). The CTT can also contribute to autoinhibition of the kinase domain of RTK (7). The lack of available crystallographic information on the CTT region of EGFR/ErbB family RTKs led us to examine whether these proteins lack a stable secondary and/or tertiary structure.
Intrinsically disordered regions (IDRs) represent an emerging area of interest in medicine. IDRs are regions within proteins that exhibit high flexibility and may lack a secondary or tertiary structure but are still able to carry out important biological functions (8–15). Algorithm prediction methods indicate that around 25–30% of eukaryotic proteins can be categorized as having disordered regions (16). These disordered regions provide certain advantages in protein-protein interactions, including a larger hydrodynamic radius (17, 18), faster on- and off-rates of binding (19), high binding specificity (20), and the ability to adopt different conformations depending on the binding partner (9, 21). Correlation studies have revealed a high propensity for disordered regions to undergo post-translational modification, particularly phosphorylation (22). Because traditional methods, such as NMR and X-ray crystallography, were developed to study stable protein structures, IDRs have been more difficult to analyze because of difficulties in obtaining high concentrations or representative protein crystals (11, 23). Correlation studies suggest a strong association between IDRs and human cancer-associated proteins (24); therefore, identifying and analyzing IDRs in cancer-related proteins is vital in understanding how they function.
Protein kinases demonstrate a high degree of specificity in facilitating phosphorylation; however, many are able to perform such interactions with multiple substrate partners (25). An analysis of the human kinome shows that as many as 83% of kinase genes contain IDRs, which could facilitate these multiple interactions. RTKs are involved in more protein-protein interactions than any other kinase group, thus potentially pointing to the involvement of IDRs in their recognition mechanisms (26). Multisequence alignment (27) of EGFR/ErbB family RTKs shows that the kinase domain sequences are highly conserved between all four family members. However, the CTT regions are highly divergent between each EGFR/ErbB family member. One previous study using circular dichroism (CD) spectroscopy indicated that the EGFR CTT is rich in α-helical and β-sheet content (6), but a later study used coarse grained modeling to show many possible conformations of the EGFR CTT based on the assumption that it is naturally disordered (28). A high degree of flexibility in the CTT would provide distinct advantages to EGFR interaction with downstream Src homology 2 (SH2) and phosphotyrosine-binding domains during signaling functions (6).
In this work, we examine the biophysical properties of the CTT of EGFR and HER3. The motivation of this study is to gather structural information on the CTT region to determine whether the tails are highly dynamic, disordered regions. First, we measured the conformational dynamics of intracellular half (ICH) constructs of EGFR/ErbB family members using amide hydrogen/deuterium exchange mass spectrometry (HDX-MS). This information is used to compare the CTTs with the kinase domains. We also expressed and purified EGFR and HER3 CTT-only constructs and demonstrated that they are functional because they are recognized and phosphorylated by EGFR family kinases and once phosphorylated can be bound by the Grb2 SH2 domain. Using these CTT constructs, we performed multiple biophysical analyses, including CD spectroscopy, size exclusion chromatography with multiangle light scattering (SEC-MALS), dynamic light scattering (DLS), analytical ultracentrifugation (AUC), and small angle X-ray scattering (SAXS). The results of these methods support the hypothesis that the EGFR and HER3 CTTs are IDRs with extended, non-globular structure in solution.
Results
Disorder Predictions on EGFR, HER2, and HER3
We used computation algorithms to provide an initial survey of disordered regions in the EGFR/ErbB family of RTKs. Predictor of natural disordered regions (PONDR) is a collection of algorithms that use an amino acid sequence to predict native disorder (29, 30). The VL-XT algorithm assigns a disorder propensity score for disorder on a residue by residue basis (29–31). The VL-XT prediction for EGFR, HER2, and HER3 can be seen in Fig. 1. PONDR scores greater than 0.5 indicate predicted disorder, and scores less than 0.5 indicate predicted order. Kinase domains of the three EGFR/ErbB family members show mostly predicted order, whereas the CTT in each shows large regions of predicted disorder. The amino acid composition of the CTT and kinase domains of both EGFR and HER3 is shown in Table 1. When comparing the CTTs with the kinase domains in both proteins, we observe that the CTTs are more enriched in polar, uncharged residues and prolines but relatively depleted in hydrophobic residues. For EGFR, polar, uncharged residues make up 30.1% of the CTT versus 14.9% of the kinase domain. Prolines make up 10.6% of the EGFR CTT versus 5.4% in the kinase domain, and hydrophobic residues (excluding tyrosine, which can become phosphorylated) are 27.4% of the EGFR CTT versus 38.9% of the kinase domain (Table 1). Similar values are seen with HER3 CTT (Table 1) despite its primary sequence divergence from EGFR CTT (22% sequence identity between EGFR and HER3 CTT; calculated with Clustal Omega) (74). The lowered hydrophobicity provides a simple explanation as to why the tails would not form a hydrophobic core and therefore be disordered in an aqueous environment. These predictions are a useful tool for gaining a general view of disordered regions in a protein sequence, but direct empirical evidence would better support the hypothesis of the CTTs as being IDRs.
FIGURE 1.
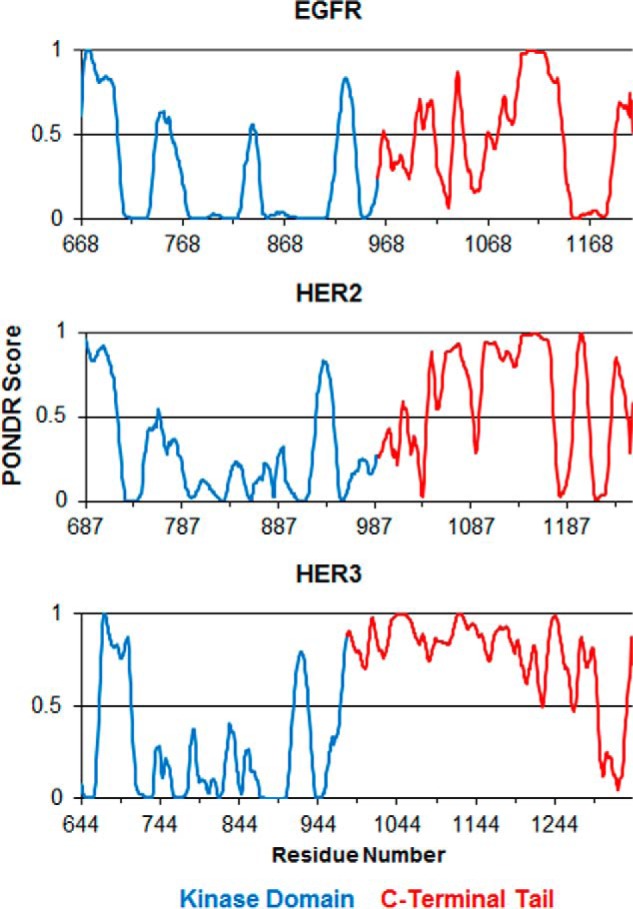
Disorder prediction by PONDR VL-XT algorithm. Shown are the disorder predictions for EGFR, HER2, and HER3 ICD construct sequences. In all three graphs, results for the kinase domain residues are colored blue, and the tail domain residues are colored in red. A score above 0.5 indicates predicted disorder, whereas a score below 0.5 indicates predicted order.
TABLE 1.
EGFR and HER3 amino acid composition analysis by region
AA, amino acid.
| AA residue | Percentage of total sequence |
|||
|---|---|---|---|---|
| C-terminal tails |
Kinase domain |
|||
| EGFR | HER3 | EGFR | HER3 | |
| % | ||||
| Polar uncharged | ||||
| Ser | 11.9 | 12.4 | 4.7 | 5.1 |
| Thr | 5.8 | 5.2 | 4.4 | 3.5 |
| Asn | 6.2 | 2.8 | 2.2 | 2.2 |
| Gln | 6.2 | 3.0 | 3.5 | 4.4 |
| Subtotal | 30.1 | 23.5 | 14.9 | 15.2 |
| Negative charge | ||||
| Asp | 8.4 | 5.0 | 4.4 | 4.4 |
| Glu | 5.3 | 10.5 | 7.9 | 6.6 |
| Subtotal | 13.7 | 15.5 | 12.3 | 11.1 |
| Positive charge | ||||
| Arg | 3.5 | 6.9 | 7.0 | 7.0 |
| His | 2.7 | 3.6 | 2.5 | 3.2 |
| Lys | 2.7 | 1.7 | 7.6 | 6.3 |
| Subtotal | 8.8 | 12.2 | 17.1 | 16.5 |
| Hydrophobic (excluding Tyr) | ||||
| Ala | 6.6 | 5.8 | 5.7 | 6.3 |
| Val | 4.9 | 3.9 | 7.3 | 7.9 |
| Ile | 3.5 | 0.6 | 7.9 | 5.7 |
| Leu | 6.2 | 9.7 | 11.1 | 12.0 |
| Met | 1.3 | 3.0 | 2.8 | 3.2 |
| Phe | 4.4 | 0.8 | 2.2 | 2.5 |
| Trp | 0.4 | 0.3 | 1.9 | 2.2 |
| Subtotal | 27.4 | 24.0 | 38.9 | 39.9 |
| Tyrosine | ||||
| Tyr | 4.0 | 3.6 | 3.5 | 3.5 |
| Special | ||||
| Cys | 1.3 | 1.7 | 1.9 | 1.3 |
| Gly | 4.0 | 9.4 | 6.0 | 7.3 |
| Pro | 10.6 | 10.2 | 5.4 | 5.4 |
Hydrogen/Deuterium Exchange Mass Spectrometry of EGFR/ErbB Family RTKs
HDX-MS is a highly useful technique to identify and study IDRs (32–36). HDX-MS provides kinetic information about the exchange rate of amide hydrogen atoms along the backbone of a protein for solvent deuterons. This rate of hydrogen/deuterium exchange is affected by factors such as the presence of strong hydrogen bonds, secondary structure, and tertiary structure (37–40). IDRs show very rapid hydrogen/deuterium exchange rates on the order of milliseconds to seconds (32–36). HDX-MS provides local information about different regions of the protein and readily distinguishes folded, globular regions from IDRs. We performed HDX-MS experiments by exposing the protein to deuterium oxide (D2O) over two ranges of time points, 108–2333 ms using quench flow and 5 s–2 h using manual labeling. HDX was quenched by lowering pH and rapidly cooling. This was followed by digesting the protein with immobilized pepsin and measuring the mass increase that results from incorporation of deuterons into peptic peptides by MS. In this study, we present HDX data as %nex, a percentage of the maximum observable mass increase. %nex is based on the number of amide hydrogens available for exchange and the H2O:D2O ratio during the labeling step.
We show that hydrogen/deuterium exchange occurs more rapidly in peptides from the CTT than those from the kinase domain (Fig. 2). Many peptides from the kinase domains (shown in blue) do not reach a maximum exchange plateau even within 2 h of exchange. However, peptides from the CTTs (shown in red) exchanged much more rapidly, often reaching a plateau before 5 s of exchange. This behavior was observed for all three ErbB ICH constructs analyzed. This indicates that the CTT region has faster hydrogen/deuterium exchange kinetics than the kinase domain with amide hydrogen protection from exchange being much weaker in the tail.
FIGURE 2.

Example HDX-MS uptake curves for EGFR, HER2, and HER3. Selected peptides from the kinase domains or C-terminal tail of each protein are shown in blue and red, respectively. Deuterium uptake reaches a plateau more quickly for the C-terminal tail as compared with the kinase domain. %nex is calculated as the percentage of exchange relative to the maximum number of exchangeable amide hydrogens in each peptide normalized by the H2O:D2O labeling ratio used in both labeling time course sets of experiments. Each data point represents the mean %nex value determined from triplicate measurements with error bars representing the S.D. at each point.
Comparisons between HDX data and the domain and secondary structure of EGFR, HER2, and HER3 are shown in Fig. 3. The heat map was constructed by measuring deuterium uptake in individual peptides and then on a residue by residue basis across the entire sequence, averaging the %nex measurement of all peptides covering specific amino acid residues. Residues and time points with no acquired peptide coverage and proline residues are shown as gaps in the data. This averaging method simplifies the observation of variations in deuterium exchange protection for localized protein regions. In the EGFR heat map, for example, we observe low exchange across much of the kinase domain, indicated by blue coloring. Exceptions to this observation include regions within the αC-helix and activation loop (A-loop), which show more rapid exchange within the millisecond time scale of exchange, indicated by yellow colors. The most striking result can be seen within the EGFR CTT where we observe that exchange reaches a plateau very rapidly, often within 5 s of exchange, and the %nex values are indicated by orange and red colors on the heat map. This rapid exchange indicates that the tail is unprotected from exchange when compared with the kinase domain. The observed rapid exchange behavior is characteristic of a highly dynamic and/or frequently exchange-competent conformational state, meaning amide hydrogen bonding between residues is transient or weak in these regions.
FIGURE 3.

HDX-MS heat maps for EGFR, HER2, and HER3 ICH constructs. Residue-specific average deuterium uptake data, 〈%nex〉, is mapped against respective EGFR, HER2, and HER3 sequences. Deuterium uptake reaches a plateau much sooner in C-terminal tail regions when compared with the more structured kinase domain. The EGFR sequence includes the 24-residue signal peptide in the numbering scheme. Similarly, the HER3 sequence numbering includes the 19-residue signal peptide. The secondary structure of the kinase domain as determined by X-ray crystallography is shown plotted against the sequence (Protein Data Bank codes 3PP0 for HER2, 3KEX for HER3, and 2GS2 for EGFR) (63–65). The location of αC-helices and activation loops are also identified.
Expression of EGFR and HER3 C-terminal Tails
To further characterize the conformational and physical state of the EGFR and HER3 CTTs, we expressed and purified EGFR and HER3 CTT-only constructs in a bacterial Escherichia coli system. EGFR residues 961–1186 with a C-terminal His tag were purified using nickel-nitrilotriacetic acid column chromatography and gel filtration. Estimated purity by SDS-PAGE was >95% (Fig. 4A) with a concentration of 5 mg ml−1. The HER3 CTT, HER3 residues 981–1342, was also expressed in E. coli and purified to a concentration of 2 mg ml−1 with >95% purity in the same manner (Fig. 4B).
FIGURE 4.

Purification and phosphorylation of CTT constructs. A, Coomassie-stained SDS-polyacrylamide gel of EGFR CTT purification. Lanes 1 and 4 are molecular weight markers labeled respectively. Lane 2 is transformed cell lysate preinduction with IPTG. Lane 3 is lysate postinduction. Lane 5 is purified EGFR CTT following Ni2+-chelating chromatography and gel filtration. EGFR CTT purity was determined to be >95%. B, Coomassie-stained SDS-polyacrylamide gel of HER3 CTT purification. This gel uses similar lane arrangement as in A for HER3 CTT expression. C, EGFR CTT is recognized and phosphorylated by the EGFR kinase domain. Phosphospecific antibodies to EGFR Tyr(P)-1068 and EGFR Tyr(P)-1173 were used to detect CTT phosphorylation. D, Western blotting analysis of HER3 CTT phosphorylation. The HER3 ICH serves as a positive control showing C-terminal tail phosphorylation via interaction with EGFR KD or HER2 KD. HER3 CTT is recognized and phosphorylated by the EGFR and HER2 kinase domains just as it would be as if it was part of its ICH construct. Phosphospecific antibodies to HER3 Tyr(P)-1289 and general anti-phosphotyrosine PY20 were used to detect HER3 CTT phosphorylation. E, phosphorylated EGFR CTT binds to the Grb2 SH2 domain. GSH-agarose beads were preloaded with GST-tagged Grb2 SH2 domain protein. Phosphorylated or unphosphorylated EGFR CTT was then incubated with the beads for 2 h at 4 °C, flow-through (FL) was collected and washed, and then the beads were boiled in sample buffer (Bound). Fractions were separated by SDS-PAGE and then transferred to nitrocellulose. Anti-His6-HRP antibody was used to detect EGFR CTT in the FL and bound fractions.
Confirmation of EGFR C-terminal Tail Function
Phosphorylation of the CTT region and subsequent binding of SH2 domain-containing proteins are essential steps in EGFR signaling. We first validated the ability of our CTT constructs to be both recognized and phosphorylated at specific tyrosine sites by separate EGFR family kinase domains in solution just as they would be if they were part of their respective ICH constructs. To test whether the E. coli-expressed EGFR CTT construct is functional, we incubated it with recombinant EGFR kinase domain and measured tail phosphorylation using reaction with ATP and blotting with phosphospecific antibodies. We show, through Western blotting identification of specific phosphorylated tyrosine residues (EGFR Tyr-1068 and Tyr-1173) in the EGFR CTT (Fig. 4C), that the EGFR CTT is recognized and phosphorylated by the EGFR kinase domain even when these two domains are expressed as separated constructs. Furthermore, we used anti-HER3 Tyr(P)-1289 and general PY20 antibodies to verify that the HER3 CTT was phosphorylated by both the EGFR kinase domain and HER2 kinase domain via Western blotting (Fig. 4D).
Next, we tested whether EGFR CTT can be bound by an SH2 domain-containing protein. We recombinantly expressed GST-tagged Grb2 SH2 domain and performed a GST pulldown assay (Fig. 4E). Incubating EGFR CTT with EGFR kinase domain produced a phosphorylated CTT that appeared as multiple bands of higher molecular weight, and this phosphorylated EGFR CTT did not bind on its own to glutathione-agarose (Fig. 4E, flow-through in lane 1 and bound in lane 2). Similarly, incubating unphosphorylated EGFR CTT with the GST-Grb2 SH2 domain construct did not result in binding, and EGFR CTT eluted in the flow-through (lane 3). However, once phosphorylated, EGFR CTT was bound by GST-Grb2-SH2 and was pulled down by glutathione-agarose (Fig. 4E, lane 6). These experiments provide evidence for the functionality of the EGFR and HER3 CTT constructs.
Analysis of EGFR CTT by CD Spectroscopy
Far-UV CD spectroscopy provides information about the secondary structure characteristics of protein molecules in solution based on the absorption of circularly polarized light by amide bonds in the polypeptide backbone. Because secondary structure types absorb circularly polarized light differently and each amide bond contributes to the UV absorption, the resulting spectrum will reflect a global average of the secondary structure content in a protein. Due to a lack of α-helix and β-sheet content, mostly or fully disordered and unfolded proteins will have CD spectra that are distinct from highly ordered proteins. The CD spectra between 190- and 260-nm wavelengths for EGFR CTT and HER3 CTT are shown in Fig. 5, A and B, respectively. Distinctive features expected for a mostly β-strand protein would include a positive ellipticity maximum at 195 nm with a negative ellipticity minimum near 215 nm. For a mostly α-helical protein, one would expect two minima, one at 208 nm and the other at 224-nm wavelength, along with a pronounced maximum near 192 nm (15, 41). However, the CD spectrum observed for EGFR CTT shows a minimum between 195 and 200 nm, which is a characteristic of disordered proteins (Fig. 5A). However, the slightly negative ellipticity at 222 nm indicates the possibility of residual secondary structure. Analysis via the CDSSTR algorithm, an algorithm used to assign secondary structure composition to CD spectra, shows 77% unordered character in the EGFR CTT spectrum (Fig. 5C). These results suggest that much of the EGFR CTT construct has an unfolded conformation in solution. The CD spectrum for HER3 CTT shows similar features as the EGFR CTT with a minimum between 195 and 200 nm and slightly negative ellipticity at 222 nm (Fig. 5B). The HER3 CTT has mostly unordered content (60%) but slightly higher β-sheet content than EGFR CTT: regular β-strand (βR) = 15% and distorted β-strand (βD) = 7% for HER3 CTT as compared with βR = 6% and βD = 3% for EGFR CTT (Fig. 5C).
FIGURE 5.

Circular dichroism spectroscopy. A, the CD spectrum for the EGFR CTT construct does not show prominent spectral features of α-helices and β-sheets. B, similarly, the HER3 CTT CD spectrum shows high unordered content with slightly higher calculated β-sheet content than in the EGFR CTT spectrum. C, secondary structure assignment was assigned the following symbols: regular α-helix (αR), distorted α-helix (αD), regular β-strand (βR), distorted β-strand (βD), turns (T), and unordered (U). deg, degrees.
Size Exclusion Chromatography with Multiangle Light Scattering
Another property of intrinsically disordered regions is a higher than expected apparent molecular mass during gel filtration separations (42). SEC can be used to separate proteins based on their hydrodynamic size, which is increased in IDRs depending on the degree of conformational extension. Proteins with higher hydrodynamic size elute earlier than smaller proteins, and accurate, absolute measurement of molecular weight can be obtained by coupling SEC to a MALS detector (43).
Fig. 6A shows the SEC elution chromatograms for both the EGFR CTT and carbonic anhydrase with the coupled MALS detection inserted above. Our analysis shows that EGFR CTT and carbonic anhydrase share similar molecular masses at 26 and 29 kDa, respectively, and both are monomeric under these conditions. EGFR CTT elutes from the SEC column before 13 ml of buffer volume. Despite having a lower molecular weight, the EGFR CTT elutes earlier than carbonic anhydrase, which elutes just after 15 ml of buffer. The expected Stokes radius of carbonic anhydrase is 2.4 nm (44). This earlier elution indicates that the EGFR CTT conformational ensemble is more extended than that of carbonic anhydrase, which has a globular structure. Fig. 6B shows the SEC-MALS chromatogram profiles for HER3 CTT and EGFR kinase domain. The molecular masses, determined by MALS, of the HER CTT and EGFR kinase domain were 42 and 38 kDa, respectively. HER3 CTT eluted at about 14 ml of buffer, which is earlier than the EGFR kinase domain, which eluted between 16 and 17 ml of buffer. As with EGFR CTT, no multimeric peaks were observed in the HER3 CTT chromatogram.
FIGURE 6.

Hydrodynamic properties of EGFR and HER3 CTT. A, elution chromatograms and molecule weight determination of EGFR CTT by SEC-MALS. EGFR CTT data are shown in red, and carbonic anhydrase data are shown in blue. Despite similar molecular weights, EGFR CTT elutes sooner, indicating a larger hydrodynamic size. B, SEC-MALS analysis of HER3 CTT. HER3 CTT data are shown in red, and EGFR kinase domain data are shown in blue. Again, the two molecules share similar molecular weights, but HER3 CTT elutes sooner than the EGFR KD. C, hydrodynamic radius determination of the EGFR CTT and BSA by dynamic light scattering. The measured DLS hydrodynamic radius distribution for EGFR CTT is shown as yellow bars. The measured hydrodynamic radius of EGFR CTT is 5.3 nm. The histogram for BSA (monomeric fraction obtained by gel filtration) is shown as red bars. The Rh of monomeric BSA is 4.8 nm. D, hydrodynamic radius determination of the HER3 CTT. The measured DLS hydrodynamic radius for HER3 CTT is 6.4 nm. E, frictional coefficient, frictional ratio, and molecular weight determined by analytical ultracentrifugation. The sedimentation coefficient was used to measure an apparent molecular mass, Mf, of 23.5 kDa. The frictional ratio for the EGFR CTT is 1.77, indicating a large Stokes radius relative to globular proteins. F, frictional coefficient, frictional ratio, and molecular weight determined by analytical ultracentrifugation. The measured apparent molecular mass of HER3CTT is 31.6 kDa. The frictional ratio is 1.52.
Dynamic Light Scattering
The DLS technique is used to analyze hydrodynamic properties of proteins in solution. Using the hydrodynamic radius, or Stokes radius, derived from measurements of translational diffusion coefficients, we are able to differentiate between a protein ensemble with mostly compact, globular conformations and one with more extended conformations as would be seen for IDRs (45). DLS is complementary to the static light scattering used in SEC-MALS in that DLS provides information on molecular size, whereas MALS provides a molecular weight and verification of a monomeric state. A histogram showing a distribution of measured hydrodynamic radii within the EGFR CTT and bovine serum albumin (BSA) populations is shown in Fig. 6C. The EGFR CTT, which has a molecular mass of 26 kDa, was determined to have a hydrodynamic radius of 5.3 nm. For a comparison, we also analyzed BSA, a globular protein with a molecular mass of 66 kDa, and the hydrodynamic radius of BSA was measured to be 4.8 nm. This demonstrates that EGFR CTT has a larger hydrodynamic radius than a globular protein more than 2.5 times its size. Similarly, HER3 CTT has a very large hydrodynamic radius (Fig. 6D). The hydrodynamic radius (RH) of HER3 CTT is 6.4 nm, although it has higher polydispersity than EGFR CTT.
Analytical Ultracentrifugation
We used sedimentation velocity AUC to determine the sedimentation coefficient, s, of EGFR CTT, which provides information about the molecular weight of the tail, Mf, as it relates to its RH (Fig. 6E). We also determined that our sample is homogenous due to the absence of secondary peaks in Fig. 6C as secondary populations or impurities would have different sedimentation velocities. From these AUC measurements, we obtained the frictional coefficient of the tail, f, which can be used to relate its molecular shape to that of a globular protein. The frictional ratio, f/fmin, provides an indication of the shape of a macromolecule based on the ratio of its measured frictional coefficient to a theoretical minimum frictional coefficient for a particle with minimum Stokes radius at a given molar mass (46). For a globular protein, the expected f/fmin would fall between 1.15 and 1.3 (47–49). We determined the f/fmin value for EGFR CTT to be much greater, at 1.77, which is consistent with this being an IDR. Similarly, HER3 CTT has a f/fmin value of 1.52, which is also greater than the expected frictional ratio for globular proteins (Fig. 6F).
Small Angle X-ray Scattering
Small angle X-ray scattering methods have been particularly useful in analyzing conformation ensemble properties of IDRs (50). In small angle X-ray scattering analyses, we measured the isotropic scattering intensity, I(s), as a function of the momentum transfer, s, of monochromatic X-ray light diffracted by protein macromolecules in solution. In protein structure studies, SAXS has been used to characterize structure and dynamics of monodisperse molecular species with a dominating structural configuration. IDRs show larger average sizes compared with globular proteins that contain tightly packed cores. By comparing the measured Rg and Dmax from SAXS with other known structured globular proteins, together with Kratky plots (I(s)·s2 as a function of s), which characterizes the flexibility state of the proteins, one is able to identify the flexibility of the IDRs (51). Fig. 7A shows the scattering profiles for EGFR CTT and BSA. In Fig. 7B, Guinier plot regions of EGFR CTT in three different concentrations are shown. Rg values calculated by AutoRg (52) are 46.6 ± 2.2 (EGFR CTT) and 33.6 ± 0.6 Å (BSA), and the molecular masses for each of them are 25.6 ± 0.7 and 69.0 ± 2.0 kDa, respectively (Table 2). The Kratky plot (Fig. 7C) of BSA shows a parabolic shape with a well defined peak. In contrast, EGFR CTT shows a hyperbolic shape on the Kratky plot, indicating an intrinsically disordered region (Fig. 7C). Fig. 7D shows the pair distribution function (P(r)) of EGFR CTT generated by Datgnom (52). The Dmax of EGFR CTT is 156 ± 6 Å, which is much larger than that of BSA (Table 2). Thus, the Rg and Dmax measurements, Kratky plot, and P(r) distribution of EGFR CTT indicate that the conformational ensemble adopted by EGFR CTT is extended and flexible.
FIGURE 7.
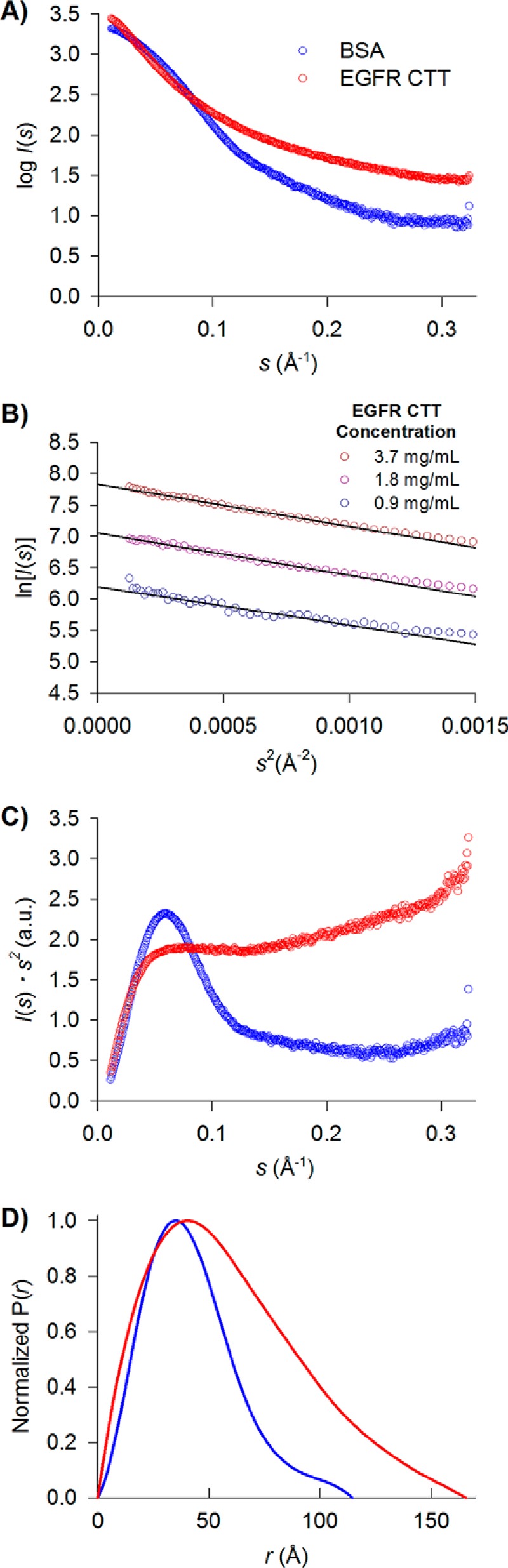
Small angle X-ray scattering analysis of EGFR CTT. A, scattering profiles of EGFR CTT (red) and BSA (blue). B, Guinier plots of EGFR CTT in different concentrations (0.9, 1.8, and 3.7 mg ml−1). C, the data are represented in a Kratky plot with arbitrary units. In folded proteins like BSA (blue), the Kratky plot shows parabolic features, whereas in the EGFR CTT Kratky plot (red), a hyperbolic shape indicates intrinsically disordered protein character. D, pair distribution functions for the EGFR CTT (red) and BSA (blue) scattering profiles generated using Datgnom (52).
TABLE 2.
Structural parameters obtained by SAXS
| Construct | Predicted molecular massa | Number of amino acids | Measured molecular massb | Rg | Dmax |
|---|---|---|---|---|---|
| kDa | kDa | Å | Å | ||
| EGFR CTTc | 26.2 | 235 | 25.6 ± 0.7 | 46.6 ± 2.2 | 156 ± 6d |
| HER3 CTTe | 40.5 | 371 | 41.5 ± 0.8 | 45.4 ± 0.4 | 153 ± 9d |
| HER3 CTT + ureae | 40.5 | 371 | 36 ± 4 | 50.5 ± 1.2 | 164 ± 10d |
| BSA | 66.5 | 583 | 69 ± 2 | 33.6 ± 0.6 | 114 ± 5 |
| EGFR kinase domain (Protein Data Bank code 1XKK)f | 41.1 | 352 | 41.1 | 20 | 64 |
a Values were calculated based on protein amino acid sequences.
b Measured molecular mass was calculated from the X-ray scattering profile.
c Buffer for EGFR CTT was 20 mm phosphate, pH 8.0, 150 mm NaCl.
d Characteristics of intrinsically disordered proteins are shown in the Kratky plots for these constructs.
e Buffer for HER3 CTT was 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 5% glycerol, +/−4 m urea as indicated above.
f Values of EGFR kinase domain were calculated from a theoretical scattering profile generated by the CRYSOL program using the crystal structure of EGFR kinase domain (Protein Data Bank code 1XKK).
When we analyzed the HER3 CTT with SAXS, we also included samples containing urea to determine whether a chemical denaturant would have an effect on the conformational ensemble. It is also important to note that, unlike in EGFR CTT buffer, 5% glycerol was also included in the HER3 CTT buffer. The glycerol was necessary to maintain protein solubility and stability of the HER3 CTT, but it may also act to stabilize conformations with diminished extendedness. We do not directly compare derived Rg values between EGFR CTT and HER3 CTT because of this difference in buffer conditions. The scattering profiles for HER3 CTT without urea and with 4 m urea are shown in Fig. 8A, and Guinier plot regions are shown in Fig. 8B. From these data, we calculated an Rg of 45.4 ± 0.4 Å in the absence of urea, and that increases slightly to 50.5 ± 1.2 Å upon the addition of 4 m urea (Table 2). The derived Kratky profiles for these two conditions are shown in Fig. 8C. Without urea present, the HER3 CTT produces a slightly parabolic Kratky profile, with a defined local maximum observed, at lower momentum transfer values. Moving toward greater momentum transfer values then gives a more hyperbolic increase in I(s)·s2. For HER3 CTT with 4 m urea, the Kratky profile has a hyperbolic shape indicative of disorder, similar to the profile observed for the EGFR CTT (Fig. 7C). The difference in Kratky plots between HER3 CTT with or without urea, together with increased Rg, indicates that HER3 CTT possesses characteristics of IDRs but not of random coil structures when in solution.
FIGURE 8.
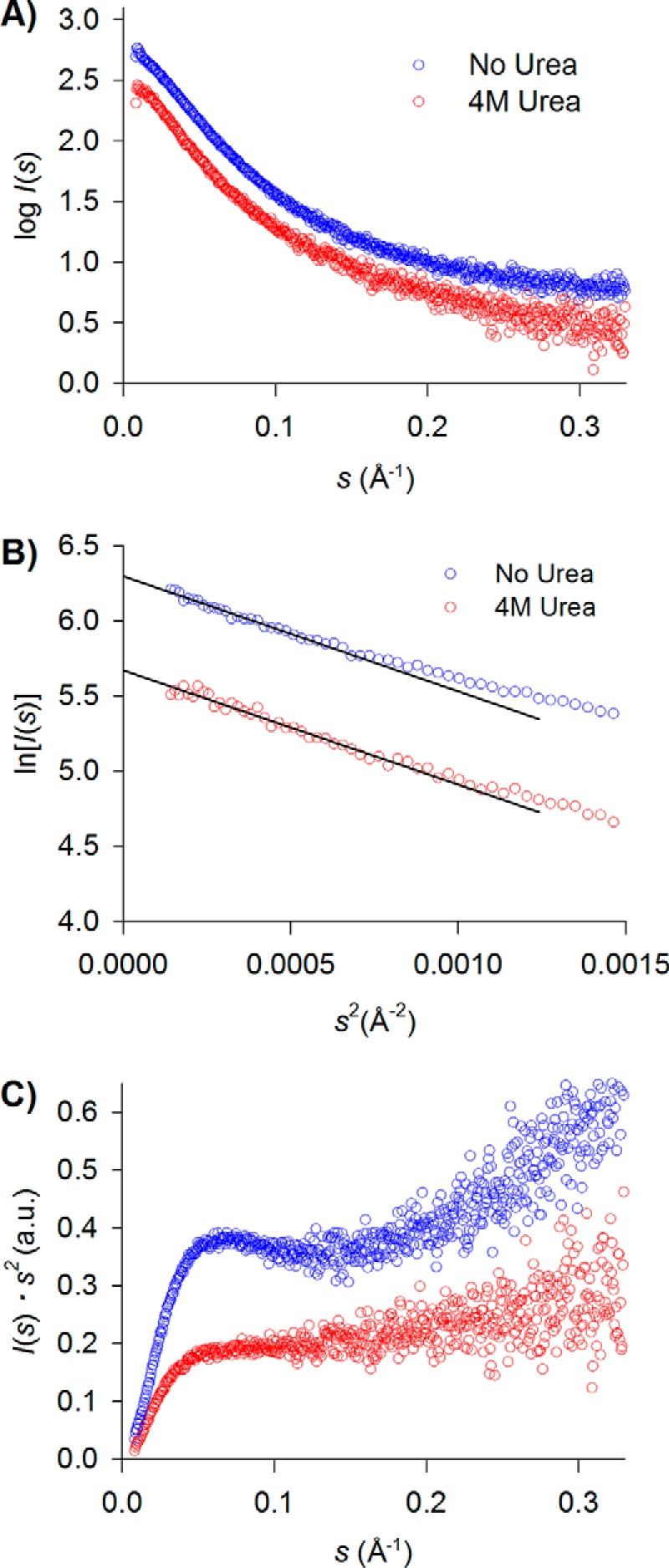
Small angle X-ray scattering analysis of HER3 CTT in solution ± urea. A, scattering profiles of HER3 CTT in solution without urea (blue) and with 4 m urea (red). B, Guinier plots of HER3 CTT with or without the presence of 4 m urea. C, the data are represented in a Kratky plot with arbitrary units. The Kratky plot for HER3 CTT with 4 m urea added shows a more hyperbolic profile, indicating greater disorder with the addition of chemical denaturant.
Discussion
We have shown that the CTT domains in EGFR family proteins are intrinsically disordered regions that have an extended, highly dynamic conformational state. HDX-MS demonstrates that the CTT domains have a highly dynamic and exchange-competent conformation. CD spectroscopy shows that they have high unordered character as expected for IDRs. SEC and DLS demonstrate that hydrodynamic size of the CTT is larger than protein standards of comparable molecular weight, which is another property of IDRs. AUC and the Kratky plot from SAXS demonstrate that the CTTs are IDRs, and Rg and Dmax measurements from SAXS demonstrate that the CTTs have an extended conformation. The Rg and Dmax measurements of the EGFR kinase domain were estimated from crystal structure (Protein Data Bank code 1XKK), and we observed that the EGFR CTT had Rg and Dmax values that were ∼2.4 times larger than those of the EGFR kinase domain.
A striking finding on the EGFR and HER3 CTTs is that despite their low primary sequence homology (22% sequence identity) the structural features of the CTTs are strongly conserved. This implies that the intrinsic disorder of these tails is important for their function, and the multiple conformers that a disordered region can adopt could reduce the thermodynamic barriers for protein binding and thereby increase both the on- and off-rates for binding of SH2 domain-containing proteins. Furthermore, the extended conformation of the CTT could increase the capture radius for recruitment of these signaling proteins (53, 54). Both of these mechanisms would facilitate signal propagation by these receptor tyrosine kinases.
The intrinsically disordered properties of these CTTs also explain why these regions are mostly absent from crystallographic studies on EGFR (7, 55). Portions of the EGFR tail located close to the kinase domain have been shown to be responsible for EGFR autoinhibition (56). The deletion of residues 982–1054 in the EGFR vIVb mutant is of particular importance in downstream signaling and kinase activation and has been shown to promote tumor formation. This region has been characterized via crystallography as containing an AP2-helix and other secondary structure elements, which appear to interact with the kinase domain in a manner that inhibits activation (4, 55, 56). In a study by Kovacs et al. (55), serial deletions of tail regions revealed specific regions important for autoinhibition as they relate to the oncogenic vIVb mutation. We observe increased protection from deuterium exchange for the EGFR 982–1054 region in the HDX-MS heat map (Fig. 3) relative to the rest of the C-terminal tail. The application of HDX-MS allows us to locally characterize conformational properties for protein regions that may not be easily analyzable with crystallography. In conclusion, these methods demonstrate that the EGFR and HER3 CTTs are IDRs with extended, non-globular structure in solution, and this finding may have important implications for the recruitment of downstream signaling proteins and signal propagation from these RTKs.
Experimental Procedures
EGFR/ErbB Family Protein Expression and Purifications
The following protein constructs were prepared for HDX-MS experiments and/or Western blotting analyses: EGFR WT ICH (668–1210; 644–1186 without signal peptide numbering), HER2 V956R mutant ICH (676–1255), HER3 WT ICH (664–1342; 645–1323 without signal peptide numbering), EGFR WT kinase domain (696–1022; 672–998 without signal peptide numbering), and HER2 WT kinase domain (704–1029). The protein expression procedure was adapted from protocols described previously (57, 58). Briefly, DNA encoding for each construct was cloned into separate pFastBacHT vectors (Invitrogen). Site-directed mutagenesis was conducted using the QuikChange II procedure (Agilent). Construct integrity was confirmed by DNA sequencing. The Bac-to-Bac protocol (Invitrogen) was used to make recombinant baculovirus for subsequent one multiplicity of infection of Spodoptera frugiperda Sf9 cells. Cells were harvested 48 h postinfection. Lysis of pelleted cells, purification of protein constructs via nickel-nitrilotriacetic acid resin, and gel filtration via Superdex-200 were repeated as reported previously (57, 58). ICH constructs were purified into buffer containing 20 mm Tris, 150 mm sodium chloride, 10% (w/v) glycerol, 0.01% Tween 20, 1 mm DTT, pH 8.0.
Hydrogen/Deuterium Exchange Mass Spectrometry
In HDX-MS experiments, we used a database of pepsin-digested peptide fragments found and identified via mass spectrometry to measure localized deuterium uptake in a protein. EGFR, HER2, and HER3 ICH construct stock solutions were each transferred into 200 μl of 0.1% TFA to a concentration of 1 μm and passed through an immobilized pepsin column (59). 30 μl of each collected digested protein eluent was analyzed by electrospray tandem mass spectrometry (MS/MS) using an LTQ FT Ultra mass spectrometer (Thermo Finnigan). A Mascot peptide search was used to build three peptic peptide databases, one for each protein, for subsequent MS analyses.
In the HDX experiments, EGFR, HER2, and HER3 ICH stocks were diluted into an H2O solvent protein storage buffer of 20 mm HEPES, 150 mm NaCl, pH 7.4. The concentration of protein prior to labeling was between 0.1 and 0.2 mg ml−1 for all experiments. A matching D2O solvent labeling buffer with 20 mm HEPES, 150 mm NaCl, pD 7.4, was used to label the protein samples for a set exposure time at room temperature.
HDX samples were prepared in triplicate as two sets. The first set was prepared via quench flow (QF) using a 1:5 volume ratio between protein sample in H2O solvent protein storage buffer and D2O labeling buffer with D2O exposure times between 108 and 2333 ms (36). For the second set of samples with D2O exposure times between 5 and 7200 s, protein samples were diluted in a 1:20 ratio of 2 μl of H2O protein storage buffer and 40 μl of D2O labeling buffer via manual labeling (ML).
In each sample, following the measured exposure time for exchange, a quench solution of equal volume to the D2O buffer of 0.4% formic acid and 3 m urea was used to lower the sample pH to about 2.6 to halt the forward exchange reaction and minimize back-exchange of deuterium-labeled amides for solvent hydrogen. Quenched samples were then flash frozen in liquid nitrogen and stored at −80 °C until we were ready to run online digestion and HPLC separation. Pepsin digestion was performed via an immobilized pepsin column. Separation of peptides was accomplished using a water/acetonitrile gradient across a Poroshell 120 EC-C18 2.1 × 50-mm analytical column (Agilent Technologies) prior to mass spectrometry analysis on a Bruker MaXis 4G electrospray ionization quadrupole time-of-flight mass spectrometer (Bruker Corp.). All chromatography solvents contained 0.1% formic acid.
HDX-MS data were analyzed using HDExaminer software (Sierra Analytics). A measurement of deuterium uptake, Δm, was made by calculating the average mass for a specific peptide's isotopic envelope at each HDX time point, mt, and subtracting the average mass of an undeuterated control's isotopic envelope, mund. Samples were analyzed in triplicate, and mass increase measurements presented throughout this work are the average uptake measurements for available data from these triplicate sample runs, Δmavg. The maximum number of exchangeable amide hydrogens, namide, was calculated for each observed peptic peptide in the peptide databases. The first two residues are not included among the exchangeable amide hydrogens due to rapid back-exchange of the deuterium label for these residues (32, 60, 61). Similarly, proline residues are excluded because of their lack of peptide chain amide hydrogen.
Because of the 1:5 H2O:D2O labeling ratio, the maximum observable exchange would be 83.3% of namide for samples prepared with QF. Similarly, the maximum observable exchange would be 95.2% namide for samples prepared through ML. HDX data collected by MS were normalized to a maximum observable exchange, nex, in both QF (Equation 1) and ML (Equation 2) data sets based on these maximum observable exchange values.
| (Eq. 1) |
| (Eq. 2) |
A deuterium uptake heat map for EGFR, HER2, and HER3 was generated using the residue by residue average, 〈%nex〉, of representative peptides' %nex measurements at each time point (Fig. 3) (36, 62). This residue-specific averaging shows more localized HDX information across entire ICH protein sequences and accounts for overlapping peptide data. Peptides were not weighted based on their respective lengths. The sequences of EGFR, HER2, and HER3 were each aligned to crystallographic secondary structure assignments of respective kinases domains. The crystal structures used were Protein Data Bank codes 3PP0 for HER2, 3KEX for HER3, and 2GS2 for EGFR (63–65).
Expression and Purification of EGFR CTT and HER3 CTT
DNA encoding residues 961–1186 of the human EGFR were cloned into a pET30b vector using NdeI and XhoI restriction sites in-frame with a C-terminal His tag in the vector. The E. coli strain BL21(DE3) was used for expression of the construct. Transformed cells were grown in Luria-Bertani medium containing 50 μg ml−1 kanamycin at 37 °C until an OD600 of between 0.5 and 0.8 was reached. Expression was induced through the addition of 0.5 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3 h at 37 °C. Bacterial cells were centrifuged, and the pellets were frozen. Cell lysis was performed at 4 °C with a probe sonicator on frozen pellets using 50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 20 mm imidazole, 1 mm phenylmethylsulfonyl fluoride (PMSF), 1 mm β-mercaptoethanol, Roche Applied Science protease inhibitor mixture tablets, 4 mm benzamidine·HCl. The lysate was clarified by centrifugation at 48,000 × g for 30 min at 4 °C. Purification was performed using Ni2+-chelating chromatography followed by gel filtration chromatography on a Superdex-75 column. The protein was concentrated to 5 mg ml−1 in 20 mm phosphate, 150 mm sodium chloride, pH 8.0. Aliquots were snap frozen in liquid N2 and stored at −80 °C. Protein concentration was determined using UV-visible spectroscopy with a molar extinction coefficient of EGFR CTT at 280 nm of ϵ = 18,910 m−1 cm−1. For specific subsequent applications, the addition of DTT or tris(2-carboxyethyl)phosphine to EGFR CTT samples was critical. A similar procedure was followed to express and purify a HER3 CTT construct with residues 981–1342. The HER3 CTT was purified at a concentration of 2 mg ml−1 in 20 mm Tris, 300 mm sodium chloride, 5% (w/v) glycerol, pH 8.0. HER3 CTT concentration was determined using a molar extinction coefficient at 280 nm of ϵ = 25,245 m−1 cm−1.
C-terminal Tail Function via Western Blotting Analysis
EGFR CTT was phosphorylated via reaction in 0.1 mm ATP, 0.1 mm sodium orthovanadate, 1 mm dithiothreitol, 1 mm MnCl2 catalyst. EGFR CTT concentrations of 0.25 and 0.50 μm tail were incubated in the presence of 0.50 μm EGFR kinase domain. Two negative controls were also incubated under the same conditions with the following exceptions: 0.50 μm CTT was incubated without kinase domain present, and separately 0.50 μm kinase domain was incubated without CTT present. A positive control of ATP phosphorylation of 0.50 μm EGFR ICH construct, which contains both kinase domain and CTT regions together, was also incubated.
Two EGFR tail region-specific phosphotyrosines were analyzed by Western blotting. Following the phosphorylation reaction, EGFR CTT samples were separated by SDS-PAGE on a 10% polyacrylamide gel followed by electroblotting onto nitrocellulose membrane. Two anti-EGFR phosphotyrosine primary antibodies were used in separate Western blotting analyses: 1:1000 anti-EGFR Tyr(P)-1068 and 1:5000 anti-EGFR Tyr(P)-1173. Secondary antibody was 1:5000 anti-rabbit in both blots. The results of the Western blotting analyses are shown in Fig. 4C. The positive control was also analyzed on the same two Western blots as the EGFR CTT samples. Bands from this positive control were observed on both Western blots but are not shown herein.
HER3 CTT was phosphorylated via the same buffer conditions as the EGFR CTT above. HER3 CTT at 0.50 μm was incubated in the presence of 0.50 μm EGFR or 0.50 μm HER2 kinase domains. Negative controls were incubated under the same conditions where only CTT or kinase domain was incubated in each case. Positive controls pairing HER3 ICH with EGFR kinase domain or HER2 kinase domain were also incubated. Samples were separated by SDS-PAGE and electroblotted onto nitrocellulose membrane. 1:1000 anti-HER3 Tyr(P)-1289 primary with 1:10,000 anti-rabbit secondary antibodies were used for the first blot. A 1:1000 general anti-phosphotyrosine antibody, PY20, was used for the second blot with 1:10,000 anti-mouse secondary antibody. The two Western blots are shown in Fig. 4D.
GST-Grb2-SH2 Expression and Purification
pGEX Grb2-SH2 (amino acids 58–159) was purchased from Addgene (plasmid 46440, deposited by Dr. Bruce Mayer). BL21(DE3) cells were transformed, and a colony was picked and used to inoculate a 100-ml LB + ampicillin culture. The culture was induced to midlog phase with 1 mm IPTG for 2 h at 37 °C. Pellets were collected and subsequently frozen at −80 °C. GST-Grb2-SH2 was purified using glutathione-agarose beads (Sigma). Fractions were eluted with reduced glutathione, pooled, dialyzed, and concentrated using an Amicon 10,000 molecular weight cutoff spin concentrator in 20 mm Tris-HCl, pH 8 (4 °C), 150 mm NaCl, 1 mm DTT. Aliquots were flash frozen and stored at −80 °C.
Interaction of Phosphorylated EGFR CTT and GST-Grb2-SH2 by Pulldown Assay
EGFR CTT was phosphorylated by EGFR KD in vitro for 60 min at room temperature. 10 μg of GST-Grb2-SH2 was prebound to a 10-μl bed volume of glutathione-agarose beads. 2 μg of either phosphorylated CTT or non-phosphorylated CTT was added to the beads in a volume of 100 μl of buffer and incubated for 2 h at 4 °C. Beads were then spun briefly and the flow-through (FL) was collected. This was followed by two washes with buffer. Beads (bound) and aliquots of the FL were then boiled in SDS-PAGE sample buffer. Western blotting was done on the fractions using anti-His6-HRP antibody (Thermo Fisher, MA1-21314-HRP) as a probe.
Circular Dichroism Spectroscopy
EGFR CTT protein was prepared at a concentration of 5 μm in 10 mm sodium phosphate, pH 7.4, 5 mm NaCl. HER3 CTT was also prepared at the same conditions. A 0.1-cm pathlength quartz cell was used for samples and buffer blanks. Cells were placed in a Jasco CD spectrometer set at room temperature. The cell in the CD spectrometer was allowed to equilibrate for 15 min with nitrogen flowing into the CD spectrometer before scans. The CD spectrometer scanned from 260 to 190 nm at 20 nm min−1. Five individual scans were averaged, and the buffer blank was subtracted before analysis.
The CD spectra were analyzed on the DichroWeb online analysis website (41, 66). The method chosen for this analysis used the CDSSTR algorithm with reference database 7 as the reference protein set (67–70). Secondary structure assignment was divided into six categories: regular α-helix (αR), distorted α-helix (αD), βR, βD, turns, and unordered. CDSSTR structure assignment results are presented in Fig. 5C.
Size Exclusion Chromatography with Multiangle Light Scattering
100 μl of EGFR CTT or HER3 CTT (A280 ∼ 1) was loaded onto a pre-equilibrated Superdex-75 10/300 GL (GE Healthcare) column. Elution buffer of 20 mm sodium phosphate, pH 8.0, 150 mm NaCl, 0.5 mm DTT was used for EGFR CTT, and 20 mm Tris, 300 mm sodium chloride, 5% (w/v) glycerol, pH 8.0 was used for HER3 CTT. The buffer flow rate was 0.5 ml min−1 for the EGFR CTT and carbonic anhydrase elutions. For the HER3 CTT and EGFR KD elutions, the buffer flow rate was 0.3 ml min−1. The column was coupled to a Wyatt Optilab rEX and Dawn Helios II, which are refractive index and multiangle light scatter detectors, respectively. Carbonic anhydrase was used as a globular reference for the EGFR CTT elution because of the similar molecular weights of the proteins. Similarly, the EGFR KD was used as a reference for the HER3 CTT elution. Astra (Wyatt) software was used to calculate the molar mass.
Dynamic Light Scattering
EGFR CTT samples at a concentration of 0.5 mg ml−1 in 20 mm Tris, 150 mm NaCl, 1 mm DTT, pH 8.0, were centrifuged for 5 min. 12 μl of sample was aliquoted into a clean microcuvette, which was placed into a Wyatt Dynapro MSX. The cuvette was equilibrated for 5 min at 25 °C before data acquisition. BSA was used as a standard at 1.0 mg ml−1 in the same buffer. Dynamics software was used to determine the radius of the protein.
Analytical Ultracentrifugation
Samples were dialyzed versus buffer (20 mm sodium phosphate, pH 8.0, 150 mm NaCl, 1 mm DTT) at 4 °C overnight. The samples and buffer were recovered and briefly centrifuged before loading into centrifuge cells. The cells were balanced and then placed in an eight-position rotor, which was put into a Beckman XLA ultracentrifuge. The speed was 42,000 rpm, and the temperature was 25 °C. The SEDFIT program was used to analyze the data, and the resulting c(s) analysis is shown in Fig. 6, E and F (71).
Small Angle X-ray Scattering Analysis
SAXS experiments were performed on the SIBYLS beamline 12.3.1.2 at the Advanced Light Source, a national user facility operated by the Lawrence Berkeley National Laboratory (Berkeley, CA) and supported by the Director, Office of Science, Office of Basic Energy Sciences of the United States Department of Energy under Contract DE-AC02-05CH11231 (72). To optimize the data quality and minimize radiation damage, exposure series of 0.5, 1, 2, and 5 s were performed. The concentration of EGFR CTT (20 mm phosphate, pH 8.0, 150 mm NaCl) used for these experiments ranged from 0.9 to 3.7 mg ml−1. BSA in 20 mm phosphate, pH 8.0, 150 mm NaCl was used as a standard in the range of 1.0–3.0 mg ml−1. The concentration of HER3 CTT (20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 5% glycerol) ranged from 0.9 to 1.5 mg ml−1. A minimum of three different concentrations for each protein were used for data collection, and five different concentrations were used for EGFR CTT.
The data sets were processed using standard procedures for ATSAS programs (52). At low angles, the scattered intensities of EGFR CTT and HER3 CTT were very well approximated by the Guinier law, whereas HER3 CTT showed signs of radiation damage at 5-s exposures. Before further analysis, the scattering curves, free of radiation damage, from a given type of sample with the same protein concentration but different exposure times were merged with either PRIMUS (52) or SCÅTTER (SIBYLS Beamline at Lawrence Berkeley National Laboratory) and then averaged among the same type of sample after normalization using their individual protein concentration. The average scattering curves are presented in Figs. 7A and 8A. The Rg from data sets free of radiation damage was calculated using AutoRg and is presented in Table 2. We evaluated the molecular weight of the sample by comparing the forward scattering I(0) with that from a reference solution of BSA. The Rg and Dmax values for EGFR kinase domain (Protein Data Bank code 1XKK) were calculated using the program CRYSOL (73). The pair distribution functions for EGFR CTT and BSA were generated using Datgnom (52).
Author Contributions
T. R. K. and R. B. conceived and wrote the paper. T. R. K. designed, performed, and analyzed the HDX experiments shown in Figs. 2 and 3. K. S., T. R. K., E. M. M., and J. M. designed, performed, and analyzed the experiments shown in Figs. 4, 5, and 6. K. S. purified and prepared the samples for SAXS. J. Z. designed and analyzed the SAXS experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the following researchers for assistance in instrumentation: David Weis for providing access to the quench-flow apparatus for rapid HDX, Michael Gross and the National Institutes of Health/National Center for Research Resources Mass Spectrometry Resource for MS instrumentation, Daved Fremont and Gaya Amarasinghe for providing dynamic light scatter and multiangle light scatter instrumentation, Timothy Lohman, Paolo De Bona, and Alex Kozlov for assistance in analytical ultracentrifugation, and Evan Sadler for facilitation of the small angle X-ray scatter experiments. We also thank Linda Pike, Rohit Pappu, Carl Frieden, and Elliot Elson for helpful discussions.
This work was supported by National Institutes of Health Grants R01 CA161001 (to R. B.) and 8P41 GM103422 and by the Cancer Biology Pathway Program at the Siteman Cancer Center through National Institutes of Health Grant 5T32 CA113275. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EGFR
- epidermal growth factor receptor
- HER
- human epidermal growth factor receptor
- RTK
- receptor tyrosine kinase
- CTT
- C-terminal tail
- KD
- kinase domain
- IDR
- intrinsically disordered region
- HDX
- hydrogen/deuterium exchange
- SH2
- Src homology 2
- SEC
- size exclusion chromatography
- MALS
- multiangle light scattering
- SAXS
- small angle X-ray scattering
- AUC
- analytical ultracentrifugation
- PONDR
- predictor of natural disordered regions
- DLS
- dynamic light scattering
- ICH
- intracellular half
- βR
- regular β-strand
- βD
- distorted β-strand
- RH
- hydrodynamic radius
- Rg
- radius of gyration
- Dmax
- maximum interatomic distance
- QF
- quench flow
- ML
- manual labeling
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- FL
- flow-through
- αR
- regular α-helix
- αD
- distorted α-helix.
References
- 1. Lemmon M. A., Schlessinger J., and Ferguson K. M. (2014) The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 6, a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lemmon M. A., and Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Franklin M. C., Carey K. D., Vajdos F. F., Leahy D. J., de Vos A. M., and Sliwkowski M. X. (2004) Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328 [DOI] [PubMed] [Google Scholar]
- 4. Wood E. R., Truesdale A. T., McDonald O. B., Yuan D., Hassell A., Dickerson S. H., Ellis B., Pennisi C., Horne E., Lackey K., Alligood K. J., Rusnak D. W., Gilmer T. M., and Shewchuk L. (2004) A unique structure for epidermal growth factor receptor bound to GW572016 (lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 64, 6652–6659 [DOI] [PubMed] [Google Scholar]
- 5. Dankort D. L., Wang Z., Blackmore V., Moran M. F., and Muller W. J. (1997) Distinct tyrosine autophosphorylation sites negatively and positively modulate neu-mediated transformation. Mol. Cell. Biol. 17, 5410–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee N. Y., Hazlett T. L., and Koland J. G. (2006) Structure and dynamics of the epidermal growth factor receptor C-terminal phosphorylation domain. Protein Sci. 15, 1142–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gajiwala K. S. (2013) EGFR: tale of the C-terminal tail. Protein Sci. 22, 995–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dunker A. K., Silman I., Uversky V. N., and Sussman J. L. (2008) Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764 [DOI] [PubMed] [Google Scholar]
- 9. Uversky V. N., Oldfield C. J., and Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 [DOI] [PubMed] [Google Scholar]
- 10. Bae S.-H., Dyson H. J., and Wright P. E. (2009) Prediction of the rotational tumbling time for proteins with disordered segments. J. Am. Chem. Soc. 131, 6814–6821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uversky V. N., and Dunker A. K. (2010) Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tantos A., Han K.-H., and Tompa P. (2012) Intrinsic disorder in cell signaling and gene transcription. Mol. Cell. Endocrinol. 348, 457–465 [DOI] [PubMed] [Google Scholar]
- 13. Zhou H.-X., Pang X., and Lu C. (2012) Rate constants and mechanisms of intrinsically disordered proteins binding to structured targets. Phys. Chem. Chem. Phys. 14, 10466–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uversky V. N. (2013) The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 587, 1891–1901 [DOI] [PubMed] [Google Scholar]
- 15. Habchi J., Tompa P., Longhi S., and Uversky V. N. (2014) Introducing protein intrinsic disorder. Chem. Rev. 114, 6561–6588 [DOI] [PubMed] [Google Scholar]
- 16. Oldfield C. J., Cheng Y., Cortese M. S., Brown C. J., Uversky V. N., and Dunker A. K. (2005) Comparing and combining predictors of mostly disordered proteins. Biochemistry 44, 1989–2000 [DOI] [PubMed] [Google Scholar]
- 17. Uversky V. N., and Ptitsyn O. B. (1994) “Partly folded” state, a new equilibrium state of protein molecules: four-state guanidinium chloride-induced unfolding of β-lactamase at low temperature. Biochemistry 33, 2782–2791 [DOI] [PubMed] [Google Scholar]
- 18. Uversky V. N., and Ptitsyn O. B. (1996) Further evidence on the equilibrium “pre-molten globule state”: four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J. Mol. Biol. 255, 215–228 [DOI] [PubMed] [Google Scholar]
- 19. Uversky V. N., Oldfield C. J., and Dunker A. K. (2005) Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18, 343–384 [DOI] [PubMed] [Google Scholar]
- 20. Liu J., Faeder J. R., and Camacho C. J. (2009) Toward a quantitative theory of intrinsically disordered proteins and their function. Proc. Natl. Acad. Sci. U.S.A. 106, 19819–19823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oldfield C. J., Meng J., Yang J. Y., Yang M. Q., Uversky V. N., and Dunker A. K. (2008) Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9, Suppl. 1, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xie H., Vucetic S., Iakoucheva L. M., Oldfield C. J., Dunker A. K., Obradovic Z., and Uversky V. N. (2007) Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 6, 1917–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Redfield C. (2004) Using nuclear magnetic resonance spectroscopy to study molten globule states of proteins. Methods 34, 121–132 [DOI] [PubMed] [Google Scholar]
- 24. Iakoucheva L. M., Brown C. J., Lawson J. D., Obradović Z., and Dunker A. K. (2002) Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584 [DOI] [PubMed] [Google Scholar]
- 25. Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 26. Kathiriya J. J., Pathak R. R., Clayman E., Xue B., Uversky V. N., and Davé V. (2014) Presence and utility of intrinsically disordered regions in kinases. Mol. Biosyst. 10, 2876–2888 [DOI] [PubMed] [Google Scholar]
- 27. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 28. Koland J. G. (2014) Coarse-grained molecular simulation of epidermal growth factor receptor protein tyrosine kinase multi-site self-phosphorylation. PLoS Comput. Biol. 10, e1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Romero P., Obradovic Z., and Dunker A. K. (1997) Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inform. 8, 110–124 [PubMed] [Google Scholar]
- 30. Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., and Dunker A. K. (2001) Sequence complexity of disordered protein. Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- 31. Li X., Romero P., Rani M., Dunker A. K., and Obradovic Z. (1999) Predicting protein disorder for N-, C-, and internal regions. Genome Inform. 10, 30–40 [PubMed] [Google Scholar]
- 32. Keppel T. R., Howard B. A., and Weis D. D. (2011) Mapping unstructured regions and synergistic folding in intrinsically disordered proteins with amide H/D exchange mass spectrometry. Biochemistry 50, 8722–8732 [DOI] [PubMed] [Google Scholar]
- 33. Rob T., Liuni P., Gill P. K., Zhu S., Balachandran N., Berti P. J., and Wilson D. J. (2012) Measuring dynamics in weakly structured regions of proteins using microfluidics-enabled subsecond H/D exchange mass spectrometry. Anal. Chem. 84, 3771–3779 [DOI] [PubMed] [Google Scholar]
- 34. Goswami D., Devarakonda S., Chalmers M. J., Pascal B. D., Spiegelman B. M., and Griffin P. R. (2013) Time window expansion for HDX analysis of an intrinsically disordered protein. J. Am. Soc. Mass. Spectrom. 24, 1584–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keppel T. R., and Weis D. D. (2013) Analysis of disordered proteins using a simple apparatus for millisecond quench-flow H/D exchange. Anal. Chem. 85, 5161–5168 [DOI] [PubMed] [Google Scholar]
- 36. Keppel T. R., and Weis D. D. (2015) Mapping residual structure in intrinsically disordered proteins at residue resolution using millisecond hydrogen/deuterium exchange and residue averaging. J. Am. Soc. Mass. Spectrom. 26, 547–554 [DOI] [PubMed] [Google Scholar]
- 37. Hvidt A., and Nielsen S. O. (1966) Hydrogen exchange in proteins. Adv. Protein Chem. 21, 287–386 [DOI] [PubMed] [Google Scholar]
- 38. Englander S. W., and Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655 [DOI] [PubMed] [Google Scholar]
- 39. Zhang Z., and Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Konermann L., Pan J., and Liu Y.-H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 [DOI] [PubMed] [Google Scholar]
- 41. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 42. Uversky V. N. (2002) What does it mean to be natively unfolded? Eur. J. Biochem. 269, 2–12 [DOI] [PubMed] [Google Scholar]
- 43. Wyatt P. J. (1993) Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 272, 1–40 [Google Scholar]
- 44. Sapir T., Eisenstein M., Burgess H. A., Horesh D., Cahana A., Aoki J., Hattori M., Arai H., Inoue K., and Reiner O. (1999) Analysis of lissencephaly-causing LIS1 mutations. Eur. J. Biochem. 266, 1011–1020 [DOI] [PubMed] [Google Scholar]
- 45. Receveur-Bréchot V., Bourhis J.-M., Uversky V. N., Canard B., and Longhi S. (2006) Assessing protein disorder and induced folding. Proteins 62, 24–45 [DOI] [PubMed] [Google Scholar]
- 46. Laue T. M., Stafford W. F. 3rd (1999) Modern applications of analytical ultracentrifugation. Annu. Rev. Biophys. Biomol. Struct. 28, 75–100 [DOI] [PubMed] [Google Scholar]
- 47. Cantor C. R., and Schimmel P. R. (1980) Biophysical Chemistry: Part II: Techniques for the Study of Biological Structure and Function, W. H. Freeman, Co, Oxford [Google Scholar]
- 48. Tcherkasskaya O., and Uversky V. N. (2001) Denatured collapsed states in protein folding: example of apomyoglobin. Proteins 44, 244–254 [DOI] [PubMed] [Google Scholar]
- 49. Manon F., and Ebel C. (2010) Analytical ultracentrifugation, a useful tool to probe intrinsically disordered proteins, in Instrumental Analysis of Intrinsically Disordered Proteins, pp. 431–449, John Wiley & Sons, Inc., Hoboken, N. J. [Google Scholar]
- 50. Bernadó P., and Svergun D. I. (2012) Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 8, 151–167 [DOI] [PubMed] [Google Scholar]
- 51. Kikhney A. G., and Svergun D. I. (2015) A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 589, 2570–2577 [DOI] [PubMed] [Google Scholar]
- 52. Petoukhov M. V., Konarev P. V., Kikhney A. G., and Svergun D. I. (2007) ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40, s223–s228 [Google Scholar]
- 53. Shoemaker B. A., Portman J. J., and Wolynes P. G. (2000) Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 97, 8868–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang Y., and Liu Z. (2009) Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J. Mol. Biol. 393, 1143–1159 [DOI] [PubMed] [Google Scholar]
- 55. Kovacs E., Das R., Wang Q., Collier T. S., Cantor A., Huang Y., Wong K., Mirza A., Barros T., Grob P., Jura N., Bose R., and Kuriyan J. (2015) Analysis of the role of the C-terminal tail in the regulation of the epidermal growth factor receptor. Mol. Cell. Biol. 35, 3083–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pines G., Huang P. H., Zwang Y., White F. M., and Yarden Y. (2010) EGFRvIV: a previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene 29, 5850–5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Monsey J., Shen W., Schlesinger P., and Bose R. (2010) Her4 and Her2/neu tyrosine kinase domains dimerize and activate in a reconstituted in vitro system. J. Biol. Chem. 285, 7035–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Collier T. S., Diraviyam K., Monsey J., Shen W., Sept D., and Bose R. (2013) Carboxyl group footprinting mass spectrometry and molecular dynamics identify key interactions in the HER2-HER3 receptor tyrosine kinase interface. J. Biol. Chem. 288, 25254–25264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang L., Pan H., and Smith D. L. (2002) Hydrogen exchange-mass spectrometry: optimization of digestion conditions. Mol. Cell. Proteomics 1, 132–138 [DOI] [PubMed] [Google Scholar]
- 60. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Connelly G. P., Bai Y., Jeng M.-F., and Englander S. W. (1993) Isotope effects in peptide group hydrogen exchange. Proteins 17, 87–92 [DOI] [PubMed] [Google Scholar]
- 62. Xia Y., DiPrimio N., Keppel T. R., Vo B., Fraser K., Battaile K. P., Egan C., Bystroff C., Lovell S., Weis D. D., Anderson J. C., and Karanicolas J. (2013) The designability of protein switches by chemical rescue of structure: mechanisms of inactivation and reactivation. J. Am. Chem. Soc. 135, 18840–18849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang X., Gureasko J., Shen K., Cole P. A., and Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 64. Jura N., Shan Y., Cao X., Shaw D. E., and Kuriyan J. (2009) Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. U.S.A. 106, 21608–21613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aertgeerts K., Skene R., Yano J., Sang B.-C., Zou H., Snell G., Jennings A., Iwamoto K., Habuka N., Hirokawa A., Ishikawa T., Tanaka T., Miki H., Ohta Y., and Sogabe S. (2011) Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Whitmore L., and Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Compton L. A., and Johnson W. C. Jr. (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 155, 155–167 [DOI] [PubMed] [Google Scholar]
- 68. Manavalan P., and Johnson W. C. Jr. (1987) Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 167, 76–85 [DOI] [PubMed] [Google Scholar]
- 69. Sreerama N., Venyaminov S. Y., and Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 287, 243–251 [DOI] [PubMed] [Google Scholar]
- 70. Sreerama N., and Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 71. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hura G. L., Menon A. L., Hammel M., Rambo R. P., Poole F. L. 2nd, Tsutakawa S. E., Jenney F. E. Jr., Classen S., Frankel K. A., Hopkins R. C., Yang S.-j., Scott J. W., Dillard B. D., Adams M. W., and Tainer J. A. (2009) Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 6, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 74. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]