

SUMMARY
Diets rich in saturated fatty acids (SFAs) produce a form of tissue inflammation driven by “metabolically activated” macrophages. We show that SFAs, when in excess, induce a unique transcriptional signature in both mouse and human macrophages that is enriched for a subset of ER stress markers, particularly IRE1α and many adaptive downstream target genes. SFAs also activate the NLRP3 inflammasome in macrophages, resulting in IL-1β secretion. We found that IRE1α mediates SFA-induced IL-1β secretion by macrophages, and that its activation by SFAs does not rely on unfolded protein sensing. We show instead that the ability of SFAs to stimulate either IRE1α activation or IL-1β secretion can be specifically reduced by preventing their flux into phosphatidylcholine (PC) or by increasing unsaturated PC levels. IRE1α is thus an unrecognized intracellular PC sensor critical to the process by which SFAs stimulate macrophages to secrete IL-1β, a driver of diet-induced tissue inflammation.
eTOC Blurb
Excessive saturated fat consumption promotes tissue inflammation driven by “metabolically activated” macrophages. Here, Robblee et al. use transcriptomic profiling to identify the ER stress sensor IRE1α as a key component of metabolic activation that senses phospholipid saturation to mediate inflammatory activation in macrophages exposed to saturated fat.
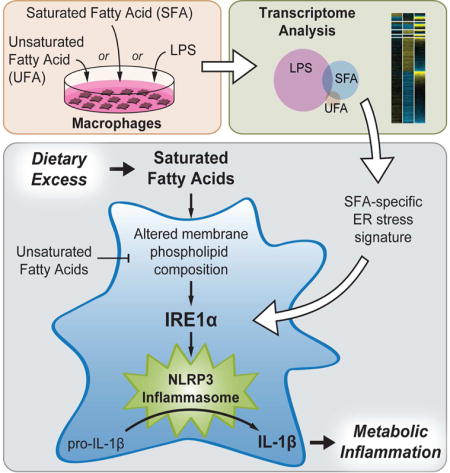
INTRODUCTION
Chronic consumption of diets rich in fat, particularly saturated fat, is associated with the accumulation of immune cells such as macrophages and dendritic cells in metabolic tissues like the white adipose. Subsets of these accumulating myeloid cells (MCs) express inflammatory markers and secrete pro-inflammatory cytokines that also comprise the response to lipopolysaccharide (LPS) stimulation (Lumeng et al., 2007; Weisberg et al., 2003), and targeting inflammatory pathways in these cell types has alleviated diet-induced insulin resistance in animal models (Yuan et al., 2001; Solinas et al., 2007). More recent work shows that adipose tissue macrophages (ATMs) from obese mice have a pattern of “metabolic activation” (MMe) that is distinct from that induced by LPS (MLPS) or other danger- and pathogen-associated molecular patterns (DAMPs and PAMPs; Xu et al., 2013; Kratz et al., 2014). However the molecular details and functional consequences of MMe polarization are poorly understood.
Treating cultured bone marrow-derived macrophages or dendritic cells (BMDMs and BMDCs) with saturated fatty acids (SFAs) recapitulates many features of MMe polarization that are seen in the ATMs of mice consuming diets high in saturated fat (Nguyen et al., 2007; Suganami et al., 2007; Kratz et al., 2014). These include not only the secretion of NF-κB-dependent MLPS cytokines such as IL-6 and TNF (Shi et al., 2006), but also activation of the NLRP3 inflammasome (Wen et al., 2011), an intracellular protein complex that assembles in response to DAMPs and PAMPs and catalyzes the cleavage and maturation of the cytokines IL-1β and IL-18.
Because circulating IL-1β levels are elevated in diet-induced obesity (DIO) and targeting IL-1β, its receptor, or components of the NLRP3 inflammasome protects obese mice from glucose intolerance and other metabolic consequences of DIO (Osborn et al., 2008; Stienstra et al., 2010; Wen et al., 2011), there is interest in understanding how SFAs activate the NLRP3 inflammasome. Prior studies have implicated reactive oxygen species accumulation due to impairment of AMPK-regulated autophagy in this process (Wen et al., 2011). Others have pointed to a stimulatory role for ceramide production (Schilling et al., 2012), but recent work suggests that de novo ceramide synthesis does not contribute to SFA-induced NLRP3 inflammasome activation (Camell et al., 2015). As such, the question remains unresolved.
SFA-treated MCs also display endoplasmic reticulum (ER) stress and activate the unfolded protein response (UPR), a key component of which is triggered by activation of the ER stress sensor inositol-requiring enzyme 1-alpha (IRE1α). Recognition of unfolded proteins in the ER lumen stimulates the endoribonuclease activity of IRE1α, which splices Xbp1 mRNA to its mature form for translation into the transcription factor X-box-binding protein 1 (XBP1). XBP1 promotes transcription of genes comprising the “adaptive UPR” that together promote restoration of ER homeostasis. More severe or sustained ER stress hyperactivates IRE1α and relaxes its endoribonuclease specificity, leading to degradation of a number of ER-associated transcripts through a process termed regulated IRE1α-dependent decay (RIDD). Engagement of the RIDD pathway promotes cell death by apoptosis and is a component of the “terminal UPR” (reviewed by Maly and Papa, 2014).
Recently, SFA treatment was demonstrated to activate IRE1α by a mechanism independent of unfolded protein recognition (Volmer et al., 2013) and that does not involve the extensive oligomerization of IRE1α that occurs in response to unfolded proteins (Kitai et al., 2013). The relative impact of this novel mode of IRE1α activation on the terminal and adaptive arms of the UPR is unknown, as is the functional significance of SFA-induced IRE1α activation in MMe polarization. Specifically, although IRE1α was shown to mediate NLRP3 inflammasome activation via the RIDD pathway in pancreatic beta cells responding to unfolded protein accumulation (Lerner et al., 2012), its role in SFA-induced NLRP3 inflammasome activation in MCs is unknown.
Here, we identify that the transcriptional program defining SFA-induced MMe polarization is distinct from that of MLPS polarization and is marked by a prominent ER stress signature, which preferentially includes targets of IRE1α-dependent Xbp1 splicing in both mouse and human macrophages. We show in mice that excess dietary SFA consumption induces both Xbp1 splicing and NLRP3 inflammasome activation within the ATM-rich compartment of the white adipose tissue (WAT). Indeed, we demonstrate that IRE1α is a critical mediator of SFA-induced NLRP3 inflammasome activation in macrophages, and that SFAs activate IRE1α by increasing the saturation of membrane phospholipids.
RESULTS
SFA treatment produces a transcriptional signature distinct from LPS treatment and defined by the induction of genes associated with the ER and UPR
We used microarray and functional enrichment analysis to probe the transcriptional response of mouse macrophages to treatment with specific fatty acid (FA) species at a concentration known to reproduce the MMe polarization of ATMs (Kratz et al., 2014). BMDMs were treated for 5 or 20 hours with either stearic acid (SA, 250 μM), a C18:0 SFA, or its C18:1 monounsaturated counterpart oleic acid (OA, 250 μM) in complex with bovine serum albumin (BSA). Responses captured by microarray were compared to those following a 5-hour treatment with BSA alone (control) or low-dose LPS (1 ng/mL). Lists of genes significantly up- or down-regulated by each treatment in comparison to BSA are provided in Table S1.
LPS treatment resulted in a pro-inflammatory transcriptional signature defined by the up-regulation of 1193 genes, enriched as expected in those encoding factors involved in immune responses, cytokine and chemokine activity, and pattern recognition receptor signaling. However, SA treatment at either time point induced only 8% of the genes induced by LPS (Figure 1A), and additional analyses revealed striking differences between the transcriptional patterns induced by LPS and SA. For example, although biological replicates within conditions were tightly grouped, principal components analysis (PCA) showed each individual treatment condition induced an overall transcriptional response that was unique (Figure 1B). The responses to LPS and SA (20-hour) defined the first two principal components, as they were robustly separated both from the BSA-treated baseline and from each other. The response to OA treatment, by comparison, separated weakly from control along the first two principal components, although somewhat further along the third (Figure 1B). These findings together indicate that whereas OA treatment produces a relatively mild transcriptional response, both SA and LPS treatments produce strong transcriptional responses that are remarkably different from one another despite the fact that each induces BMDMs to secrete pro-inflammatory cytokines.
Figure 1. SFA and LPS treatments induce distinct patterns of gene expression in BMDMs.
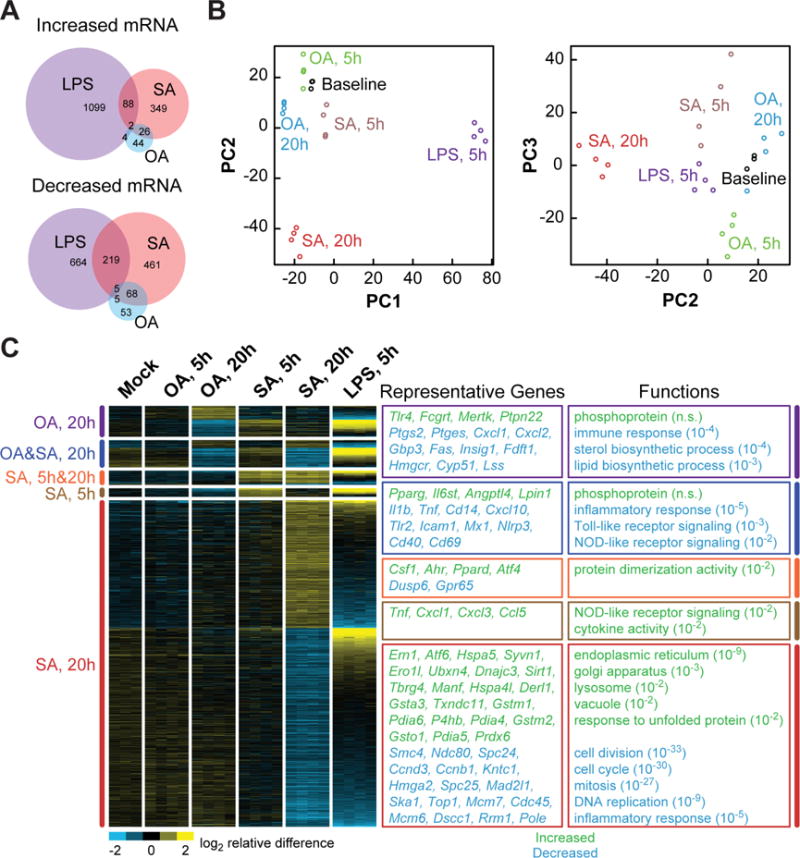
(A) Venn diagrams, depicting the distinct and intersecting nature of gene sets whose transcription is significantly regulated by treatment of BMDMs with LPS (5 h), stearic acid (SA, 5 h or 20 h), or oleic acid (OA, 5 h or 20 h) as compared to baseline vehicle control. (B) Principal components analysis, showing that the treatment conditions above produce distinct transcriptional responses. Each circle represents one biological replicate of the indicated treatment (n=3–4). (C) Heat map, showing categories of genes significantly up- or down-regulated in response one or more of the treatments (indicated at left). Each column represents one biological replicate of the treatment indicated at the top. Boxes at right show enrichment of functional categories (with Benjamini Hochberg-corrected P values in parentheses) and representative genes that were either significantly increased (green) or decreased (blue) in expression in response to one or more treatments. See also Figure S1.
The distinct responses of BMDMs to FAs and LPS are also clear when significant gene expression differences are grouped and visualized as a heat map (Figure 1C). Functional enrichment analysis showed that although 5 hours of SA treatment up-regulated the expression of genes encoding several chemokines and cytokines, more prolonged SA treatment actually reduced the mRNA levels of genes involved in immune responses and inflammatory pathways. By contrast, the set of 433 genes induced by 20 hours of SA treatment was most profoundly enriched in ER, UPR, Golgi apparatus, and lysosome functional groups, a pattern that was not seen in response to LPS treatment. On the other hand, both LPS and SA (20-hour) treatments reduced the expression of genes involved in functions related to cell division. BMDMs treated with OA for 20 hours had a relatively anti-inflammatory transcriptional profile, as seen in the heat map by an opposing gene expression pattern compared to LPS and down-regulation of genes involved in immune responses (Figure 1C).
The distinction between the transcriptional response of BMDMs to SFA and LPS treatments was further underscored by our analysis of BMDCs lacking Toll-like receptor 4 (TLR4), which binds LPS. On one hand, TLR4 deficiency abolished the ability of both LPS and long-chain SFAs, in this case C16:0 palmitic acid (PA), to increase mRNA levels of the MLPS inflammatory marker Nos2 (Figure S1A). However, whereas TLR4 deficiency abolished LPS-dependent secretion of the MLPS cytokine TNF, it only minimally reduced PA-induced TNF secretion (Figure S1B). Furthermore, treating BMDCs with the FA uptake inhibitor sulfo-n-succinimidyl oleate (SSO) markedly reduced PA-induced TNF secretion but did not alter the response to LPS treatment (Figures S1C and D). Taken together, these findings indicate that whereas the LPS-induced inflammatory response of BMDCs is entirely TLR4-dependent, a large component of the low-grade inflammatory response to SFAs is TLR4-independent and instead requires intracellular FA uptake.
SFA treatment preferentially induces adaptive, but not terminal, IRE1α signaling in both mouse and human macrophages
The ER and UPR-enriched transcriptional signature induced by SA treatment in BMDMs included an upregulation of genes targeted by each of the three mammalian ER stress-sensing pathways (IRE1α, PERK, and ATF6). Of these, we noted that IRE1α itself (Ern1) and several XBP1 target genes were particularly induced by SA treatment. Indeed, the upregulated ATF6 target genes were largely shared targets of XBP1. We therefore asked whether SA preferentially induced either the XBP1-dependent adaptive aspects of IRE1α signaling, or the IRE1α-dependent pro-apoptotic RIDD pathway. Genes marking the XBP1-dependent adaptive and RIDD responses, respectively, were compiled from published studies (Lee et al., 2003; Acosta-Alvear et al., 2007; So et al., 2012; Hollien et al., 2009; So et al., 2012; Maurel et al., 2014). Those genes that were identified in at least two of the studies we examined are listed in Figure 2A. Consistent with the reported stimulation of Xbp1 splicing by SFAs in MCs and other cell types, we found that all probes corresponding to the 25 “consensus” XBP1 target genes were numerically increased in BMDMs treated with SA for 20 hours, and 9 of these genes were significantly up-regulated. By contrast, SA treatment did not significantly decrease the expression of any consensus genes known to be downregulated by the RIDD pathway (Figure 2A).
Figure 2. SFA treatment preferentially induces adaptive, but not terminal, IRE1α signaling in both mouse and human macrophages.
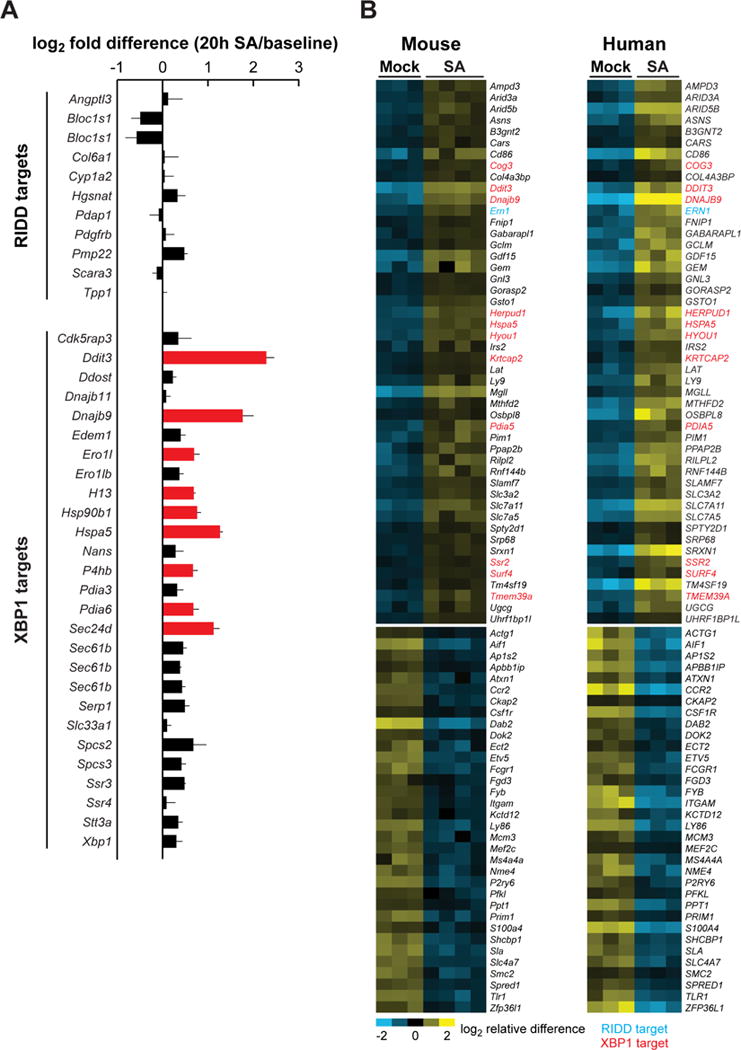
(A) Changes in the expression level of genes associated with the adaptive UPR (XBP1 targets) and the terminal UPR (RIDD targets). Differences between the average log2 expression values for each sample group are shown, with red bars indicating significantly up-regulated genes. Error bars, SD. (B) Heat map, showing the genes significantly up- or down-regulated in response to SA in both mouse BMDMs (this study, 20 h treatment) and human monocyte-derived macrophages (reanalysis of data from Xue, et al. 2014, 24 h treatment). XBP1 targets are shown in red and RIDD targets in blue. See also Figure S2.
In validating our microarray data, we also measured the expression of six RIDD targets by quantitative RT-PCR (qPCR) in BMDCs treated for 4–72 h with a higher dose of SA (500 μM) or thapsigargin (positive control). Whereas thapsigargin treatment tended to reduce mRNA levels of several of these genes, only one (Hgsnat) showed a RIDD-like response to SA treatment (Figure S2A–F). Consistent with this relative lack of RIDD activity, treating BMDCs with 500 μM PA or SA produced relatively little caspase-3/7 activation or cell death as compared to classical UPR inducers or staurosporine, a pro-apoptotic positive control (Figure S2G and H). Together, these data indicate that SFAs activate predominantly the adaptive components of IRE1α signaling and suggest that the SFA-induced UPR may not transition to the terminal state that is triggered by the unremitting accumulation of unfolded proteins.
To test whether the SA-induced IRE1α-XBP1 macrophage signature we identified is conserved between mice and humans, we performed a meta-analysis comparing the results of our 20-hour SA treatment with those of a published bead-array on SA-treated human monocyte-derived macrophages (Xue et al., 2014). The 48 genes induced by SA in both species, shown in a heat map (Figure 2B), were significantly enriched by genes associated with the ER, including 12 XBP1 targets. By contrast, there were no RIDD-responsive genes downregulated by SA treatment in both species (Figure 2B). Our meta-analysis further confirms that SFA treatment selectively induces adaptive IRE1α signaling in macrophages, and suggests that this response is prominent in both mice and humans.
IRE1α is progressively and reversibly activated in mice consuming excess saturated fat
We next examined IRE1α activation in mice consuming a diet rich in saturated fat. Transgenic mice expressing a luciferase reporter coupled to the IRE1α-dependent splicing of Xbp1 (ERAI-Luc; Iwawaki et al., 2009) were longitudinally monitored while being fed standard chow or a high saturated fat diet (HFD; 42 kcal% fat; 62% saturated). HFD consumption for 1–6 months progressively increased whole-body ERAI-Luc reporter activity compared to age-matched chow-fed mice, with the highest signal intensity coming from the abdomen and proximal lower limbs (Figures 3A and B). ERAI-Luc activity was evident within 5 days of HFD initiation and despite up to 6 months of HFD, was reversible within 5 days of replacing the HFD with standard chow (Figures 3C and D). The changes in reporter activity we observed support the concept that IRE1α activation in some tissues may be rapidly responsive to changes in dietary fat consumption and not a consequence of obesity per se.
Figure 3. IRE1α is progressively and reversibly activated in mice consuming excess saturated fat.
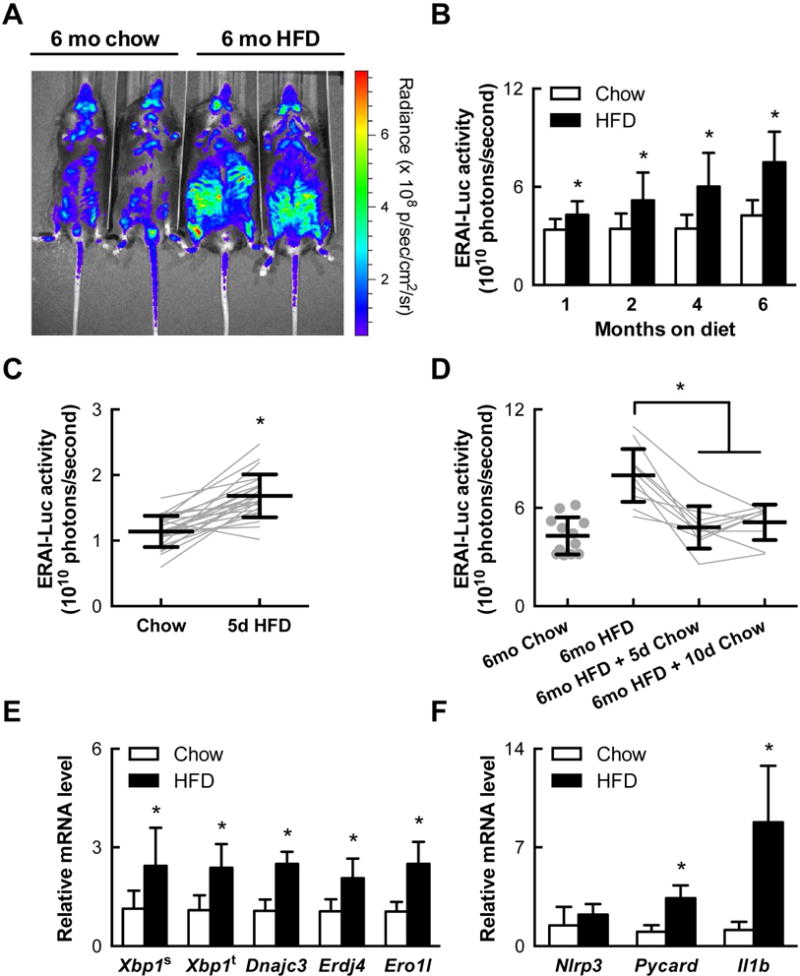
(A) IRE1α-mediated Xbp1 splicing activity as measured by a luciferase reporter (ERAI-Luc) in live mice fed chow or a high fat diet (HFD) for 6 months (representative images). (B) Quantification of whole-body luciferase activity in ERAI-Luc mice (n=9–24) fed chow or HFD and imaged as in (A). (C) Whole-body IRE1α reporter activity in mice before and after 5 days of HFD (n=24). (D) Reversal of IRE1α reporter activity in mice switched to chow for 5–10 days after 6 months of HFD. (E) Expression of total and spliced Xbp1 and XBP1-target genes Erdj4, Ero1l, and Dnajc3 as measured by quantitative PCR in the stromal vascular fraction (SVF) of visceral adipose tissue (VAT) of mice fed chow (n=15–18) or HFD for 3 months (n=8–10). (F) Expression of NLRP3 inflammasome-related genes Il1b, Nlrp3, and Pycard in VAT SVF as in (E). Error bars, SD. *, p<0.05 vs. vehicle.
Focusing specifically on the WAT, we found that feeding mice a HFD for 3 months increased the mRNA levels of spliced Xbp1 and XBP1-dependent target genes in the ATM-containing stromal vascular fraction (SVF) of visceral WAT depots (Figure 3E). Interestingly, HFD-induced XBP1-dependent signaling in SVF cells was concurrent with a significant increase in the mRNA levels of the gene encoding IL-1β (Il1b) and the NLRP3 inflammasome component Pycard (Figure 3F). These data suggest that IRE1α activation in the visceral ATMs of HFD-fed mice is accompanied by NLRP3 inflammasome activation, as it is in MCs treated with SFAs in vitro.
SFAs act intracellularly to activate IRE1α and the NLRP3 inflammasome
We examined the mechanism by which SFAs activate IRE1α in BMDCs from ERAI-Luc mice. PA treatment induced ERAI-Luc reporter activity in a dose- and time-dependent manner, with high PA concentrations producing responses on par with those induced by treatment with the classical UPR activators thapsigargin, dithiotreiol (DTT), and tunicamycin (Figure 4A). Moreover, this response was highly specific to long-chain SFAs (PA and SA) as opposed to medium-chain SFAs or either long-chain monounsaturated or polyunsaturated FAs (Figure 4B). Similar SFA-induced ERAI-Luc reporter responses were seen in BMDMs (Figure S3A).
Figure 4. Long-chain SFAs activate IRE1α and the NLRP3 inflammasome through an intracellular mechanism.
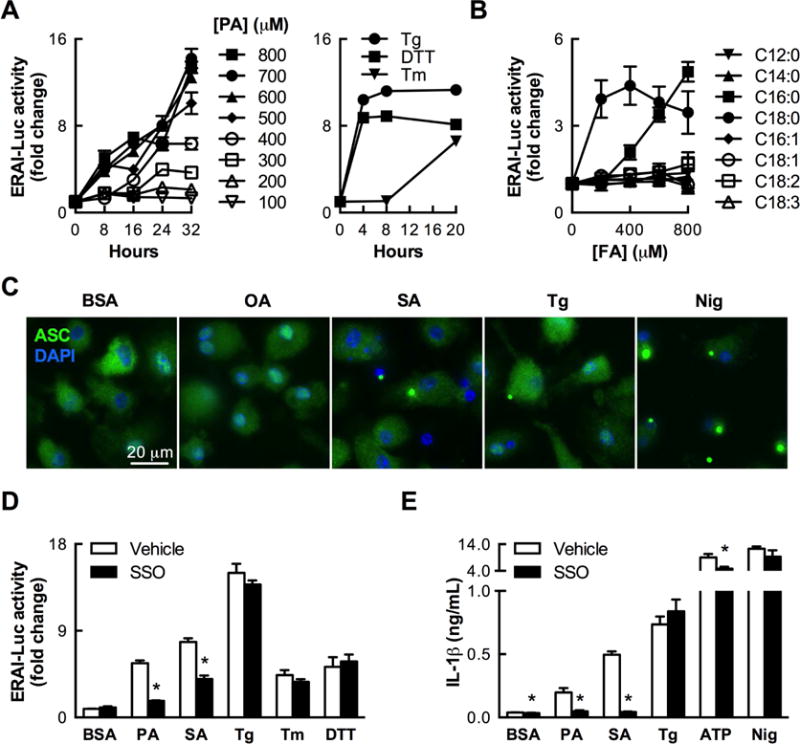
(A) Time course of IRE1α-mediated Xbp1 splicing activity, as measured by a luciferase reporter in mouse BMDCs (n=2–3) treated with palmitic acid (PA) or the classical ER stress inducers thapsigargin (Tg, 100 nM), dithiothreiol (DTT, 5 mM), and tunicamycin (Tm, 5 μg/mL). (B) Long-chain SFAs PA (C16:0) and SA (C18:0) specifically induce IRE1α reporter activity in BMDCs treated for 24 h with a variety of FAs (n=3). (C) Formation of ASC puncta indicative of NLRP3 inflammasome activation in BMDCs treated for 20–24 h with SA (500 μM), thapsigargin (200 nM), or nigericin (positive control), but not OA (500 μM) or BSA (vehicle control for FA treatments). (D) Inhibiting FA uptake with sulfo-n-succinimidyl oleate (SSO, 500 μM) reduces IRE1α reporter activity (n=6) stimulated by SFAs (1 mM PA, 500 μM SA) but not classical ER stress inducers (200 nM Tg, 5 μg/mL Tm, 2.5 mM DTT). (D) SSO specifically abrogates IL-1β secretion induced by SFAs (500 μM) but not other stimuli (n=4). In all experiments measuring IL-1β secretion, BMDCs were primed with LPS (200 ng/mL, 3 h) prior to stimulation. Error bars, SD. *, p<0.05 vs. vehicle. See also Figure S3.
Building on work showing that SFAs stimulate myeloid cells to secrete IL-1β, an NLRP3 inflammasome-dependent cytokine (Wen et al., 2011), we found that treating LPS-primed BMDCs with either SA or thapsigargin induced formation of intracellular ASC puncta, indicating inflammasome activation, whereas OA treatment did not do so (Figure 4C). SA and PA, but not OA, also induced NLRP3 inflammasome-dependent IL-1β secretion in LPS-primed BMDMs (Figure S3B).
On the other hand, LPS priming only minimally impacted SA-induced ERAI-Luc reporter activity (Figure S3C), and LPS treatment alone only modestly stimulated this indicator of Xbp1 splicing in BMDCs (Figure S3D). Whereas TLR4 deficiency essentially eliminated the capacity of LPS to induce Xbp1 splicing in BMDCs, it had a much smaller impact on the corresponding response to SA treatment (Figure S3D).
SSO treatment, however, partially abrogated Xbp1 splicing induced by PA or SA without impacting the responses to classical UPR activators (Figure 4D), indicating that intracellular entry, rather than TLR4, is required for SFAs to induce IRE1α-dependent Xbp1 splicing. SSO treatment also abolished IL-1β secretion from LPS-primed BMDCs stimulated with PA or SA, whereas it did not alter IL-1β secretion in response to thapsigargin or the canonical NLRP3 inflammasome activators ATP and nigericin (Figure 4E). Treatment with PA or SA was notably insufficient to induce IL-1β secretion by BMDCs without LPS priming (data not shown), likely due to an inability of SFAs to induce substantial Il1b transcription on their own.
IRE1α endoribonuclease activity is required for SFA-induced NLRP3 inflammasome activation
IRE1α activation has been linked to NLRP3 inflammasome activity in beta cells (Lerner et al., 2012). This, and our finding that intracellular SFAs induce both IRE1α and NLRP3 inflammasome activation, prompted us to consider whether IRE1α mediates SFA-induced NLRP3 inflammasome activation in MCs. To test this, we manipulated IRE1α (Ern1) expression by treating BMDCs from mice expressing a conditional allele of Ern1 (Ern1fl/fl) with a cell-permeable Cre recombinase. This treatment reduced Ern1 mRNA levels by 63% (Figure 5A) and similarly reduced SFA-induced IL-1β secretion without altering the response to ATP, which stimulates IL-1β secretion without inducing ER stress (Figure 5B). We also crossed Ern1fl/fl mice with mice that preferentially express Cre in MCs (Cd11b-Cre). Primary BMDCs cultured from Cd11b-Cre+/Ern1fl/fl mice secreted less IL-1β in response to PA treatment than did similarly treated BMDCs harvested from Cd11b-Cre−/Ern1fl/fl mice (Figure 5C), although examination of Ern1 mRNA expression suggested low Cre-LoxP excision efficiency in this model (data not shown). Overall, our findings indicate that IRE1α is required for SFA treatment to activate the NLRP3 inflammasome.
Figure 5. IRE1α endoribonuclease activity is required for SFA-induced NLRP3 inflammasome activation.
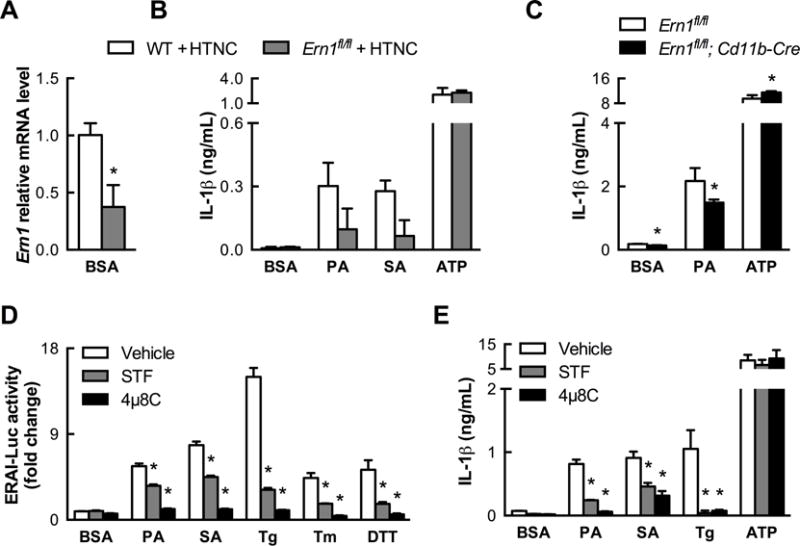
(A) Extent of in vitro deletion of IRE1α (Ern1) by treating Ern1fl/fl BMDCs with the cell-permeable Cre recombinase HTNC, as measured by quantitative PCR (n=3). (B) IRE1α-deficient BMDCs from (A) exhibit reduced IL-1β secretion when stimulated by SFAs (500 μM, 20 h) but not by ATP (n=2–3). (C) Partial deletion of IRE1α in Cd11b-Cre+/Ern1fl/fl BMDCs also modestly diminishes IL-1β secretion stimulated by PA (500 μM, 24 h) but not by ATP (n=4). (D) Efficacy of IRE1α endonuclease inhibitors STF083010 (200 μM) and 4μ8C (50 μM) in reducing IRE1α reporter activation by 24 h treatment with PA (1 mM), SA (500 μM), or classical ER stress inducers (n=6). (E). STF083010 (200 μM) and 4μ8C (200 μM) abrogate IL-1β secretion stimulated by SFAs (500 μM, 24 h) but not ATP (n=3). Error bars, SD. *, p<0.05 vs. WT + HTNC (A,B), Ern1fl/fl (C), or vehicle (D,E). See also Figure S4.
IRE1α is a bifunctional protein with both kinase and endoribonuclease activities. To determine which activity is responsible for NLRP3 inflammasome activation, we treated WT BMDCs with STF083010 and 4μ8C, two specific IRE1α endoribonuclease inhibitors. Remarkably, both inhibitors markedly decreased the extent to which SFA treatment induced either Xbp1 splicing or consequent IL-1β secretion (Figures 5D and E). Moreover, they also reduced the ability of other ER stress inducers (e.g. thapsigargin) to activate the NLRP3 inflammasome without altering the ability of ATP to do so. By contrast, treating BMDCs with APY29, which is both an ATP-competitive kinase inhibitor and an allosteric endoribonuclease activator of IRE1α (Wang et al., 2012), failed to reduce SFA-induced IL-1β secretion and was sufficient to induce IL-1β secretion on its own (Figure S4A). Thus, the endoribonuclease activity of IRE1α in MCs specifically mediates NLRP3 inflammasome activation in response to SFAs and other inducers of ER stress.
Models of ER stress resulting from unfolded protein accumulation showed that IRE1α promotes the RIDD-dependent induction of TXNIP, which associates with NRLP3 in the presence of reactive oxygen species (ROS) and promotes inflammasome activation (Lerner et al. 2012; Oslowski et al., 2012)). As predicted by the relative lack of RIDD activity seen in SFA-treated MCs (Figures 2 and S2), Txnip mRNA levels in BMDCs were not increased by PA or SA treatment for durations of time over which both thapsigargin and tunicamycin induced Txnip to peak levels (Figure S4B). Indeed, SA did not increase Txnip at any point over a 72-hour treatment (Figure S4C). Furthermore, the antioxidants N-acetylcysteine and ebselen reduced IL-1β secretion in response to treatment with thapsigargin, but not PA or SA (Figure S4D). Our studies focused on TXNIP and ROS together suggest that IRE1α mediates NLRP3 inflammasome activation by distinct mechanisms when activated by SFAs versus classical UPR activators.
SFAs and classical UPR activators induce Xbp1 splicing by distinct mechanisms
In addition to engaging stimulus-specific downstream effectors of NLRP3 inflammasome activation, we found evidence indicating that SFAs and chemical inducers of ER stress also activate IRE1α by different mechanisms. In this effort, we studied MCs co-treated with unsaturated fatty acids (UFAs), which are can protect many cell types against several effects of SFA exposure (Welters et al., 2004; L’homme et al., 2013). Co-treating BMDCs with monounsaturated or polyunsaturated UFAs dose-dependently abolished Xbp1 splicing in response to PA without affecting responsiveness to thapsigargin, tunicamycin, or DTT (Figure 6A). Prolonged pretreatment with OA, even when stopped prior to PA treatment, could reproduce the protective effect of UFA co-treatment, but adding OA to BMDCs after PA treatment was initiated could not reverse Xbp1 splicing even if the PA was removed from the medium (Figure S5A).
Figure 6. Co-treatment with UFAs specifically mitigates SFA-induced IRE1α and NLRP3 inflammasome activation.
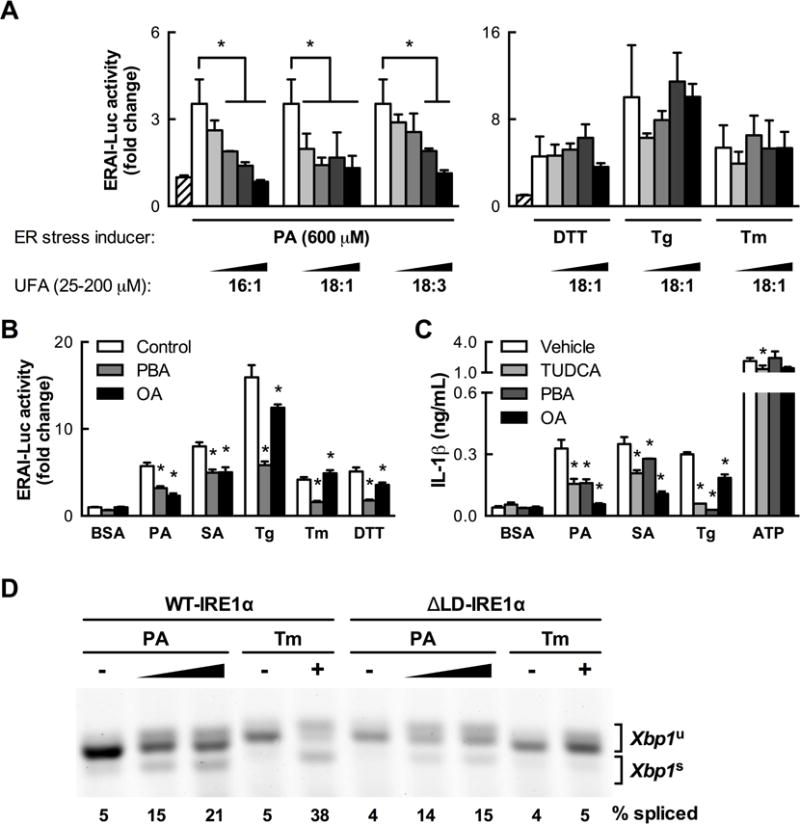
(A) UFA co-treatment of BMDCs dose-dependently reduces IRE1α reporter activity stimulated by PA but not by classical ER stress inducers (2.5 mM DTT, 100 nM Tg, 5 μg/mL Tm, 24 h, n=3). (B) In contrast to the SFA-specific protective effect of OA co-treatment (100 μM, 24 h), co-treatment with the chemical chaperone 4-phenylbutyric acid (PBA, 1 mM) mitigates IRE1α reporter activity induced by both SFAs (1 mM PA, 500 μM SA) and classical ER stress inducers (n=6). (C) Reduction of IL-1β secretion induced by SFAs (500 μM, 24 h, n=3–4) and other stimuli by co-treatment with the chemical chaperone tauroursodeoxycholic acid (TUDCA, 1 mM), PBA or OA as in (B). (D) Xbp1 splicing measured by RT-PCR and gel electrophoresis in mouse embryonic fibroblasts expressing only wild type (WT-IRE1α) or mutant IRE1α lacking the luminal domain (ΔLD-IRE1α) treated with PA (0.5–1 mM) or Tm (5 μg/ml). PA and Tm lanes were taken from separate sections of the same gel. Error bars, SD. *, p<0.05 vs. control unless otherwise indicated. See also Figures S5, S6, S7, and S8.
The ability of UFAs to block Xbp1 splicing induced by SFAs but not classical UPR activators suggests that SFAs activate IRE1α through a mechanism at least partly independent of unfolded protein accumulation. To test this possibility, we treated BMDCs with the chemical chaperones 4-phenylbutyrate (PBA) and tauroursodeoxycholic acid (TUDCA). PBA reduced Xbp1 splicing induced by classical UPR activators by 62–65% but only diminished SFA-induced Xbp1 splicing by 38–45% (Figure 6B). Similarly, PBA and TUDCA abolished IL-1β secretion in response to thapsigargin, but only partially reduced SFA-induced IL-1β secretion (Figure 6C). Finally, combining PBA or TUDCA with OA co-treatment did not add to the ability of OA to abrogate SFA-induced Xbp1 splicing or IL-1β secretion (Figure S5B and C).
Together, these data suggest that although SFA treatment may induce some degree of unfolded protein accumulation, SFAs induce IRE1α activation through a mechanism that is at least partially distinct from that by which unfolded protein accumulation activates IRE1α. To test this, we built on emerging evidence that IRE1α can be activated through an ability to sense increases in ER membrane lipid saturation (Volmer et al., 2013). We found that mouse embryonic fibroblasts that only express a mutant form of IRE1α incapable of recognizing unfolded proteins could still splice Xbp1 in response to PA treatment, even though their ability to do so in response to tunicamycin was abolished (Figure 6D).
Protein kinase RNA-like endoplasmic reticulum kinase (PERK) is another sensor of ER stress and has also been implicated in membrane lipid sensing (Volmer et al., 2013). Consistently, we found that the kinetics with which SA treatment of BMDCs phosphorylated PERK was similar to those with which it phosphorylated IRE1α (Figure S6A). By contrast, ATF6α is an ER stress sensor that has not been implicated in lipid sensing, and we found that SA treatment of BMDCs induced little proteolytic processing or nuclear translocation of ATF6α (Figure S6B). Taken together, these data support the idea that SFAs can activate IRE1α and PERK in BMDCs through changes in ER membrane composition but that ATF6α activation requires unfolded protein accumulation at levels beyond those induced by SFA treatment. Unlike the impact of inhibiting IRE1α endoribonuclease activity however, PERK inhibition did not reduce SFA-induced IL-1β secretion (Figure S6C).
Intracellular SFAs flux in a way that is distinct from UFAs and not controlled by UFA levels
Intracellular UFAs are proposed to mitigate the lipotoxic effects of SFAs by promoting the flux of SFAs into triacylglycerol (TG; Listenberger et al., 2003), which would potentially reduce their availability to increase ER membrane lipid saturation and thus stimulate IRE1α activation (Volmer et al., 2013). We treated BMDCs with radiolabeled FAs and found that intracellular OA accumulates preferentially in TG and cholesterol ester storage pools, whereas PA fluxes predominantly into diacylglycerol, phospholipid, and sphingolipid pools (Figures S7A and B). However to our surprise, we did not find that co-treating BMDCs with OA affects the degree to which PA fluxes into TG or any other lipid compartment, precluding the possibility that OA exerts its protective effects in BMDCs by boosting the incorporation of PA into TG (Figures S7C and D).
Furthermore, altering the capacity of macrophages to form TG had little impact on the ability of SFA treatment to activate either IRE1α or the NLRP3 inflammasome. For example, BMDCs from WT mice lacking the TG-synthetic enzyme DGAT1 (Dgat1−/−), or transgenic mice overexpressing DGAT1 in MCs (aP2-Dgat1) did not substantially differ in their vulnerability to SFA-induced Xbp1 splicing (Figure S8A). Moreover, neither genetic deficiency nor pharmacologic inhibition of DGAT1 could impair either the capacity of SFA treatment to induce IL-1β secretion or the protective effects of OA in SFA-treated BMDCs (Figures S8B–E). These findings support the concept that rather than altering the flux of SFAs into TG, the ability of UFAs to protect against SFA-induced IRE1α activation and subsequent NLRP3 inflammasome activation is a function of their own flux into phospholipids or other lipid species that may counteract SFA-induced changes in membrane lipid composition and saturation.
The flux of SFAs into phosphatidylcholine contributes to IRE1α and NLRP3 inflammasome activation
If SFA-induced IRE1α activation is a function of effects on ER membrane lipid composition and saturation, as our data suggests, then blocking the flux of SFAs into membrane lipids such as phospholipids and sphingolipids should mitigate IRE1α activation and resultant NLRP3 inflammasome activation. Indeed, inhibiting the first step of de novo sphingolipid and ceramide synthesis with myriocin was shown to reduce IL-1β secretion in macrophages co-treated with PA and LPS (Schilling et al., 2012). However, more recent work has called this mechanism into question (Camell et al., 2015), and we observed that myriocin treatment did not significantly reduce IRE1α activation or IL-1β secretion in response to PA (Figure S9A and B).
Phosphatidylcholine (PC) is the most abundant phospholipid in mammalian cellular membranes, including those of the ER, and we saw that BMDCs form PC to a greater degree upon treatment with PA than OA (Figures S7A and B). As such, we sought to determine the impact of limiting PC synthesis in BMDCs treated with SFAs. The synthetic alkylphospholipids miltefosine and edelfosine inhibit CDP:phosphocholine cytidylyltransferase (CCT), which catalyzes the rate-limiting step of PC biosynthesis. Treatment with either miltefosine or edelfosine reduced the ability of both PA and SA to induce Xbp1 splicing and IL-1β secretion, suggesting that incorporation of SFAs into PC contributes to the activation of IRE1α (Figures 7A and B).
Figure 7. Limiting the ability of SFA treatment to increase phospholipid saturation blocks consequent IRE1α and NLRP3 inflammasome activation.
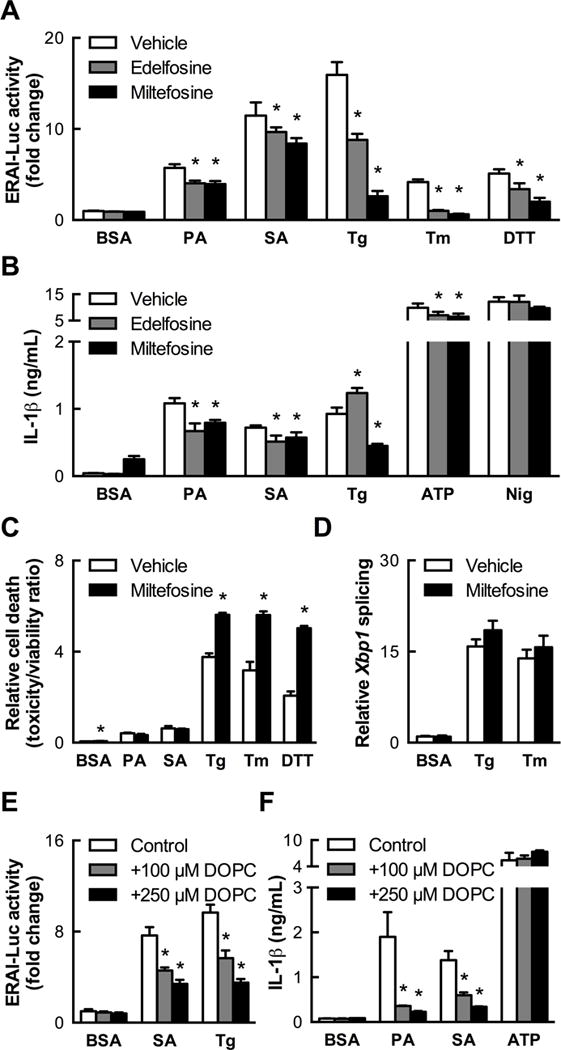
(A) Phosphatidylcholine biosynthesis inhibitors edelfosine (25 μM) and miltefosine (100 μM) reduce IRE1α reporter activity induced by SFAs (1 mM PA, 500 μM SA, 24 h, n=6). (B) Edelfosine and miltefosine co-treatment as in (A) diminishes IL-1β secretion induced by SFAs (500 μM, 24 h, n=4). (C) Miltefosine co-treatment potentiates cell death due to classical ER stress inducers but not SFAs (500 μM, 24 h, n=4). (D) Tg- and Tm-induced Xbp1 splicing is unaffected by miltefosine co-treatment when measured by quantitative PCR (n=4), indicating that reductions in IRE1α reporter activity in BMDCs co-treated with inhibitors and classical ER stress inducers in (A) are secondary to cell death. (E) Reduction of SA- and Tg-induced IRE1α reporter activity in BMDCs co-treated with dioleoylphosphatidylcholine (DOPC) liposomes for 24 h (n=5). (F) Abrogation of SFA-induced, but not ATP-induced, IL-1β secretion by DOPC co-treatment of BMDCs (n=3–4) as in (E). Error bars, SD. *, p<0.05 vs. vehicle (A–D) or control (E,F). See also Figure S9.
Interestingly, whereas CCT inhibition was tolerated by untreated BMDCs and those treated with SFAs, it caused pervasive cell death and reduced ERAI-Luc reporter activity in cells treated with thapsigargin, tunicamycin, or DTT (Figure 7A and C). Moreover unlike its effect on SFA treatment, miltefosine did not reduce Xbp1 splicing induced by thapsigargin and tunicamycin when measured by qPCR (Figure 7D). This finding suggests that the miltefosine-induced reduction in ERAI-Luc reporter activity seen in the context of severe unfold protein accumulation is secondary to cell death and highlights the potential importance of PC biosynthesis in the ability of BMDCs to contend with ER stress under such conditions.
Our findings support the concept that SFA flux into PC leads to IRE1α activation and that UFAs counteract this activation through their own flux into phospholipids. We therefore reasoned that enriching MC membranes with unsaturated PC would be sufficient to reproduce the protective effects of UFA co-treatment. BMDCs were co-treated with SFAs and liposomes composed of dioleoylphosphatidylcholine (DOPC). Remarkably, DOPC liposomes (100–250 μM) abrogated SA-induced Xbp1 splicing (Figure 7E) to a degree similar to that observed with OA co-treatment at comparable concentrations (Figure 6B and C) and also reduced Xbp1 splicing in response to thapsigargin (Figure 7E). Together these data strongly support the concept that reducing the saturation status of membrane phospholipids prevents activation of IRE1α by SFA excess and decreases the sensitivity of IRE1α to unfolded protein accumulation.
Importantly, and in correspondence with their effect on limiting IRE1α activation, DOPC liposomes also protected against IL-1β secretion induced by SFAs or thapsigargin but not by ATP (Figure 7F). Thus, shifting the equilibrium of cellular phospholipids to favor higher unsaturation specifically abolishes the ability of SFAs to induce IL-1β secretion without impairing the responsiveness to DAMPs that are indicative of infection or tissue damage.
DISCUSSION
Although ATMs chronically activated by DIO share features in common with macrophages responding acutely to pathogens, such as secretion of pro-inflammatory cytokines, key distinctions exist that could potentially be exploited therapeutically to combat inflammation-related metabolic dysfunction. Here we show that macrophages activated by SFA treatment display a pronounced transcriptional signature of ER stress, in striking contrast to the inflammatory signature induced by stimulation with LPS. This ER stress response, driven by the flux of SFAs to PC and resultant IRE1α activation, has functional consequences. In particular, we found that SFA-induced IRE1α endoribonuclease activity mediates activation of the NLRP3 inflammasome and secretion of IL-1β, a pro-inflammatory cytokine associated with insulin resistance. These findings were corroborated in vivo, as we could progressively increase tissue IRE1α activity by feeding mice a diet high in saturated fat. Indeed, such a diet activated both IRE1α and the NLRP3 inflammasome in the ATM-rich compartment of the WAT. Thus, IRE1α activation among tissue MCs may be important in promoting inflammation and metabolic disease in DIO.
SFAs are thought to elicit pro-inflammatory TLR4 signaling in MCs on account of their similarity to the saturated acyl chains of LPS (Lee et al., 2001; Suganami et al., 2007), which bind specifically to TLR4 (Park et al., 2009). However, direct binding of SFAs to TLR4 has never been demonstrated (Schaeffler et al., 2009), though an adaptor protein, Fetuin A, has been proposed (Pal et al., 2012). It has become increasingly clear that SFAs do not merely mimic LPS (Erridge and Samani, 2009; Xu et al., 2013; Kratz et al., 2014), and our microarray analysis underscores this point. Though SFAs do stimulate MLPS inflammatory pathways and the secretion of associated cytokines such as TNF from BMDCs, the magnitude of this response is small compared to that induced by LPS or other PAMPs, and SFAs are insufficient to substitute for LPS in priming the NLRP3 inflammasome. Furthermore, we found that many of the responses to SFA treatment are independent of TLR4 and instead require FA uptake, suggesting that additional intracellular pathways mediate SFA-induced macrophage activation.
Indeed, we demonstrate that activation of IRE1α via the flux of SFAs into phospholipids and resulting induction of the UPR are defining features of MC activation by SFAs. Previous transcriptional and proteomic analyses of macrophages treated with SFAs have revealed programs of lysosomal biogenesis and intracellular lipid metabolism (Xu et al., 2013; Kratz et al., 2014), but a predominant ER stress signature has not been described. Differences in methodology may account for these varying results. Nonetheless, our meta-analysis against SA-treated human macrophages strongly confirms our findings in mouse MCs and suggests that activation of IRE1α by SFAs is a conserved process.
Our transcriptome analysis also reveals that SFA treatment disproportionately induces the XBP1-dependent adaptive arm of IRE1α signaling. Despite stimulating robust IRE1α activation, evidenced by similar levels of Xbp1 splicing to those elicited by thapsigargin and other UPR inducers, SFA treatment does not result in an extensive RIDD response. This bias towards adaptive IRE1α signaling may outweigh the pro-apoptotic pathways associated with the PERK arm of the UPR, as we observed little apoptosis or cell death in BMDCs treated with SFAs despite a significant level of PERK phosphorylation.
In models of prolonged or irremediable ER stress due to unfolded protein accumulation, IRE1α undergoes a “switch” from adaptive to terminal signaling outputs, which is mediated by a transition to higher-order oligomerization of IRE1α that promotes RIDD (reviewed by Maly and Papa, 2014). Our results lead us to speculate that activation of IRE1α in response SFAs does not lead to extensive oligomerization of IRE1α. Consistent with this hypothesis, SFA-treated cells expressing fluorescently tagged IRE1α do not form the large IRE1α oligomeric puncta that are seen following treatment with classical ER stress inducers (Kitai et al., 2013). Thus, SFA treatment may represent a paradigm in which progressive, dose-dependent Xbp1 splicing does not trigger the typical switch-like behavior of IRE1α. This SFA-induced mode of IRE1α activation appears to mediate NLRP3 inflammasome activation by a different mechanism than that which does so under conditions of excess unfolded proteins, and the molecular features defining this mechanism need to be explored. Based on this, our data suggest that disrupting either Xbp1 splicing or XBP1-dependent transcriptional activity may reveal new ways to control NLRP3 inflammasome activation in the context of SFA excess and should be a focus of future studies.
Though IRE1α is best known as a sensor of unfolded proteins, its ability to directly sense changes in membrane lipid composition in mammalian cells was recently appreciated (reviewed by Volmer and Ron, 2015). This ability of IRE1α to recognize and respond to ER membrane perturbations may be critically linked to its role in regulating lipid homeostasis. Activation of IRE1α promotes phospholipid synthesis in order to expand the capacity of the ER during times of increased protein folding demand. The importance of this adaptive response for cell survival was strikingly evident in our studies, as BMDCs in which PC biosynthesis was pharmacologically impaired did not survive treatment with classical ER stress inducers. By contrast, inhibiting PC biosynthesis did not cause such cell death in response to SFA treatment, suggesting that the unfolded protein burden associated with SFA-stimulated IRE1α activation does not necessitate ER expansion to the degree that is required following treatment with traditional UPR inducers.
Though we clearly show that the ability of IRE1α to recognize unfolded proteins is not needed for its activation by SFAs, we cannot completely rule out the possibility that unfolded proteins may play some role in this process. For example, many chaperones that facilitate protein folding require calcium, which is depleted from the ER following SFA exposure (Wei et al., 2009; Egnatchik et al., 2014). ER luminal calcium levels are maintained by the sarco/endoplasmic reticulum calcium ATPase (SERCA), the activity of which is impaired by obesity-induced changes in the PC/PE ratio of the ER (Fu et al., 2011) or by enrichment of the ER with saturated PC (Li et al., 2004). Given the partial protection we saw using chemical chaperones, it is plausible that unfolded proteins could accumulate secondary to SFA-induced changes in ER membrane composition. However, any unfolded protein accumulation due to SFA treatment is likely mild, as it did not appear to engage the ATF6α arm of the UPR in our studies.
The ability of UFAs to mitigate the lipotoxic effects of SFAs are well appreciated (Welters et al., 2004; L’homme et al., 2013) and have been proposed to do so by facilitating the incorporation of SFAs into TG and away from potentially deleterious metabolic pathways (Listenberger et al., 2003; Coll et al., 2008; Leamy et al., 2014). Consistent with this, we previously showed that increasing TG synthesis capacity in MCs by overexpressing DGAT1 was anti-inflammatory against SFAs in vitro or HFD in vivo (Koliwad et al., 2010). Here, however, we found that OA co-treatment did not enhance PA flux to TG, and that altering DGAT1 activity or expression did not affect SFA-induced IRE1α or NLRP3 inflammasome activation.
Instead, we found that co-treatment with DOPC reproduces the protective effect of OA co-treatment, supporting the concept that the flux of OA itself into PC and/or other phospholipids counteracts the effect of SFA treatment on membrane saturation. Furthermore, DOPC also protected against IRE1α activation stimulated by thapsigargin, suggesting that increased membrane fluidity may also limit the activation of IRE1α in response to unfolded proteins. OA co-treatment does not have this effect, but it may not alter ER membrane fluidity to the same degree as direct supplementation with DOPC.
What are the physiological consequences of SFA-induced IRE1α activation in MCs? We show a progressive rise in IRE1α activity in tissues of mice consuming a diet rich in saturated fat, including the ATM compartment of WAT, and our findings imply that MCs exposed to SFAs undergo a sustained form of IRE1α activation that fails to resolve, yet does not transition to a terminal program. As this unremitting form of IRE1α activation is coupled with NLRP3 inflammasome activation both in vitro and in the ATMs of mice fed a HFD, our findings could help explain what promotes MCs in metabolic tissues to persistently propagate metabolic inflammation in the context of DIO. Additionally, our findings suggest that features of the chronic, low-level inflammation seen in DIO may be cell-autonomous, and targeting the intracellular pathways driving these programs could be effective in limiting metabolic inflammation and its consequences.
However, prior efforts to block inflammatory pathways in the context of DIO have been hampered by a lack of specificity that is problematic, as general immunosuppression can result in an unacceptable vulnerability to infection. Importantly, we show here that inhibiting IRE1α, or the membrane perturbations that lead to its activation, abolishes IL-1β secretion stimulated by SFAs without affecting NLRP3 inflammasome activation in response to other triggers associated with infection or tissue damage. Though there are other implications of targeting this pathway that require further study, we have uncovered a mechanism for SFA-induced activation of IRE1α and the NLRP3 inflammasome with translational implications relevant to combating metabolic inflammation without impairing acute inflammatory responses to pathogens.
EXPERIMENTAL PROCEDURES
Isolation of primary mouse bone marrow-derived MCs
All procedures were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. Wild type, ERAI-Luc (Iwawaki et al., 2009), Tlr4−/− (Poltorak et al., 1998), Dgat1−/− (Chen et al., 2002a), and aP2-Dgat1 (Chen et al., 2002b) mice on a C57Bl/6 background and Cd11b-Cre/Ern1fl/fl mice (Boillée et al., 2006; Iwawaki et al., 2009) on a mixed 129/C57Bl/6 background were group-housed and fed a standard chow diet (LabDiet 5053) in a barrier facility with a 12-hour light-dark cycle. Bone marrow cells were isolated from mice euthanized with Avertin and differentiated for 6–8 days in RPMI 1640 (Gibco) containing 10% FBS (Atlanta Biologicals), penicillin/streptomycin, and either recombinant GM-CSF (for BMDCs) or M-CSF (for BMDMs; 10 ng/mL; Peprotech).
Microarray sample preparation
Replicate wells (n = 3–4) of WT BMDMs were treated with LPS (1 ng/mL), SA or OA (250 μM), or BSA (125 μM) for 5 or 20 hours as indicated. Purified DNase-treated total RNA was analyzed for quality using chip-based capillary electrophoresis (Bioanalyzer, Agilent, Inc) and quantity and purity was determined with a NanoDrop spectrometer. The NuGEN Pico V2, based on Ribo-SPIA technology, was used for amplification, fragmentation, and biotin-labeling of ~10 ng input RNA. The labeled cDNA was hybridized to GeneChip Mouse Gene 1.0 ST microarrays (Affymetrix, Santa Clara, CA). Signal intensities of fluorescent images produced during GeneChip hybridizations were read by an Affymetrix Model 3000 Scanner and converted into GeneChip probe results files (CEL) using Command and Expression Console software (Affymetrix). Array data are available in the NCBI Gene Expression Omnibus (GEO) under the accession number GSE77104.
Microarray data analysis
Data were quantile normalized and median centered across samples as described (Baccarella et al., 2013). Differentially expressed genes were identified using Significance Analysis of Microarrays (Tusher et al., 2001) using two-class unpaired comparisons with 3% false discovery rate (FDR) and 1.5-fold change cutoffs. Clustering was performed with xcluster (Gollub and Sherlock, 2006) and visualized as heatmaps using Java Treeview (Saldanha, 2004). Gene set intersections were determined using custom Perl scripts. Principal component analysis was performed in R. Functional enrichment analysis utilized DAVID (Huang et al., 2009). Human monocyte-derived macrophage datasets were downloaded from GEO (Accession number GSE46903) and processed identically to our mouse datasets. Only data from GM-CSF-derived macrophages treated for 24 hours with 150 μM SA and corresponding baseline controls were used in our analysis. The intersection of mouse and human genes significantly induced by SA was identified using custom scripts.
In vivo luminescence imaging
Male ERAI-Luc mice (Iwawaki et al., 2009) fed chow or HFD (Harlan Teklad TD.88137) starting at 4–6 weeks of age were imaged in the IVIS Spectrum (PerkinElmer) under isoflurane anesthesia 10 minutes after intraperitoneal injection of luciferin (150 mg/kg, GoldBio). Whole body radiance was quantified using Living Image software (PerkinElmer).
Cytokine measurements
Cytokine levels in conditioned media were measured by Ready-Set-Go ELISA kits (Ebioscience). For all IL-1β secretion measurements, BMDCs were primed with 200 ng/mL LPS (Sigma Aldrich L4391) for 3 hours prior to FA treatment. Pharmacological inhibitors, when used, were present during both priming and treatment periods. ATP (Sigma Aldrich A7699, 5 mM, 45–120 minutes) and nigericin (Calbiochem 481990, 5 μM, 1 hour) were used as positive controls.
In vitro deletion of Ern1 and expression of mutant IRE1α
Ern1fl/fl and control WT BMDCs were treated with HTNC (Excellgen RP-7) as described (Krahmer et al., 2011). Ern1−/− MEFs were rescued with full-length human IRE1α or human IRE1α lacking its luminal domain (ΔLD-IRE1α) by retroviral transduction as described (Volmer et al., 2013).
Statistics
Data are represented as mean ± SD and considered statistically significant at P < 0.05. Outliers were removed according to Grubbs’ test (α = 0.05). Analyses were performed in GraphPad Prism using two-tailed Student’s t test or ANOVA with Holm-Sidak or Dunnett’s post-tests in the case of multiple comparisons.
Supplementary Material
Highlights.
Saturated fatty acids induce an ER stress transcriptional signature in macrophages
Flux of saturated fatty acids into phospholipids activates the ER stress sensor IRE1α
IRE1α mediates saturated fatty acid-induced activation of the NLRP3 inflammasome
Acknowledgments
We thank Hui Wang and the Gladstone Institutes Genomics and Bioinformatics Cores for technical assistance, Dr. Joachim Schultze and his laboratory for making available their human macrophage array data, and the laboratory of David Ron, University of Cambridge, for reagents and advice. This work was supported by NIH grants 5K08DK080174 and R01DK103175 (to S.K.K.), an NSF Graduate Research Fellowship (to M.M.R.), The UCSF Diabetes Research Center (DRC), and The UCSF Nutrition and Obesity Research Center (NORC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
M.M.R. and S.K.K. conceived and designed the research, analyzed data, and wrote the manuscript with feedback from M.V. and J.P.A. M.M.R. performed the majority of experiments with contributions from M.V. (immunoblotting and immunofluorescence), K.L.M.S. (SVF analysis), and R.V. (expression of mutant IRE1α). J.P.A. conducted the microarray and performed preliminary analyses, along with M.S. and M.M.R. C.C.K. performed final bioinformatic analyses of the array data and created the resulting figures. I.K. provided the ERAI-Luciferase mice.
References
- Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Baccarella A, Fontana MF, Chen EC, Kim CC. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun. 2013;81:4431–4442. doi: 10.1128/IAI.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Camell CD, Nguyen KY, Jurczak MJ, Shulman GI, Shadel GS, Dixit VD. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin-resistance in obesity. J Biol Chem. 2015;290:29402–29413. doi: 10.1074/jbc.M115.680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Ladha Z, Farese RV. Deficiency of acyl coenzyme a:diacylglycerol acyltransferase 1 increases leptin sensitivity in murine obesity models. Endocrinology. 2002a;143:2893–2898. doi: 10.1210/endo.143.8.8941. [DOI] [PubMed] [Google Scholar]
- Chen HC, Stone SJ, Zhou P, Buhman KK, Farese RV. Dissociation of obesity and impaired glucose disposal in mice overexpressing acyl coenzyme a:diacylglycerol acyltransferase 1 in white adipose tissue. Diabetes. 2002b;51:3189–3195. doi: 10.2337/diabetes.51.11.3189. [DOI] [PubMed] [Google Scholar]
- Coll T, Eyre E, Rodríguez-Calvo R, Palomer X, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M. Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem. 2008;283:11107–11116. doi: 10.1074/jbc.M708700200. [DOI] [PubMed] [Google Scholar]
- Egnatchik RA, Leamy AK, Jacobson DA, Shiota M, Young JD. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol Metab. 2014;3:544–553. doi: 10.1016/j.molmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. 2009;29:1944–1949. doi: 10.1161/ATVBAHA.109.194050. [DOI] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollub J, Sherlock G. Clustering microarray data. Meth Enzymol. 2006;411:194–213. doi: 10.1016/S0076-6879(06)11010-1. [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitai Y, Ariyama H, Kono N, Oikawa D, Iwawaki T, Arai H. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells. 2013;18:798–809. doi: 10.1111/gtc.12074. [DOI] [PubMed] [Google Scholar]
- Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, Naylor S, Rao M, Hubbard B, Farese RV., Jr DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest. 2010;120:756–767. doi: 10.1172/JCI36066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, Newman HW, Schmidt-Supprian M, Vance DE, Mann M, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011;14:504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metab. 2014;20:614–625. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’homme L, Esser N, Riva L, Scheen A, Paquot N, Piette J, Legrand-Poels S. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res. 2013;54:2998–3008. doi: 10.1194/jlr.M037861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamy AK, Egnatchik RA, Shiota M, Ivanova PT, Myers DS, Brown HA, Young JD. Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J Lipid Res. 2014;55:1478–1488. doi: 10.1194/jlr.M050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- Lerner AG, Upton JP, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. IRE1alpha; Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ge M, Ciani L, Kuriakose G, Westover EJ, Dura M, Covey DF, Freed JH, Maxfield FR, Lytton J, et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J Biol Chem. 2004;279:37030–37039. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- Maly DJ, Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014;10:892–901. doi: 10.1038/nchembio.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. 2014;39:245–254. doi: 10.1016/j.tibs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Nguyen MTA, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- Osborn O, Brownell SE, Sanchez-Alavez M, Salomon D, Gram H, Bartfai T. Treatment with an Interleukin 1 beta antibody improves glycemic control in diet-induced obesity. Cytokine. 2008;44:141–148. doi: 10.1016/j.cyto.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–1285. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Schaeffler A, Gross P, Buettner R, Bollheimer C, Buechler C, Neumeier M, Kopp A, Schoelmerich J, Falk W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-κB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126:233–245. doi: 10.1111/j.1365-2567.2008.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling JD, Machkovech HM, He L, Sidhu R, Fujiwara H, Weber K, Ory DS, Schaffer JE. Palmitate and LPS trigger synergistic ceramide production in primary macrophages. J Biol Chem. 2012;288:2923–2932. doi: 10.1074/jbc.M112.419978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, et al. Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16:487–499. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM, et al. JNK1 in Hematopoietically Derived Cells Contributes to Diet-Induced Inflammation and Insulin Resistance without Affecting Obesity. Cell Metab. 2007;6:386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Stienstra R, Joosten LAB, Koenen T, van Tits B, van Diepen JA, van den Berg SAA, Rensen PCN, Voshol PJ, Fantuzzi G, Hijmans A, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, Kotani H, Yamaoka S, Miyake K, Aoe S, et al. Role of the Toll-like Receptor 4/NF- B Pathway in Saturated Fatty Acid-Induced Inflammatory Changes in the Interaction Between Adipocytes and Macrophages. Arterioscler Thromb Vasc Biol. 2007;27:84–91. doi: 10.1161/01.ATV.0000251608.09329.9a. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R, Ron D. Lipid-dependent regulation of the unfolded protein response. Curr Opin Cell Biol. 2015;33:67–73. doi: 10.1016/j.ceb.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Perera BGK, Hari SB, Bhhatarai B, Backes BJ, Seeliger MA, Schürer SC, Oakes SA, Papa FR, Maly DJ. Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat Chem Biol. 2012;8:982–989. doi: 10.1038/nchembio.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem. 2009;331:31–40. doi: 10.1007/s11010-009-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welters HJ, Tadayyon M, Scarpello JHB, Smith SA, Morgan NG. Monounsaturated fatty acids protect against beta-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett. 2004;560:103–108. doi: 10.1016/S0014-5793(04)00079-1. [DOI] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MTH, Brickey WJ, Ting JPY. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18:816–830. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.