


Abstract
The trinuclear platinum agent BBR3464, a representative of a new class of anticancer drugs, is more potent than conventional mononuclear cisplatin [cis-diamminedichloroplatinum(II)]. BBR3464 retains significant activity in human tumor cell lines and xenografts that are refractory or poorly responsive to cisplatin, and displays a high activity in human tumor cell lines that are characterized by both wild-type and mutant p53 gene. In contrast, on average, cells with mutant p53 are more resistant to the effect of cisplatin. It has been hypothesized that the sensitivity or resistance of tumor cells to cisplatin might be also associated with cell cycle control and repair processes that involve p53. DNA is a major pharmacological target of platinum compounds and DNA binding activity of the p53 protein is crucial for its tumor suppressor function. This study, using gel-mobility-shift assays, was undertaken to examine the interactions of active and latent p53 protein with DNA fragments and oligodeoxyribonucleotide duplexes modified by BBR3464 in a cell free medium and to compare these results with those describing the interactions of these proteins with DNA modified by cisplatin. The results indicate that structurally different DNA adducts of BBR3464 and cisplatin exhibit a different efficiency to affect the binding affinity of the modified DNA to p53 protein. It has been suggested that different structural perturbations induced in DNA by the adducts of BBR3464 and cisplatin produce a differential response to p53 protein activation and recognition and that a ‘molecular approach’ to control of downstream effects such as protein recognition and pathways of apoptosis induction may consist in design of structurally unique DNA adducts as cell signals.
INTRODUCTION
Multinuclear platinum complexes represent a new class of anticancer drugs that contain two reactive platinum centers linked by a variable-length alkanediamine chain and are characterized by different DNA binding profile with respect to that of their mononuclear counterparts (1,2). [{trans-PtCl(NH3)2}2μ-trans-Pt(NH3)2{H2N(CH2)6NH2}2]4+ (BBR3464, Figure 1) is the first example of this class to enter clinical trials (3). It is more potent than cisplatin [cis-diamminedichloroplatinum(II)], and retains significant activity in human tumor cell lines and xenografts that are refractory or poorly responsive to cisplatin.
Figure 1.

Structures of platinum compounds.
The molecular mechanisms by which BBR3464 is able to overcome cisplatin resistance, are not fully understood. However, its ability to induce DNA adducts such as long-range delocalized intra- and interstrand cross-links (CLs), which are not produced by conventional mononuclear platinum compounds (4–9) suggest that BBR3464 may escape, at least in part, the classical mechanism of cisplatin resistance that is related to DNA damage recognition and repair. Moreover, due to its ability to modify DNA in a manner different from cisplatin, BBR3464 could distinctly evoke different pathways of cellular response to DNA damage such as triggering of the apoptotic pathway.
It has been demonstrated recently that BBR3464 displays high activity in human tumor cell lines characterized by both wild-type and mutant p53 gene (10). In contrast, on average, cells with mutant p53 are more resistant to the effect of cisplatin (11). It has been hypothesized that sensitivity or resistance of tumor cells to cisplatin might also be associated with the processes that involve p53 (12,13). Evidence of p53 implication in BBR3464 cytotoxicity is indicated by the fact that transfer of functional p53 into p53-null SAOS osteosarcoma cells actually reduced cellular sensitivity to BBR3464 compared with the parent p53-null line, but caused moderate chemosensitization of cisplatin (10). Similarly, in a cisplatin-resistant ovarian cancer cell line (OAW42MER) the presence of a wild-type and functional p53 protein was considered as an important determinant of cellular sensitivity to cisplatin but the presence of a non-functional p53 (as indicated by the lack of p21waf1 induction for a p53-dependent apoptotic pathway) was not detrimental to the cytotoxicity of BBR3464 (14). These results have clinical relevance as partial response in a PhaseII clinical trial of BBR3464 in cisplatin-relapsed ovarian cancer showed the respondents to be p53 wild type whereas the one respondent in a cisplatin-refractory arm was p53 mutant (3,15). As the tumor suppressor function of p53 protein is crucially related to its DNA binding activity (16,17) and DNA is considered as a major pharmacological target of platinum compounds (18), it is of great interest to understand the interactions of p53 protein with platinum-modified DNA. It is of particular importance to ascertain the differences that exist between structurally different compounds (e.g. structurally unique adducts) as exemplified by the cisplatin and polynuclear (BBR3464) class.
The active protein p53 is a nuclear phosphoprotein that consists of 393 amino acids and contains four major functional domains (19). Active p53 binds as a tetramer to ∼50 different response elements that occur naturally in the human genome and shows functionality (20). Free DNA in the segments corresponding to the consensus sequence is already intrinsically bent toward the major groove and the bends are mostly localized at two C(a/t)|(a/t)G tetramers (21,22). It has been suggested that this localized intrinsic bending contributes in a fundamental way to the stability of tetrameric p53–DNA complex (21).
In our recent work (23), we investigated DNA interactions of active wild-type human p53 protein with DNA that is modified by cisplatin and its clinically ineffective trans isomer (transplatin). We have found that DNA adducts of cisplatin reduce binding affinity of the consensus DNA sequence to p53, whereas adducts of transplatin do not. This result has been interpreted to mean that the precise steric fit, that is required for the formation and stability of the tetrameric complex of p53 with the consensus sequence in DNA cannot be attained, as a consequence of additional conformational perturbations induced by cisplatin adducts in the consensus sequence. The results also demonstrated an increase of the binding affinity of p53 to DNA that lacks the consensus sequence and modified by cisplatin, but not by transplatin. In addition, only major 1,2-GG intrastrand CLs of cisplatin were found to be responsible for this enhanced binding affinity of p53. The distinctive structural features of 1,2-intrastrand CLs of cisplatin have been proposed to play a unique role for this adduct in the binding of p53 to DNA that lacks the consensus sequence (23). Thus, the intriguing mechanism by which cisplatin adducts modulate biological effects of p53 might also be some manifestation of the hijacking model that involves the p53 protein of the mechanism underlying the antitumor effects of this drug (24,25). A comparison of the binding of active and latent p53 to DNA fragments modified by cisplatin has been performed as well (26). Modifications of DNA that lacks the consensus sequence by cisplatin enhanced the affinity of both forms of p53. The difference in the binding affinity to cisplatin-modified and unmodified DNA is even more pronounced in the case of the latent form. In addition, the p53 core domain appeared to be the primary site in active p53 of its sequence-specific binding to non-modified DNA while the C-terminus of p53 interacted selectively with DNA that lacks consensus sequence and modified by cisplatin (26).
This study was undertaken to examine interactions of active and latent p53 protein with DNA fragments and oligodeoxyribonucleotide duplexes modified by BBR3464 in a cell free medium and to compare these results with those describing interactions of these proteins with DNA modified by cisplatin.
MATERIALS AND METHODS
DNA
For the consensus DNA response element (CDRE) binding of the p53 protein we used a small PvuII (474 bp) fragment of pPGM1 plasmid that is derived from pBluescript SK II− DNA (2961 bp, Stratagene) by cloning the p53 20 bp CDRE 5′-AGACATGCCTAGACATGCCT-3′/5′-AGGCATGTCTAGGCATGTCT-3′ into the HindIII site. pPGM1 and pBluescript SK II+ DNAs were purified using QIAGEN kits (QIAGEN, GmbH, Germany). Ethanol precipitated plasmids were resuspended in TE buffer [10 mM Tris–HCl, 0.1 mM Na3EDTA (pH 7.5)] and stored at 4°C. The synthetic oligodeoxyribonucleotide duplexes (Figure 2B) were purchased from IDT, Inc. (Coralville, IA) and purified as described previously (27); in the present work their molar concentrations are related to the whole duplexes.
Figure 2.

Binding of active p53 protein to the PvuII fragment of pPMG1 DNA 474 bp long containing CDRE. Gel-mobility retardation assay was performed in the presence of the unplatinated 2513 bp nonspecific competitor (PvuII fragment of pPMG1 lacking CDRE) in 1% agarose gel; concentrations of the 474 and 2513 bp fragments were 10 and 57 μg/ml (3.3 and 3.5 × 10−8 M), respectively. (A) The fragment was unplatinated (lanes 1, 9 and 17), globally modified by cisplatin (lanes 2–4 and 10–12), transplatin (lanes 5 and 13) or BBR3464 (lanes 6–8 and 14–16). Concentration of the p53 protein was 0 (lanes 1–8) or 3.14 × 10−8 M (lanes 9–17). rb values: 0 (lanes 1, 9 and 17); 0.02 (lanes 2,6,10 and 14); 0.04 (lanes 3,7,11 and 15) and 0.06 (lanes 4,5,8,12,13 and 16). Lane 17: the same as in lane 9, but MAb DO-1 was added at the molar ratio MAb/p53 = 3. For other details, see the experimental part. (B) The fragment was unplatinated (lanes 1 and 5), globally modified by BBR3464 (lanes 2–4 and 6–7). Concentration of the p53 protein was 0 (lanes 1–4) or 3.14 × 10−8 M (lanes 5–8). rb values: 0 (lanes 1 and 5); 0.003 (lanes 2 and 6); 0.005 (lanes 3 and 7); 0.01 (lanes 4 and 8).
Purification of the active wild-type human p53 protein
The human active p53 protein was expressed in baculovirus-infected recombinant Sf9 insect cells. The latent p53 was isolated from Escherichia coli BL21/DE3 strain harboring pT7-7Hup53. The details of the purification and characterization were described previously (26,28). The protein concentration was determined by the Bradford method. In the present paper the concentration of the p53 protein is related to tetrameric protein units.
Platination reactions
Cisplatin and transplatin were purchased from Sigma (Prague, Czech Republic). BBR3464 (Figure 1) was prepared by standard methods. Short PvuII fragments of pPGM1 and pBluescript II SK+ plasmid DNAs (474 and 448 bp, respectively) and oligodeoxyribonucleotide duplex (oligo–CDRE) 5′-AGACATGCCTAGACATGCCT-3′/5′-AGGCATGTCTAGGCATGTCT-3′ were incubated with platinum compounds in 10 mM NaClO4 at 37°C for 48 h in the dark. The values of rb (the rb value is defined as the number of the molecules of the platinum compound bound per nucleotide) were determined by flameless atomic absorption spectrophotometry or differential pulse polarography (29).
Preparation of DNA–protein complexes
Formation of the complexes of the active and latent p53 protein with the 474 or 448 bp-long PvuII fragments of pPGM1 or pBluescript SK II−, respectively, that were unplatinated or modified by platinum compounds, was examined in a buffer that contained 5 mM Tris–HCl, pH 7.6, 0.5 mM Na3EDTA, 50 mM KCl and 0.01% Triton X-100 in a total volume of 15 μl. In the experiments with active p53, the nonmodified or platinated 474 or 448 bp fragment was mixed with nonmodified 2513 bp-long fragment of pPGM1. The final amounts of the short and long fragments in these reactions were 150 and 850 ng, respectively (the molar ratio of these fragments was ∼1). The molar ratio of p53 to 474 or 448 bp fragment was 0–6. In the experiments with latent p53, the final amounts of the short and long fragments in the reactions were 60 and 340 ng, respectively (the molar ratio of these fragments was also ∼1). The molar ratio of latent p53 to 448 bp fragment was 5. Samples with p53 protein were incubated in ice for 30 min. After the incubation was completed 3 μl of the loading buffer (50% glycerol, 50 mM Na3EDTA and 2% bromophenol blue) was added, the samples were loaded on the 1% agarose gel that was precooled to 4°C and electrophoresed in 0.5× TBE buffer [TBE buffer = 0.09 M Tris–borate and 2 mM Na3EDTA (pH 8.0)]. The gel was finally stained by ethidium bromide.
Formation of the complexes of p53 with the oligonucleotide duplex was examined in the same buffer as that used for analysis of the complexes of p53 with the plasmid fragments (see above) in a total volume of 12 μl. The nonmodified or platinated duplexes were mixed with the nonmodified 2513 bp-long fragment of pPGM1. The final amounts of the duplexes and long fragment in the reactions were 20 and 120 ng, respectively. The molar ratio p53/duplex was 0–3. Samples with p53 were incubated in ice for 30 min. After the incubation was completed 3 μl of the loading buffer (50% glycerol, 50 mM Na3EDTA and 2% bromophenol blue) was added, the samples loaded on the native 5% polyacrylamide gel [mono:bis(acrylamide) ratio = 29:1] precooled to 4°C in 0.5× TBE buffer. The radioactivity associated with the bands was quantified by means of a molecular dynamics PhosphorImager (Storm 860 system with ImageQuant software).
The primary p53 monoclonal antibody (MAb) DO-1 [purified and characterized as described in (30)] was also added to the p53–DNA complex (molar ratio of MAb/p53 tetramer was 3), the mixture was incubated for an additional 30 min at 20°C and the resulting p53–DNA–MAb complexes were loaded onto the gels.
Other chemicals
T4 polynucleotide kinase and restriction endonucleases were purchased from New England Biolabs (Beverly, MA). [γ-32P]ATP used for 5′-end radioactive labeling of the top strands of the oligonucleotide duplexes were from Amersham (Arlington Heights, IL, USA). Acrylamide, bis(acrylamide), agarose and urea were from Merck KgaA (Darmstadt, Germany).
RESULTS
Binding of active p53 protein to platinated DNA containing the consensus response element
The pPGM1 plasmid was cleaved by PvuII (blunt end-forming enzyme that cuts twice within the pPGM1). This cleavage produced 474 and 2513 bp fragments that contained and lacked CDRE, respectively. The two fragments were separated on the agarose gel, extracted and purified. The 474 bp fragment was further globally modified by BBR3464 or cisplatin at rb = 0.02–0.06. In order to further characterize these platinated fragments, they were cleaved by HindIII. This cleavage produced the 20 bp CDRE which was separated on the 12% native polyacrylamide gel, extracted, purified and the amount of platinum bound to the CDRE was determined by flameless atomic absorption spectrophotometry. It was verified in this way that the average amount of the molecules of cisplatin and BBR3464 bound to the CDRE that was incorporated in the 474 bp fragment pPGM1 plasmid was identical at all rb values used. The platinated 474 bp fragments were then mixed with the nonspecific competitor, which was the unplatinated 2513 bp fragment. This mixture was incubated with various amounts of active p53 (at molar ratios of p53/474 bp fragment in the range of 0–6) and analyzed using agarose gel electrophoresis. The incubation of the unplatinated 474 bp fragment with increasing amount of active p53 resulted in the appearance of a new, more slowly migrating species with a concomitant decrease of the intensity of the band corresponding to the 474 bp fragment incubated in the absence of p53 (shown for p53–474 bp fragment ratio of 0.95 in Figure 2A, lane 9). This result was in agreement with the previously published reports and demonstrated the formation of a sequence-specific complex between DNA and active p53 protein (23,26,31). Importantly, the addition of DO-1 MAb (which maps to the N-terminal domain of p53) produced supershifted complexes that migrated still more slowly than the p53–474 bp complex (Figure 2A, lane 17) which confirms the presence of p53 in the more slowly migrating species. In contrast, incubation of the 474 bp fragment that was modified by BBR3464 or cisplatin at rb = 0.02 – 0.06 with active p53 (in the presence of the unplatinated 2513 bp fragment) considerably reduced the yield of the species that migrated more slowly in the agarose gel [shown for p53/474 bp fragment ratio of 0.95 in Figure 2A, the fragment modified by cisplatin (lanes 10–12) and by BBR3464 (lanes 14–16)]. Importantly, for instance 50% reduction of the yield of the species migrating more slowly in the gel was observed for the modification by cisplatin at rb = ∼0.02 (Figure 2A, lane 10), which was ∼8 times higher than that for the modification by BBR3464 at rb = ∼0.003 (Figure 2B, lane 6). This result is consistent with the hypothesis that modification of DNA through covalent adduct formation by both BBR3464 and cisplatin reduces the binding affinity of active p53 to the CDRE and that BBR3464 adducts are considerably more efficient in this regard.
Further studies were performed using a short (20 bp) oligodeoxyribonucleotide duplex, oligo–CDRE (for its nucleotide sequence, see Materials and Methods), whose sequence follows the consensus sequence pattern (16). The duplex was globally modified by BBR3464 or cisplatin to rb in the range of 0.0125-0.05 and the unplatinated PvuII fragment of pPGM1 that was 2513 bp long (containing no CDRE) was added as the nonspecific competitor. These mixtures were incubated with active p53 at various p53/duplex molar ratios (0.1–3) and analyzed by using native PAGE (Figure 3A). Incubation of the unplatinated oligo–CDRE with increasing amounts of active p53 resulted in the appearance of the new, more slowly migrating species with a concomitant decrease in the intensity of the band corresponding to the 20 bp duplex that was incubated in the absence of p53 (shown for p53/duplex ratio of 0.3 in Figure 3A, lane 1). This result confirmed the formation of the complex between oligo-CDRE and active p53. In contrast, the incubation of oligo–CDRE that was modified by BBR3464 and cisplatin at rb = 0.0125-0.05 with active p53 reduced the yield of the species migrating more slowly in the gel, BBR3464 again being considerably more effective (Figure 3B). For instance, the modification of oligo–CDRE by BBR3464 at the level, which corresponded to 1 molecule of the platinum compound fixed per duplex (rb = 0.025) inhibited almost completely the formation of the complex between this duplex and active p53, whereas this inhibition due to the modification by cisplatin at the same rb was markedly less efficient (Figure 3B).
Figure 3.
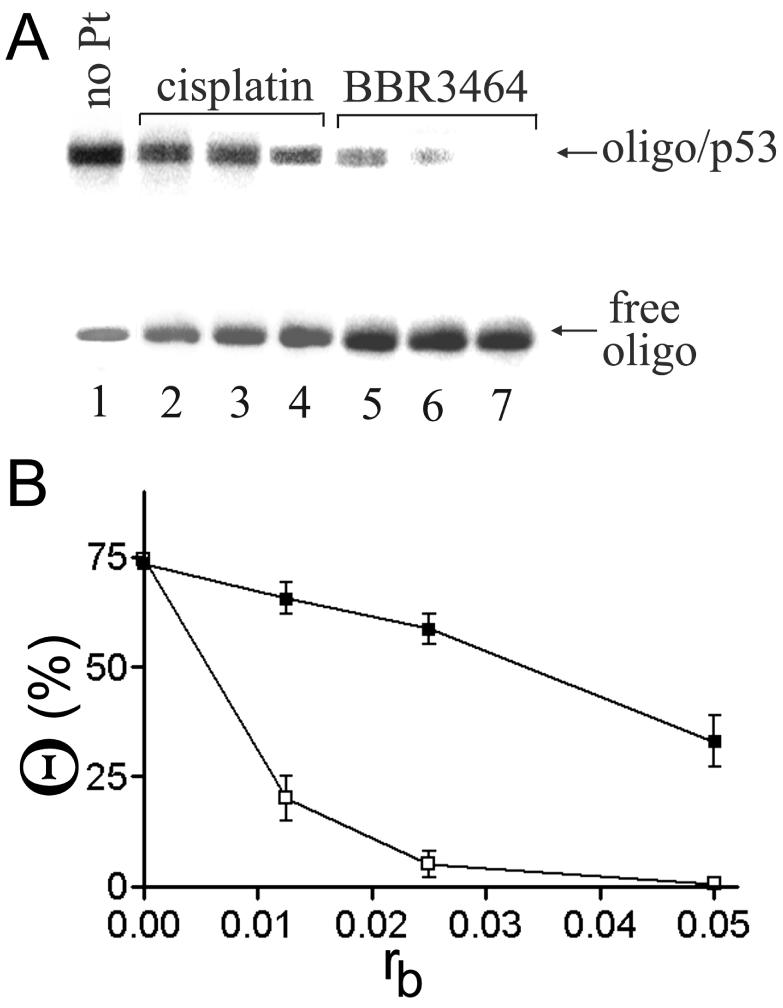
Binding of active p53 protein to the 20 bp duplex containing CDRE (see Figure 2B for its sequence). The duplex was unplatinated (lane 1), globally modified by cisplatin (lanes 2–4) or BBR3464 (lanes 5–7). Gel-mobility retardation assay was performed in the presence of the unplatinated 2513 bp nonspecific competitor (PvuII fragment of pPMG1 lacking CDRE) in 5% native polyacrylamide gel; concentrations of the oligonucleotide duplex and 2513 bp fragment were 1.6 and 10 μg/ml (1.26 × 10−7 and 6 × 10−9 M), respectively and concentration of p53 was 3.9 × 10−8 M. rb values: 0 (lane 1); 0.0125 (lanes 2 and 5); 0.025 (lanes 3 and 6); 0.05 (lanes 4 and 7). The oligonucleotide duplex was radioactively labeled at the 5′-end of the top strand. For other details, see the experimental part. (A) Autoradiogram. (B) The plot of the amount of the oligonucleotide duplex in the complex with p53 protein on the amount of the platinum complex bound per one molecule of the duplex. Cisplatin, filled squares; BBR3464, empty squares.
Binding of active p53 protein to platinated DNA lacking the consensus response element
We next investigated the binding of p53 to the 448 bp fragment of the pBluescript II SK+ plasmid lacking CDRE, but modified by BBR3464 or cisplatin. The plasmid was cleaved by PvuII, which yielded the 448 and 2513 bp fragments. The longer fragment was identical to that produced by the PvuII cleavage of pPGM1, whereas the shorter fragment only differed from that produced by PvuII cleavage of pPGM1 by lacking the 26 bp CDRE. The 448 bp fragment was globally modified by BBR3464 or cisplatin at rb = 0.01–0.08. After the 448 bp fragment was platinated, the free unplatinated 2513 bp fragment was added as the nonspecific competitor. These mixtures were incubated with p53 at various p53/448 bp fragment molar ratios (in the range of 0.5–6) and analyzed by using agarose gel electrophoresis. Incubation of the unplatinated PvuII fragments with increasing amount of p53 did not result in any changes in the migration of these fragments, demonstrating no effect on formation of the complex between p53 and DNA lacking CDRE (not shown). On the other hand and consistent with our recent work (23), a new species migrating considerably more slowly in the gel was observed when the 448 bp fragment modified by cisplatin at rb = 0.01–0.08 was analyzed (not shown). This result demonstrated the formation of a complex between p53 and DNA lacking CDRE but modified by cisplatin (31). Importantly, supershifted complexes were noticed as a consequence of addition of MAb DO-1 to the complex of p53 with 448 bp fragment modified by cisplatin (not shown). In contrast, in the same experiments with the 448 bp fragment globally modified by the more potent BBR3464, no slowly migrating species were noticed at rb = 0.08 and p53/DNA ratio of 6 (not shown). Thus, these results indicate that the binding affinity of p53 to DNA lacking CDRE is enhanced selectively by modification by cisplatin but not by BBR3464.
Binding of latent p53 protein to platinated DNA lacking consensus response element
Posttranslationally unmodified latent p53 (such as the protein isolated from the bacterial expression system) displays a poor affinity to the CDRE (26). On the other hand, it may exhibit well-pronounced DNA structure-specific binding (26,31,32). For example, the affinity of DNA lacking CDRE modified by cisplatin to p53 protein is noticeably enhanced if the latent form is used instead of its active form (26). Therefore, we have also examined the binding of latent p53 to the 448 bp fragment of the pBluescript II SK+ plasmid lacking CDRE, but modified by BBR3464 or cisplatin in a similar manner to the experiments using the active form of p53 (vide supra). Incubation of the unplatinated PvuII fragment with latent p53 added in the amount corresponding to p53/448 bp fragment ratio of 5 resulted in no changes in the migration of these fragments demonstrating no effect on formation of the complex between latent p53 and DNA lacking CDRE (Figure 4, lane 1). On the other hand and consistent with our recent work (26), a new species migrating in the gel considerably more slowly was observed if the 448 bp fragment modified by cisplatin at rb = 0.05 was analyzed (shown for p53/448 bp fragment ratio of 1 in Figure 4, lane 2). When the fragment modified by BBR3464 at the same rb was analyzed for its interaction with latent p53 under identical conditions, no more slowly migrating species was noticed (shown for p53/448 bp fragment ratio of 1 in Figure 4, lane 3). Thus, neither the active nor the latent form of p53 protein exhibit affinity to the DNA sequence lacking CDRE and modified by BBR3464. These results represent a fundamental difference in the processing by critical cellular components of the DNA adducts formed by BBR3464 and cisplatin.
Figure 4.
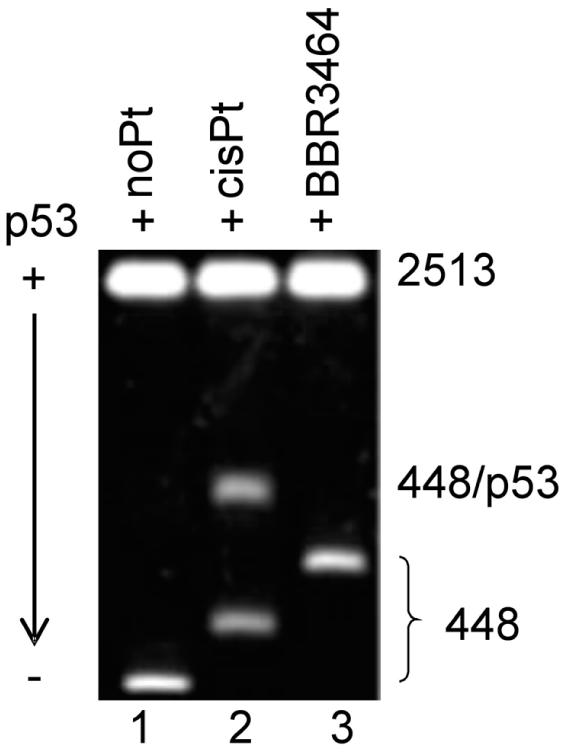
Binding of latent p53 to the PvuII fragment of pBluescript SK II+ DNA 448 bp long lacking CDRE. The fragment was unplatinated (lane 1), globally modified at rb = 0.05 by cisplatin (lane 2) or BBR3464 (lane 3). For other details, see the text.
DISCUSSION
Whereas cells with mutant p53 are, on average, more resistant to the effect of cisplatin (11), BBR3464 maintains activity in several tumors with mutant p53 with both acquired and inherent resistance to cisplatin (10). In line with these observations, the p53 status of tumor cells appears to affect their sensitivity to BBR3464 and cisplatin in a different way. As stated, this may be an important determinant of the outcome in the clinical response to BBR3464. A number of factors may be responsible for these differences. DNA is a major pharmacological target of platinum compounds and DNA binding activity of p53 protein is crucial for its tumor suppressor function. Hence, a plausible explanation for the different sensitivity of human tumors with wild-type and mutant p53 to BBR3464 and cisplatin may lie in a different efficiency of the adducts of these drugs to affect the binding affinity of the modified DNA to p53 protein. Interestingly, the adducts of cisplatin and the clinically ineffective transplatin affect recognition of DNA by active p53 protein in a distinctly different way (23). The present results, in comparing the affinity with the p53 protein of DNA containing or lacking consensus sequence modified by cisplatin and BBR3464, confirm that structurally different (but both antitumor active) drugs exhibit distinctly different protein recognition patterns.
The results of the present work demonstrate that the efficiency of the inhibition of binding of active p53 to the DNA consensus sequence is markedly more efficient for adducts of BBR3464 than the adducts of cisplatin (Figure 2). Sequence-dependent conformational variability of response elements plays a critical role in the sequence-specific binding of p53 to DNA and the stability of the resulting complex. Extraordinary demands for this binding specificity and selectivity of p53 are closely related to its tetrameric association with CDRE in which the precise steric fit is extremely important (21).
The consensus sequences investigated in the present work contained several sites at which bifunctional adducts of both cisplatin and BBR3464 are formed. In the CDREs investigated in the present work, cisplatin forms bifunctional adducts, which strongly disturb its secondary structure. For instance, the structure of major 1,2-GG intrastrand CLs determined by NMR methods has revealed (33–35) that these adducts induce the overall helix bend of 40-78° towards major groove, DNA unwinding of 25–27° and severe perturbation of hydrogen-bonding within the 5′-coordinated GC base pair. It has been proposed (23) that the result of these perturbances is that the precise steric fit required for the formation and stability of the tetrameric complex of p53 with the consensus nucleotide sequence, cannot be attained so that p53 binds to its CDRE with a reduced affinity. In contrast, clinically ineffective transplatin also forms various types of adducts in DNA, but these lesions induce relatively subtle structural perturbations in DNA, which have no substantial effect on the formation of the tetrameric complex of p53 with the CDRE (36,37).
The general outline of DNA modification by BBR3464 includes significantly faster binding in comparison with neutral cisplatin (4). In a 14mer sequence that contained sites of interstrand CL formation as well as potential ‘cisplatin-like’ intrastrand adducts, the interstrand CL was preferred (38) and thus, it is likely that the interstrand CL is also formed in the CDRE. The adducts of BBR3464 are mainly long-range delocalized intrastrand and interstrand CLs between guanine residues, which produce an unwinding angle similar to that found for the adducts of cisplatin and bend DNA significantly less than the CLs of cisplatin (5,6). Following the argument used for cisplatin, these less severe structural distortions induced in DNA by BBR3464, may suggest a smaller inhibition effect on the formation of the tetrameric complex of p53 with the CDRE. The structural analysis of the long-range interstrand CLs of BBR3464 [formed in DNA with a relatively high frequency (20%) (4–9)] has shown that the central tetraamine linker of this CL is situated in or very close to the minor groove of DNA (9,39). It is reasonable to expect that this location of the linker could sterically block the binding of the p53 protein to DNA [since the protein residues are also localized in DNA minor groove (40)]. In addition, the location of the linker in the minor groove could restrict its narrowing, a feature also required for p53 binding (41).
A plausible explanation of the different sensitivity of human tumors with wild-type or mutant p53 to BBR3464 and cisplatin may also lie in the different efficiency of their adducts to increase the binding affinity of DNA lacking the consensus sequence to p53. In contrast to the adducts of cisplatin, the adducts of BBR3464 do not increase the binding affinity of p53 to DNA lacking the consensus sequence (Figure 4). This enhancement is specific for cisplatin-modified DNA. Moreover, among the DNA adducts of cisplatin only the 1,2-intrastrand CLs are responsible for this increase in the DNA binding of p53 (23). Hence, 1,2-intrastrand CLs distort DNA in a specific way producing a structural motif that is recognized by p53. It has been proposed (23) that directional and stable bending of DNA due to formation of the 1,2-intrastrand CLs of cisplatin affords a structural element exhibiting this specific affinity to p53. In this way a stable flexure of DNA by 1,2-intrastrand CL provides an opportunity for more stable contacts between p53 and DNA. In other words, lesions such as the 1,2-intrastrand CLs that efficiently induce the directional and fixed bend in DNA towards the major groove, thus providing a stable prebent site on DNA to p53, serve as a structural motif for DNA recognition and binding by p53. The distinctive structural features of the 1,2-intrastrand CLs of cisplatin suggest a unique role for this adduct in the enhancement of the binding of p53 to platinated DNA segments lacking CDRE which is, however, weaker than the binding of active p53 to unplatinated CDRE (23).
The CLs of BBR3464 distort DNA in a way that apparently produces a structural motif considerably different from that afforded by the 1,2-GG intrastrand CL and recognized by p53. The structural features of the adducts of BBR3464 are considerably different—for instance, long-range intrastrand CLs of BBR3464 induce in DNA a flexible joint rather than a rigid directional bending (5). Similarly, the bends induced by the interstrand CLs of this trinuclear platinum compound are only 15–21° toward the major groove (6), i.e. the value considerably lower than that observed for the CLs of cisplatin. Thus, it is reasonable to conclude that different structural perturbations produce differential responses to p53 protein activation and recognition. Similar to our results on comparative binding of high mobility group (HMG) proteins (6,27) the current results suggest a ‘molecular approach’ to control of downstream effects such as protein recognition and pathways of apoptosis induction by design of structurally unique DNA adducts as cell signals.
Recognition of platinated DNA by p53 protein and other DNA-binding proteins remains an exciting and potentially significant area of research that is aimed at further understanding mechanisms that underlie resistance to platinum compounds or the unique antitumor properties of this new class of polynuclear anticancer agents. Further, the differential responses of structurally different anticancer agents may be extendable to clinical practice and utilized to predict more suitable and appropriate cancer drug treatments for those ∼50% of cases with diagnosed p53 mutations.
Acknowledgments
ACKNOWLEDGEMENTS
This research was supported by the Grant Agency of the Academy of Sciences of the Czech Republic (Grants 5004101, B5004301 and S5004009) and the Grant Agency of the Czech Republic (Grant 204/02/0734). The research was also supported by active grants from The National Institutes of Health (CA-78754). J.K. is the international research scholar of the Howard Hughes Medical Institute. J.K. and V.B. acknowledge that their participation in the EC COST Chemistry Action D20 enabled them to exchange regularly the most recent ideas in the field of platinum anticancer drugs with several European colleagues.
REFERENCES
- 1.Farrell N., Qu,Y., Bierbach,U., Valsecchi,M. and Menta,E. (1999) Structure-activity relationship within di- and trinuclear platinum phase I clinical agents. In Lippert,B. (ed.), Cisplatin. Chemistry and Biochemistry of a Leading Anticancer Drug. VHCA, WILEY-VCH, Zurich, Weinheim, pp. 479–496. [Google Scholar]
- 2.Farrell N. (2000) Polynuclear charged platinum compounds as a new class of anticancer agents: Toward a new paradigm. In Kelland,L.R. and Farrell,N.P. (eds), Platinum-based Drugs in Cancer Therapy. Humana Press Inc, Totowa, NJ, pp. 321–338. [Google Scholar]
- 3.Farrell N. (2004) Polynuclear platinum drugs. In Sigel,A. and Sigel,H. (eds), Metal Ions in Biological Systems. Marcel Dekker, Inc., New York, Basel, Hong Kong, Vol. 41, pp. 251–296. [PubMed] [Google Scholar]
- 4.Brabec V., Kasparkova,J., Vrana,O., Novakova,O., Cox,J.W., Qu,Y. and Farrell,N. (1999) DNA modifications by a novel bifunctional trinuclear platinum Phase I anticancer agent. Biochemistry, 38, 6781–6790. [DOI] [PubMed] [Google Scholar]
- 5.Zehnulova J., Kasparkova,J., Farrell,N. and Brabec,V. (2001) Conformation, recognition by high mobility group domain proteins, and nucleotide excision repair of DNA intrastrand cross-links of novel antitumor trinuclear platinum complex BBR3464. J. Biol. Chem., 276, 22191–22199. [DOI] [PubMed] [Google Scholar]
- 6.Kasparkova J., Zehnulova,J., Farrell,N. and Brabec,V. (2002) DNA interstrand cross-links of the novel antitumor trinuclear platinum complex BBR3464. Conformation, recognition by high mobility group domain proteins, and nucleotide excision repair. J. Biol. Chem., 277, 48076–48086. [DOI] [PubMed] [Google Scholar]
- 7.Davies M.S., Thomas,D.S., Hegmans,A., Berners-Price,S.J. and Farrell,N. (2002) Kinetic and equilibria studies of the aquation of the trinuclear platinum phase II anticancer agent [{trans-PtCl(NH3)2}2{μ-trans-Pt(NH3)2(NH2(CH2)6NH2)2}]4+ (BBR3464). Inorg. Chem., 41, 1101–1109. [DOI] [PubMed] [Google Scholar]
- 8.McGregor T.D., Bousfield,W., Qu,Y. and Farrell,N. (2002) Circular dichroism study of the irreversibility of conformational changes induced by polyamine-linked dinuclear platinum compounds. J. Inorg. Biochem., 91, 212–219. [DOI] [PubMed] [Google Scholar]
- 9.Qu Y., Scarsdale,N.J., Tran,M.C. and Farrell,N.P. (2003) Cooperative effects in long-range 1,4 DNA–DNA interstrand cross-links formed by polynuclear platinum complexes: an unexpected syn orientation of adenine bases outside the binding sites. J. Biol. Inorg. Chem., 8, 19–28. [DOI] [PubMed] [Google Scholar]
- 10.Pratesi P., Righetti,S.C., Supino,R., Pollizi,D., Manzotti,C., Giuliania,F.C., Pezzoni,G., Tognella,S., Sinelli,S., Perego,P.et al. (1999) High antitumor activity of a novel multinuclear platinum complex against cisplatin-resistant p53 mutant human tumors. Brit. J. Cancer, 80, 1912–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Connor P.M., Jackman,J., Bae,I., Myers,T.G., Fan,S., Mutoh,M., Scudiero,D.A., Monks,A., Sausville,E.A., Weinstein,J.N., Friend,S.et al. (1997) Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res., 57, 4285–4300. [PubMed] [Google Scholar]
- 12.Jordan P. and Carmo-Fonseca,M. (2000) Molecular mechanisms involved in cisplatin cytotoxicity. Cell. Mol. Life Sci., 57, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riva C.M. (2000) Restoration of wild-type p53 activity enhances the sensitivity of pleural metastasis to cisplatin through an apoptotic mechanism. Anticancer Res., 20, 4463–4471. [PubMed] [Google Scholar]
- 14.Calvert A.H., Thomas,H., Colombo,N., Gore,M., Earl,H., Sena,L., Camboni,G., Liati,P. and Sessa,C. (2001) Phase II clinical study of BBR 3464, a novel, bifunctional platinum analogue, in patients with advanced ovarian cancer. Eur. J. Cancer, 37 (Suppl. 6), S260. [Google Scholar]
- 15.Orlandi L., Colella,G., Bearzatto,A., Abolafio,G., Manzotti,C., Daidone,M.G. and Zaffaroni,N. (2001) Effects of a novel trinuclear platinum complex in cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell lines: interference with cell cycle progression and induction of apoptosis. Eur. J. Cancer, 37, 649–659. [DOI] [PubMed] [Google Scholar]
- 16.El-Deiry W.S., Kern,S.E., Pietenpol,J.A., Kinzler,K.W. and Vogelstein,B. (1992) Definition of a consensus binding site for p53. Nature Genet., 1, 45–49. [DOI] [PubMed] [Google Scholar]
- 17.Ahn J. and Prives,C. (2001) The C-terminus of p53: the more you learn the less you know. Nature Struct. Biol., 8, 730–732. [DOI] [PubMed] [Google Scholar]
- 18.Johnson N.P., Butour,J.-L., Villani,G., Wimmer,F.L., Defais,M., Pierson,V. and Brabec,V. (1989) Metal antitumor compounds: The mechanism of action of platinum complexes. Prog. Clin. Biochem. Med., 10, 1–24. [Google Scholar]
- 19.May P. and May,E. (1999) Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene, 18, 7621–7636. [DOI] [PubMed] [Google Scholar]
- 20.Tokino T., Thiagalingam,S., Eldeiry,W.S., Waldman,T., Kinzler,K.W. and Vogelstein,B. (1994) p53 tagged sites from human genomic DNA. Hum. Mol. Genet., 3, 1537–1542. [DOI] [PubMed] [Google Scholar]
- 21.Nagaich A.K., Zhurkin,V.B., Durell,S.R., Jernigan,R.L., Appella,E. and Harrington,R.E. (1999) p53-induced DNA bending and twisting: p53 tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proc. Natl Acad. Sci. USA, 96, 1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lebrun A., Lavery,R. and Weinstein,H. (2001) Modeling multi-component protein-DNA complexes: the role of bending and dimerization in the complex of p53 dimers with DNA. Protein Eng., 14, 233–243. [DOI] [PubMed] [Google Scholar]
- 23.Kasparkova J., Pospisilova,S. and Brabec,V. (2001) Different recognition of DNA modified by antitumor cisplatin and its clinically ineffective trans isomer by tumor suppressor protein p53. J. Biol. Chem., 276, 16064–16069. [DOI] [PubMed] [Google Scholar]
- 24.Kartalou M. and Essigmann,J.M. (2001) Recognition of cisplatin adducts by cellular proteins. Mutat. Res., 478, 1–21. [DOI] [PubMed] [Google Scholar]
- 25.Brabec V. (2002) DNA modifications by antitumor platinum and ruthenium compounds: their recognition and repair. In Moldave,K. (ed.), Prog. Nucleic Acid Res. Mol. Biol. Academic Press Inc, San Diego, CA, Vol. 71, pp. 1–68. [DOI] [PubMed] [Google Scholar]
- 26.Fojta M., Pivonkova,H., Brazdova,M., Kovarova,L., Palecek,E., Pospisilova,S., Vojtesek,B., Kasparkova,J. and Brabec,V. (2003) Recognition of DNA modified by antitumor cisplatin by ‘latent’ and ‘active’ protein p53. Biochem. Pharmacol., 65, 1305–1316. [DOI] [PubMed] [Google Scholar]
- 27.Kasparkova J., Farrell,N. and Brabec,V. (2000) Sequence specificity, conformation, and recognition by HMG1 protein of major DNA interstrand cross-links of antitumor dinuclear platinum complexes. J. Biol. Chem., 275, 15789–15798. [DOI] [PubMed] [Google Scholar]
- 28.Hupp T.R. and Lane,D.P. (1995) Two distinct signaling pathways activate the latent DNA binding function of p53 in a casein kinase II-independent manner. J. Biol. Chem., 270, 18165–18174. [DOI] [PubMed] [Google Scholar]
- 29.Kim S.D., Vrana,O., Kleinwächter,V., Niki,K. and Brabec,V. (1990) Polarographic determination of subnanogram quantities of free platinum in reaction mixture with DNA. Anal. Lett., 23, 1505–1518. [Google Scholar]
- 30.Vojtesek B., Bartek,J., Midgley,C.A. and Lane,D.P. (1992) An immunochemical analysis of the human nuclear phosphoprotein p53—New monoclonal-antibodies and epitope mapping using recombinant p53. J. Immunol. Meth., 151, 237–244. [DOI] [PubMed] [Google Scholar]
- 31.Mazur S.J., Sakaguchi,K., Appella,E., Wang,X.W., Harris,C.C. and Bohr,V.A. (1999) Preferential binding of tumor suppressor p53 to positively or negatively supercoiled DNA involves the C-terminal domain. J. Mol. Biol., 292, 241–249. [DOI] [PubMed] [Google Scholar]
- 32.Brazdova M., Palecek,J., Cherny,D.I., Billova,S., Fojta,M., Pecinka,P., Vojtesek,B., Jovin,T.M. and Palecek,E. (2002) Role of tumor suppressor p53 domains in selective binding to supercoiled DNA. Nucleic Acids Res., 30, 4966–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamieson E.R. and Lippard,S.J. (1999) Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev., 99, 2467–2498. [DOI] [PubMed] [Google Scholar]
- 34.Gelasco A. and Lippard,S.J. (1998) NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) adduct d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry, 37, 9230–9239. [DOI] [PubMed] [Google Scholar]
- 35.Takahara P.M., Frederick,C.A. and Lippard,S.J. (1996) Crystal structure of the anticancer drug cisplatin bound to duplex DNA. J. Am. Chem. Soc., 118, 12309–12321. [Google Scholar]
- 36.Brabec V., Sip,M. and Leng,M. (1993) DNA conformational distortion produced by site-specific interstrand cross-link of trans-diamminedichloroplatinum(II). Biochemistry, 32, 11676–11681. [DOI] [PubMed] [Google Scholar]
- 37.Keck M.V. and Lippard,S.J. (1992) Unwinding of supercoiled DNA by platinum ethidium and related complexes. J. Am. Chem. Soc., 114, 3386–3390. [Google Scholar]
- 38.Berners-Price S.J., Davies,M.S., Cox,J.W., Thomas,D.S. and Farrell,N. (2003) Competitive reactions of interstrand and intrastrand DNA-Pt adducts: A dinuclear-platinum complex preferentially forms a 1,4-interstrand cross-link rather than a 1,2 intrastrand cross-link on binding to a GG 14mer duplex. Chem. Eur. J., 9, 713–725. [DOI] [PubMed] [Google Scholar]
- 39.Hegmans A., Berners-Price,S.J., Davies,M.S., Thomas,D., Humphreys,A. and Farrell,N. (2004) Long range 1,4 and 1,6-interstrand cross-links formed by a trinuclear platinum complex. Minor groove pre-association affects kinetics and mechanism of cross-link formation as well as adduct structure. J. Am. Chem. Soc., 126, 2166–2180. [DOI] [PubMed] [Google Scholar]
- 40.Cho Y., Gorina,S., Jeffrey,P.D. and Pavletich,N.P. (1994) Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science, 265, 346–355. [DOI] [PubMed] [Google Scholar]
- 41.Nagaich A.K., Zhurkin,V.B., Sakamoto,H., Gorin,A.A., Clore,G.M., Gronenborn,A.M., Appella,E. and Harrington,R.E. (1997) Architectural accommodation in the complex of four p53 DNA binding domain peptides with the p21/waf1/cip1 DNA response element. J. Biol. Chem., 272, 14830–14841. [DOI] [PubMed] [Google Scholar]