

Abstract
MicroRNAs (miRNAs) constitute a large family of noncoding RNAs that function as guide molecules in diverse gene silencing pathways. Current efforts are focused on the regulatory function of miRNAs, while little is known about how these unusual genes themselves are regulated. Here we present the first direct evidence that miRNA genes are transcribed by RNA polymerase II (pol II). The primary miRNA transcripts (pri-miRNAs) contain cap structures as well as poly(A) tails, which are the unique properties of class II gene transcripts. The treatment of human cells with α-amanitin decreased the level of pri-miRNAs at a concentration that selectively inhibits pol II activity. Furthermore, chromatin immunoprecipitation analyses show that pol II is physically associated with a miRNA promoter. We also describe, for the first time, the detailed structure of a miRNA gene by determining the promoter and the terminator of mir-23a∼27a∼24-2. These data indicate that pol II is the main, if not the only, RNA polymerase for miRNA gene transcription. Our study offers a basis for understanding the structure and regulation of miRNA genes.
Keywords: cap, Drosha, microRNA, promoter, transcription
Introduction
Hundreds of microRNA (miRNA) genes have been found in animals, plants, and viruses (Bartel, 2004; Murchison and Hannon, 2004; Pfeffer et al, 2004), making them one of the largest gene families miRNAs are defined as single-stranded RNAs ∼22 nt in length (ranging 19–25 nt) generated from endogenous transcripts that can form local hairpin structures in silico (Ambros et al, 2003). MiRNAs act as guide molecules, by base-pairing with the target mRNAs leading to translational repression and/or mRNA cleavage.
Recent studies revealed the key roles of miRNAs in diverse regulatory pathways, including development timing control, hematopoietic cell differentiation, apoptosis, cell proliferation, and organ development (Bartel, 2004). MiRNAs and their targets seem to constitute remarkably complex regulatory networks since a single miRNA can bind to and regulate many different mRNA targets and, conversely, several different miRNAs can bind to and cooperatively control a single mRNA target (Lewis et al, 2003). To dissect these complex networks operated by miRNAs, it would be critical to understand how miRNA genes themselves are regulated.
The majority of miRNA genes are located in intergenic regions or in antisense orientation to annotated genes (Lagos-Quintana et al, 2001; Lau et al, 2001; Lee and Ambros, 2001; Mourelatos et al, 2002), indicating that they form independent transcription units (Lee et al, 2002). Most of the other miRNA genes are found in intronic regions, which may be transcribed as part of the annotated genes. In animals, miRNAs are transcribed as long primary transcripts (pri-miRNAs), which are cropped into the hairpin-shaped pre-miRNAs by nuclear RNase III Drosha (Lee et al, 2003; Kim, 2004). This cleavage event is important because it predetermines mature miRNA sequence and generates optimal substrate for the subsequent events (Lee et al, 2003; Lund et al, 2004). The processing intermediate, pre-miRNA, is exported out of the nucleus by exportin-5 (Exp5), member of the Ran-dependent nuclear transport receptor family (Yi et al, 2003; Bohnsack et al, 2004; Lund et al, 2004). Pre-miRNA is subsequently cleaved by cytoplasmic RNase III Dicer (Bernstein et al, 2001; Grishok et al, 2001; Hutvagner et al, 2001; Ketting et al, 2001; Knight and Bass, 2001) into ∼22-nt miRNA duplex. One strand of this short-lived duplex is degraded by an unknown nuclease, while the other strand remains as a mature miRNA. Which strand to select is determined by the relative internal stability of the two ends of the duplex, that is, the strand with the less stable 5′ end (for instance, G:U pair versus G:C pair) usually survives (Khvorova et al, 2003; Schwarz et al, 2003).
Our knowledge on miRNA biogenesis has been significantly advanced in recent years. However, little is known about transcription of miRNA genes although it is likely to be the key regulatory step in miRNA biogenesis. To understand the mechanism of miRNA gene regulation, the basic machinery for miRNA transcription needs first to be identified.
RNA polymerase III (pol III) was initially believed to mediate miRNA transcription because it transcribes most small RNAs such as tRNAs and U6 snRNA. However, several circumstantial evidences suggest otherwise. Firstly, pri-miRNAs are sometimes over several kilobases long and contain stretches of more than four U's, which would have terminated transcription by pol III (Lee et al, 2002). In addition, a number of chimeric transcripts containing miRNA sequences and pieces of adjacent mRNAs have been found from EST analyses (Smalheiser, 2003). Many of these ESTs contain poly(A) tails and are occasionally spliced, suggesting that these transcripts are produced by RNA polymerase II (pol II), although it is not clear from this computational analysis whether these chimeric transcripts are indeed involved in miRNA biogenesis. Secondly, the expression profiles of miRNAs suggest the class II gene-like control over miRNA genes. Insertion of a pol II enhancer can induce miRNA in the case of bantem RNA in Drosophila (Brennecke et al, 2003). Temporal regulation of let-7 RNA in Caenorhabditis elegans is dependent on an enhancer element, termed temporal regulatory element (TRE) (Johnson et al, 2003). Thirdly, the analysis of the noncoding RNAs containing miRNA sequence (miR-155 (BIC) and miR-172 (EAT)) showed that they are polyadenylated and spliced (Tam, 2001; Aukerman and Sakai, 2003). The miR-172 precursor, EAT, in Arabidopsis was suggested to contain the cap structure because the RACE protocol used in the study favors capped RNAs (Aukerman and Sakai, 2003).
However, direct experimental evidence is still missing as to (1) whether pol II is indeed responsible for miRNA gene transcription and (2) how miRNA genes are structured. Here we present the first direct evidence that miRNA genes are transcribed by pol II.
Results
In order to address these questions directly, we decided to analyze the 5′- and 3′-end structures of various pri-miRNAs. First, RNAs containing 7-methyl guanosine cap were selectively enriched from total RNA by affinity purification using eIF4E, the high-affinity cap-binding protein, fused to glutathione-S-transferase (GST) (Figure 1). As a control, hnRNP A1, which is a generic pre-mRNA-binding protein, was included (Burd and Dreyfuss, 1994). RNase protection assay (RPA) was carried out then to detect pri-miR-23a from the bound (B) and unbound (UB) fractions. The fragment of 69 nt representing pri-miR-23a was detected from the GST-eIF4E-bound fraction but not from the GST-hnRNP A1-bound fraction (Figure 1A). Pre-miR-23a and mature miR-23a was not retained in the eIF4E-bound fraction, indicating that only pri-miR-23a, but not its processed products, contains the cap structure.
Figure 1.
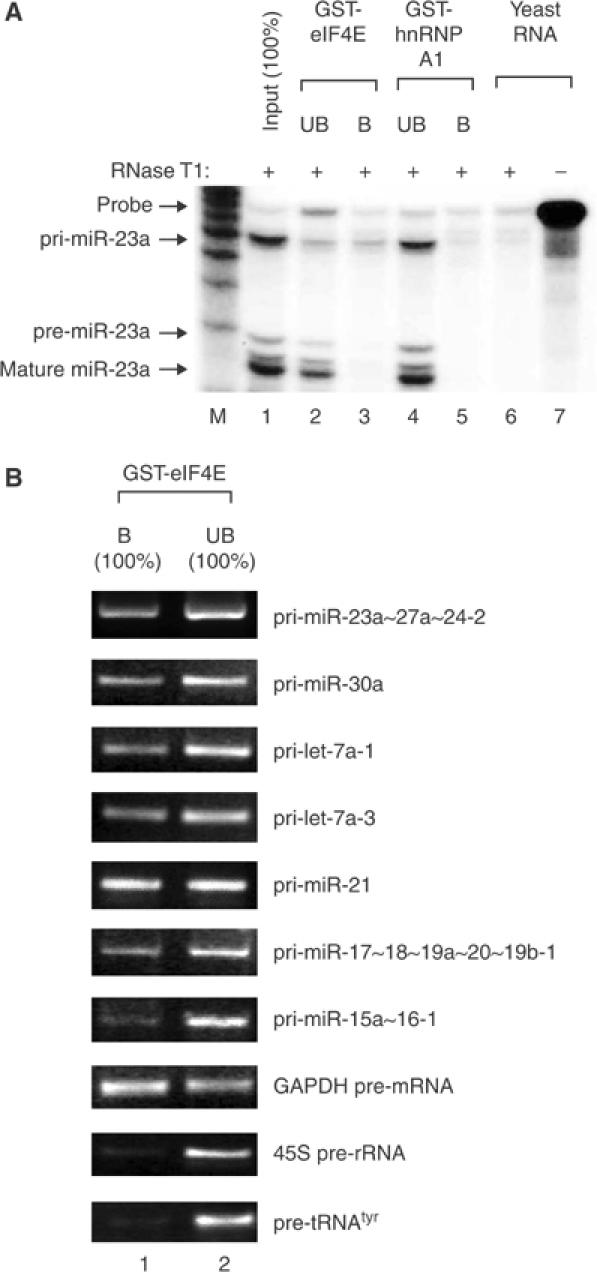
Pri-miRNAs contain the 5′ cap structures. (A) Affinity purification of cap-containing RNA followed by RPA. Total RNA from HeLa cells was used for selective enrichment using the cap-binding protein eIF4E. RNA was extracted from the bound (B) or unbound (UB) fraction. Because the whole RNA from each fraction was used for the assay without further normalization, it corresponds to 100% of the input. (B) RT–PCR demonstrating that cap is present not only in pri-miR-23a∼27a∼24-2 but also in pri-let-7a-1, pri-let-7a-3, pri-miR-30a, pri-miR-21, pri-miR-17∼18∼19a∼20∼19b-1, and pri-miR-15a∼16-1. GAPDH pre-mRNA, 45S pre-rRNA, and pre-tRNAtyr were used as controls. The whole RNA extracted from each fraction (100% of the bound (B) or 100% of the unbound (UB) fraction) was used for the assay.
To test whether the 5′ cap is a general property of pri-miRNAs, we randomly chose seven different pri-miRNAs (miR-23a∼27a∼24-2, miR-30a, let-7a-1, let-7a-3, miR-21, miR-17∼18∼19a∼20∼19b-1, and miR-15a∼16-1) and analyzed them by RT–PCR (Figure 1B). Their genomic loci relative to the closest annotated genes are shown in Supplementary figure. Reverse transcription was carried out with gene-specific primers instead of oligo-dT primer. GAPDH pre-mRNA, 45S pre-rRNA, and pre-tRNAtyr were amplified as controls representing transcripts produced by RNA pol II, I, and III, respectively. All the pri-miRNAs tested in this study showed affinity to the GST-eIF4E column, indicating that they contain the cap structures. The affinities appear to vary, with pri-miR-15a∼16-1 being the lowest. Assuming that all the GAPDH pre-mRNA molecules are capped, approximately 5–50% of pri-miRNAs seem to contain the cap structures, which implicates that additional processing event(s) may exist prior to Drosha-mediated cropping step and removes the cap from the primary transcript.
A similar approach was then taken to verify the presence of poly(A) tail at the 3′ end of pri-miRNA (Figure 2). Following selective enrichment of polyadenylated RNA using oligo-d(T) cellulose beads, RNA was extracted and analyzed by RPA (Figure 2A) and RT–PCR (Figure 2B). The results show that all of the pri-miRNAs tested in our study contain poly(A) tails although some of these signals may have come from internal priming at A stretches in the transcripts.
Figure 2.
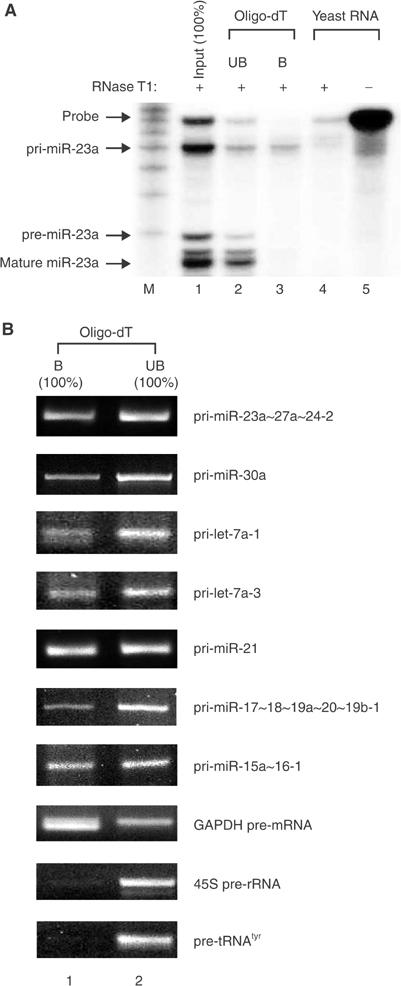
Pri-miRNAs are polyadenylated. (A) Selective enrichment of polyadenylated RNA from HeLa total RNA using oligo-dT cellulose, followed by RPA. RNA was extracted from the bound (B) or unbound (UB) fraction. Because the whole RNA from these fractions was used for the assay without further normalization, it corresponds to 100% of the input. (B) RT–PCR showing that poly(A) tail is present not only in pri-miR-23a∼27a∼24-2 but also in pri-let-7a-1, pri-let-7a-3, pri-miR-30a, pri-miR-21, pri-miR-17∼18∼19a∼20∼19b-1, and pri-miR-15a∼16-1. GAPDH pre-mRNA, 45S pre-rRNA, and pre-tRNAtyr were used as controls. The whole RNA extracted from each fraction (100% of the bound (B) or 100% of the unbound (UB) fraction) was used for the assay.
Next, we examined the sensitivity of miRNA transcription to α-amanitin, which is often used to distinguish among the activities of pol I, II, and III. Pol II is highly sensitive and therefore the treatment of mammalian cells in culture at a concentration of 50 μg/ml for 5–9 h results in the selective inhibition of RNA pol II (Figure 3). The levels of all tested pri-miRNAs were reduced (compare lanes 3 and 4), whereas those of pre-tRNAtyr and 45S rRNA precursor showed no change, indicating that miRNA transcription is carried out by pol II. The degrees of inhibition vary among different miRNAs, which may indicate the differential processing rates among pri-miRNAs. The RNAs with higher turnover rates will disappear more rapidly than others when transcription is blocked. The GAPDH pre-mRNA level was affected more than the levels of miRNAs, possibly due to the fact that pre-mRNA splicing takes place more rapidly than pri-miRNA processing. However, we do not exclude the possibility from these results that the effect of α-amanitin on pri-miRNA is indirect and that a minor fraction of pri-miRNAs can be transcribed by other RNA polymerase(s).
Figure 3.
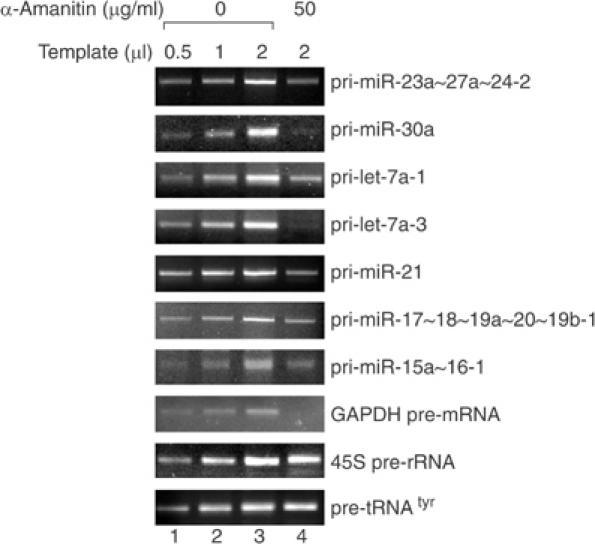
Transcription of miRNA genes is sensitive to α-amanitin. HeLa cells were treated with α-amanitin at a concentration of either 0 or 50 μg/ml for 9 h. Total RNA was prepared from these cells and 5 μg of total RNA was subjected to cDNA synthesis in 20 μl reaction. The levels of pri-miRNAs were determined by RT–PCR using gene-specific primers. Different amounts (0.5, 1, or 2 μl) of cDNA template from the untreated cells (lanes 1–3) were used for PCR to see if the PCR amplification is in a quantitative range. GAPDH pre-mRNA, 45S pre-rRNA, and pre-tRNAtyr were used as controls.
In order to gain further insights into miRNA transcription, we analyzed the gene structure of miR-23a∼27a∼24-2. Primer extension analysis was carried out to determine the transcription start site, using a primer that binds to the site between 13 and 42 bp downstream from the 3′ end of mature miR-23a (Figure 4A). Initial attempts failed due to the low steady-state level of pri-miR-23a∼27a∼24-2. This problem was circumvented by RNAi against Drosha, which otherwise rapidly cleaves pri-miRNA into smaller pre-miRNAs. Synthetic siRNA against Drosha mRNA was transfected to HeLa cells and total RNA was used for primer extension analysis. The major band of 123 nt and the minor band of 133 nt were detected from the Drosha-depleted cells (Figure 4B), indicating that transcription may start mainly at 59 bp upstream of the 5′ end of miR-23a. The fact that this band was detected only after Drosha depletion indicates that this extension product was derived from the authentic precursor of miRNA.
Figure 4.
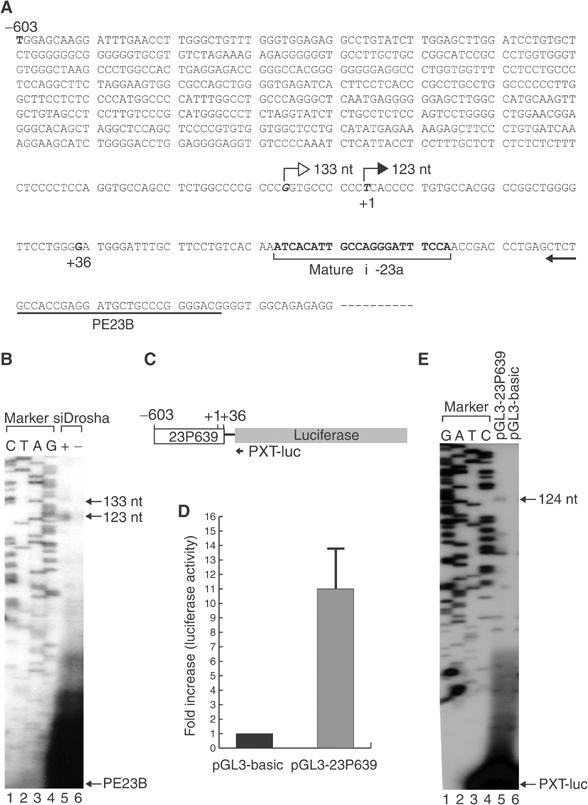
The 5′-terminal structure of the miRNA gene cluster miR-23a∼27a∼24-2. (A) Sequences around the transcription start site of miR-23a∼27a∼24-2 gene. The sequences that bind to the primer used for primer extension (PE23B) are indicated. The filled arrow indicates the major transcription start site as determined by primer extension, while the empty arrow shows the minor start site. (B) Primer extension analysis to determine the transcription initiation site. The extension product was detectable only after the cell was treated with siRNA against Drosha (+siDrosha, lane 5). (C) Schematic diagram of the reporter construct. The segment ranging from −603 to +36 bp relative to the transcription initiation site was placed at the upstream of the luciferase gene (pGL3-23P639). (D) Relative activities of the firefly luciferase are presented after normalization against the cotransfected Renilla luciferase activity (internal control). pGL3-basic is the promoter-less construct used as the backbone to generate pGL3-23P639. (E) Primer extension analysis. The transcription initiation site from the reporter construct (pGL3-23P639) is identical to that from the endogenous miR-23a∼27a∼24-2 gene. Note that the primer (PXT-luc) used for extension is complimentary to the luciferase mRNA.
To confirm that the site measured by primer extension is the transcription initiation site of the miR-23a∼27a∼24-2 gene, we constructed a reporter plasmid by inserting the region surrounding the extension termination site (between –603 and +36 bp) into pGL3-basic (Promega) that contains a promoter-less luciferase gene (Figure 4C). The resulting reporter plasmid, pGL3-23P639, was transfected into HEK293T cells along with Renilla luciferase expression vector, pRL-CMV, as an internal control. The luciferase activity from the reporter plasmid was elevated about 11-fold compared to that from the promoter-less pGL3-basic (Figure 4D). The transcription start site from pGL3-23P639 was determined by primer extension analysis using a primer that binds to the luciferase mRNA (PXT-luc; Figure 4C). If the transcription initiation site is identical to that from the endogenous miR-23a∼27a∼24-2 gene, primer extension is expected to result in a 124-nt product. Indeed, the primer was extended to 124 nt, demonstrating that the region covering –603 and +36 bp contains the genuine promoter of miR-23a∼27a∼24-2 (Figure 4E). However, we do not exclude the possibility of additional alternative promoter(s) further upstream of this promoter.
The promoter region of miR-23a∼27a∼24-2 was further analyzed by constructing a series of reporter constructs containing different regions of the promoter (Figure 5). Expression levels of luciferase from the region covering −2010 to +36, −1519 to +36, −1014 to +36, and −806 to +36 bp were approximately five- to seven-fold higher compared to that from the promoter-less construct. Similar level of expression was observed with −403 to +36 and −203 to +36 bp, while gene expression from −603 to +36 bp was significantly higher than others, suggesting that there may be negative control element(s) between −806 and −603 bp. Positive control element(s) may be present between −603 and −403 bp and between −74 and −42 bp. Further deletion to −42 bp abolished the luciferase expression, indicating that this region between −74 and −42 bp contains critical element(s) required for transcriptional initiation.
Figure 5.
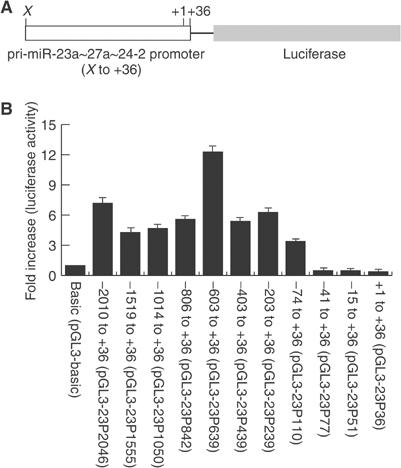
Analysis of the mir-23a∼27a∼24-2 gene promoter. (A) Schematic diagram of the reporter construct. The 5′ end (X) of the promoter varies from −2010 to +1, while the 3′ end corresponds to +36 relative to the transcription start site as determined in Figure 4. (B) Relative activities of luciferase expressed from the reporter constructs. The plasmids were cotransfected with Renilla luciferase construct into HEK293T cells and enzyme assay was carried out 48 h after transfection. Relative activities of the firefly luciferase are presented after normalization against the Renilla luciferase activity.
To test if the promoter of miR-23a∼27a∼24-2 is recognized and used by RNA pol II, HEK293T cells were transfected with pGL3-23P639 and treated with α-amanitin at a concentration of 50 μg/ml for 6 h (Figure 6). Luciferase expression from the miR-23a∼27a∼24-2 promoter was strongly suppressed under this condition, confirming that the region covering –603 to +36 bp contains a promoter that is dependent on pol II.
Figure 6.
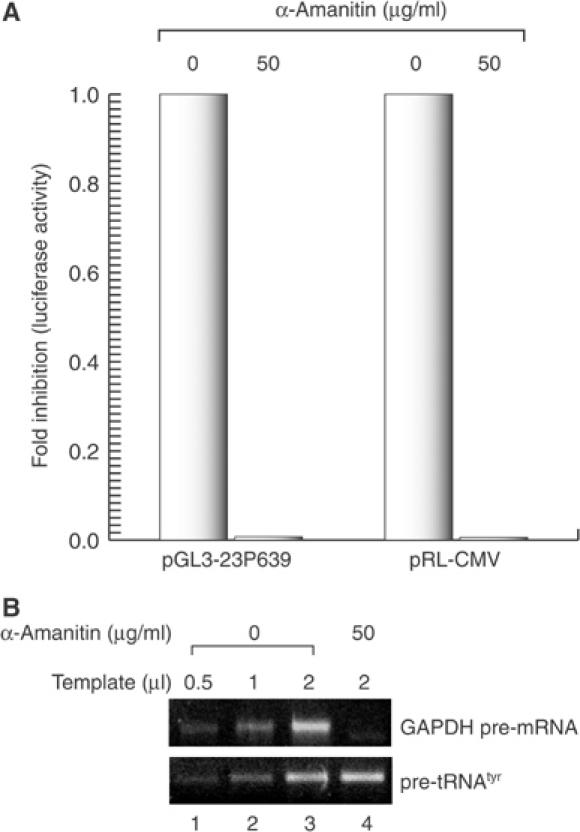
Transcription of mir-23a∼27a∼24-2 gene cluster is dependent on pol II. (A) Luciferase assay was performed using the reporter plasmid pGL3-23P639 or pRL-CMV. HEK293T cells were transfected with the reporter plasmids. Right after adding the plasmids, the cells were incubated in 0 or 50 μg/ml of α-amanitin for 6 h before the cells were harvested for assay. (B) RT–PCR to show that pol II activity (GAPDH pre-mRNA) is selectively reduced under this condition while pol III activity (pre-tRNAtyr) is not affected.
Next, we examined the physical presence of pol II on this promoter by chromatin immunoprecipitation (ChIP) (Figure 7). Immunoprecipitation was performed with either IgG against pol II (anti-pol II; 8WG16), phosphorylated form of pol II (anti-pol II-P; H5), or normal mouse IgG. Phosphorylation of the C-terminal domain of the largest subunit of pol II triggers the polymerase to shift from initiation to elongation mode. In fact, unphosphorylated pol II binds much more tightly to the promoter region than does the phosphorylated form. The promoter region of miR-23a∼27a∼24-2 was precipitated with anti-pol II antibody but not with anti-phosphorylated pol II antibody or preimmune mouse IgG. Thus, this result demonstrates that transcription initiation by pol II occurs within this region.
Figure 7.
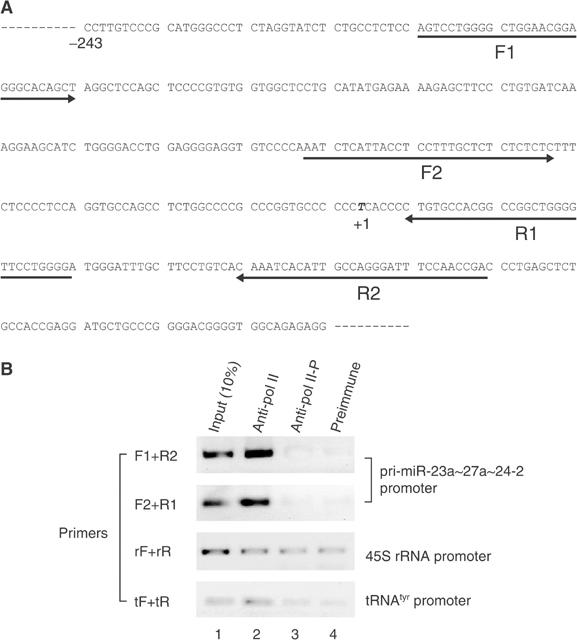
RNA pol II is physically associated with the promoter of mir-23a∼27a∼24-2 gene. (A) Sequences of the promoter region. The arrows indicate the primer sequences used for PCR. ‘+1' represents the major transcription start site. (B) ChIP analyses of HeLa cell cultures are shown. ChIP analyses were performed with monoclonal antibody (anti-pol II; 8WG16) against the C-terminal domain of the largest RNAP II subunit RPB1 or monoclonal antibody against the phosphorylated form of RPB1 (anti-pol II-P; H5). Normal mouse serum (preimmune) was also used as a negative control. Chromatin immunoprecipitates were analyzed by PCR with primer pairs flanking the promoter regions of miR-23a∼27a∼24-2, 45S rRNA promoter, or tRNAtyr gene. PCR products were resolved by agarose gel electrophoresis, revealed by staining with ethidium bromide, and presented as the reverse images.
A putative polyadenylation signal (AAUAAA) was found at +1752 bp downstream from the 3′ end of mature miR-24-2 (Figure 8A). From database searches, we could not find any ESTs containing miR-23a∼27a∼24-2 cluster itself. Interestingly, however, we found an mRNA (AL832183) and several ESTs (Unigene Hs.460763 and EC gene H19C3262.1) containing the sequences from right downstream of mature miR-24-2 (from +1 to +1771 relative to the 3′ end of miR-24-2). We hypothesized that these transcripts may have been derived from a longer primary transcript that is cleaved by Drosha. To test this possibility, we determined the actual cleavage site created by Drosha by directional cloning of pre-miR-24-2. Pre-miR-24-2 was prepared by in vitro processing and ligated to the 5′ and 3′ adapters as described previously (Lee et al, 2003). After cloning and sequencing of the ligated RNA, we found that Drosha cleaves at the 3′ end of miR-24-2 and generates exactly the same 5′ end as that of AL832183 (Figure 8B). This was confirmed by RT–PCR using three different reverse primers that bind to the sequences nearby the putative polyadenylation site (Figure 8A and C). The band of ∼2.2 kb was detected only when Drosha was depleted (Figure 8C). The intensity of PCR band was dramatically diminished, when the reverse primers (primers B and C) bind to the downstream sequences from the predicted polyadenylation site, confirming that the polyadenylation of pri-miR-23a∼27a∼24-2 occurs mainly at +1772 relative to the 3′ end of miR-24-2.
Figure 8.
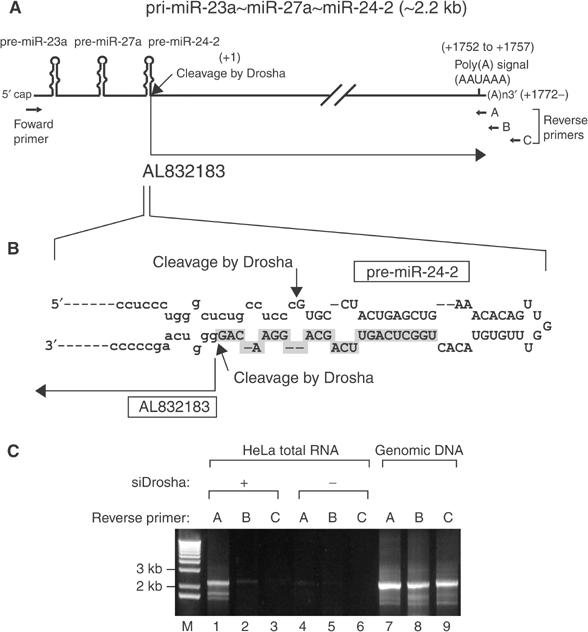
The 3′-terminal structure of the miRNA gene cluster mir-23a∼27a∼24-2. (A) Schematic representation of pri-miR-23a∼27a∼24-2. A putative polyadenylation signal (AAUAAA) was found at +1751 bp downstream from the 3′ end of mature miR-24-2. From database searches, we found an mRNA (AL832183) containing the sequences from right downstream of mature miR-24-2 (from +1 to +1771 relative to the 3′ end of miR-24-2). (B) AL832183 appears to be the by-product of Drosha-mediated processing because Drosha cleaves at the 3′ end of miR-24-2 and generates exactly the same 5′ end as that of AL832183. The cleavage sites generated by Drosha are indicated by arrows. (C) RT–PCR using three different reverse primers that bind to the sequences nearby the putative polyadenylation site as indicated in (A). The band of ∼2.2 kb was clearly detected when reverse primer A was used (lane 1). Note that this signal can be seen only when Drosha was depleted by transfecting siRNA against Drosha (siDrosha) (lanes 1–3). Genomic DNA was used as the template in lanes 7–9 to qualify the PCR primer pairs.
Discussion
Our data demonstrate that miRNA genes are transcribed by pol II and they can be classified as class II genes along with all protein-coding genes and some of the noncoding genes such as U1, U2, U4, and U5 snRNA genes. Pol II-dependent transcription of miRNA genes may allow elaborate control of miRNA biogenesis during various regulatory pathways.
The overall gene structure and transcription of miR-23a∼27a∼24-2 resemble that of protein-coding genes, except that the gene does not contain a long open reading frame. The short open reading frames found in this gene do not encode for any proteins that are homologous to known proteins. This raises a question concerning the role of the long flanking sequences in pri-miRNA and the fate of the processing products. Are they just simply ‘junk' sequences? Most of the flanking sequences are actually dispensable for miRNA biogenesis itself while only about 25–100 nucleotides surrounding both the ends of the stem–loops are required for pri-miRNA processing (Zeng et al, 2002; Lee et al, 2003; Chen et al, 2004). Chimeric ESTs containing both miRNA and mRNA sequences have been previously found (Smalheiser, 2003). Some of these chimeric ESTs possess long open reading frames that may code for proteins. It is not clear, however, if any of these proteins are actually expressed from these transcripts. It remains to be seen if a single primary transcript can be used to generate both miRNA and protein. A related question to this is if there is any fundamental difference between pri-miRNAs and protein-coding pre-mRNAs. Also important is if miRNA pathway and mRNA (protein synthesis) pathway affect each other, which may provide a new way of gene regulation. Extensive analyses on miRNA gene structure and processing will be necessary to fully answer these questions.
Our results suggest that a considerable amount of a given pri-miRNA does not contain 5′ cap or poly(A) tail. This can be interpreted such that RNA polymerase(s) other than pol II may be partly involved in transcription. Alternatively, the primary transcripts are processed in a way that the original 5′- and 3′-end structures are lost during the process. For intronic miRNAs such as miR-15a∼16-1, splicing may separate the 5′ cap and the poly(A) tail from the miRNA sequence. For others, it would be interesting to ask if there is any unidentified processing event(s) prior to Drosha-mediated cleavage.
The promoter of miR-23a∼27a∼24-2 gene determined in this study seems to lack the known common promoter elements that are required for transcription initiation complex, including the TATA box, the initiator element, the downstream promoter element (DPE), or the TFIIB recognition element (BRE) (Smale and Kadonaga, 2003). Less common promoter elements such as the downstream core element (DCE), the DPE found in the human glial fibrillary acidic protein, and the MED-1 (multiple start site element downstream) are also missing in this promoter (Smale and Kadonaga, 2003). The miR-23a∼27a∼24-2 promoter is not similar to the promoters of RNA pol II-transcribed U snRNA genes because we could not find the proximal sequence element (PSE) that is highly conserved in U snRNA genes. Possibly, the only exception may be the GC boxes. Relatively GC-rich region can be found between −530 and −410 bp, which contains at least two GC box consensus sequences. These may be functional GC boxes because the deletion of this region (−603 through −404 bp) resulted in a moderate (∼2-fold) but reproducible reduction in luciferase expression (Figure 5B).
Further analysis on the promoters of various miRNA genes would be necessary to draw a general conclusion about miRNA promoters and to fully understand the mechanism of transcriptional initiation of miRNA genes. The information on miRNA-specific element(s) would be invaluable for the prediction of miRNA genes in genomic context and for dissecting the miRNA-operated regulatory networks.
Materials and methods
RT–PCR analysis
The first-strand cDNA synthesis was carried out using SUPERSCRIPT II (Invitrogen) and the gene-specific primers. For pri-miR-23a∼27a∼24-2, 5′-AACCCCACCCACCACATCCCTCCTCCAGAC-3′ was used for the first-strand cDNA synthesis, and 5′-CCCTGTTCCTGCTGAACTGAGCCAGTGTAC-3′ (reverse) and 5′-CGCCCGGTGCCCCCCTCACCCCTGTGCCAC-3′ (forward) were used for PCR amplification. For pri-miR-30a, 5′-TTCAGCTTTGTAAAAATGTATCAAAGAGAT-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-ATTGCTGTTTGAATGAGGCTTCAGTACTTT-3′ (forward) were used for PCR amplification. For pri-miR-21, 5′-ACCAGACAGAAGGACCAGAGTTTCTGATTA-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-GTTCGATCTTAACAGGCCAGAAATGCCTGG-3′ (forward) were used for PCR amplification. For pri-let-7a-1, 5′-TTTCTATCAGACCGCCTGGATGCAGACTTT-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-GATTCCTTTTCACCATTCACCCTGGATGTT-3′ (forward) were used for PCR amplification. For pri-let-7a-3, 5′-TCTGTCCACCGCAGATATTACAGCCACTTC-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-CGGAGTCCCATCGGCACCAAGACCGACTGC-3′ (forward) were used for PCR amplification. For pri-miR-15a∼16-1, 5′-ATATACATTAAAACACAACTGTAGAGTATG-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-CCTTGGAGTAAAGTAGCAGCACATAATG-3′ (forward) were used for PCR amplification. For pri-17∼18∼19a∼20∼19b-1, 5′-GGGGTTTGAGTTTCCCTTACTTTTGTACAG-3′ was used for the first-strand cDNA synthesis, and 5′-CACTACCACAGTCAGTTT-3′ (reverse) and 5′-TGCTGAATTTGTATGGTT-3′ (forward) were used for PCR amplification. For GAPDH pre-mRNA, 5′-CGCCCCACTTGATTTTGGAGGGATCTCGCCTACCG-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-CCCATCACCATCTTCCAGGAGTGAGTGGAAGAC-3′ (forward) were used for PCR amplification. For pre-tRNAtyr, 5′-AAAAAACCGCACTTGTCTCCTTCG-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-CCTTCGATAGCTCAGCTGGTAGAG-3′ (forward) were used for PCR amplification. For 45S rRNA precursor, 5′-TGTACCGGCCGTGCGTACTTAGAC-3′ was used for the first-strand cDNA synthesis, and this primer (reverse) and 5′-CCGTCCGTCCGTCGTCCTCCTCGC-3′ (forward) were used for PCR amplification. For detection of pri-miR-23a∼27a∼24-2 of ∼2.2 kb, the first-strand cDNA synthesis was carried out using oligo-dT primers and the following primers were used for PCR amplification. Forward primer sequences are 5′-CGCCCGGTGCCCCCCTCACCCCTGTGCCAC-3′ and reverse primer (A, B, and C) sequences are 5′-GAAGTCAGGGCAACTTTTATTTAC-3′, 5′-TGAATCACAGACCAGACCTTCAGG-3′ and 5′-GATGGGCAGGCAGGAGCCCGGAGTGCCAGG-3′.
Affinity purification of 5′ capped RNA
Purified GST-eIF4E (150 μg) was incubated with glutathione agarose beads in PBS for 1 h at 4°C with consistent rotation and the beads were washed four times with RSB-100 (10 mM Tris–HCl pH 7.4, 100 mM NaCl, 2.5 mM MgCl2). The immobilized GST-eIF4E protein was then mixed with 100 μg of HeLa total RNA and 80 μg of yeast tRNA in RSB-100 for 1 h at room temperature with consistent rotation and washed six times with RSB-100. RNA retained in the beads was extracted by phenol extraction. The unbound fraction was also collected and subjected to RNA extraction. As a control, the same amount of GST-hnRNP A1 protein was used for the identical procedure.
Affinity purification of polyadenylated RNA
The purification of poly(A)-containing RNA using oligo-dT cellulose (Ambion) was carried out following the method described by Molecular Cloning laboratory manual (Sambrook and Russell, 3rd edition). A 100 μg portion of HeLa total RNA was used for this experiment.
Ribonuclease protection assay
RPA was carried out as previously described (Lee et al, 2002). Briefly, the whole RNA (100%) extracted from the bound or unbound fraction from the affinity purification was used for annealing to the radiolabled probe. RNase T1 was used for digestion instead of RNase A/T1 mix.
Cloning and sequencing of pre-miR-24-2
Pre-miR-24-2 was prepared by in vitro processing as previously described (Lee et al, 2003). To do this, pri-miR-23a∼27a∼24-2 was transcribed in vitro and processed using the immunoprecipitated FLAG-Drosha for 90 min at 37°C. Cloning was carried out as originally described by Elbashir et al (2001) with some modification. The 3′ and 5′ adapters used for the cloning are 5′-pUUUaaccgcgaattccagidT-3′ (uppercase, RNA; lowercase, DNA; p, phosphate; idT, inverted deoxythymidine) and 5′-acggaattcctcactAAA-3′ (uppercase, RNA; lowercase, DNA), respectively. Reverse transcription was performed using reverse transcription primer (5′-actggaattcgcggttaaa-3′). Forward primer (5′-cagccaacggaattcctcctcactaaa-3′) and reverse primer (reverse transcription primer) were used for PCR amplification. PCR products were subcloned into pGEM-T-easy (Promega) and nine clones were sequenced.
Primer extension analysis
Total RNA (100 μg) from normal or Drosha-depleted HeLa cells was used for annealing to the 5′-end-labeled primer (PE23B) at 42°C for 16 h and subsequently for extension using SUPERSCRIPT II at 50°C for 1 h. A 50 μg portion of RNA was used in the cases of the RNA samples from HEK293T cells transfected with pGL3-basic or pGL3-23P639. For this experiment, the primer PXT-luc was employed instead. Sequences of the primers, PE23B and PXT-luc, are 5′- CGTCCCCGGGCAGCATCCTCGGTGGCAGAG-3′ and 5′-GGGCCTTTCTTTATGTTTTTGGCGTCTTCC-3′, respectively.
RNA interference
An siRNA duplex targeting Drosha mRNA was transfected to HeLa cells using Oligofectamine Reagent (Invitrogen) as described (Lee et al, 2003). Target sequences are 5′-AACGAGUAGGCUUCGUGACUU-3′.
Promoter analysis
DNA fragments from X to +36 bp relative to the transcription initiation site of pri-miR-23a∼27a∼24-2 were inserted into pGL3-basic (Promega) using KpnI and XhoI sites. These plasmids (1.5 μg) were cotransfected with Renilla luciferase expression vector (pRL-CMV) (0.5 μg) to HEK293T cells grown in six-well plates. After 2 days, cells were harvested and luciferase assay was carried out using the Dual-luciferase assay kit (Promega) as described in the manufacturer's manual.
Chromatin immunoprecipitation assay
The ChIP assay was carried out as previously described (Baek et al, 2002). Briefly, HeLa cells were grown to 95% confluence in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. Cells were washed with PBS twice and crosslinked with 1% formaldehyde in PBS at room temperature for 15 min. Cells were washed with ice-cold PBS twice and collected into 100 mM Tris–HCl (pH 9.4) and 10 mM DTT and centrifuged for 5 min at 6000 r.p.m. Cells were washed sequentially with 1 ml of ice-cold PBS, buffer I (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5), and buffer II (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5). Cells were then resuspended in 0.3 ml of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1, 1 × protease inhibitor cocktail) and sonicated four times for 15 s each at the submaximal setting (Branson sonifier 450, output control 4) followed by centrifugation for 15 min at 13 000 r.p.m. The supernatants were collected and diluted in buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl, pH 8.1). Immunoprecipitation was performed for 6 h or overnight at 4°C with either 2 μg of IgG against RNA pol II (8WG16) (Berkeley antibody company, MMS-126R), phosphorylated form of RNA pol II (H5) (Berkeley antibody company, MMS-129R), or normal mouse IgG. After immunoprecipitation, 50 μl of protein A/G–Sepharose (50 μl of 50% slurry in 10 mM Tris–HCl, pH 8.1, 1 mM EDTA) and 2 μg of salmon sperm DNA were added and the incubation was continued for another 2 h. Precipitates were washed sequentially for 5 min each in TSE I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 150 mM NaCl), TSE II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl), and buffer III (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1). Precipitates were then washed three times with 1 ml of TE buffer and extracted two times with 150 μl each of 1% SDS and 0.1 M NaHCO3. Eluates were pooled and heated at 65°C for 6 h to overnight to reverse the formaldehyde crosslinking. DNA fragments were purified with a QIAquick spin kit (Qiagen). For PCR, 1 μl from a 30 μl DNA extraction and 25–30 cycles of amplification were used. The primers used were the following. For pri-miR-23a∼27a∼24-2 promoter, forward primers were 5′-AGTCCTGGGGCTGGAACGGAGGGCACAGCT-3′ (F-1) and 5′-AATCTCATTACCTCCTTTGCTCTCTCTCTC-3′ (F-2), and reverse primers were 5′-CCCCAGGAACCCCAGCCGGCCGTGGCACAG-3′ (R-1), 5′-TCGGTTGGAAATCCCTGGCAATGTGATTTG-3′ (R-2). For pol I promoter, 45S rRNA promoter, forward primer was 5′-GGCGCTCCGTGTGTGGCTGCGATG-3′ (rF) and reverse primer was 5′-CTAGCCGGGTCACCGGTAGGCCAG-3′ (rR). For pol III promoter, tRNAtyr promoter, forward primer was 5′-GGGACGCCGACACACGTACACGTC-3′ (tF) and reverse primer was 5′-AAAAAACCGCACTTGTCTCCTTCG-3′ (tR).
Supplementary Material
Supplementary figure
Acknowledgments
We are grateful to members of our laboratory and to Drs Kyriacos Mitrophanous, Jin Mo Park, and Young Joon Kim for their critical reading of this manuscript and helpful discussion. We thank Samchully Pharm. Co. Ltd for the gift of siRNAs. This work was supported by a grant (R02-2004-000-10173-0) from the Basic Research Program of the Korea Science & Engineering Foundation and by the BK21 Research Fellowship from the Ministry of Education and Human Resources Development.
References
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, Matzke M, Ruvkun G, Tuschl T (2003) A uniform system for microRNA annotation. RNA 9: 277–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15: 2730–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell 110: 55–67 [DOI] [PubMed] [Google Scholar]
- Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366 [DOI] [PubMed] [Google Scholar]
- Bohnsack MT, Czaplinski K, Gorlich D (2004) Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 10: 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM (2003) bantam Encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113: 25–36 [DOI] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G (1994) RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J 13: 1197–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CZ, Li L, Lodish HF, Bartel DP (2004) MicroRNAs modulate hematopoietic lineage differentiation. Science 303: 83–86 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T (2001) RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev 15: 188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106: 23–34 [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838 [DOI] [PubMed] [Google Scholar]
- Johnson SM, Lin SY, Slack FJ (2003) The time of appearance of the C. elegans let-7 microRNA is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Dev Biol 259: 364–379 [DOI] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH (2001) Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev 15: 2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209–216 [DOI] [PubMed] [Google Scholar]
- Kim VN (2004) MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol 14: 156–159 [DOI] [PubMed] [Google Scholar]
- Knight SW, Bass BL (2001) A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 293: 2269–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T (2001) Identification of novel genes coding for small expressed RNAs. Science 294: 853–858 [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862 [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294: 862–864 [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419 [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21: 4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U (2004) Nuclear export of microRNA precursors. Science 303: 95–98 [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G (2002) miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16: 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison EP, Hannon GJ (2004) miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr Opin Cell Biol 16: 223–229 [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, Tuschl T (2004) Identification of virus-encoded microRNAs. Science 304: 734–736 [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199–208 [DOI] [PubMed] [Google Scholar]
- Smale ST, Kadonaga JT (2003) The RNA polymerase II core promoter. Annu Rev Biochem 72: 449–479 [DOI] [PubMed] [Google Scholar]
- Smalheiser NR (2003) EST analyses predict the existence of a population of chimeric microRNA precursor–mRNA transcripts expressed in normal human and mouse tissues. Genome Biol 4: 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam W (2001) Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene 274: 157–167 [DOI] [PubMed] [Google Scholar]
- Yi R, Qin Y, Macara IG, Cullen BR (2003) Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: 3011–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Wagner EJ, Cullen BR (2002) Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 9: 1327–1333 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure