

Abstract
The murine c-fms (Csf1r) gene encodes the macrophage colony-stimulating factor receptor, which is essential for macrophage development. It is expressed at a low level in haematopoietic stem cells and is switched off in all non-macrophage cell types. To examine the role of chromatin structure in this process we studied epigenetic silencing of c-fms during B-lymphopoiesis. c-fms chromatin in stem cells and multipotent progenitors is in the active conformation and bound by transcription factors. A similar result was obtained with specified common myeloid and lymphoid progenitor cells. In developing B cells, c-fms chromatin is silenced in distinct steps, whereby first the binding of transcription factors and RNA expression is lost, followed by a loss of nuclease accessibility. Interestingly, regions of de novo DNA methylation in B cells overlap with an intronic antisense transcription unit that is differently regulated during lymphopoiesis. However, even at mature B cell stages, c-fms chromatin is still in a poised conformation and c-fms expression can be re-activated by conditional deletion of the transcription factor Pax5.
Keywords: antisense RNA, c-fms locus, chromatin, gene silencing, Pax5
Introduction
During haematopoietic differentiation, haematopoietic stem cells (HSCs) become gradually restricted in their differentiation potential. The balanced formation of the different blood cell types therefore requires the activation of genes in appropriate cells as well as the silencing of genes in cells in which expression is undesired (Hu et al, 1997; Miyamoto et al, 2002; Smale, 2003). Blood cell growth and differentiation are regulated by specific cytokines, which act on cells expressing particular combinations of lineage-specific cytokine receptors. Because the cytokine requirement for precursor cells is different from that of mature cells, it is important to render cells of one lineage unresponsive to the cytokines regulating alternative lineage cells. Cell type-specific inhibitors of cytokine receptor signalling have been characterized (reviewed in Fujimoto and Naka, 2003), but little is known about how cytokine receptor gene expression is silenced at the epigenetic level.
Once a cell is committed to differentiate towards a particular lineage, it responds to lineage-specific signals and generally cannot alter its cell fate. However, certain types of apparently committed cells still have the potential to differentiate into cells of another lineage (reviewed in Graf, 2002). The importance of cytokine signals in lineage determination was emphasized by a series of experiments using transgenic mice expressing the human interleukin (IL)-2 receptor β or granulocyte–macrophage colony-stimulating factor (GM-CSF) receptor in haematopoietic progenitors (Kondo et al, 2000; Iwasaki-Arai et al, 2003). These experiments showed that purified common lymphoid progenitors (CLPs) and pro-T cells from these mice can be converted by alternative cytokine signalling into myeloid cells. Lineage switching was also found in murine pro-B cells ectopically expressing the human macrophage colony-stimulating factor (CSF-1) receptor (c-fms) gene (Borzillo et al, 1990). Such manipulated cells can differentiate to macrophages in response to CSF-1. This alternative differentiation is suppressed by IL-7 signalling, which suggests that signals through the CSF-1 and IL-7 receptors can play an instructive role in myeloid and lymphoid differentiation, respectively.
Expression of the c-fms gene is tightly controlled. c-fms mRNA is detected in HSCs at a low level and is upregulated during macrophage differentiation. Receptor protein expression on the surface of the cells is only found on committed macrophage precursors (Tagoh et al, 2002). Tissue-specific mRNA expression of c-fms is regulated by well-studied promoter and intronic enhancer elements (Figure 1). The promoter used in macrophages is a TATA-less myeloid promoter, with multiple purine-rich elements bound by Ets family transcription factors, notably PU.1 (Yue et al, 1993; Ross et al, 1998). Tissue-restricted expression of the gene is dependent on the c-fms intron regulatory element or FIRE (Himes et al, 2001; Sasmono et al, 2003). We have previously examined the mechanism of upregulation of c-fms expression during macrophage differentiation. We showed that the c-fms promoter is already occupied by transcription factors at the stage of common myeloid progenitors (CMPs) where only a low level of transcripts is detected. During macrophage differentiation, c-fms expression is regulated by the coordinated assembly and disassembly of transcription factor complexes on FIRE (Tagoh et al, 2002). c-fms cis-elements in macrophages show a high level of histone acetylation, but associate with both positive and negative chromatin modification activities (Follows et al, 2003).
Figure 1.
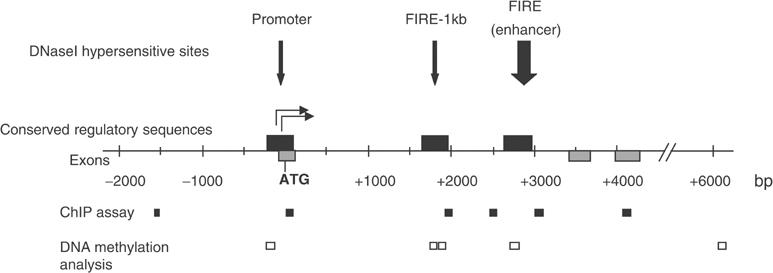
Map of the c-fms regulatory region. DHSs present in macrophages are shown as vertical arrows. Numbers indicate the nucleotide position relative to the ATG start codon. Grey boxes represent exons, and black boxes show regions conserved between human and mouse. The main transcription start sites are indicated as horizontal arrows. Amplicons for ChIP assays and DNA methylation analysis are indicated.
c-fms expression is silenced in non-macrophage cells. Silencing of c-fms mRNA in B lymphocytes is dependent on the presence of the transcription factor Pax5 (Nutt et al, 1999). Interestingly, conditional inactivation of Pax5 in committed B cell precursors leads to derepression of the c-fms gene (Mikkola et al, 2002). This is also observed after overexpression of the myeloid transcription factors C/EBP α and β in B cells (Xie et al, 2004). However, the chromatin structure of c-fms in B cells with which these transcription factors interact and the mechanism of derepression are essentially unknown. Silencing of macrophage-specific genes during B-lymphopoiesis is not well understood and may not be straightforward, as B cells and macrophages share a number of different transcription factors such as PU.1. To address this issue, we have examined the chromatin alterations occurring during the silencing of c-fms expression in B cell development. We show that the chromatin at cis-regulatory regions of the c-fms locus is in an active conformation and is bound by transcription factors in stem cells and early precursor cells. Epigenetic silencing of c-fms during B-lymphopoiesis occurs in distinct steps, but even in mature B cells c-fms chromatin is still in a poised conformation. This is confirmed by our finding that de novo DNA methylation is increased in T cells, but is delayed in B cell development whereby the promoter and FIRE remain unmethylated throughout. We show that such a poised chromatin conformation correlates with the potential to re-activate c-fms expression even in purified mature B cells by conditional deletion of Pax5. An important finding of this study is that regions of de novo DNA methylation in lymphoid cells overlap with an intronic antisense (AS) transcription unit that is active in committed B cells and macrophages, but not in cells where c-fms is completely shut down.
Results
The c-fms promoter and FIRE bind transcription factors in pluripotent stem cells and common lymphoid progenitor cells but not in differentiated B cell populations
Expression of c-fms mRNA is detectable in HSCs and CMPs (Miyamoto et al, 2002; Tagoh et al, 2002), but is absent in pro-B cells (Nutt et al, 1999). To define the first steps in c-fms silencing, we asked whether transcription factor occupancy on c-fms in stem cells is complete and thus indicative of a primed chromatin structure. Secondly, because restricted lymphoid progenitor cells (CLPs) were shown to possess latent myeloid differentiation potential (Kondo et al, 2000; Iwasaki-Arai et al, 2003), we examined whether c-fms was still occupied by transcription factors. We addressed these questions by studying purified Lin−Sca1+c-Kithi (LSK) cells, which were highly enriched for HSCs and short-term reconstituting stem cells (Geiger et al, 1998; Adolfsson et al, 2001), CLPs (Kondo et al, 1997) and CMPs (Akashi et al, 2000; Tagoh et al, 2002). As a control, we examined purified pro-B cells from the bone marrow of RAG2−/− mice and mature bone marrow-derived macrophages. The purity of each population was confirmed by surface marker analysis, Giemsa staining, colony assays and mRNA expression analysis (Figure 2 and Supplementary Figures 1 and 2; data not shown). Purified cells displayed distinct in vitro differentiation potentials (Figure 2A). As described previously, LSKs and CMPs mostly generated myeloid colonies (Akashi et al, 2000). CLPs generated lymphoid colonies containing B and NK cells but no myeloid cells, as confirmed by FACS analysis of CD19, NK1.1 and CD11b expression (data not shown). The CLP fraction formed in average one GM colony in 1000 cells, which was probably derived from a low-level contamination with myeloid precursors. Clonogenicity of CLPs under lymphoid assay conditions was much lower than that of myeloid progenitors under myeloid assay conditions, the latter regularly produced at least 250 colonies from 1000 cells (Tagoh et al, 2002 and this study). The expression profile of selected genes in purified restricted progenitors confirmed the identity of these cells (Akashi et al, 2000; DeKoter and Singh 2000; Supplementary Figure 1). mRNA for the myeloid-specific lysozyme M gene was only detected at trace levels in CLPs (Supplementary Figure 3A), confirming that this cell population had negligible myeloid contamination.
Figure 2.
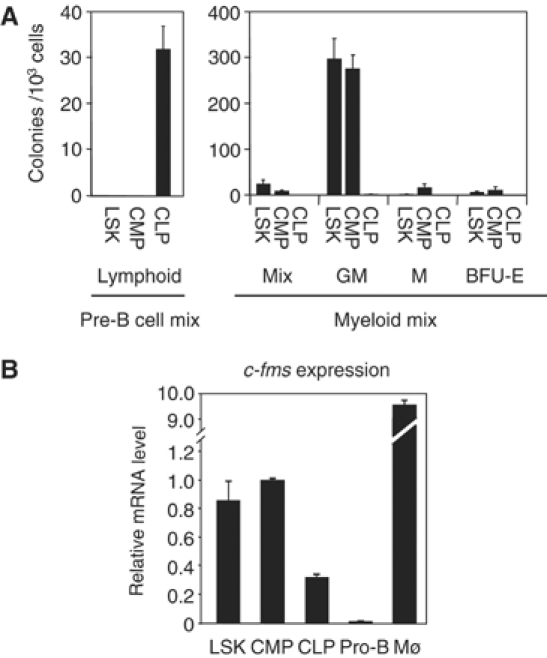
Characterization of purified LSKs, CLPs and CMPs. (A) Colony-forming activities of LSKs, CLPs and CMPs. Sorted cells were seeded at a density of 1000 cells/ml in methylcellulose medium containing IL-7, SCF and VEGF for lymphoid assays (pre-B cell mix), and IL-3, IL-6, SCF and Epo for myelo-erythroid assays (GM, M, Mix, BFU-E). Colonies were scored at days 8–10. The results are represented as the mean value of four independent experiments. (B) Expression of c-fms mRNA in purified LSKs, CLPs, CMPs, pro-B cells and macrophages (Mø). Gene expression was measured by RT–PCR. Arbitrary units were calculated relative to the expression level in CMPs. The bars represent the mean value of two independent experiments.
The result of our c-fms expression studies is shown in Figure 2B. c-fms transcripts were found around the detection limit in purified primary pro-B cells. In contrast, all other precursor cell types (LSKs, CLPs and CMPs) expressed low but clearly detectable levels of c-fms RNA. The low level of GM colony formation of the CLP fraction (less than 1% compared with CMPs) was not reflected at the level of c-fms expression (20% of that of CMPs). These results were mirrored by transcription factor-binding site occupancy as revealed by in vivo DMS footprinting analysis with these cell populations. As shown in Figure 3, the PU.1 consensus sequences within the promoter and FIRE were occupied in LSKs, CLPs and CMPs. The footprints seen at the promoter were weaker in CLPs than in LSKs, CMPs or macrophages, whereas the footprints at FIRE were similar in both CLPs and CMPs. In contrast, the Ets/AML1/Sp1 cluster in FIRE was only occupied in CLPs, CMPs and mature myeloid cells. To further rule out the possibility of myeloid contamination, we performed a DMS footprinting experiment on a sample consisting of a mixture of 95% wild-type pro-B cells (lacking footprints) and 5% monocytic cells (with fully occupied transcription factor-binding sites). RNA prepared from this cell mixture contained a much higher level of lysozyme M RNA than CLPs (Supplementary Figure 3A), but showed no footprints at the promoter and FIRE (Supplementary Figure 3B and C). This confirms that the threshold for detecting alterations of DMS reactivity in mixed cell populations is significantly higher than any possible myeloid contamination. In summary, although purified CLPs were functionally distinct from CMPs and could not generate myeloid cells, c-fms chromatin in these cells is accessible to transcription factor binding, resulting in low level but clearly detectable c-fms mRNA expression. No footprints on any of the c-fms cis-regulatory elements were seen in pro-B cells, confirming that the loss of steady-state mRNA is accompanied by a loss of stable transcription factor binding in the majority of cells.
Figure 3.
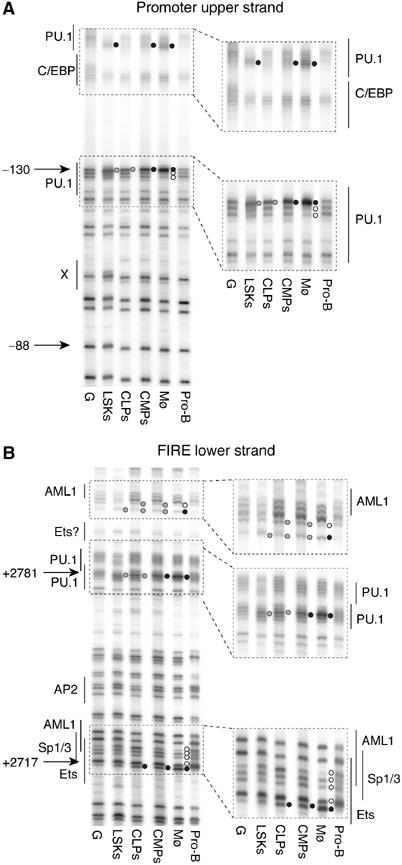
Transcription factor binding to the c-fms promoter and FIRE is lost in pro-B cells. DMS footprinting analysis of promoter (upper strand) (A) and FIRE (lower strand) (B). The numbers on the left indicate the nucleotide position relative to the ATG codon. Transcription factor-binding sites and their nature as determined by ChIP assays and in vitro DNA–protein interaction studies (Tagoh et al, 2002; Follows et al, 2003) are indicated. Black circles indicate hypermethylated guanines, and open circles indicate hypomethylated guanines compared with DMS-treated naked DNA (G). Grey circles indicate weaker footprints. From left to right: DMS-treated naked DNA (G), purified cells (LSKs, CLPs, CMPs, bone marrow macrophages (Mø), freshly purified pro-B cells from RAG2−/− mice).
c-fms chromatin in B cells is still in a poised conformation
Experiments from our laboratory showed that genes destined for activation undergo a number of defined chromatin alterations during cell differentiation, which start long before the onset of gene expression (Kontaraki et al, 2000; Tagoh et al, 2004). If such a stepwise process could be linked to a specific chromatin fine structure of given genes during gene silencing, this would enable the definition of windows of opportunity for the reversion of specific gene expression programmes. We therefore examined different chromatin features of c-fms during B-lymphopoiesis as well as in other non-myeloid cell types and compared them with those of myeloid cells.
A hallmark of active chromatin is its enhanced accessibility to digestion with nucleases. We measured differential nuclease accessibility in various cell types using DNaseI and micrococcal nuclease (MNase). Digestion products were visualized at single nucleotide resolution by using two different types of ligation-mediated PCR (LM-PCR). DNaseI digestion followed by amplifying single-strand molecules measures the number and position of nicks in one DNA strand, thus assaying both DNA accessibility and DNA topology. MNase digestion was followed by the selective amplification of double-strand breaks, which usually occur in nucleosomal linker regions. The appearance of specific bands in such an assay is interpreted as an indication for a specifically positioned nucleosome. To perform these assays we isolated B220+ bone marrow B cells, consisting mainly of early B cell precursors and CD19+ splenic B cells consisting mostly of mature B cells, as indicated in Supplementary Figure 3. In addition, we assayed primary embryonic fibroblasts, mature macrophages and purified T cells (Supplementary Figure 3).
Figure 4A shows an MNase digestion experiment examining nuclease accessibility at the c-fms promoter. To control for equal digestion efficiency, we also examined the GAPDH promoter, which is active in every cell type. With fibroblasts we obtained a rather diffuse cleavage pattern except for one slightly stronger band upstream of the PU.1-binding sites at −130 bp just upstream of the main transcription start sites. Macrophages showed a different digestion pattern. Here we observed a strong MNase hypersensitive site (MNase HS) downstream of the main transcription start sites. Interestingly, we saw the same MNase HS in immature B cells, although the upstream band was also present. The same was true for mature B cells, although the intensity of the MNase HS was reduced compared to the upstream band. In contrast, the pattern in T cells resembled more that of fibroblasts, and the MNase HS had almost disappeared. However, as the naked DNA control showed some preferential digestion of the MNase HS sequence, we confirmed by restriction enzyme accessibility assay using PvuII that this region was indeed differentially accessible to nuclease digestion in macrophage and fibroblast nuclei (Himes et al, 2001; data not shown).
Figure 4.
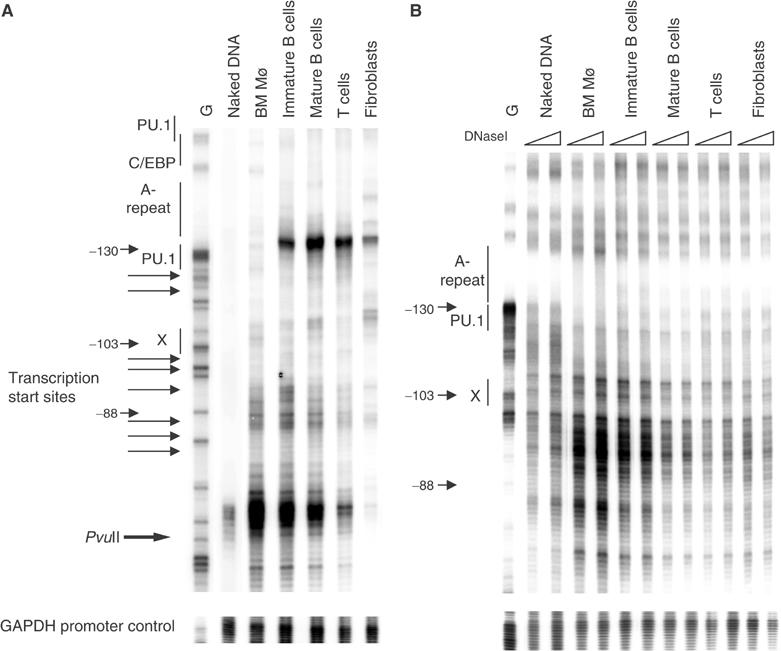
Chromatin at the c-fms promoter is in a partially active conformation in B cells. (A) From left to right: In vivo MNase footprinting experiment with naked DNA and chromatin prepared from the indicated cell populations using primers specific for the c-fms promoter (upper panel) or the GAPDH promoter (lower panel). Horizontal arrows indicate the position of transcription start sites. The PvuII site is at position −66 bp. (B) In vivo DNaseI footprinting experiment with naked DNA and the indicated cell populations using increasing amounts of DNaseI (20 and 40 U) G: G-reaction of naked DNA. For further description, see Figure 3.
In DNaseI hypersensitive site (DHS) mapping experiments, which assay double-strand cuts produced by DNaseI, we have previously shown that the c-fms promoter is strongly DNaseI hypersensitive in macrophages but not in fibroblasts (Himes et al, 2001). More importantly, no DHS was seen in an IL-7-dependent pro-B cell line (data not shown). Figure 4B shows the result of a high-resolution DNaseI digestion experiment with the GAPDH promoter as control. c-fms chromatin in macrophages was highly accessible across the entire promoter region, and a pattern was generated that was similar but not identical to that generated by naked DNA. DNaseI accessibility in immature B cells was only slightly reduced as compared to macrophages and was progressively reduced in mature B cells, T cells and fibroblasts.
Taken together, our experiments demonstrate that although transcription factors are no longer stably bound, the chromatin structure at the c-fms promoter in immature B cells is very similar to that of myeloid cells. Moreover, even in mature B cells, the region downstream of the main transcription start sites was still highly MNase accessible. No cell population exhibited a DNaseI hypersensitive region at the exact position of the MNase HS, which therefore most likely marks the position of linker DNA. A reasonable hypothesis is therefore that the c-fms promoter is occupied by a nucleosome that adopts alternative average positions in c-fms-expressing and nonexpressing cells. In B cells, this nucleosome maintains a position identical to that of myeloid cells and is only destabilized later in B cell development, along with gradually increasing chromatin compaction.
Histone modifications at the c-fms locus are altered in a complex fashion during B-lymphopoiesis
Chromatin activation and silencing are regulated by different enzymatic activities that modify the N-terminal tails of histones (Fischle et al, 2003). Histone acetylation is a hallmark of active chromatin, whereas the methylation of histone H3 lysine 9 (K9) is indicative of inactive chromatin. Moreover, transcription leaves a trail of histone modification (histone H3 lysine 4 (K4) trimethylation) behind, which has been interpreted as a ‘memory' of recent transcriptional events and can be maintained through several cell generations (Ng et al, 2003). We examined the level of histone H3 modification at c-fms by chromatin immunoprecipitation (ChIP) assays using crosslinked chromatin digested with MNase (Figure 5). Each amplicon was represented at equal levels in the input DNA (data not shown). We have previously shown that the activation of c-fms expression in macrophages is accompanied by hyperacetylation of histone H3 at each regulatory region (Follows et al, 2003). These data were obtained with sonicated chromatin and could be reproduced by the experiment shown in Figure 5A, thus validating our experimental approach. The analysis includes examination of a second conserved region of the c-fms intron, which is annotated as FIRE-1kb. This sequence is also DNaseI hypersensitive in macrophages and has enhancer activity in transient transfection assays (Himes et al, 2001). An elevated level of H3K9 acetylation was only seen in macrophages and monocytes, whereas in all other cell types acetylation levels were low, but were still slightly elevated in immature B cells. H3K9 dimethylation levels across c-fms were high in fibroblasts, but lower in all other cell types (Figure 5B). However, the c-fms intronic region but not the promoter and downstream genomic regions showed increased H3K9 dimethylation levels in B-lineage cells as compared to myeloid cells. The result of the analysis of H3K4 trimethylation was surprising (Figure 5C). Here we saw a high signal at the promoter and downstream regulatory regions not only in macrophages and monocytes but also in B cell precursors. The H3K4 trimethylation level in the intronic region was as high as in macrophages.
Figure 5.
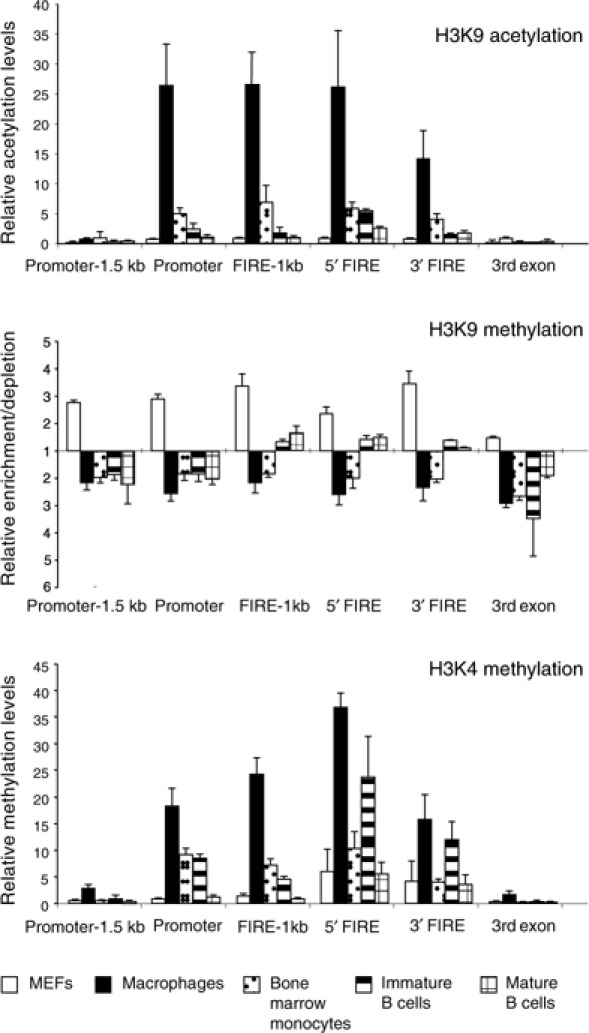
Alterations of histone H3 modifications inside and outside the c-fms regulatory region during B-lymphopoiesis. ChIP assays using antibodies specific for acetylated H3K9 (A), dimethylated H3K9 (B) and trimethylated H3K4 (C). The region-specific enrichment by ChIP was examined by real-time PCR using primers indicated in Figure 1. DNA enrichment was calculated as described in Materials and methods. Bars represent the mean±s.d. of quantifications from two to four separate immunoprecipitations analysed in duplicate.
CpGs at c-fms cis-regulatory regions are unmethylated in early progenitors and myeloid cells and are differentially methylated during B- and T-lymphocyte differentiation
Methylation of CpGs is a hallmark of silent chromatin (Bird, 2002). We therefore examined the DNA methylation status of selected CpG motifs centrally located within each of the c-fms cis-elements at the different stages of myeloid cell and B cell differentiation by using methylation-sensitive restriction enzymes. This analysis was performed with LSKs, CMPs, CLPs pro-B cells, sorted splenic B and thymic T cells as well as embryonic fibroblasts (Figure 6B and Supplementary Figure 4). The c-fms regions investigated were the promoter, FIRE-1kb, FIRE and a downstream region ((C); Figure 6A), which did not harbour a DHS and is not conserved between human and mouse. The promoter of the silent α-fetoprotein gene served as additional control. Both genomic regions were fully methylated in every cell type examined (Figure 6B).
Figure 6.
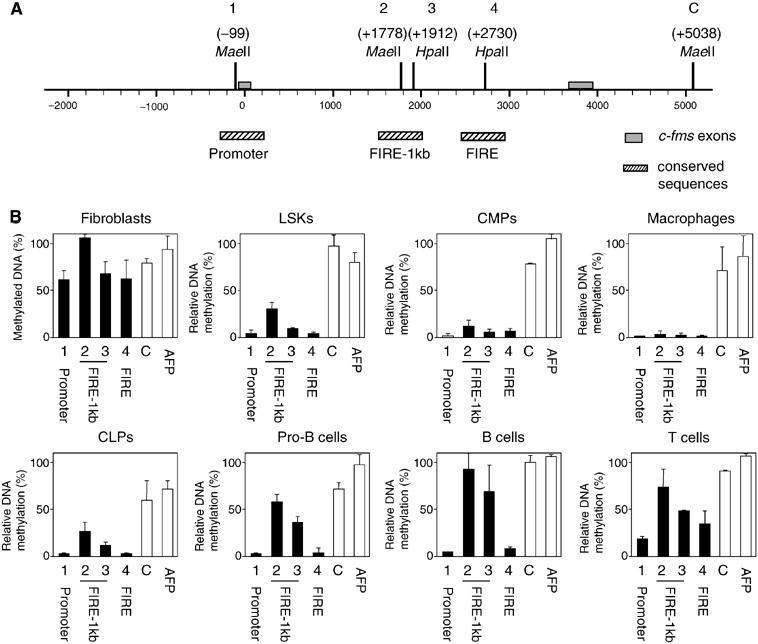
DNA methylation at specific c-fms cis-regulatory elements during haematopoietic differentiation. (A) Schematic representation of the position of the recognition sites of the differentially methylation-sensitive restriction enzymes MaeII and HpaII in the c-fms promoter and first intron (1–4: regulatory regions; C: downstream control region). Relevant DNA sequences and the position of transcription factor-binding sites are depicted in Supplementary Figure 4. (B) DNA methylation status at specific c-fms cis-regulatory (black bars) and control regions (white bars) in the indicated cell types. After HpaII or MaeII digestion of genomic DNA from each cell type, the amount of undigested DNA was measured by real-time PCR. The bars represent the mean value±s.d. of two to four independent experiments analysed in duplicate. AFP: α-fetoprotein promoter.
The CpGs at all c-fms cis-elements analysed were highly methylated in embryonic fibroblasts and completely unmethylated in macrophages (Figure 6B). CpGs at the promoter (site 1) and FIRE (site 4) showed a similarly low methylation level in LSKs, CMPs and CLPs. However, the methylation level of CpGs at the intronic FIRE-1kb (sites 2 and 3) was elevated in CLPs and was further increased with progressing B cell differentiation. CpG methylation at the promoter and FIRE remained at low levels during B cell development. In contrast, CpG methylation levels at the promoter and FIRE were increased in T cells.
Regions of DNA methylation and increased histone H3K9 dimethylation overlap with a differentially regulated antisense transcription unit
As shown above, levels of H3K4 trimethylation were elevated throughout the intronic regulatory region in B cells. Two explanations were possible for our findings. One was that H3K4 trimethylation was highly stable and was still present in B cells, although transcription had ceased. However, H3K4 trimethylation levels at some positions of the c-fms locus were almost as high in B cells as in macrophages. An alternative explanation was that the c-fms intronic region was subject to intragenic transcription. Small RNA molecules and in particular AS RNAs have been implied in gene silencing and the initiation of heterochromatin formation (Grewal and Moazed, 2003). To this end, we examined the different cell types for the presence of AS c-fms RNA using real-time RT–PCR analysis (Figure 7). To ensure that only AS RNA molecules were detected, we used biotinylated primers for cDNA synthesis, bound the reaction products to magnetic beads and removed excess RNA and contaminating genomic DNA. To get a first idea of the position of a putative AS transcription unit and to quantify accurately RNA levels, we amplified specific regions by real-time PCR using different sets of primers. All cDNA synthesis reactions were further controlled by (i) including GAPDH primers to test for RNA quality, (ii) performing real-time PCR experiments on samples without reverse transcriptase (−RT) and (iii) verifying the correct fragment size by gel electrophoresis (data not shown). AS transcription was only found in macrophages and B cells, but not in fibroblasts and T cells. The level of AS RNA was low, but similar in macrophages and B cells, whereas significant levels of spliced sense transcripts were only detected in macrophages (Figure 7B). This indicates that AS transcription in B cells is genuine and does not result from contamination with myeloid cells. AS RNA was only found around FIRE. To confirm this finding, the start site of the AS transcript at FIRE was determined by reverse transcriptase-terminal transferase-dependent PCR (RT–TDPCR). This method uses terminal transferase tailing and linker ligation to amplify cDNA and to identify the start site of low abundant transcripts (Chen et al, 2000). Two major transcription start sites within FIRE at +2760 and +2706 were detected in macrophages and immature as well as mature B cells, but not in any other cell types tested (Figure 7C).
Figure 7.
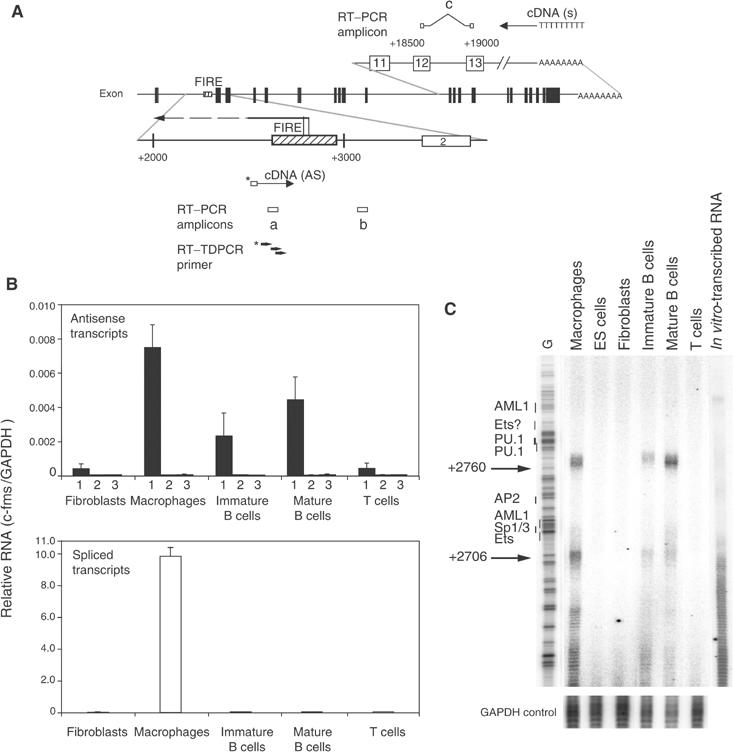
Antisense (AS) RNA starting at FIRE is expressed in macrophages and B cells. (A) Schematic representation of the position of primers used to carry out cDNA synthesis as well as real-time PCR amplicons. cDNA specific for AS RNA was synthesized from a biotinylated primer (indicated below the map), whereas an oligo (dT) primer was used to detect spliced transcripts (indicated above the map). Primer sets (a) (FIRE06) and (b) (3′-FIRE) were used to detect AS RNA transcribed from FIRE and from downstream of FIRE, respectively. Primer set (a) (c-fms QPCR), which is located between exons 12 and 13, was used to detect spliced sense transcripts. Black boxes in the map and numbered white boxes in the top and bottom map represent c-fms exons. Dashed boxes in the bottom row represent FIRE sequences. The numbers indicate the nucleotide position relative to the ATG start codon. The localization of the AS transcript is indicated as a horizontal arrow and the two AS transcription start sites mapped in (C) are indicated. (B) Expression of sense and AS RNA at the c-fms locus as assayed by real-time PCR. The top panel represents signals obtained with primer pair (a) measuring AS RNA originating from FIRE (1), signals from genomic DNA contamination (2) (assays with primer pair (a) but without reverse transcriptase) and signals obtained with primer pair (b) (3). The bottom panel represents spliced sense transcripts detected by primer pair (c). Bars represent mean value±s.d. of two to four independent experiments analysed in duplicate. (C) Determination of the start site of the AS RNA within FIRE. RT–TDPCR was performed as described in Materials and methods using RNA prepared from the indicated cell types and from in vitro-synthesized RNA as a control for cDNA synthesis and amplification artefacts. An RT–TDPCR reaction examining the GAPDH gene was performed as internal control. A sequence reaction was run on the gel alongside the RT–TDPCR samples and the positions of transcription factor-binding sites within FIRE are indicated. This result was confirmed in two independently performed experiments.
Epigenetic c-fms silencing is reversible throughout B-lymphopoiesis
Our observation of a poised chromatin structure and incomplete DNA methylation of c-fms in mature B cells raised the possibility that c-fms chromatin could be reprogrammed into the active state in mature B cells. Conditional deletion of the B cell commitment gene Pax5 was previously shown to re-activate c-fms expression in pro-B cells (Mikkola et al, 2002). However, it is not known whether the loss of Pax5 at later stages of B cell development could also lead to derepression of the c-fms gene. To address this question, we have taken advantage of the CD19-cre line, which efficiently deletes a floxed (F) Pax5 allele only in mature B cells (Horcher et al, 2001). Conditional Pax5 inactivation in mature B cells of Pax5F/− CD19-cre mice leads to the loss of Pax5 function and concomitant upregulation of CD25 in contrast to the B cells of control Pax5F/+CD19-cre mice (Horcher et al, 2001). We therefore sorted mature Pax5-deficient (Pax5Δ/−) B cells as CD25+IgM+ cells from the lymph nodes of Pax5F/−CD19-cre mice after MACS depletion of non-B cells, whereas control Pax5Δ/+ B cells were isolated as IgM+IgD+ cells from Pax5F/+CD19-cre mice (Supplementary Figure 5). RT–PCR analysis confirmed that the floxed Pax5 allele was deleted in all sorted Pax5Δ/− B cells, leading to the downregulation of the Pax5 target gene CD19 (Figure 8A) as previously published (Horcher et al, 2001). Figure 8A clearly shows that the loss of Pax5 led to re-expression of the c-fms gene in mature Pax5Δ/− B cells in contrast to the control Pax5/+ B cells. Real-time PCR quantification furthermore revealed that the c-fms gene was re-activated to the same expression level seen in Pax5−/− bone marrow pro-B cells (Figure 8B). These results unequivocally demonstrate that c-fms silencing in B lymphocytes requires the continuous presence of Pax5 even at late stages of B cell differentiation.
Figure 8.
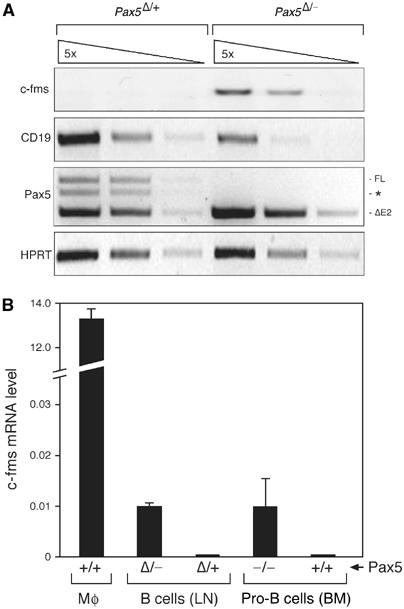
Epigenetic silencing of c-fms is reversible in mature B cells. (A) Mature control and Pax5-deficient B cells were isolated free of contaminating non-B cells from the lymph nodes (LN) of Pax5F/+ CD19-cre mice (abbreviated as Pax5Δ/+) or Pax5F/−CD19-cre (Pax5Δ/−) mice (Horcher et al, 2001) prior to RNA preparation and cDNA synthesis as described in Supplementary Figure 5 and Materials and methods. The cDNA of both cell types was normalized for equal expression of the hypoxanthine phosphoribosyltransferase (HPRT) gene followed by analysis of the indicated transcripts by semiquantitative RT–PCR of five-fold serial cDNA dilutions. Pax5 transcripts were amplified from exons 1A to 5. ΔE2 denotes the truncated transcript of the Pax5Δ allele lacking exon 2 in contrast to the full-length transcript (FL). A PCR artefact consisting of an FL/ΔE2 cDNA hybrid is indicated by an asterisk. All PCR fragments had the correct size, expect for the respective spliced mRNA. (B) Real-time PCR quantification of c-fms mRNA levels in macrophages (Mø), sorted lymph node (LN) B cells and in vitro-cultured bone marrow (BM) pro-B cells of the indicated Pax5 genotypes. The c-fms mRNA level of the CMP was used as reference, which was arbitrarily assigned a value of 1.
Discussion
c-fms cis-regulatory elements are occupied in pluripotent and restricted haematopoietic precursor cells
In this paper, we present for the first time in vivo footprinting experiments with CLPs and with a cell population (LSKs) that was highly enriched for HSCs and short-term reconstituting stem cells. LSKs show basically the same degree of transcription factor occupancy at the c-fms promoter as CMPs, thus providing direct confirmation of the hypothesis that the chromatin of most, and not just a subset, of stem cells and early progenitors is primed and accessible to transcription factor binding. Another important result of our study is that the c-fms gene is still expressed at low levels in CLPs. This is in apparent contrast to previous studies that have not detected c-fms mRNA expression in single CLPs that were purified by the same method (Miyamoto et al, 2002). This may be because the level of c-fms expression is below the detection limit of single-cell PCR. More convincingly, we observed that c-fms transcription factor-binding sites in CLPs were still occupied by transcription factors, although the signal was weaker than in CMPs and LSKs. We excluded the possibility that the signal originated from the small population of myeloid precursor cells in the sorted population or from a different level of transcription factors in these cells. Instead, we believe that transcription factor binding in CLPs is becoming destabilized, in contrast to cells which are functionally committed to the myeloid lineage. Support for the idea that transcription factor action can be dynamic comes from lineage tracing experiments in which mice carrying a Cre recombinase gene knocked into the myeloid-specific lysozyme M gene were crossed with ROSA26-EYFP reporter mice (Ye et al, 2003). These experiments revealed EYFP expression in non-myeloid cells, indicating that pluripotent cells giving rise to all myeloid and lymphoid cell types express the recombinase at a low level at some point in time. Transplantation experiments with sorted EYFP-negative cells from such mice showed restoration of EYFP activity, demonstrating that the expression of Cre recombinase under the control of the mouse lysozyme M gene is an infrequent but dynamic event.
Silent chromatin formation at the c-fms locus during B cell differentiation is a gradual process
Taken together, our results show that epigenetic silencing of the c-fms locus during lymphopoiesis is a slow process that begins at the CLP stage. c-fms cis-regulatory regions first lose transcription factor binding and DNaseI hypersensitivity. In parallel, mRNA expression from the c-fms promoter ceases and H3K9 hyperacetylation at the c-fms regulatory region is reduced. These processes are completed at the pro-B cell stage. However, at the same stage, not all c-fms chromatin features have reversed to an inactive state. In contrast to T cells, B cell precursors and even mature B cells showed certain chromatin features characteristic of an active locus. In immature B cells, DNaseI accessibility at the promoter was still high and levels of H3K4 trimethylation throughout the intronic regulatory region were elevated. In all B cell types studied, levels of H3K9 dimethylation were low compared to fibroblasts, and the region immediately downstream of the transcription start sites was still MNase hypersensitive. Our studies of DNA methylation confirmed this idea. Using a randomly integrated transgene, it was recently shown that DNA methylation is the last step in gene silencing and slowly increases during different cell generations (Mutskov and Felsenfeld, 2004). A similar observation was made with the mouse Dntt gene that is shut down during the maturation of immature thymocytes into mature T cells (Su et al, 2004). The situation at the c-fms locus appears to be more complex. c-fms cis-regulatory elements exhibited low DNA methylation levels in all precursor types and macrophages. However, although in T-lymphopoiesis DNA at all c-fms elements became significantly methylated, it was differentially methylated during B-lymphopoiesis. CpGs between FIRE and the promoter were progressively methylated, whereas CpGs at the cores of the c-fms promoter and FIRE stayed unmethylated throughout. Experiments from our laboratory indicate that CpGs located in the centre of critical transcription factor-binding sites but not in the flanking regions of myeloid-specific genes are the first to be demethylated during haematopoiesis (Tagoh et al, 2004). A possible role of transcription factors in protecting DNA from methylation has been reported and may be caused by an interference of these transcription factors with maintenance methylation after DNA synthesis (Mummaneni et al, 1998; Kress et al, 2001; Thomassin et al, 2001). Such transient interaction cannot be detected by DMS in vivo footprinting (Lefevre et al, 2003). Therefore, it is tempting to speculate that differential methylation of the c-fms promoter and FIRE in B cells and T cells may be caused by transcription factors (such as PU.1), which are expressed both in macrophages and B cells.
The c-fms locus contains a differentially regulated antisense transcription unit
Our experiments revealed the presence of an AS RNA within the coding region of the c-fms gene starting at the FIRE that is expressed at low levels in macrophages and B cells. AS RNAs have been shown to play a role in the regulation of X-inactivation and genomic imprinting (Lee et al, 1999; Sleutels et al, 2002). For the human α-globin locus, it was recently shown that the insertion of an AS promoter next to a CpG island leads to gene silencing and DNA methylation (Tufarelli et al, 2003). We do not yet know whether AS RNA expression is strictly required for c-fms silencing. However, our extensive analyses of c-fms chromatin and expression are fully consistent with a regulatory role of the AS transcription unit. We found that AS transcripts were upregulated by agonists (LPS, phorbol esters, CSF-1) that repress c-fms mRNA expression in macrophages (Himes and Hume, manuscript in preparation). Here, we show that the balance between sense and AS RNA expression in the c-fms intronic region is dynamically regulated in different cell types. AS RNA is expressed at similar levels in both macrophages and B cells, whereas sense RNA is only expressed in macrophages. The ratio of sense to AS expression therefore correlates with c-fms activity. This is in agreement with our previous observation that FIRE is a dynamic element, which differentially binds transcription factors in development and associates with histone acetylases as well as histone deacetylases (Tagoh et al, 2002; Follows et al, 2003). The data presented here show most convincingly that the AS transcription unit overlaps with a region of increased de novo DNA methylation as well as with areas of elevated H3K4 trimethylation and H3K9 dimethylation. This raises the interesting possibility that small double-stranded RNA molecules are formed that target gene silencing complexes to the c-fms intronic region, as shown in yeast and Drosophila (Schramke and Allshire, 2003; al-Bhadra et al, 2004; Verdel et al, 2004). The level of AS transcript was low as compared to the sense transcript. This is reminiscent of reports from intergenic transcripts within the β-globin locus that were found at similar low levels, as they were only expressed at a specific point within the cell cycle (Gribnau et al, 2000). AS transcription was absent in T cells and fibroblasts where c-fms chromatin was shut down. This could indicate a requirement for AS transcription to initiate epigenetic silencing, but not for its maintenance.
c-fms silencing is reversible in B cells
Our chromatin structure studies exclude some possible mechanisms by which Pax5 represses c-fms in B cells. The presence of Pax5 in wild-type B-lineage cells does not cause remodelling of the promoter nucleosome into the inactive conformation, it does not induce locus-wide histone H3K9 hypermethylation and it does not induce competitive binding of Pax5 to cis-regulatory elements and other conserved regions of the c-fms gene (this study and unpublished observations). Our experiments point to a mechanism by which Pax5 shifts the balance between activating and repressing activities recruited to the c-fms locus, and it is likely that this balance needs to be continuously re-established during B-lymphopoiesis. In support of this idea, we show here that c-fms expression can be re-activated not only in pro-B cells (Mikkola et al, 2002) but also in mature B cells, indicating that the repression of c-fms requires the continuous presence of Pax5 throughout B cell differentiation. Our finding that the c-fms locus is not completely assembled into silent chromatin in mature B cells provides a molecular explanation for the c-fms re-activation in response to Pax5 loss and C/EBP overexpression (Xie et al, 2004). Importantly, the derepression of the c-fms gene in mature Pax5-deficient cells of the Pax5F/-CD19-cre genotype resulted in a similar level of c-fms mRNA expression as in Pax5−/− pro-B cells, strongly suggesting that re-activation events occur in the majority of the mature Pax5-deficient B cells.
Our data link the plasticity of the epigenetic state of the c-fms gene to a specific chromatin state that is depleted of activating histone marks (acetylated histones) and even contains partly methylated DNA, but is still in a poised conformation. Not only c-fms but also a number of other lineage-specific genes were activated upon the conditional inactivation of Pax5 in mature B cells (A Schebesta and M Busslinger, unpublished observations). It will be interesting to see whether these genes adopt similar intermediate epigenetic states in development.
Materials and methods
Purification and RT–PCR analysis of mature Pax5-deficient B cells
Lymph node cells from Pax5F/+ CD19-cre or Pax5F/−CD19-cre mice (Horcher et al, 2001) were stained with PE-anti-CD8 (53-6.7), CD4 (L3T4), DX5 (DX5), CD11c (HL3), Mac-1 (M1/70), Gr-1 (RB6-8C5), CD43 (S7) and Ter119 (TER119) antibodies, and non-B cells were eliminated by magnetic cell sorting with anti-PE MACS beads. Mature B cells of the Pax5F/+ CD19-cre genotype were subsequently isolated by FACS sorting as IgM+IgD+ B cells, and Pax5-deficient mature B cells of the Pax5F/− CD19-cre genotype were sorted as IgM+CD25+ B cells (Horcher et al, 2001). Flow cytometric reanalysis indicated that the sorted cells were free of contaminating non-B cells (Supplementary Figure 5). RNA was prepared from the sorted cells and analysed by RT–PCR with previously described primers (Horcher et al, 2001) except for the c-fms primers.
Detection of antisense RNA
cDNA was synthesized from 2 μg of DNaseI-treated total RNA by using 400 U of M-MLV reverse transcriptase using biotinylated primers specific for c-fms (2 pmol) and GAPDH (0.2 pmol) in one reaction. Synthesized cDNA was immobilized on Dynabeads (Dynal, M-270). RNA and trace amounts of genomic DNA were removed by alkaline denaturation and serial washing (Chen et al, 2000). cDNA was eluted by heating the beads suspension in 0.1 × TE for 15 min at 99°C and was followed by real-time quantitative PCR. Relative activity was calculated using genomic DNA as a standard. Input and efficiency of cDNA synthesis was normalized against GAPDH signals. The amount of spliced transcripts was calculated using cDNA evaluated as equivalent amount to genomic DNA.
RT–TDPCR
RT–TDPCR reaction was performed as described previously (Chen et al, 2000). First strand cDNA was synthesized as described above. After the immobilization on Dynabeads, cDNA was tailed with three guanines and ligated to the linker, which contains three cytosines at the end. The ligated products were PCR-amplified using linker primer and nested gene-specific primer and visualized by primer extension reaction using a radiolabelled nested primer. In vitro-transcribed RNA was prepared from c-fms fragment (+849 to +3656) cloned into pBluescript™II KS+ using T7 RNA polymerase.
Further previously published methods can be found as Supplementary material. All primer sequences are described in Supplementary Table 1.
Supplementary Material
Suppl. Table 1
Suppl. Figure 1
Suppl. Figure 2
Suppl. Figure 3
Suppl. Figure 4
Suppl. Figure 5
Supplementary materials and methods
Acknowledgments
C Bonifer's laboratory is supported by grants from the Leukaemia Research Fund, Yorkshire Cancer Research and the Wellcome Trust. M Busslinger's research is supported by Boehringer Ingelheim. We thank Elisabeth Straszynski for cell sorting. H Tagoh is a recipient of a Kay Kendall Leukaemia Fund Fellowship.
References
- Adolfsson J, Borge OJ, Bryder D, Theilgaard-Monch K, Astrand-Grundstrom I, Sitnicka E, Sasaki Y, Jacobsen SE (2001) Upregulation of Flt3 expression within the bone marrow Lin−Sca1+c-Kit+ stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 15: 659–669 [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL (2000) A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404: 193–197 [DOI] [PubMed] [Google Scholar]
- al-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SC (2004) Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303: 669–672 [DOI] [PubMed] [Google Scholar]
- Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 [DOI] [PubMed] [Google Scholar]
- Borzillo GV, Ashmun RA, Sherr CJ (1990) Macrophage lineage switching of murine early pre-B lymphoid cells expressing transduced fms genes. Mol Cell Biol 10: 2703–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HH, Castanotto D, LeBon JM, Rossi JJ, Riggs AD (2000) In vivo, high-resolution analysis of yeast and mammalian RNA–protein interactions, RNA structure, RNA splicing and ribozyme cleavage by use of terminal transferase-dependent PCR. Nucleic Acids Res 28: 1656–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill PN (2000) Identification of DNaseI hypersensitive sites within nuclei. Methods Mol Biol 130: 29–46 [DOI] [PubMed] [Google Scholar]
- DeKoter RP, Singh H (2000) Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 288: 1439–1441 [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172–183 [DOI] [PubMed] [Google Scholar]
- Follows GA, Tagoh H, Lefevre P, Morgan GJ, Bonifer C (2003) Differential transcription factor occupancy but evolutionarily conserved chromatin features at the human and mouse M-CSF (CSF-1) receptor loci. Nucleic Acids Res 31: 5805–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Naka T (2003) Regulation of cytokine signaling by SOCS family molecules. Trends Immunol 24: 659–666 [DOI] [PubMed] [Google Scholar]
- Geiger H, Sick S, Bonifer C, Mueller AM (1998) Globin gene expression is reprogrammed in chimeras generated by injecting adult hematopoietic stem cells into mouse blastocysts. Cell 93: 1055–1065 [DOI] [PubMed] [Google Scholar]
- Graf T (2002) Differentiation plasticity of hematopoietic cells. Blood 99: 3089–3101 [DOI] [PubMed] [Google Scholar]
- Grewal SIS, Moazed D (2003) Heterochromatin and epigenetic control of gene expression. Science 301: 798–802 [DOI] [PubMed] [Google Scholar]
- Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P (2000) Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol Cell 5: 377–386 [DOI] [PubMed] [Google Scholar]
- Himes SR, Tagoh H, Goonetilleke N, Sasmono T, Oceandy D, Clark R, Bonifer C, Hume DA (2001) A highly conserved c-fms gene intronic element controls macrophage-specific and regulated expression. J Leukoc Biol 70: 812–820 [PubMed] [Google Scholar]
- Horcher M, Souabni A, Busslinger M (2001) Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity 14: 779–790 [DOI] [PubMed] [Google Scholar]
- Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, Enver T (1997) Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev 11: 774–785 [DOI] [PubMed] [Google Scholar]
- Huber MC, Bosch FX, Sippel AE, Bonifer C (1994) Chromosomal position effects in chicken lysozyme gene transgenic mice are correlated with suppression of DNase I hypersensitive site formation. Nucleic Acids Res 22: 4195–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki-Arai J, Iwasaki H, Miyamoto T, Watanabe S, Akashi K (2003) Enforced granulocyte/macrophage colony-stimulating factor signals do not support lymphopoiesis, but instruct lymphoid to myelomonocytic lineage conversion. J Exp Med 197: 1311–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Scherer DC, Miyamoto T, King AG, Akashi K, Sugamura K, Weissman IL (2000) Cell-fate conversion of lymphoid-committed progenitors by instructive actions of cytokines. Nature 407: 383–386 [DOI] [PubMed] [Google Scholar]
- Kondo M, Weissman IL, Akashi K (1997) Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91: 661–672 [DOI] [PubMed] [Google Scholar]
- Kontaraki J, Chen HH, Riggs A, Bonifer C (2000) Chromatin fine structure profiles for a developmentally regulated gene: reorganization of the lysozyme locus before trans-activator binding and gene expression. Genes Dev 14: 2106–2122 [PMC free article] [PubMed] [Google Scholar]
- Kress C, Thomassin H, Grange T (2001) Local DNA demethylation in vertebrates: how could it be performed and targeted? FEBS Lett 494: 135–140 [DOI] [PubMed] [Google Scholar]
- Lee JT, Davidow LS, Warshawsky D (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21: 400–404 [DOI] [PubMed] [Google Scholar]
- Lefevre P, Melnik S, Wilson N, Riggs AD, Bonifer C (2003) Developmentally regulated recruitment of transcription factors and chromatin modification activities to chicken lysozyme cis-regulatory elements in vivo. Mol Cell Biol 23: 4386–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3: 662–673 [DOI] [PubMed] [Google Scholar]
- Mikkola I, Heavey B, Horcher M, Busslinger M (2002) Reversion of B cell commitment upon loss of Pax5 expression. Science 297: 110–113 [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Iwasaki H, Reizis B, Ye M, Graf T, Weissman IL, Akashi K (2002) Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell 3: 137–147 [DOI] [PubMed] [Google Scholar]
- Mummaneni P, Yates P, Simpson J, Rose J, Turker MS (1998) The primary function of a redundant Sp1 binding site in the mouse aprt gene promoter is to block epigenetic gene inactivation. Nucleic Acids Res 26: 5163–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutskov V, Felsenfeld G (2004) Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J 14: 138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M (1999) Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401: 556–562 [DOI] [PubMed] [Google Scholar]
- Ross IL, Yue X, Ostrowski MC, Hume DA (1998) Interaction between PU1 and another Ets family transcription factor promotes macrophage-specific Basal transcription initiation. J Biol Chem 273: 6662–6669 [DOI] [PubMed] [Google Scholar]
- Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA (2003) A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101: 1155–1163 [DOI] [PubMed] [Google Scholar]
- Schramke V, Allshire R (2003) Hairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencing. Science 301: 1069–1074 [DOI] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, Barlow DP (2002) The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415: 810–813 [DOI] [PubMed] [Google Scholar]
- Smale ST (2003) The establishment and maintenance of lymphocyte identity through gene silencing. Nat Immunol 4: 607–615 [DOI] [PubMed] [Google Scholar]
- Su RC, Brown KE, Saaber S, Fisher AG, Merkenschlager M, Smale ST (2004) Dynamic assembly of silent chromatin during thymocyte maturation. Nat Genet 36: 502–506 [DOI] [PubMed] [Google Scholar]
- Sudo T, Nishikawa S, Ohno N, Akiyama N, Tamakoshi M, Yoshida H (1993) Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc Natl Acad Sci USA 90: 9125–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagoh H, Himes R, Clarke D, Leenen PJ, Riggs AD, Hume D, Bonifer C (2002) Transcription factor complex formation and chromatin fine structure alterations at the murine c-fms (CSF-1 receptor) locus during maturation of myeloid precursor cells. Genes Dev 16: 1721–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagoh H, Melnik S, Lefevre P, Chong S, Riggs A, Bonifer C (2004) Dynamic reorganization of chromatin structure and selective DNA demethylation prior to stable enhancer complex formation during differentiation of primary hematopoietic cells in vitro. Blood 103: 2950–2955 [DOI] [PubMed] [Google Scholar]
- Thomassin H, Flavin M, Espinas ML, Grange T (2001) Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J 20: 1974–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufarelli C, Stanley JA, Garrick D, Sharpe JA, Ayyub H, Wood WG, Higgs DR (2003) Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 34: 157–165 [DOI] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D (2004) RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303: 672–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B cells into macrophages. Cell 117: 663–676 [DOI] [PubMed] [Google Scholar]
- Ye M, Iwasaki H, Laiosa CV, Stadtfeld M, Heck S, Clausen B, Akashi K, Graf T (2003) Hematopoietic stem cells expressing the myeloid lysozyme gene retain long-term, multilineage repopulation potential. Immunity 19: 689–699 [DOI] [PubMed] [Google Scholar]
- Yue X, Favot P, Dunn TL, Cassady AI, Hume DA (1993) Expression of mRNA encoding the macrophage colony-stimulating factor receptor (c-fms) is controlled by a constitutive promoter and tissue-specific transcription elongation. Mol Cell Biol 13: 3191–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Table 1
Suppl. Figure 1
Suppl. Figure 2
Suppl. Figure 3
Suppl. Figure 4
Suppl. Figure 5
Supplementary materials and methods