

ABSTRACT
Human immunodeficiency virus type 1 (HIV-1) entry into cells is mediated by the viral envelope glycoproteins (Env), a trimer of three gp120 exterior glycoproteins, and three gp41 transmembrane glycoproteins. The metastable Env is triggered to undergo entry-related conformational changes when gp120 binds sequentially to the receptors, CD4 and CCR5, on the target cell. Small-molecule CD4-mimetic compounds (CD4mc) bind gp120 and act as competitive inhibitors of gp120-CD4 engagement. Some CD4mc have been shown to trigger Env prematurely, initially activating Env function, followed by rapid and irreversible inactivation. Here, we study CD4mc with a wide range of anti-HIV-1 potencies and demonstrate that all tested CD4mc are capable of activating as well as inactivating Env function. Biphasic dose-response curves indicated that the occupancy of the protomers in the Env trimer governs viral activation versus inactivation. One CD4mc bound per Env trimer activated HIV-1 infection. Envs with two CD4mc bound were activated for infection of CD4-negative, CCR5-positive cells, but the infection of CD4-positive, CCR5-positive cells was inhibited. Virus was inactivated when all three Env protomers were occupied by the CD4mc, and gp120 shedding from the Env trimer was increased in the presence of some CD4mc. Env reactivity and the on rates of CD4mc binding to the Env trimer were found to be important determinants of the potency of activation and entry inhibition. Cross-sensitization of Env protomers that do not bind the CD4mc to neutralization by an anti-V3 antibody was not evident. These insights into the mechanism of antiviral activity of CD4mc should assist efforts to optimize their potency and utility.
IMPORTANCE The trimeric envelope glycoproteins of human immunodeficiency virus type 1 (HIV-1) mediate virus entry into host cells. Binding to the host cell receptors, CD4 and CCR5, triggers changes in the conformation of the HIV-1 envelope glycoprotein trimer important for virus entry. Small-molecule CD4-mimetic compounds inhibit HIV-1 infection by multiple mechanisms: (i) direct blockade of the interaction between the gp120 exterior envelope glycoprotein and CD4; (ii) premature triggering of conformational changes in the envelope glycoproteins, leading to irreversible inactivation; and (iii) exposure of cryptic epitopes to antibodies, allowing virus neutralization. The consequences of the binding of the CD4-mimetic compound to the HIV-1 envelope glycoproteins depends upon how many of the three subunits of the trimer are bound and upon the propensity of the envelope glycoproteins to undergo conformational changes. Understanding the mechanistic factors that influence the activity of CD4-mimetic compounds can help to improve their potency and coverage of diverse HIV-1 strains.
KEYWORDS: antibody, conformation, envelope glycoprotein, inhibitor, neutralization, sensitization, stoichiometry, virus entry
INTRODUCTION
The entry of human immunodeficiency virus type 1 (HIV-1) into target cells is mediated by the trimeric envelope glycoprotein (Env) complex, which consists of three gp120 exterior envelope glycoproteins and three gp41 transmembrane envelope glycoproteins (1). Binding of gp120 to the receptor, CD4, on the target cell surface induces major conformational changes in the envelope glycoproteins (2–5). These changes allow gp120 to bind the viral coreceptor, either CXCR4 or CCR5 (6–13). CD4 binding also induces the formation of a gp41 prehairpin intermediate, in which three hydrophobic grooves on the surface of a coiled coil formed by the heptad repeat 1 (HR1) region of gp41 are exposed (14–17). These hydrophobic groves are subsequently occupied by helices from the gp41 heptad repeat 2 (HR2) region during the formation of an energetically stable six-helix bundle that is thought to drive the fusion of the viral and target cell membranes (18–23).
Molecules that mimic CD4 have been developed as potential inhibitors of HIV-1 infection. They include soluble CD4 (sCD4), enhanced CD4 (eCD4), sCD4 miniproteins, and small-molecule CD4-mimetic compounds (CD4mc) (24–41). All of these inhibitors bind gp120 near the natural binding site for CD4, a well-conserved surface element adjacent to a pocket located at the interface of the gp120 inner domain, outer domain, and bridging sheet (32–39). A phenylalanine residue at position 43 (Phe 43) of CD4 fills the opening of this gp120 pocket, and thus, the pocket has been designated the Phe 43 cavity (42, 43). Small-molecule CD4mc bind within the Phe 43 cavity of gp120, penetrating deeper into the gp120 interior than CD4 (32–39). Soluble CD4 and CD4mc inhibit HIV-1 infection by multiple mechanisms: (i) competitive inhibition of gp120 interaction with target cell CD4 and (ii) premature activation of Env, followed by apparently irreversible inactivation (30, 32, 38, 44). Consistent with the latter mechanism, in some experimental systems, sCD4 and some CD4mc can enhance HIV-1 entry into CD4-negative cells expressing CCR5 (31, 32, 44). Under some circumstances, sCD4 binding leads to the detachment or shedding of gp120 from the Env trimer (45, 46).
CD4mc were initially identified in a screen for small-molecule blockers of gp120-CD4 binding (30). The hits from this screen, NBD-556 and the closely related NBD-557, exhibited only weak antiviral activity against a very limited number of natural HIV-1 variants (30–32). Iterative, structure-based modifications of these prototypic CD4mc have led to significant improvements in the potency and breadth of their anti-HIV-1 activity (32–41). Of particular importance was the replacement of the chemically intractable tetramethylpiperidine ring of NBD-556 and NBD-557 with an indane ring that allowed multiple substitutions (37, 38). The indane-containing analogues (DMJ-II-121, JP-III-48, and BNM-III-170) exhibited progressive increases in potency and breadth as various moieties were added to the indane ring at specific locations (37–39).
The prototypic CD4mc, NBD-556 and NBD-557, efficiently activated HIV-1 infection of CD4− CCR5+ target cells (31, 32). Interestingly, as the potency of the CD4mc in inhibiting HIV-1 infection of CD4+ CCR5+ cells increased, the activation of HIV-1 infection of CD4− CCR5+ target cells diminished (35–39). In the assay used to detect activation, the CD4mc was incubated at 37°C with a primary HIV-1 isolate, HIV-1YU2, for 30 min, and then the virus-compound mixture was added to CD4-negative Cf2Th-CCR5 cells. Because the diffusion of virus to the target cells is a slow process that can take several hours (47, 48), activation events, particularly those that eventually lead to the decay of infectivity, can be missed by the assay. As an understanding of the mechanism of inhibition is important to attempts to improve and utilize the CD4mc, we adapted our assays to allow greater sensitivity in detecting transient activation of HIV-1 Env function. Here, we use these assays to evaluate the antiviral mechanism of CD4mc exhibiting a range of potencies against HIV-1 strains that differ in susceptibility to inhibition by CD4-mimetic molecules. The results of these studies provide important insights into the interaction of CD4mc with the functional HIV-1 Env trimer.
RESULTS
Susceptibility of selected HIV-1 variants to CD4mc.
For these studies, we selected five CD4mc that exhibited a range of anti-HIV-1 potencies and three HIV-1 variants that demonstrated a range of sensitivities to sCD4 and CD4-mimetic compounds (Table 1). We tested the ability of the CD4mc to inhibit HIV-1 infection by incubating the compound with recombinant, luciferase-expressing HIV-1 for 30 min at 37°C and then adding the compound-virus mixture to Cf2Th-CD4/CCR5 target cells. Measurement of the luciferase activity in the target cells 48 h later provided an indication of the level of infection. Under the conditions of the assay, virus activation by the CD4mc is minimal because of the short half-life of the activated state and the slow diffusion of the virus to the target cell (42, 46, 49). As previously observed (39), BNM-III-170, a recently developed analogue, exhibited dramatically greater antiviral activity against the three HIV-1 strains than the prototypic CD4mc, NBD-556 (Table 1). Of the three HIV-1 strains tested, HIV-1YU2 was the variant most sensitive and HIV-1JR-FL was the variant least sensitive to the CD4mc. The sCD4 sensitivities of the three HIV-1 strains did not correlate with CD4mc sensitivity (Table 1). The antiviral activity of the CD4mc was specific for viruses with HIV-1 Envs, as minimal inhibition of HIV-1 pseudotyped with the amphotropic murine leukemia virus (AMLV) Env was observed.
TABLE 1.
IC50s of soluble CD4 and CD4mc for Envs from different HIV-1 strains or AMLV
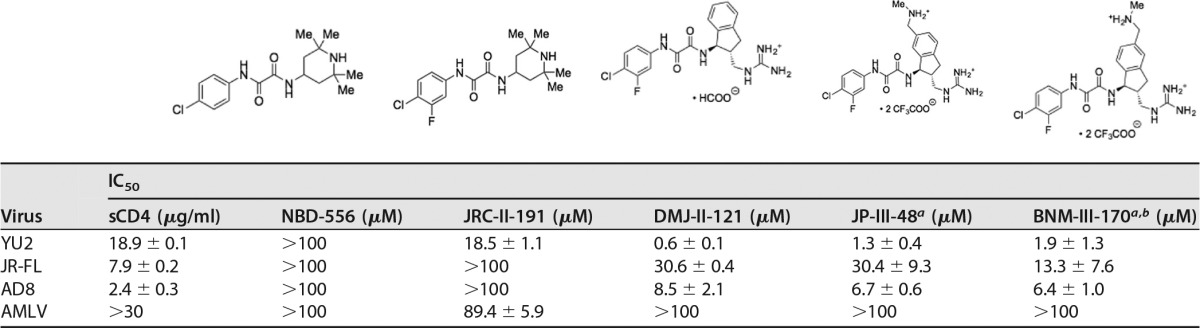
a The cytotoxicity of JP-III-48 and BNM-III-170 for Cf2Th-CD4/CCR5 cells was tested with the CellTiter-Glo luminescent cell viability assay. No cytotoxicity was detected at JP-III-48 and BNM-III-170 concentrations up to 300 μM.
b In a pilot experiment, we varied the amount of the HIV-1JR-FL Env-expressing plasmid transfected into the 293T cells producing recombinant luciferase-expressing HIV-1 up to 100-fold. No significant effect of the amount of Env on the IC50 of BNM-III-170 was observed (data not shown). We determined that the IC50 for BNM-III-170 inhibition of a single-round infection of recombinant HIV-1 with the SF162-P3 Env ranged from 14.7 to 19.8 μM. These IC50s are relevant to the inhibition of replication-competent immunodeficiency viruses. For example, we found that the replication of a simian-human immunodeficiency virus with the SF162-P3 Env in rhesus macaque peripheral blood mononuclear cells was completely inhibited at 50 to 100 μM concentrations of BNM-III-170 (data not shown).
Activation of Env-mediated virus entry into CD4− CCR5+ target cells.
To investigate the activating/inactivating mechanism of Env inhibition by the CD4mc, we utilized experimental systems in which competitive inhibition of CD4 binding does not contribute to the antiviral effects of these compounds. One such experimental system measures the infection of CD4-negative Cf2Th-CCR5 target cells by recombinant HIV-1 with different Envs in the absence or presence of increasing concentrations of CD4mc. In these assays, after incubation of the CD4mc and virus at 37°C for different lengths of time, the virus-compound mixtures were spinoculated onto the Cf2Th-CCR5 cells, which were further cultivated and assessed for infection 48 h later. The spinoculation step allows detection of activation events that might be missed in assays dependent on the slow process of virus diffusion to the target cell (47, 48). In the absence of CD4mc, the infection of the Cf2Th-CCR5 cells by the HIV-1 strains tested was near the assay background, which was established by using recombinant HIV-1 lacking Env. The prototypic CD4mc NBD-556 generally activated infection by all three HIV-1 strains, with increased levels of activation at the highest tested concentration of the compound (Fig. 1). This observation suggests that, at least for a low-affinity CD4mc like NBD-556, the level of HIV-1 activation is usually limited by the concentration of the compound. With prolonged incubation (4 h), the level of NBD-556 activation diminished or even decreased for HIV-1JR-FL and HIV-1AD8, respectively. As discussed further below, at higher concentrations and after long incubation periods, NBD-556 may achieve sufficiently high stoichiometry of Env trimer binding to trigger some inactivation.
FIG 1.

Activation of HIV-1 infection of CD4− CCR5+ target cells. Recombinant luciferase-expressing HIV-1 vectors with Envs from HIV-1YU2, HIV-1AD8, and HIV-1JR-FL were incubated with increasing concentrations of the CD4mc at 37°C for the indicated times. The virus-CD4mc mixture was then spinoculated onto Cf2Th-CCR5 cells at 500 × g for 1 h at 25°C. For the 0-min time point, the virus-CD4mc mixture was immediately spinoculated onto Cf2Th-CCR5 cells after mixing. The virus-cell mixtures were cultured for 48 h, after which the cells were lysed and luciferase activity was measured in the cell lysates. The level of viral infection is reported as a percentage of the level seen in the absence of added CD4mc. The results are reported as the means of triplicate measurements and standard deviations derived from 2 to 4 independent experiments.
In contrast to the NBD-556 dose-response curves, the dose-response curves for the more potent CD4mc were biphasic (Fig. 1). In the lower concentration ranges of DMJ-II-121, JP-III-48, and BNM-III-170, activation of infection of Cf2Th-CCR5 cells increased with increasing concentrations of the CD4mc. At higher concentrations of these compounds, less infection of the Cf2Th-CCR5 cells occurred as the concentration of CD4mc increased. In several cases, particularly for longer incubation of the compound with the virus, no infection was detected at the highest concentration of CD4mc tested. These observations can be explained if saturated Env trimers (with all three protomers occupied by a CD4mc) are much less able to support infection of the Cf2Th-CCR5 cells than Envs with lower CD4mc occupancy.
Comparison of the dose-response curves for CD4mc activation of different viruses in CD4− CCR5+ target cells and virus inhibition in CD4+ CCR5+ target cells can provide insight into the stoichiometry of CD4mc-Env trimer binding required for these processes. The concentration of the CD4mc and the affinity of the compound for the Env trimer determine the CD4mc occupancy of the Env protomers on the trimer. At a given CD4mc occupancy, the percentage of the Env trimers with a given number of bound CD4mc can be estimated based on a binomial distribution (see Materials and Methods). By postulating various stoichiometric requirements for virus activation and inhibition, the hypothetical dose-response curves for these processes can be predicted (Fig. 2A and B). Both the activation and inhibition curves for each virus variant are dictated by Env trimer occupancy. If the same virus incubated with a given concentration of CD4mc is used in both activation and inhibition assays, the Env trimer occupancy will be identical. A comparison of the experimental dose-response curves for the activation and inhibition assays, therefore, greatly constrains the possible models. For example, for each model, the observed CD4mc concentration associated with optimal virus activation and the 50% viral inhibitory concentration (IC50) are both functions of the Kd (dissociation constant), which is a fixed property of a CD4mc for a given virus variant. Thus, the underlying stoichiometric models of activation and inhibition by CD4mc can be supported or refuted by analyzing the experimental dose-response curves.
FIG 2.

Analysis of dose-response curves for CD4mc activation and inhibition. (A) Hypothetical relationship between the probability (P) of a CD4mc occupying an Env protomer and HIV-1 infection of CD4-negative, CCR5-expressing cells (top) and HIV-1 infection of CD4-positive, CCR5-positive cells (bottom) based on a binomial probability distribution. The curves shown are based on models in which the Env trimers with the indicated numbers of bound CD4mc (red ovals) are activated for infection (top) or neutralized (bottom). The protomers of the Env trimer bound by the CD4mc are colored black. (B) The hypothetical relationships in panel A are shown as a function of the free CD4mc concentration. The observation that CD4mc can sensitize HIV-1 to neutralization by antibodies (57–60) renders the model in which Env trimers with one, two, or three bound CD4mc are all neutralized improbable; nonetheless, the hypothetical curve is shown for comparison. (C) Relationship between the IC50 associated with the inhibition of infection of CD4-positive, CCR5-positive cells and the total [CD4mc] associated with maximal activation of infection of CD4-negative, CCR5-expressing cells. The Spearman rank correlation coefficient rS = 0.9074; two-tailed P = 0.000046. (D) The indicated concentrations of JP-III-48 were added to recombinant luciferase-expressing HIV-1 with the HIV-1YU2, HIV-1AD8, and HIV-1JR-FL Envs. The virus-CD4mc mixture was immediately split into two portions, with one portion added to Cf2Th-CD4/CCR5 cells (bottom) and the other portion spinoculated onto Cf2Th-CCR5 cells at 500 × g for 1 h at 25°C (top). The virus-cell mixtures were returned to 37°C, and the level of infection was determined by measuring luciferase activity in the target cells 48 h later and is shown as a percentage of the level seen in the absence of added JP-III-48. The means and standard deviations of triplicate measurements are shown. Note the correspondence between the IC50 associated with inhibition of HIV-1 infection of Cf2Th-CD4/CCR5 cells and the JP-III-48 concentration associated with maximal activation of infection of CD4-negative Cf2Th-CCR5 cells.
We applied this analysis to the activation/inhibition data of the HIV-1YU2, HIV-1AD8, and HIV-1JR-FL variants treated with the more potent CD4mc. In every case, the concentration of CD4mc associated with the optimal level of activation in CD4− CCR5+ target cells was similar to the IC50 of the compound for inhibition of infection by the same virus in CD4+ CCR5+ cells. The correlation between the CD4mc concentration resulting in maximal activation and the IC50 of inhibition is shown in Fig. 2C. We confirmed this relationship when the same preparation of HIV-1YU2, HIV-1AD8, and HIV-1JR-FL was incubated with JP-III-48 and then used for infection of CD4− CCR5+ cells and for infection of CD4+ CCR5+ cells (Fig. 2D). For all three viruses, the optimal JP-III-48 concentration for activation of virus infection in CD4− CCR5+ cells was closely related to the IC50 for infection of CD4+ CCR5+ cells. These observations are compatible with the following models: (i) under the conditions of our assay measuring inhibition of infection of CD4+ CCR5+ cells, Envs with two and three CD4mc bound per trimer are inactivated; (ii) under the conditions of our assay measuring the infection of CD4− CCR5+ cells, Envs with one or two CD4mc bound can be activated.
Assuming that the total CD4mc concentration reflects the free CD4mc concentration, the Kd values of JP-III-48 for the functional Env trimers of HIV-1YU2, HIV-1AD8, and HIV-1JR-FL are approximately 6 to 10 μM, 9 to 10 μM, and 40 to 50 μM, respectively.
The stoichiometry of CD4mc inhibition of HIV-1 infection was investigated further by studying recombinant viruses with differing proportions of Envs that are sensitive and resistant to the CD4mc. The S375W change in HIV-1 Env fills the Phe 43 cavity and renders HIV-1 resistant to the effects of CD4mc (32, 50). During the course of our studies, we found that the I309A change in the gp120 V3 variable region resulted in viruses that were more sensitive to BNM-III-170 than the wild-type HIV-1JR-FL; the I309A mutant virus infected Cf2Th-CD4/CCR5 cells with an efficiency similar to that of the wild-type HIV-1JR-FL (data not shown). Because the results of the mixing experiment are interpreted at saturating concentrations of CD4mc, we used the I309A mutant to facilitate achieving saturation with BNM-III-170. Recombinant HIV-1JR-FL with differing proportions of the S375W (CD4mc-resistant) Env and I309A (CD4mc-sensitive) Env were incubated with increasing concentrations of BNM-III-170 and then used to infect Cf2Th-CD4/CCR5 cells. The levels of infection observed at saturating concentrations (typically 30 μM and above) of BNM-III-170 are compared in Fig. 3 with the hypothetical curves expected for the different models of BNM-III-170 inhibition stoichiometry. The results support a model in which Envs with either two or three of the protomers occupied by BNM-III-170 are neutralized.
FIG 3.

Inhibition of viruses with mixed Env variants by a CD4mc. Recombinant HIV-1 containing different ratios of the I309A mutant Env and the S375W mutant Env were produced in 293T cells by transfecting the appropriate molar quantities of the Env expressor plasmids. The infectivities of the I309A and S375W mutant viruses for Cf2Th-CD4/CCR5 target cells were comparable to that of the wild-type HIV-1 (data not shown). The I309A change in the gp120 V3 region slightly increases HIV-1 sensitivity to BNM-III-170 inhibition. The S375W change fills the gp120 Phe 43 cavity, rendering HIV-1 resistant to the CD4mc BNM-III-170. (A) Recombinant luciferase-expressing HIV-1 strains with different ratios of I309A-S375W Envs were used to infect Cf2Th-CD4/CCR5 cells in the presence of increasing concentrations of BNM-III-170. The viruses were incubated with BNM-III-170 for 30 min at 37°C and then added to the Cf2Th-CD4/CCR5 cells. The level of infection 48 h later was determined by measuring luciferase activity in the target cells. The level of infection is reported as a percentage of the level of infection observed for a given virus in the absence of the CD4mc. Recombinant HIV-1 pseudotyped with the AMLV Env was included as a control virus. WT, wild type. The error bars indicate standard deviations. (B) Hypothetical inhibition curves showing the level of HIV-1 infection expected at saturating concentrations of the CD4mc BNM-III-170 as a function of the percentage of the CD4mc-resistant S375W Env in the virus population. The magenta curve represents the hypothetical levels of infection expected if only Envs with all three protomers bound by CD4mc are neutralized, the green curve depicts the hypothetical levels of infection expected if only Envs with two or three protomers bound by CD4mc are neutralized, and the red curve shows the levels expected if the Env is neutralized by any bound CD4mc. The blue squares represent the actual experimental data obtained for mixtures of HIV-1JR-FL S375W and I309A Envs at saturating concentrations (typically 30 μM) of BNM-III-170. Each data point represents the results of a single experiment like that shown in panel A and indicates the percentage of the level of infection observed for a given virus at saturating BNM-III-170 concentrations relative to the level of infection seen in the absence of the CD4mc. The results of multiple independent experiments with different virus preparations are shown.
Env reactivity influences sensitivity to CD4mc.
In the experimental system discussed above, the CD4mc concentration associated with the peak level of activation in CD4− CCR5+ cells and with the IC50 in CD4+ CCR5+ cells is closely related to the affinity of the compound for the functional Env trimer of the particular HIV-1 strain. The CD4mc apparently interact with the Env trimer in the rank order HIV-1YU2 > HIV-1AD8 > HIV-1JR-FL. As the CD4mc binding site on the HIV-1 gp120 glycoprotein is well conserved (32–34, 37–39), what accounts for the differences in sensitivity of these HIV-1 strains? CD4mc, like CD4, induce conformational changes in the Env trimer that potentially contribute to binding and its consequences (44). Envs from different HIV-1 strains can vary in their propensities to undergo conformational changes, a property that has been designated Env reactivity (51, 52). We hypothesized that differences in Env reactivities among HIV-1 strains might contribute to variation in sensitivity to CD4mc. Some changes in the HIV-1 gp120 V2 region have been shown to result in an increase in Env reactivity (53–56). We introduced additional changes in the HIV-1JR-FL gp120 V2 region to identify Env mutants with increased reactivity. Such mutants typically exhibit increased sensitivity to soluble CD4, CD4mc, and antibodies against CD4-induced gp120 epitopes; many HIV-1 Env mutants with increased reactivity exhibit decreased sensitivity to blockers of conformational change, like BMS-806 (17, 51, 54, 56). We identified HIV-1JR-FL V2 mutants that exhibited phenotypes consistent with those associated with increased Env reactivity (Table 2) (56). Of note, both syncytium formation and virus infection mediated by the Y177R, P183Y, and L193A Env mutants were inhibited more efficiently by BNM-III-170 than the corresponding processes mediated by wild-type HIV-1JR-FL Env. These results are consistent with the hypothesis that changes in Env reactivity can alter HIV-1 susceptibility to inhibition by CD4mc.
TABLE 2.
Phenotypes of HIV-1JR-FL Env mutants
| HIV-1JR-FL Env variant | Cell-cell fusiona | Inhibition of cell-cell fusion (IC50) (μM)b |
Relative infectivitya | Inhibition of virus infection (IC50)c |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BMS-806 | BNM-III-170 | BMS-806 (nM) | BNM-III-170 (μM) | sCD4 (μg/ml) | 19b (μg/ml) | 17b (μg/ml) | 902090 (μg/ml) | VRC01 (μg/ml) | |||
| Wild type | 1.00 | 0.80 | 160 | 1.00 | 7.8 | 15 | 7.3 | >30 | >30 | >30 | 0.23 |
| D107R | 0.29 | 0.48 | 28 | 0.012 | 11 | 0.72 | 0.44 | 0.151 | 1.5 | 15 | 0.22 |
| R166G | 0.43 | 1.1 | 208 | 0.38 | 14 | 6.5 | 2.6 | 26 | >30 | >30 | 0.21 |
| D167G | 0.32 | 0.43 | 160 | 0.27 | 14 | 8.3 | 3.6 | 18 | >30 | >30 | 0.20 |
| D167Q | 0.67 | 0.23 | 183 | 0.22 | 4.4 | 8.7 | 2.4 | >30 | >30 | >30 | 0.21 |
| D167R | 0.92 | 0.28 | 150 | 0.17 | 6.0 | 4.8 | 3.8 | >30 | >30 | >30 | 0.21 |
| E168G | 0.96 | 0.26 | 186 | 0.20 | 9.8 | 9.7 | 3.4 | >30 | >30 | >30 | 0.19 |
| Y177R | 0.90 | >30 | 168 | 0.32 | 82 | 1.4 | 0.51 | 0.22 | 2.3 | >30 | 0.48 |
| Y177R + D180E | 0.30 | >30 | 152 | 0.14 | >100 | 0.87 | 0.46 | 0.16 | 0.18 | >30 | 4.4 |
| P183Y | 0.45 | 1.2 | 23 | 0.025 | 38 | 1.2 | 0.41 | 2.9 | >30 | >30 | 0.36 |
| I184K | 0.22 | 0.19 | 26 | 0.0060 | 9.4 | 1.2 | 0.87 | 10.8 | >30 | 16 | 0.25 |
Cell-cell fusion and relative infectivity were measured as described in Materials and Methods, with the values normalized to those of the wild-type HIV-1JR-FL Env. The values represent the means of those obtained in at least three independent experiments. Values that differ more than 5-fold from the value of the wild-type HIV-1JR-FL Env are shown in boldface.
Cell-cell fusion mediated by the indicated HIV-1JR-FL variant was tested for inhibition by BMS-806, a blocker of conformational change (17, 56), and a CD4mc, BNM-III-170. The values represent the means of those obtained in at least two independent experiments. Values in boldface differ from the value of the wild-type HIV-1JR-FL by more than 5-fold.
Recombinant HIV-1 pseudotyped with the indicated HIV-1JR-FL Env was tested for inhibition by the indicated Env ligands. The 19b antibody recognizes the gp120 V3 region, the 17b antibody recognizes a CD4-induced gp120 epitope, 902090 recognizes a gp120 V2 region epitope, and VRC01 recognizes the CD4-binding site of gp120. The values represent the means of those obtained in at least two independent experiments. Values that differ from the value of the wild-type HIV-1JR-FL Env by more than 5-fold are shown in boldface.
We also evaluated the effects of increased Env reactivity on CD4mc activation of HIV-1 infection of CD4-negative cells expressing CCR5. We compared the abilities of the wild-type HIV-1JR-FL and three V2 mutants (Y177R, P183Y, and L193A) with increased Env reactivity to infect Cf2Th-CCR5 cells in the absence and presence of the CD4mc DMJ-II-121 and JP-III-48. We also included two control V2 mutants, D167G and E168G, that exhibit phenotypes consistent with Env reactivities close to that of the wild-type HIV-1JR-FL Env. The viruses were incubated with the CD4mc for 30 min at 37°C and then spinoculated onto Cf2Th-CCR5 cells. As seen above, infection by the wild-type HIV-1JR-FL was activated by JP-III-48, with maximal activation occurring at a compound concentration of ∼30 μM (Fig. 4A). Although the overall level of activation by JP-III-48 was less for the E168G mutant, and much lower for the D167G mutant, the maximal activation of these mutants also occurred at a JP-III-48 concentration of 30 μM. Thus, the apparent affinities of JP-III-48 for the wild-type, D167G, and E168G Envs are comparable. The levels of infection of Cf2Th-CCR5 cells by the Y177R, P183Y, and L193A mutants following incubation with JP-III-48 were much lower than those of the wild-type HIV-1JR-FL. Importantly, the maximal level of activation of these mutants occurred at concentrations of JP-III-48 significantly lower than that of the wild-type HIV-1JR-FL. Similar results were observed for this panel of HIV-1JR-FL mutants with DMJ-II-121 (Fig. 4B). For both JP-III-48 and DMJ-II-121, the CD4mc concentration that resulted in maximal activation of Cf2Th-CCR5 infection exhibited the rank order L193A < Y177R < P183Y < wild-type HIV-1JR-FL, D167G, and E168G. Apparently, the Env mutants with increased reactivity bind CD4mc and undergo activation/inactivation at much lower concentrations of the compounds, likely reflecting a higher affinity of the CD4mc for these Env trimers.
FIG 4.

Influence of Env reactivity on HIV-1 sensitivity to CD4mc. The HIV-1JR-FL variants were incubated with the indicated concentrations of CD4mc, and the virus-compound mixture was immediately spinoculated onto Cf2Th-CCR5 cells. After 48 h at 37°C, the cells were lysed, and the cell lysates were assayed for luciferase activity. The results represent the mean values from triplicate data points and standard deviations and are reported as the level of luciferase activity relative to that seen in the absence of the CD4mc. The results of a typical experiment are shown; the experiment was repeated with similar results.
Infection of CD4-negative target cells by the wild-type HIV-1JR-FL is activated inefficiently by the prototypic CD4mc NBD-556 (Fig. 1). To evaluate whether a more reactive Env would be more susceptible to activation by NBD-556, we compared the infection of Cf2Th-CCR5 cells by the wild-type HIV-1JR-FL and L193A mutant in the absence or presence of NBD-556. As a control, we also tested the more potent CD4mc, BNM-III-170, in the assay. Infection of the CD4-negative cells by the wild-type HIV-1JR-FL progressively increased with increasing BNM-III-170 concentrations up to 10 μM (Fig. 4C). As was seen for JP-III-48 and DMJ-II-121 (see above), the L193A mutant exhibited a reduced level of infection that peaked at a very low BNM-III-170 concentration. NBD-556 had minimal effect on the infection of the wild-type HIV-1JR-FL. In striking contrast, incubation with NBD-556 resulted in a dramatic increase in infection of the Cf2Th-CCR5 cells by the L193A mutant. Thus, an increase in Env reactivity can lead to enhanced susceptibility to activation of CD4-independent infection by a weak CD4mc. These results are consistent with the concept that HIV-1 responsiveness to a CD4mc depends upon the propensity of Env to undergo conformational changes.
Sensitization of HIV-1 activated by CD4mc to neutralizing antibodies.
During infection of CD4-positive cells, HIV-1 exposed to subneutralizing concentrations of CD4mc becomes susceptible to neutralization by a number of antibodies that recognize the CD4-bound Env conformation (57–60). We wished to determine if the virus activated for infection of CD4− CCR5+ cells by CD4mc is susceptible to neutralization by these antibodies. For this purpose, we incubated HIV-1JR-FL with 25 μM JP-III-48 for 15 min at 37°C and then incubated the virus-CD4mc mixture with anti-Env antibodies or sCD4 for an additional 15 min at 37°C. The virus-CD4mc-antibody mixtures were then spinoculated onto Cf2Th-CCR5 cells, and the efficiency of infection was measured (Table 3).
TABLE 3.
Antibody neutralization of JP-III-48-activated HIV-1JR-FL infection of CD4-negative Cf2Th-CCR5 cellsa
| Antibody/Env ligand | gp120 specificity | IC50 (nM) |
||
|---|---|---|---|---|
| HIV-1JR-FL + 25 μM JP-III-48 | AMLV |
|||
| +25 μM JP-III-48 | −25 μM JP-III-48 | |||
| 17b | CD4 induced | <3.3 | >200 | >200 |
| 19b | V3 | <3.3 | >200 | >200 |
| 2G12 | Outer domain glycan | <3.3 | >200 | >200 |
| F105 | CD4-binding site | >200 | >200 | >200 |
| VRC01 | CD4-binding site | <3.3 | >200 | >200 |
| VRC03 | CD4-binding site | <3.3 | >200 | >200 |
| PG9 | V2 quaternary | >200 | >200 | >200 |
| sCD4 | CD4-binding site | >200 | >200 | >200 |
HIV-1 pseudotyped with the HIV-1JR-FL or AMLV Env was incubated with 25 μM JP-III-48 for 5 min at 37°C and then for 15 min at 37°C with different concentrations of antibodies or sCD4. The virus–JP-III-48–antibody mixtures were then spinoculated onto Cf2Th-CCR5 cells. After 48 h at 37°C, the cells were lysed, and the cell lysates were assayed for luciferase activity. The mean values for luciferase activity were derived from triplicate samples. The level of luciferase activity relative to that seen in the absence of antibody or sCD4 was used to calculate the level of infection. The IC50s of the antibodies and sCD4 are reported.
In the absence of added JP-III-48, the infection of the Cf2Th-CCR5 cells by HIV-1JR-FL was very inefficient, as expected. This low basal level of CD4-independent HIV-1JR-FL infection was minimally affected by the addition of antibodies (data not shown). After incubation with 25 μM JP-III-48, HIV-1JR-FL infection of Cf2Th-CCR5 cells was dramatically enhanced, and this activated infection was efficiently inhibited by the 17b, 19b, 2G12, VRC01, and VRC03 antibodies (Table 3). JP-III-48-activated HIV-1JR-FL infection was not inhibited by the F105 antibody, a weakly neutralizing antibody directed against the gp120 CD4-binding site. JP-III-48-activated infection of the Cf2Th-CCR5 cells was also not inhibited by the PG9 antibody, which does not recognize HIV-1JR-FL (61). These results demonstrate that the CD4mc-activated infection of CD4− CCR5+ cells by HIV-1 can be efficiently neutralized by broadly neutralizing antibodies like 2G12, VRC01, and VRC03, as well as poorly neutralizing antibodies like 17b and 19b that recognize gp120 epitopes induced by CD4mc and CD4.
To investigate whether the binding of a CD4mc to one Env protomer might expose neutralizing epitopes on adjacent protomers, we tested the ability of the CH23 antibody to inhibit the infection of Cf2Th-CCR5 cells by viruses with I309A-S375W and F317A-S375W Env mixtures. The F317A virus has a change in the gp120 V3 region that makes it resistant to neutralization by the CH23 antibody, which recognizes a V3 epitope (62). The I309A change in V3 does not affect sensitivity to the CH23 antibody, so the I309A Env was included as a control in these experiments. Unlike the S375W mutant, the I309A and F317A mutants can be activated by CD4mc. In the experiments, we tested viruses with a high ratio of the S375W Env relative to the I309A or F317A Env. Under these conditions, nearly all of the viruses capable of infecting CD4-negative Cf2Th-CCR5 cells should contain mixed Envs with at least one CD4mc-activatable, CH23-resistant Env protomer and at least one CD4mc-resistant, CH23-sensitive Env protomer. Therefore, if the binding of a CD4mc to one Env protomer contributes to the exposure of the CH23 epitope on an adjacent Env protomer, a substantial fraction of the viruses should be neutralized by the CH23 antibody. The design of the experiment is summarized in Fig. 5A and B, and the results are shown in Fig. 5C. The infection of the Cf2Th-CCR5 cells by the S375W mutant was activated by sCD4, but not by the CD4mc BNM-III-170, as expected (data not shown). Infection of the Cf2Th-CCR5 cells by the BNM-III-170-activated I309A mutant was blocked by the CH23 antibody and by another V3-directed antibody, 19b. In contrast, infection of the Cf2Th-CCR5 cells by the BNM-III-170-activated F317A mutant virus was not inhibited by the CH23 antibody. Notably, the CH23 antibody did not inhibit the BNM-III-170-activated infection of Cf2Th-CCR5 cells by viruses with high proportions of the S375W mutant relative to the F317A mutant Envs. In contrast, the CH23 antibody neutralized the infection of Cf2Th-CCR5 cells by viruses with mixtures of the S375W and I309A Envs. Therefore, in this experimental setting, we did not find evidence for the binding of BNM-III-170 to an Env protomer sensitizing an adjacent Env protomer to CH23 binding and virus neutralization.
FIG 5.

Test of CD4mc cross-protomer sensitization of recombinant HIV-1 to antibodies. (A) Env mixing strategy to test whether the binding of a CD4mc to one protomer of the Env trimer induces the exposure of the gp120 V3 region on an adjacent protomer. Mixed Env trimers were prepared from the S375W mutant, which does not bind the CD4mc BNM-III-170 but can be recognized by the CH23 anti-V3 antibody, and the F317A mutant, which can bind the CD4mc but is resistant to the CH23 antibody. (B) Mixed Envs with the indicated proportions of S375W and F317A mutant Envs are predicted to have the percentages of each Env mixture shown. The predicted percentage of the virus fraction that can potentially infect CD4-negative Cf2Th-CCR5 cells after BNM-III-170 treatment and that can potentially be neutralized by the CH23 antibody is shown in the right-hand column. This percentage is based on the assumption that binding of a single CH23 antibody neutralizes the HIV-1 Env trimer. (C) Recombinant viruses with the indicated ratio of I309A-S375W Envs or F317A-S375W Envs were incubated with 30 μM BNM-III-170 and increasing concentrations of the CH23 or 19b anti-V3 antibody for 30 min at 37°C and then added to Cf2Th-CCR5 cells. Recombinant viruses with the wild-type HIV-1JR-FL Env or I309A, F317A, and S375W Envs were tested in parallel. The level of infection after 48 h was determined by measuring luciferase activity in the target cells. The level of infection was normalized to that seen in the absence of antibody, and the IC50 of the antibody was calculated. *, no infection was detected above the assay background.
Activation of Env-mediated syncytium formation with CD4− CCR5+ target cells.
We investigated the activating/inactivating mechanism of Env inhibition by the CD4mc in a second experimental system where competitive inhibition of CD4 binding does not contribute to the antiviral effects of the compounds. In this experimental system, HIV-1 Env-expressing cells are incubated with CD4mc and then assessed for the ability to fuse with CD4-negative cells expressing CCR5. 293T cells transiently expressing different HIV-1 Envs were cocultivated with CD4-negative Cf2Th-CCR5 cells in the absence or presence of increasing concentrations of various CD4mc. In the absence of CD4mc, no syncytium formation was detected, consistent with the CD4 dependence of the HIV-1 Envs tested. All of the CD4mc tested exhibited some ability to activate Env function in the assay (Fig. 6A). The indane analogues (DMJ-II-121, JP-III-48, and BNM-III-170), which are more potent inhibitors of HIV-1 infection than the piperidine analogues (NBD-556 and JRC-II-191), generally enhanced cell-cell fusion more efficiently. The HIV-1JR-FL Env exhibited minimal levels of activation by the low-potency CD4mc, NBD-556 and JRC-II-191, but was activated by the potent CD4mc, BNM-III-170 and JP-III-48, even more efficiently than HIV-1YU2 and HIV-1AD8 Envs. The dose-response curves for activation of syncytium formation were biphasic; this implies that at lower levels of occupancy of the Env trimer, activating events predominate, whereas at a higher occupancy level, events detrimental to Env function occur.
FIG 6.

Activation by CD4mc of Env-mediated syncytium formation with CD4− CCR5+ cells. (A) 293T cells expressing the HIV-1 Env and the α peptide of β-galactosidase were cocultivated with CD4-negative, CCR5-positive Cf2Th-CCR5 cells expressing the ω peptide of β-galactosidase in medium containing the indicated concentrations of CD4mc. After 6 h at 37°C, the cells were lysed, and the lysates were assayed for β-galactosidase activity. The results are reported as the percentage of β-galactosidase activity compared with that seen in the absence of added CD4mc. The results of a single experiment, with each point done in duplicate/triplicate, are shown. The experiment was repeated with comparable results. The error bars indicate standard deviations. (B) 293T cells expressing the HIV-1JR-FL Env were radiolabeled with [35S]cysteine and [35S]methionine in medium alone or in the presence of DMSO, 20 μg/ml sCD4, or different concentrations of BNM-III-170 for 24 h at 37°C. The cell supernatants were then used for precipitation by a polyclonal antiserum from an HIV-1-infected individual. The precipitated gp120 was analyzed by SDS-PAGE, autoradiography, and densitometry. The background shedding of gp120 in the untreated control was subtracted from all of the other values. *, value less than or equal to the background of the assay.
One potential explanation for the biphasic activation curves is that, as Env occupancy increases at higher concentrations, the CD4mc induce shedding of gp120 from the Env trimer. To test this hypothesis, we incubated cells expressing the HIV-1JR-FL Envs with different concentrations of BNM-III-170 at 37°C for 24 h. As a positive control, the cells were incubated with 20 μg/ml sCD4 at 37°C for 24 h. The cell supernatants were collected and precipitated with a polyclonal mixture of sera from HIV-1-infected individuals. The precipitates were analyzed by SDS-PAGE, followed by autoradiography (Fig. 6B). Compared with the dimethyl sulfoxide (DMSO) negative control, incubation with sCD4 resulted in an increase in the gp120 concentration in the cell supernatants. A dose-dependent increase in the amount of gp120 in the supernatant was observed for BNM-III-170. These results suggest that BNM-III-170 increases gp120 shedding from the Env trimer.
Kinetics of CD4mc interaction with Env.
In the experiments testing the activation of HIV-1 infection of CD4-negative Cf2Th-CCR5 cells (Fig. 1), we also examined the effect of the length of the 37°C incubation of the CD4mc with the virus. In general, longer incubation times affected the infection efficiencies similarly to increasing the concentration of CD4mc. In most cases, longer incubation of the CD4mc with virus resulted in lower levels of infection (Fig. 1). However, in some cases (e.g., NBD-556 incubation with HIV-1JR-FL), longer incubation times were associated with increased levels of infection. These results can be understood in light of the proposed biphasic relationship between the amount of CD4mc bound per Env trimer and the level of activation of infection of the CD4-negative Cf2Th cells. When the affinity of the CD4mc for the Env trimer is low (as in the cases of NBD-556 and HIV-1JR-FL), longer incubation times would favor increased occupancy of the Env trimer and greater activation of infection. When the affinity of the CD4mc for the Env trimer is higher, the increased levels of trimer occupancy resulting from longer incubation times can result in decreases in the level of infection (e.g., DMJ-II-121 or JP-III-48 and HIV-1AD8).
The observed differences in the degree of activation for longer incubation periods could reflect differences in the on rates of binding of the CD4mc to the Env trimer. We used the activation of cell-cell fusion by the CD4mc to examine this possibility. Cells expressing the wild-type HIV-1JR-FL, HIV-1JR-FL Y177R, HIV-1JR-FL P183Y, and AMLV Envs were incubated at 37°C for various lengths of time with different CD4mc or the control DMSO. The cells were then washed and cocultivated with Cf2Th-CCR5 cells, and syncytia were scored. Thus, the assay requires the CD4mc to bind Env within the period of incubation and to maintain Env binding through the wash. Under the conditions of the assay, the cell-cell fusion activities of the wild-type HIV-1JR-FL and HIV-1JR-FL P183Y Envs were minimally activated. In contrast, the more reactive HIV-1JR-FL Y177R Env exhibited marked activation of syncytium-forming ability by all of the CD4mc. The level of activation of Y177R Env was greater for the more potent CD4mc, DMJ-II-121, JP-III-48, and BNM-III-170, than for the less potent NBD-556 and JRC-II-191 (Table 4). Of note, the level of activation increased with longer incubation periods of NBD-556 and JRC-II-191, whereas the more potent CD4mc achieved high levels of activation within the 5-min incubation period. These results are consistent with higher on rates for the more potent CD4mc.
TABLE 4.
Activation of fusion between Env-expressing cells and CD4-negative, CCR5-positive cells
| Compound | Incubation time (min) | Area under the curve (arbitrary units) for HIV-1 Enva: |
|||
|---|---|---|---|---|---|
| AD8 | JR-FL | JR-FL Y177R | JR-FL P183Y | ||
| NBD-556 | 5 | <10 | <10 | 36 | <10 |
| 10 | <10 | <10 | 23 | <10 | |
| 30 | <10 | <10 | 106 | <10 | |
| 60 | <10 | <10 | 105 | <10 | |
| JRC-II-191 | 5 | <10 | <10 | 140 | <10 |
| 10 | <10 | <10 | 272 | <10 | |
| 30 | <10 | <10 | 411 | <10 | |
| 60 | <10 | <14 | 285 | <10 | |
| DMJ-II-121 | 5 | 47 | 161 | 1,139 | 354 |
| 10 | 94 | 171 | 659 | 335 | |
| 30 | 32 | 19 | 679 | 146 | |
| 60 | <10 | <10 | 168 | 12 | |
| JP-III-48 | 5 | 59 | 59 | 877 | 12 |
| 10 | 69 | 12 | 884 | 29 | |
| 30 | 25 | 30 | 1,175 | 19 | |
| 60 | 20 | 29 | 746 | 15 | |
| BNM-III-170 | 5 | 47 | 74 | 1,567 | 76 |
| 10 | 37 | 77 | 1,095 | 90 | |
| 30 | 66 | 27 | 1,334 | 58 | |
| 60 | 14 | 27 | 872 | 60 | |
Different concentrations (0 to 100 μM) of the CD4mc were incubated with 293T cells expressing HIV-1 Env variants and the α peptide of β-galactosidase at 37°C for the indicated times. The cells were then washed and cocultivated with CD4-negative CCR5-positive Cf2Th-CCR5 cells expressing the ω peptide of β-galactosidase at 37°C in medium without the CD4mc. After 6 h, the extent of cell-cell fusion was assessed by measuring β-galactosidase activity in the cell lysates. The β-galactosidase activity was plotted as a function of the CD4mc concentration, and the area under the curve was measured for each time point. The results of a typical experiment out of two independent experiments are shown.
DISCUSSION
In this work, we investigated the mechanisms underlying the interactions of CD4mc analogues of varying potencies with the functional Envs from different HIV-1 variants. The rank order of potency of CD4mc and the rank order of susceptibility of HIV-1 variants were similar in three different contexts: activation of Env-mediated syncytium formation, activation of virus entry into CD4− CCR5+ cells, and inhibition of HIV-1 entry into CD4+ CCR5+ cells. These observations are consistent with the affinity of the CD4mc for the Env trimer being a major determinant of the efficiency of these processes. The results also underscore the contribution of premature Env activation leading to functional inactivation as an important antiviral mechanism of all CD4mc. When competitive inhibition of CD4 binding is eliminated as a possible mechanism (for example, in experimental systems where CD4− CCR5+ target cells are used), the biphasic nature of the relationship between the concentration of CD4mc and activation of infection or syncytium formation becomes apparent. The experimental dose-response curves correspond closely to theoretical predictions of CD4mc-Env trimer binding based on a binomial probability distribution. The models suggest that occupancy of one or two protomers of the Env trimer by a CD4mc can lead to entry activation (Fig. 7). Note that in these models, Envs with two CD4mc bound per trimer are either active or inactive, depending on the assay conditions. Envs with two protomers occupied by a CD4mc can support infection if spinoculated onto CD4− CCR5+ cells. This allows the activated Env trimer to engage CCR5 efficiently. However, under conditions where the CD4mc-bound virus diffuses slowly to the target cell, such viruses may undergo inactivation. Moreover, in target cells expressing CD4 and CCR5, viruses with two CD4mc bound per trimer infect inefficiently compared with untreated viruses. Even if these viruses remain infectious upon reaching the target cell, blockade of the interaction with cellular CD4 inhibits virus entry. Although the bound CD4mc can supplant host cell CD4 with respect to Env activation, CD4mc do not anchor and orient the virus particle in the manner expected for cellular CD4.
FIG 7.

Effects of stoichiometry of CD4mc binding to the HIV-1 Env trimer on activation and inactivation. The proposed effect of binding of the CD4mc (red oval) to the subunits of the HIV-1 Env trimer is depicted. In all cases, the binding of a single CD4mc to the Env trimer leads to activation, and the binding of three CD4mc leads to inactivation. The binding of two CD4mc to the Env trimer leads to virus activation of infection in CD4-negative, CCR5-positive cells but results in virus inactivation during infection of CD4-positive, CCR5-positive cells.
Occupancy of all three Env protomers by a CD4mc does not typically lead to successful infection of CD4− CCR5+ cells and apparently is inactivating under these experimental conditions. A previous study has shown that, when HIV-1 is distant from a potential target cell, Env inactivation by even modestly potent CD4mc occurs rapidly, at a rate comparable to that which follows the binding of sCD4 (44). The transient opening and spontaneous closure of the gp41 HR1 hydrophobic groove has been shown to be correlated temporally with Env inactivation by CD4mc (44), supporting a model in which Env conformations induced by CD4mc are functional only transiently. Moreover, in some of the CD4mc-induced conformations, Env subunit association is apparently weakened, resulting in gp120 shedding from the Env trimer. Because the ability of CD4mc to both compete with CD4 and induce viral activation/inactivation is a function of occupancy of the protomers of the Env trimer, the affinity of the CD4mc for the Env trimer is a major determinant of the antiviral potency of these compounds.
The on rates of CD4mc binding are critical determinants of the binding affinity of these compounds for the functional Env trimer. As might be expected for any ligand-protein interaction, the enthalpic contributions of an increased number of bonds between the CD4mc and gp120 result in higher affinity and increased potency (32–41). However, because CD4mc induce conformational changes in Env in the process of binding, the CD4mc-Env binding affinity is strongly influenced by the propensity of the Env trimer to undergo conformational changes (i.e., Env reactivity) (51, 52). The HIV-1 Env trimer is maintained in its unliganded state (state 1) by multiple inter- and intrasubunit interactions, thus averting premature triggering and inactivation. Changes in particular gp120 and gp41 residues apparently release these restraints and lower the energy barriers that separate state 1 from downstream conformations (56). For example, alteration of several residues in the gp120 V2 region resulted in more reactive Envs that exhibited increased sensitivity to both activation and inhibition by CD4mc. Of note, changes in V2 residues 166 to 168, which are predicted by several available HIV-1 Env structures to be involved in key interactions at the trimer apex (63–66), exerted relatively little impact on HIV-1 phenotypes. These observations raise questions about the relationship of these structures to functional Env states, particularly state 1. Further work will be required to understand the structural restraints that maintain the HIV-1 Env trimer in state 1 and resist the binding of CD4mc and consequent induction of conformational changes. Such restraints operate much less efficiently in the context of free monomeric gp120, which binds the potent CD4mc at least 2 orders of magnitude better than our estimates for the interaction of CD4mc with the functional Env trimer (31, 35–39).
Lastly, we established assays to evaluate whether the binding of a CD4mc to one Env protomer can influence the conformation of the other Env protomers. This phenomenon has been recently documented for sCD4 (X. Ma, A. Herschhorn, J. D. Ventura, J. R. Grover, D. S. Terry, J. Gorman, X. Hong, Z. Zhou, H. Zhao, R. B. Altman, A. B. Smith III, J. Arthos, P. D. Kwong, J. Sodroski, S. C. Blanchard, W. Mothes, and J. B. Munro, unpublished data), but we did not find evidence that subneutralizing concentrations of CD4-mimetic molecules sensitize adjacent Env protomers to neutralization by antibodies (57–60). Apparently, the conformational changes induced in the HIV-1 Env trimer by sCD4 and CD4mc are not identical. An understanding of these conformational changes and how they contribute to virus entry or inhibition should help to optimize the potency and utility of the CD4mc.
MATERIALS AND METHODS
Compounds.
The CD4-mimetic compounds were synthesized and the chemical structures were characterized as described previously (31–39). The compounds were dissolved in dimethyl sulfoxide at a stock concentration of 10 to 20 mM, aliquoted, and stored at −20°C. Each compound was then diluted to 1 mM in serum-free Dulbecco's modified Eagle medium (DMEM) and used for different assays.
HIV-1 Env variants.
The wild-type HIV-1YU2, HIV-1AD8, and HIV-1JR-FL Envs were expressed by the pSVIIIenv plasmid (32). Mutations were introduced into the pSVIIIenv plasmid expressing the HIV-1JR-FL Env using the QuikChange II site-directed mutagenesis protocol (Stratagene). The presence of the desired mutations was confirmed by DNA sequencing. All Env residues are numbered according to convention based on the HXBc2 prototypic sequence (67).
Cell lines.
293T human embryonic kidney and Cf2Th canine thymocytes (ATCC) were grown at 37°C and 5% CO2 in DMEM (Invitrogen) containing 10% fetal bovine serum (Sigma) and 100 μg/ml penicillin-streptomycin (Mediatech, Inc.). Cf2Th cells stably expressing human CCR5 and CD4 were grown in medium supplemented with 0.4 mg/ml G418 and 0.2 mg/ml hygromycin (Invitrogen). Cf2Th-CCR5 cells, which stably express human CCR5, were grown in medium supplemented with 0.4 mg/ml G418. The cytotoxicity of two of the more potent CD4-mimetic compounds, JP-III-48 and BNM-III-170, for Cf2Th-CD4/CCR5 cells was tested with the CellTiter-Glo luminescent cell viability assay (Promega).
Recombinant luciferase viruses.
293T human embryonic kidney cells were cotransfected with plasmids expressing the pCMVΔP1Δenv HIV-1 Gag-Pol packaging construct, the HIV-1 envelope glycoproteins, or the envelope glycoprotein of the control AMLV and the firefly luciferase-expressing vector at a DNA ratio of 1:1:3 μg using the Effectene transfection reagent (Qiagen). For the production of the AMLV-pseudotyped viruses, a plasmid expressing HIV-1 Rev was added. Cotransfection produced recombinant luciferase-expressing viruses capable of a single round of infection. The virus-containing supernatants were harvested 36 to 40 h after transfection, spun, aliquoted, and frozen at −80°C until further use. The reverse transcriptase (RT) levels of all virus stocks were measured as described previously (68).
Infection by single-round luciferase viruses.
Cf2Th-CD4/CCR5 or Cf2Th-CCR5 target cells were seeded at a density of 6 × 103 cells/well in 96-well luminometer-compatible tissue culture plates (PerkinElmer) 24 h before infection. On the day of infection, CD4mc (0 to 100 μM) were incubated with recombinant viruses (10,000 RT units) at 37°C for different lengths of time. In the case of sensitization assays, a constant concentration of compounds was incubated with virus at 37°C for the time indicated; then, antibodies (over a range of 0 to 200 nM) were added to the virus-compound mixture and incubated for an additional period at 37°C. The mixtures were then added to the target cells; for infections of Cf2Th-CCR5 target cells, the mixtures containing the recombinant viruses were spinoculated onto the cells at 500 × g for 1 h at 25°C. The target cells were then incubated at 37°C for 48 h. Afterward, the medium was removed from each well, and the cells were lysed by the addition of 30 μl passive lysis buffer (Promega) and three freeze-thaw cycles. An EG&G Berthold Microplate Luminometer LB 96V was used to measure the luciferase activity of each well after the addition of 100 μl of luciferin buffer (15 mM MgSO4, 15 mM KPO4, pH 7.8, 1 mM ATP, and 1 mM dithiothreitol) and 50 μl of 1 mM firefly d-luciferin free acid 99% (Prolume).
Cell-cell fusion (syncytium formation) assay.
293T cells expressing the HIV-1 Env and the α peptide of β-galactosidase were cocultivated with either CD4-positive, CCR5-positive Cf2Th-CD4/CCR5 cells or CD4-negative, CCR5-positive Cf2Th-CCR5 cells expressing the ω peptide of β-galactosidase, as described previously (69). In some cases, CD4mc were added to the medium. After 6 h at 37°C, the cells were lysed, and the lysates were assayed for β-galactosidase activity.
Modeling HIV-1 activation and neutralization by CD4mc.
The dose-response curves for CD4mc activation of HIV-1 infection of CD4− CCR5+ cells and neutralization of HIV-1 infection of CD4+ CCR5+ target cells were analyzed as follows. The binding of CD4mc to the three protomers of the Env trimer was modeled using a binomial distribution:
| (1) |
where b is the binomial probability of an Env trimer having the given number of bound CD4mc, x is the total number of protomers on the Env trimer bound by a CD4mc (x = 0, 1, 2, or 3), P is the probability of the CD4mc occupying an individual Env protomer, and 3Cx is the 6/x!(3 − x)! (3C0 = 1, 3C1 = 3, 3C2 = 3, and 3C3 = 1).
This binomial distribution model assumes that each binding event of the CD4mc to the Env trimer occurs independently of the others (i.e., the binding of one CD4mc does not change the probability that a second CD4mc binds to an adjacent protomer).
We considered four different models for activation of HIV-1 infection of CD4− CCR5+ cells that differ in x. In this case, x represents the stoichiometry of the CD4mc with respect to the Env trimer that results in activation of infection of the CD4− CCR5+ cells:
| (2) |
| (3) |
| (4) |
| (5) |
Only the first three models are compatible with the biphasic dose-response curves observed, so the model in which only a fully occupied Env trimer is activated (x = 3) (equation 5) was not considered further.
We also considered three models for pure neutralization of HIV-1 infection of CD4+ CCR5+ cells that differ in x, the stoichiometry of the CD4mc required for neutralization of the Env trimer. Here, b represents the probability of infection in the presence of the CD4mc:
| (6) |
| (7) |
| (8) |
The binomial probabilities for each of the models of HIV-1 activation and neutralization are shown in Fig. 2A as a function of P, the probability of the CD4mc occupying an individual Env protomer.
P is related to the free concentration of CD4mc, [CD4mc], and the dissociation constant of the CD4mc for the Env trimer, Kd, as follows:
| (9) |
Therefore, combining equation 9 with equations 2, 3, and 4, the probability of HIV-1 infection of CD4− CCR5+ cells can be expressed as a function of the free concentration of CD4mc:
| (10) |
| (11) |
| (12) |
Likewise, combining equation 9 with equations 6, 7, and 8, the probability of HIV-1 infection of CD4+ CCR5+ cells in the presence of the CD4mc can be expressed as a function of the free concentration of CD4mc:
| (13) |
| (14) |
| (15) |
The binomial probabilities for each of the models of HIV-1 activation and neutralization as a function of the free [CD4mc] are shown in Fig. 2B. Note that, depending on the model for virus activation, maximal activation occurs at different CD4mc concentrations that represent 0.5 Kd, Kd, or 2 Kd. Likewise, depending on the model for virus neutralization, the IC50 of the CD4mc is 0.26 Kd, Kd, or 3.85 Kd. Thus, for a given virus and CD4mc, comparison of the CD4mc concentrations resulting in maximal activation of infection of CD4− CCR5+ cells and in 50% neutralization of infection of CD4+ CCR5+ cells should indicate which models best explain the experimental observations.
Should cooperative binding of CD4mc to the Env trimer occur, the dose-response curves associated with the binding of two or more CD4mc to the Env trimer would shift leftward, but the basic relationships among the curves predicted by the various models would remain the same.
Infections with recombinant-HIV-1-containing mixtures of Env mutants.
The stoichiometry of CD4mc inhibition of infection was investigated further by testing viruses with differing proportions of sensitive and resistant Envs. Recombinant HIV-1JR-FL viruses with mixtures of an Env (S375W) that is resistant to CD4mc and an Env (I309A) that is sensitive to CD4mc were produced as described above, varying the molar quantities of the plasmids expressing the Env mutants. The recombinant viruses were used to infect CD4-positive, CCR5-positive Cf2Th-CD4/CCR5 cells after incubation with saturating concentrations of BNM-III-170. In this case, the probability (b) of infection in the presence of saturating concentrations of BNM-III-170 depends upon x, the stoichiometry of the CD4mc required to inhibit virus infection, and upon z, the proportion of the resistant Env in the population. Thus, at saturating concentrations of BNM-III-170:
| (16) |
| (17) |
| (18) |
The binomial probabilities for each of these three models of HIV-1 inhibition by CD4mc as a function of z (the percentage of S375W Env in the virus population) are shown in Fig. 3B.
For studies of CD4mc-induced sensitization of HIV-1 to antibody neutralization, viruses with mixed Envs were generated as described above. Recombinant HIV-1JR-FL viruses with mixtures of an S375W Env and an F317A Env were produced. The S375W Env is resistant to CD4mc but potentially sensitive to neutralization by the CH23 anti-V3 antibody (62); the F317A Env is resistant to the CH23 antibody but can be activated by CD4mc. Recombinant viruses with various proportions of mixed Envs were incubated with 30 μM BNM-III-170 and different concentrations of the CH23 antibody for 30 min at 37°C. The virus-CD4mc-antibody mixtures were then added to CD4-negative, CCR5-positive Cf2Th-CCR5 cells. After 48 h, the cells were lysed and the lysates were assayed for luciferase activity.
ACKNOWLEDGMENTS
We thank Elizabeth Carpelan for manuscript preparation. We thank Irwin Chaiken and Wayne Hendrickson for valuable discussions and input.
This study was supported by the National Institutes of Health (GM56550 and AI24755) and the late William F. McCarty-Cooper. N.M. was supported by amfAR grant 107431-45-RFNT, NIH grant AI90682, and a Ragon Institute Innovation Award. A.H. was supported by amfAR research grant 109285-58-RKVA.
REFERENCES
- 1.Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 2.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 3.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 4.Myszka DG, Sweet RW, Hensley P, Brigham-Burke M, Kwong PD, Hendrickson WA, Wyatt R, Sodroski J, Doyle ML. 2000. Energetics of the HIV gp120-CD4 binding reaction. Proc Natl Acad Sci U S A 97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 7.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. 1996. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 8.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148. doi: 10.1016/S0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 9.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 10.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 11.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149–1158. doi: 10.1016/S0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 12.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 14.Furuta RA, Wild CT, Weng Y, Weiss CD. 1998. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol 5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 15.He Y, Vassell R, Zaitseva M, Nguyen N, Yang Z, Weng Y, Weiss CD. 2003. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J Virol 77:1666–1671. doi: 10.1128/JVI.77.3.1666-1671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koshiba T, Chan DC. 2003. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J Biol Chem 278:7573–7579. doi: 10.1074/jbc.M211154200. [DOI] [PubMed] [Google Scholar]
- 17.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB III, Sodroski JG. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci U S A 101:5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A 91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 20.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 21.Lu M, Blacklow SC, Kim PS. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol 2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 22.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol 151:413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hussey RE, Richardson NE, Kowalski M, Brown NR, Chang HC, Siliciano RF, Dorfman T, Walker B, Sodroski J, Reinherz EL. 1988. A soluble CD4 protein selectively inhibits HIV replication and syncytium formation. Nature 331:78–81. doi: 10.1038/331078a0. [DOI] [PubMed] [Google Scholar]
- 25.Fisher RA, Bertonis JM, Meier W, Johnson VA, Costopoulos DS, Liu T, Tizard R, Walker BD, Hirsch MS, Schooley RT, Flavell RA. 1988. HIV infection is blocked in vitro by recombinant soluble CD4. Nature 331:76–78. doi: 10.1038/331076a0. [DOI] [PubMed] [Google Scholar]
- 26.Smith DH, Byrn RA, Marsters SA, Gregory T, Groopman JE, Capon DJ. 1987. Blocking of HIV-1 infectivity by a soluble, secreted form of the CD4 antigen. Science 238:1704–1707. doi: 10.1126/science.3500514. [DOI] [PubMed] [Google Scholar]
- 27.Vita C, Drakopoulou E, Vizzavona J, Rochette S, Martin L, Menez A, Roumestand C, Yang YS, Ylisastigui L, Benjouad A, Gluckman JC. 1999. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc Natl Acad Sci U S A 96:13091–13096. doi: 10.1073/pnas.96.23.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang CC, Stricher F, Martin L, Decker JM, Majeed S, Barthe P, Hendrickson WA, Robinson J, Roumestand C, Sodroski J, Wyatt R, Shaw GM, Vita C, Kwong PD. 2005. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure 13:755–768. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 29.Gardner MR, Kattenhorn LM, Kondur HR, von Schaewen M, Dorfman T, Chiang JJ, Haworth KG, Decker JM, Alpert MD, Bailey CC, Neale ES Jr, Fellinger CH, Joshi VR, Fuchs SP, Martinez-Navio JM, Quinlan BD, Yao AY, Mouquet H, Gorman J, Zhang B, Poignard P, Nussenzweig MC, Burton DR, Kwong PD, Piatak M Jr, Lifson JD, Gao G, Desrosiers RC, Evans DT, Hahn BH, Ploss A, Cannon PM, Seaman MS, Farzan M. 2015. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature 519:87–91. doi: 10.1038/nature14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. 2005. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Schön A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB III, Sodroski J, Freire E. 2006. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 45:10973–10980. doi: 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madani N, Schön A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB III, Sodroski J. 2008. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 16:1689–1701. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curreli F, Kwon YD, Zhang H, Yang Y, Scacalossi D, Kwong PD, Debnath AK. 2014. Binding mode characterization of NBD series CD4-mimetic HIV-1 entry inhibitors by X-ray structure and resistance study. Antimicrob Agents Chemother 58:5478–5491. doi: 10.1128/AAC.03339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon YD, LaLonde JM, Yang Y, Elban MA, Sugawara A, Courter JR, Jones DM, Smith AB III, Debnath AK, Kwong PD. 2014. Crystal structures of HIV-1 gp120 envelope glycoprotein in complex with NBD analogues that target the CD4-binding site. PLoS One 9:e85940. doi: 10.1371/journal.pone.0085940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lalonde JM, Elban MA, Courter JR, Sugawara A, Soeta T, Madani N, Princiotto AM, Kwon YD, Kwong PD, Schon A, Freire E, Sodroski J, Smith AB III. 2011. Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg Med Chem 19:91–101. doi: 10.1016/j.bmc.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaLonde JM, Kwon YD, Jones DM, Sun AW, Courter JR, Soeta T, Kobayashi T, Princiotto AM, Wu X, Schon A, Freire E, Kwong PD, Mascola JR, Sodroski J, Madani N, Smith AB III. 2012. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J Med Chem 55:4382–4396. doi: 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lalonde JM, Le-Khac M, Jones DM, Courter JR, Park J, Schon A, Princiotto AM, Wu X, Mascola JR, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB III. 2013. Structure-based design and synthesis of an HIV-1 entry inhibitor exploiting X-ray and thermodynamic characterization. ACS Med Chem Lett 4:338–343. doi: 10.1021/ml300407y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courter JR, Madani N, Sodroski J, Schon A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, LaLonde JM, Smith AB III. 2014. Structure-based design, synthesis and validation of CD4-mimetic small molecule inhibitors of HIV-1 entry: conversion of a viral entry agonist to an antagonist. Acc Chem Res 47:1228–1237. doi: 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melillo B, Liang S, Park J, Schon A, Courter JR, LaLonde JM, Wendler DJ, Princiotto AM, Seaman MS, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB III. 2016. Small-molecule CD4-mimics: structure-based optimization of HIV-1 entry inhibition. ACS Med Chem Lett 7:330–334. doi: 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narumi T, Arai H, Yoshimura K, Harada S, Hirota Y, Ohashi N, Hashimoto C, Nomura W, Matsushita S, Tamamura H. 2013. CD4 mimics as HIV entry inhibitors: lead optimization studies of the aromatic substituents. Bioorg Med Chem 21:2518–2526. doi: 10.1016/j.bmc.2013.02.041. [DOI] [PubMed] [Google Scholar]
- 41.Ohashi N, Harada S, Mizuguchi T, Irahara Y, Yamada Y, Kotani M, Nomura W, Matsushita S, Yoshimura K, Tamamura H. 2016. Small-molecule CD4 mimics containing mono-cyclohexyl moieties as HIV entry inhibitors. Chem Med Chem 11:940–946. doi: 10.1002/cmdc.201500590. [DOI] [PubMed] [Google Scholar]
- 42.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 2000. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure 8:1329–1339. doi: 10.1016/S0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 44.Haim H, Si Z, Madani N, Wang L, Courter JR, Princiotto A, Kassa A, DeGrace M, McGee-Estrada K, Mefford M, Gabuzda D, Smith AB III, Sodroski J. 2009. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog 5:e1000360. doi: 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore JP, McKeating JA, Weiss RA, Sattentau QJ. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250:1139–1142. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- 46.Fu YK, Hart TK, Jonak ZL, Bugelski PJ. 1993. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J Virol 67:3818–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haim H, Steiner I, Panet A. 2005. Synchronized infection of cell cultures by magnetically controlled virus. J Virol 79:622–625. doi: 10.1128/JVI.79.1.622-625.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haim H, Steiner I, Panet A. 2007. Time frames for neutralization during the human immunodeficiency virus type 1 entry phase, as monitored in synchronously infected cell cultures. J Virol 81:3525–3534. doi: 10.1128/JVI.02293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richard J, Veillette M, Ding S, Zoubchenok D, Alsahafi N, Coutu M, Brassard N, Park J, Courter JR, Melillo B, Smith AB III, Shaw GM, Hahn BH, Sodroski J, Kaufmann DE, Finzi A. 2016. Small CD4 mimetics prevent HIV-1 uninfected bystander CD4 + T cell killing mediated by antibody-dependent cell-mediated cytotoxicity. EBioMedicine 3:122–134. doi: 10.1016/j.ebiom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiang SH, Kwong PD, Gupta R, Rizzuto CD, Casper DJ, Wyatt R, Wang L, Hendrickson WA, Doyle ML, Sodroski J. 2002. Mutagenic stabilization/disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus (HIV-1) gp120 envelope glycoprotein. J Virol 76:9888–9899. doi: 10.1128/JVI.76.19.9888-9899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haim H, Strack B, Kassa A, Madani N, Wang L, Courter JR, Princiotto A, McGee K, Pacheco B, Seaman MS, Smith AB III, Sodroski J. 2011. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS Pathog 7:e1002101. doi: 10.1371/journal.ppat.1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haim H, Salas I, McGee K, Eichelberger N, Winter E, Pacheco B, Sodroski J. 2013. Modeling virus- and antibody-specific factors to predict human immunodeficiency virus neutralization efficiency. Cell Host Microbe 14:547–558. doi: 10.1016/j.chom.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Rourke SM, Schweighardt B, Phung P, Fonseca DP, Terry K, Wrin T, Sinangil F, Berman PW. 2010. Mutation at a single position in the V2 domain of the HIV-1 envelope protein confers neutralization sensitivity to a highly neutralization-resistant virus. J Virol 84:11200–11209. doi: 10.1128/JVI.00790-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herschhorn A, Gu C, Espy N, Richard J, Finzi A, Sodroski JG. 2014. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat Chem Biol 10:845–852. doi: 10.1038/nchembio.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGee K, Haim H, Korioth-Schmitz B, Espy N, Javanbakht H, Letvin N, Sodroski J. 2014. The selection of low envelope glycoprotein reactivity to soluble CD4 and cold during simian-human immunodeficiency virus infection of rhesus macaques. J Virol 88:21–40. doi: 10.1128/JVI.01558-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herschhorn A, Ma X, Gu C, Castillo-Menendez L, Melillo B, Terry DS, Smith AB III, Blanchard SC, Munro JB, Mothes W, Finzi A, Sodroski J. 2016. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. MBio 7:e01598-16. doi: 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshimura K, Harada S, Shibata J, Hatada M, Yamada Y, Ochiai C, Tamamura H, Matsushita S. 2010. Enhanced exposure of human immunodeficiency virus type 1 primary isolate neutralization epitopes through binding of CD4 mimetic compounds. J Virol 84:7558–7568. doi: 10.1128/JVI.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madani N, Princiotto AM, Schon A, LaLonde J, Feng Y, Freire E, Park J, Courter JR, Jones DM, Robinson J, Liao HX, Moody MA, Permar S, Haynes B, Smith AB III, Wyatt R, Sodroski J. 2014. CD4-mimetic small molecules sensitize human immunodeficiency virus to vaccine-elicited antibodies. J Virol 88:6542–6555. doi: 10.1128/JVI.00540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Otsuki H, Hishiki T, Miura T, Hashimoto C, Narumi T, Tamamura H, Yoshimura K, Matsushita S, Igarashi T. 2013. Generation of a replication-competent simian-human immunodeficiency virus, the neutralization sensitivity of which can be enhanced in the presence of a small-molecule CD4 mimic. J Gen Virol 94:2710–2716. doi: 10.1099/vir.0.055590-0. [DOI] [PubMed] [Google Scholar]
- 60.Madani N, Princiotto AM, Easterhoff D, Bradley T, Luo K, Williams WB, Liao HX, Moody MA, Phad GE, Bernat NV, Melillo B, Santra S, Smith AB III, Karlsson Hedestam GB, Haynes B, Sodroski J. 2016. Antibodies elicited by multiple envelope glycoprotein immunogens in primates neutralize primary human immunodeficiency viruses (HIV-1) sensitized by CD4-mimetic compounds. J Virol 90:5031–5046. doi: 10.1128/JVI.03211-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Protocol G Principal Investigators, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montefiori DC, Karnasuta C, Huang Y, Ahmed H, Gilbert P, de Souza MS, McLinden R, Tovanabutra S, Laurence-Chenine A, Sanders-Buell E, Moody MA, Bonsignori M, Ochsenbauer C, Kappes J, Tang H, Greene K, Gao H, LaBranche CC, Andrews C, Polonis VR, Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Self SG, Berman PW, Francis D, Sinangil F, Lee C, Tartaglia J, Robb ML, Haynes BF, Michael NL, Kim JH. 2012. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J Infect Dis 206:431–441. doi: 10.1093/infdis/jis367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. 2013. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. 2013. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. 2014. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JH, Ozorowski G, Ward AB. 2016. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351:1043–1048. doi: 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korber B, Foley B, Kuiken C, Pillai S, Sodroski J. 1998. Numbering positions in HIV relative to HXBc2, p III102–III111. In Korber B, Kuiken C, Foley B, Hahn B, McCutchan F, Mellors J, Sodroski J (ed), Human retroviruses and AIDS. Los Alamos National Laboratory, Los Alamos, NM. [Google Scholar]
- 68.Rho HM, Poiesz B, Ruscetti FW, Gallo RC. 1981. Characterization of the reverse transcriptase from a new retrovirus (HTLV) produced by a human cutaneous T-cell lymphoma cell line. Virology 112:355–360. doi: 10.1016/0042-6822(81)90642-5. [DOI] [PubMed] [Google Scholar]
- 69.Holland AU, Munk C, Lucero GR, Nguyen LD, Landau NR. 2004. α-Complementation assay for HIV envelope glycoprotein-mediated fusion. Virology 319:343–352. doi: 10.1016/j.virol.2003.11.012. [DOI] [PubMed] [Google Scholar]