

Abstract
Translation elongation factor 1α (EF-1α, or EF-Tu in bacteria) is a highly conserved core component of the translation machinery that is shared by all cellular life. It is part of a large superfamily of GTPases that are involved in translation initiation, elongation, and termination, as well as several other cellular functions. Eukaryotic EF-1α (eEF-1α) is well studied and widely sampled and has been used extensively for phylogenetic analyses. It is generally thought that such highly conserved and functionally integrated proteins are unlikely to be involved in events such as lateral gene transfer or ancient duplication and gene sorting, which would undermine phylogenetic reconstruction. Here we describe a GTPase called EF-like (EFL), which is very similar to, but also distinct from, canonical eEF-1α. EFL is found in a wide variety of eukaryotes (dinoflagellates, haptophytes, cercozoa, green algae, choanoflagellates, and fungi), but its distribution is punctate: organisms that possess EFL are not closely related to one another, and EFL appears to be absent from the closest relatives of organisms that do possess it. Moreover, in most genomes where EFL is present, canonical eEF-1α appears to be absent. Analysis of functional divergence suggests that, whereas EFL is divergent in general, putative functional binding sites involved in translation are not significantly divergent as a whole. Altogether, it appears that EFL has replaced eEF-1α several times independently. This finding could be an indication of an ancient paralogy or, more likely, eukaryote-to-eukaryote lateral gene transfer.
Translation elongation factor 1α (EF-1α) is a highly conserved member of the GTPase superfamily that also includes protein factors involved in translation initiation and termination (1, 2). GTP-bound EF-1α (and its functional equivalent in bacteria, EF-Tu) binds aminoacyl tRNAs (aa-tRNAs) and brings them to the A site of ribosomes. The aa-tRNA is released after GTP hydrolysis, and GDP-bound (inactive) eEF-1α binds the GDP/GTP exchange factor EF-1β to recharge GTP and enter the next round of peptide elongation. eEF-1α is also known to be involved in a number of nontranslational cellular processes, interacting with cytoskeletal proteins, calmodulin, and the ubiquitin-dependent proteolytic system (3).
Because of its high level of conservation and seemingly ubiquitous distribution, eukaryotic EF-1α (eEF-1α) has been used to examine a variety of evolutionary questions. At one level, it has served as a model for molecular evolutionary processes: because it has relatively well defined functional interactions and a known crystal structure, it has been used to illustrate methods for detecting evolutionary rate variation at functional sites and covarion-like behavior (4–8). At another level, EF-1α has also served as an important molecular marker for determining phylogenetic relationships among eukaryotes (9–14). Altogether, eEF-1α is thought to be ubiquitous and to perform core cellular roles for which it is functionally integrated with many other proteins, and for these reasons it is considered to be a stable representative of organismal evolutionary history that is not prone to complications such as ancient paralogy or lateral gene transfer [except in instances where it is transferred and assumes a novel function (15)].
Here, we show that several eukaryotic lineages defy our expectations about eEF-1α: dinoflagellates, haptophytes, chlorophyte and trebouxiophyte green algae, cercozoa, and several other eukaryotic lineages appear to lack canonical eEF-1α. Instead, these groups possess a variety of expressed GTPase that is similar to canonical eEF-1α, but is clearly distinct from it, which we will refer to as EF-like (EFL). Our survey revealed that EFL is widely but sporadically distributed among eukaryotes, and organisms that possess EFL are typically closely related to other organisms that possess canonical eEF-1α and lack EFL; the currently understood distributions of EFL and canonical eEF-1α are nearly mutually exclusive. In addition, we detected no significant correlation between divergence in EFL and the binding sites of known importance for translational function in canonical eEF-1α, suggesting that EFL might plausibly perform at least these eEF-1α functions. We propose that EFL has replaced eEF-1α several times independently in eukaryotic history, and that eukaryote-to-eukaryote lateral gene transfer may be involved in the EFL evolution.
Materials and Methods
Identification and Characterization of EFL Genes. Genes encoding all proteins with high similarity to EF-1α were identified in EST projects from a chlorarachniophyte cercozoan Bigelowiella natans (strain CCMP 621); a dinoflagellate Heterocapsa triquetra (strain CCMP 449); two haptophytes, Isochrysis galbana (strain CCMP 1323) and Pavlova lutheri (strain CCMP 1325); a trebouxiophyte green alga Helicosporidium sp. (strain AT-2000); and an ulvophyte green alga Acetabularia acetabulum. All clones were isolated and resequenced on both strands, resulting in full-length cDNA sequence from all organisms except Acetabularia, where the 5′ end was missing. EFL genes were also identified in EST projects on the green algae Prototheca wickerhamii and Scenedesmus obliquus, which can be accessed at amoebidia.bcm.umontreal.ca/public/pepdb/agrm.php, and were provided by B. Lee and T. Borza (Dalhousie University, Halifax, Canada). An EFL gene was found in publicly available ESTs from a zygomycete Conidiobolus coronatus, and EST projects from chytrids Spizellomyces puntatus and Allomyces macrogynus (made available by B. F. Lang, Universitédu Montréal, Montréal). Other partial fungal EFL genes were identified in the Fungal Tree of Life Project, which can be accessed at ocid.nacse.org/research/aftol, and made available by T. James and R. Vilgalys (Duke University, Durham, NC). EFL genes were used to search other public databases by using the program blast to find previously unidentified members of this protein family. Wholegenome and EST databases were also specifically searched for both canonical EF-1α and EFL genes for eukaryotic lineages known to contain the EFL gene, or close relatives of these lineages.
Phylogenetic Analyses. Conceptual translations of all newly characterized EF-1α and EFL sequences were added to an alignment consisting of eEF-1α and archaebacterial EF-1α (aEF-1α), respectively, and two eukaryote-specific paralogues, eukaryotic release factor 3 (eRF3) and Hsp70 subfamily B suppressor 1 (HBS1). Ambiguously aligned sites were removed, resulting in 66 sequences and 373 alignment sites, which were subjected to phylogenetic analyses by using Bayesian and maximum likelihood (ML) methods.
Bayesian trees were reconstructed from the 66 sequence data set by using the program mrbayes 3.0 (16) under the Jones–Taylor–Thornton (JTT) substitution frequency matrix with among-sites rate variation (ASRV) modeled by using a discrete γ distribution with four equally probable categories. One cold and three heated Markov chain Monte Carlo (MCMC) chains with the default-chain temperatures were run for 500,000 generations, sampling loglikelihoods (lnLs), and trees at 100-generation intervals (i.e., 5,000 lnLs and trees were saved during MCMC). The likelihood plot suggested that MCMC reached the stationary phase after the first 70,000 generations (data not shown). Thus, the remaining 4,300 trees were used to obtain clade probabilities and branch-length estimates. ML bootstrap analysis (100 replicates) was conducted by using the JTT model with ASRV modeled, using a discrete γ distribution (eight equally probable categories plus the proportion of invariable sites) with the programs phyml 2.1b1 (17) and proml (18) implemented in phylip v3.6a (global rearrangement plus input sequence order randomized, but no ASRV). A second alignment of full-length EFL sequences alone (24 sequences and 411 positions) was analyzed in a similar fashion to determine relationships within the EFL clade.
Alternative positions for the EFL clade were tested by using approximately unbiased (AU) tests (19). A monophyletic EFL clade was grafted to 94 possible positions in the Bayesian tree. For each topology, the lnL at each site (site lnL) was calculated with the JTT model, with ASRV modeled by using eight equally probable categories, using the program tree-puzzle 5.1 (20). Site lnL data were reformatted by using the program puzz2lnf (J. Leigh, Dalhousie University, Halifax, Canada), and AU tests were conducted by using the program consel 0.1f (21).
Testing Functional Divergence Between Canonical eEF-1α and EFL. We created a data set including 25 aEF-1α, 25 nonmicrosporidian eEF-1α (eEF-1αNM), 3 microsporidian EF-1α (eEF-1αM), 7 EFL, and 25 eRF3 sequences (HBS1 sequences were excluded due to the extremely high rate of evolution). The addition of divergent eRF3 sequences from unicellular eukaryotes resulted in a final alignment of 335 unambiguous positions, from which the rate of change at each site [site rate (SR)] was estimated for the aEF-1α, eEF-1αNM, eEF-1αM, EFL, and eRF3 subtrees. SR was estimated by using the bivariate ML rate method (22) under the PAM001 amino acid substitution matrix with ASRV in the two subtrees described by a matrix of 25 × 25 equally separated rate categories. The upper boundary of SR was set as 6.0. The probability for each rate category was estimated from the data. The differences in SR (ΔSR) across the eEF-1αNM and other subtrees were calculated by subtracting the latter from the former, coupled with constructing the 95% confidence intervals of ΔSR. The overall differences in evolutionary rate distribution across (i) eEF-1αNM and aEF-1α subtrees, (ii) eEF-1αNM and eEF-1αM subtrees, (iii) eEF-1αNM and EFL subtrees, and (iv) eEF-1αNM and eRF3 subtrees can be represented by the sum of absolute values of ΔSR (arsum). The SR estimation of the eEF-1αM subtree may include large errors due to poor sequence sampling of microsporidia (three sequences), so we also calculated the sum of absolute values of ΔSR, of which the 95% confidence intervals do not contain 0 (arsum*). This value may represent the overall rate distance across two subtrees better than the arsum (8).
Amino acid residues putatively involved in binding to EF-1β, aa-tRNA, and GTP/GDP have been predicted from tertiary structural analyses of yeast EF-1α/EF-1β/GDP complex, and an EF-1α orthologue in bacteria crystallized with aa-tRNA and a GTP analogue (7). We will refer to the sites and the corresponding positions in eRF3 and EFL as “putative binding sites.” The 335 aligned sites contain 57 of 71 putative binding sites (Fig. 3, which is published as supporting information on the PNAS web site). To emphasize the functional divergence at the putative binding sites, arsum and arsum* were also calculated for each comparison from these sites alone.
The functional constraints at sites also can be altered across two subtrees with no significant SR change (23). Functionally divergent (FD) sites, which do not necessarily associate with significant SR, have been defined as type II FD sites. Such type II FD sites across two subtrees fall into three categories. Differently evolving sites are where the amino acid identity in one subtree is constrained to I, whereas the corresponding sites in the other subtree are varied such that I < 0.2. Absolutely differently evolving sites are where the identities are constant in both subtrees (I1 and I2), but I1 and I2 are different from each other. Finally, ΔCPS sites are where the chemical property of the amino acid side chains in one subtree is significantly different from that in the other subtree. We identified the type II FD sites across the eEF-1αNM and other (i.e., aEF-1α, eEF-1αM, EFL, or eRF3) subtrees by using the program covares v2.0 (24), and then investigated how significantly these FD sites overlap with the putative binding sites by using χ2 tests.
Results and Discussion
EFL Is a Member of the GTPase Superfamily Related to eEF-1α. EST surveys were conducted on representatives of a number of major algal groups: the cercozoan Bigelowiella (3,995 sequences), the dinoflagellate Heterocapsa (9,309 sequences), the haptophytes Isochrysis (14,234 sequences) and Pavlova (9,025 sequences), the trebouxiophyte green alga Helicosporidium (1,369 sequences), and the ulvophyte green alga Acetabularia (3,712 sequences). As a core component of the translation apparatus, eEF-1α is typically highly expressed and accordingly abundant in EST surveys. Surprisingly, however, only Acetabularia data included canonical eEF-1α transcripts. From all other organisms, EFL transcripts were abundant, but no canonical eEF-1α transcripts were identified. The EFL gene was represented in 20 transcripts from Bigelowiella, 59 transcripts from Heterocapsa, 31 transcripts from Isochrysis, 17 transcripts from Pavolva, and 9 transcripts from Helicosporidium. It is impossible to accurately translate these numbers into expression levels, but, if one compares these levels with those of a highly expressed housekeeping protein (actin, for example), they are comparable (19, 78, 22, 22, and 0, respectively, for actin). This finding suggests that EFL is relatively highly expressed, but this conclusion must be confirmed directly. The deduced EFL amino acid sequences are clearly distinguished from the previously known members of the EF-1α family. All EFL sequences identified share six insertions of various lengths and a large number of signature sequences comprising numerous unique substitutions (Fig. 4, which is published as supporting information in the PNAS web site). One insertion occurs at, or close to, a well studied animal-fungal insertion (9).
The distribution of EFL was examined by searching against existing databases for previously unrecognized members of this protein family. Full- or near-full-length EFL orthologues were found in two other dinoflagellates, Amphidinium carterae and Lingulodinium polyedrum, the zygomycete fungus Conidiobolus coronatus, the choanoflagellate Monosiga brevicollis, an environmental sample from the Sargasso Sea, an EST from the nematode Heterodera glycines (GenBank accession no. CA940117), and the green algae Chlamydomonas reinhardtii, Pleodorina sp., Prototheca wickerhamii, and Scenedesmus obliquus. Most interestingly, there is no canonical eEF-1α in the Chlamydomonas genome, which can be accessed at genome.jgi-psf.org/chlamy, or from the abundant ESTs from this organism, whereas EFL is highly represented (>100 clones in current EST data). Other short fragments of EFL genes were identified in the green alga Dunaliella salina, the chytrids Spizellomyces punctatus and Allomyces macrogynus, several other early diverging fungi, and the dinoflagellate Alexandrium tamarensis. A single, partial EST attributed to Oryza sativa was also identified (GenBank accession no. AK110624) and found to be remarkably similar to the Scenedesmus EFL sequence (they were sisters in all phylogenetic analyses; data not shown). However, the corresponding gene is not present in the Oryza genome project, so this sequence is likely a contaminant.
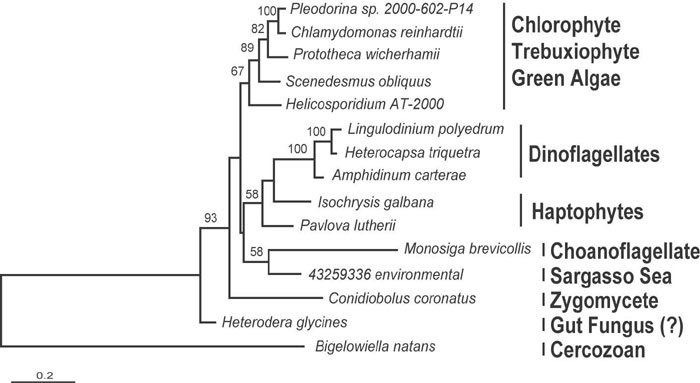
Phylogenetic Position of EFL in the EF-1α Superfamily. The full-length EFL and the most closely related members of the EF-1α superfamily were subjected to phylogenetic analyses. As expected, the EFL sequences formed a unique and well supported clade (100% bootstrap support), distinct from all other known members of the superfamily (Fig. 1). In turn, EFL, eRF3, and HBS1 formed a clade at the exclusion of eEF-1α and aEF-1α (64–81% bootstrap support), but this position is not conclusive because the AU tests failed to reject three alternative trees out of the 98 topologies tested. In these alternative topologies, the EFL clade is sister to eEF-1α, aEF-1α as a whole, or euryarchaeal EF-1α (Fig. 1, nodes A–C, respectively). AU tests reject the possibility that EFL arose from either eRF3 or HBS1. Altogether, EFL certainly forms a discrete clade, but the relationships between EFL and eEF-1α (or aEF-1α) remain uncertain: EFL may represent a unique GTPase paralogue or a highly derived EF-1α.
Fig. 1.

Phylogeny of EF-1α and related subfamilies of the GTPase translation factor superfamily. The phylogeny includes eEF-1α, eRF3, HBS1, and EFL, and is rooted by using aEF-1α. Numbers at nodes correspond to bootstrap supports from ML analyses considering ASRV by using the program phyml (top of figure) and considering no ASRV by using the program proml (bottom of figure). Bootstraps are only shown for nodes uniting major subfamilies or for nodes >50% within the EFL subtree. In the AU test, only the tree shown and three alternative tree topologies, in which the EFL clade is attached to nodes A–C, were not rejected at the 5% α-level. The P values of the AU test for tree topologies A–C are 0.532, 0.42, and 0.123, respectively.
The support for most relationships within both eEF-1α and EFL clades was very poor. Within EFL, only the dinoflagellates were highly supported, and the green algae received weak support (Fig. 1). When EFL sequences were analyzed alone to allow more positions to be used, the tree was still poorly supported and only differed in that the haptophytes were paraphyletic at the base of the dinoflagellates in some analyses (Fig. 5, which is published as supporting information on the PNAS web site). The Heterodera (nematode) EST showed weak but consistent affinity to Conidiobolus (Fig. 1), suggesting it is derived from a fungus, perhaps in the gut of the host nematode. Analyses, including the short EFL fragments of other fungi (based on only 141 positions), support this hypothesis because Heterodera branches within the chytrids (data not shown). Overall, the relationships between various subgroups were not resolved within the EFL clade.
EFL and eEF-1α Tend to Be Mutually Exclusive in Distribution. The significance of the distribution of EFL and the concomitant absence of eEF-1α are best appreciated by considering the evolutionary diversity of the organisms in question and what their close relatives are like. Fig. 2 shows a hypothetical tree of eukaryotes based on many kinds of evidence (25, 26), plotting the occurrence of known EFL. There are two significant aspects to the distribution of EFL. First, organisms with EFL are not closely related to one another. Indeed, organisms with EFL are scattered across the tree of eukaryotes, falling into four of the hypothetical supergroups shown. Second, nearly all of the EFL-encoding organisms are closely related to lineages that possess canonical eEF-1α. The same cannot be said for Bigelowiella (no other cercozoan EF data are known), but the distribution within other groups can be discerned with various levels of confidence. The choanoflagellates are related to animals and ichthyosporea (27), which both have eEF-1α but no evidence of EFL (which is supported by genomic data from several animals). Consistent with this idea, the choanoflagellate Monosiga ovata has canonical eEF-1α and no evidence of EFL from EST sequencing (H. Philippe, personal communication). In contrast, however, M. brevicollis possesses EFL and there is no evidence as to whether this organism also has eEF-1α. Likewise, EFL is present in certain zygomycete and chytrid fungi, but no EFL is present in any of the complete or partially complete genomes from ascomycetes (e.g., Saccharomyces, Candida, Schizosaccharomyces, and Neurospora) or basidiomycetes (e.g., Cryptococcus). Moreover, some chytrids and some zygomycetes have EFL whereas others have canonical eEF-1α, and one, Basidiobolus ranarum, may have both (see ocid.nacse.org/research/aftol). The origin and distribution of EFL in these organisms will be important to determine, but is presently hampered by a poor understanding of the relationships between chytrids and zygomycetes, and because most fungal eEF-1α and EFL gene fragments were acquired by PCR. Without genomic or EST data we cannot rule out the presence of both genes in any of these organisms. Accordingly, the eEF-1α/EFL distribution among chytrids and zygomycetes is tentative, with the possible exceptions of the zygomycete Conidiobolus and the chytrids Spizellomyces and Allomyces, which are based on EST sequencing that revealed no eEF-1α (B. F. Lang, personal communication).
Fig. 2.

Distribution of EFL and eEF-1α in eukaryotes. Hypothetical synthesis of eukaryotic relationships based on many molecular and structural characteristics. Groups with canonical eEF-1α are shown in black text. Groups where EFL but no canonical eEF-1α is known are highlighted by white text on black. Groups with no EF data are shown in gray text with dashed lines. Eukaryotic groups where the EF-1α/EFL distribution is supported by substantial or complete-genome data are indicated by asterisks, and support from ESTs is indicated by a cross. The expanded view of plants and green algae shows details of the distribution and the possible origin of the EFL gene in this group.
Distantly related members of both dinoflagellates and haptophytes possess expressed EFL genes and no eEF-1α transcripts are found in any of the relatively large EST projects known from these groups (two haptophytes and four dinoflagellates). Moreover, eEF-1α is known from all other related groups. In particular, complete or nearly complete genomes are known from four apicomplexa (Plasmodium, Toxoplasma, Theileria, and Cryptosporidium), two ciliates (Paramecium and Tetrahymena), and a heterokont (Thalassiosira): all encode eEF-1α, and none encode EFL (nor is it found in any of the many EST projects from these groups).
Perhaps the best case to examine the distribution of EFL is in the green algae and plants. Here the broad phylogenetic relationships are relatively well understood (28) and there is an abundance of supporting genomic and EST data from plants and some algae, including whole genomes (e.g., Arabidopsis, Oryza, Chlamydomonas, and Cyanidioschyzon). No plant has EFL (except for a probable contaminant in Oryza; see Materials and Methods), whereas at the same time the complete genome of Chlamydomonas encodes only EFL and not eEF-1α. The green algae are also particularly informative because the distribution of EFL is well refined: the trebouxiophytes (Helicosporidium and Prototheca) and chlorophytes (Chlamydomonas, Scenedesmus, Pleodorina, and Dunaliella) have EFL, whereas their sister group the ulvophytes (Acetabularia) has eEF-1α. Accordingly, the origin of EFL in the green algae appears to have occurred after the divergence of ulvophytes, but before the divergence of trebouxiophytes and chlorophytes from one another (Fig. 2).
Evidence That EFL Is Functionally Similar to eEF-1α. The essential function of EF-1α as a translation elongation factor must be fulfilled by some protein, so cells that lack canonical eEF-1α (e.g., Chlamydomonas) must have some other protein or proteins to replace it. The nearly nonoverlapping distribution of eEF-1α and EFL, together with its apparently highly expressed nature and relationship to eEF-1α, suggest that EFL may replace at least some of the functions of eEF-1α. If this conjecture is true, EFL should retain many or most of the residues known to play a significant role in translation elongation, despite its overall divergent nature. To examine this hypothesis, we have assessed functional divergence at three important binding sites defined from tertiary structure: EF-1β, aa-tRNA, and GTP/GDP. We have compared the rates of change at these sites between eEF-1αNM and EFL, aEF-1α, and eRF3. In addition, eEF-1αNM was compared with eEF-1αM, which are highly divergent and evolving in a covarion fashion (8), but have never been disputed to carry out the job of eEF-1α.
When all 335 alignable sites are considered, the arsum* value for the eEF-1αNM and EFL comparison is much larger than those from the eEF-1αNM and aEF-1α or eEF-1αNM and eEF-1αM comparisons (Table 1). This finding confirms the overall divergent nature of EFL, evident from the tree. Interestingly, however, when the arsum* was calculation based on the 57 putative binding sites, the eEF-1αNM and EFL comparison is very similar to the aEF-1α and eEF-1αM comparisons (Table 1). Regardless of the sites considered, the values from the eEF-1αNM and eRF3 comparison are much greater than those from other comparisons. These data indicate that the overall rate distribution of EFL may be different from that of canonical EF-1α, but there is no significant difference between the evolutionary tempo at the putative binding sites in EFL.
Table 1. Arsum* values calculated across the eEF-1αNM and other subtrees at the putative sites involved in EF-1α primary functions.
| eEF-1αNM vs. | 335 sites | 57 putative binding sites† |
|---|---|---|
| aEF-1α | 88.06 (208.56)‡ | 4.09 (17.58) |
| EFL | 129.15 (204.18)‡ | 6.27 (15.40) |
| eEF-1αM | 57.46 (184.96)‡ | 8.46 (23.19) |
| eRF3 | 151.97 (256.26)‡ | 16.52 (30.27) |
Putative EF-1β, aa-tRNA, and/or GDP/GTP-binding sites identified in ref. 7 for 57 of 335 analyzed positions. Arsum values are in parentheses
We also investigated type II FD sites, which associate with no significant site rate change. Of 32 type II FD sites detected, only seven correspond to putative binding sites in EF-1α (Fig. 6, which is published as supporting information on the PNAS web site), so the null hypothesis of independence was not rejected by a χ2 test (P = 0.442; Table 2). Similarly, the null hypothesis was not rejected in the eEF-1αNM and aEF-1α and eEF-1αNM and eEF-1αM comparisons (P = 0.0853 and 0.522; Table 2), although the result from the poorly sampled microsporidian subtree must be evaluated with caution. On the other hand, the significant overlap between the type II FD sites and putative binding sites was observed in a comparison between eEF-1αNM and eRF3 (P < 0.0; Table 2), probably reflecting the greater functional divergence of eRF3.
Table 2. Independence of the putative sites involved in EF-1α primary functions to type II FD sites detected across the eEF-1αNM and other subtrees.
| eEF-1αNM vs. | Type II FD sites* | Type II FD sites overlapped with the putative binding sites† | P values from χ2 tests of independence |
|---|---|---|---|
| aEF-1α | 15 | 5 | 0.0853 |
| EFL | 32 | 7 | 0.442 |
| eEF-1αM | 31 | 4 | 0.522 |
| eRF3 | 48 | 16 | 0.00115‡ |
DE, ADE, and/or ΔCPs sites identified in 335 positions by using the program covares v2.0
Putative EF-1β, aa-tRNA, and/or GDP/GTP-binding sites identified in ref. 7 for 57 of 335 analyzed positions
The null hypothesis is rejected at the 1% α level
The analyses of functional divergence across the eEF-1αNM and other subtrees show that the evolutionary tempo and mode of putative EF-1β, aa-tRNA, and GTP/GDP binding sites of EFL do not depart significantly from those of canonical EF-1α, including the archaebacterial and microsporidian orthologues. Thus, we predict that EFL may be able to perform at least these functions, even though the overall evolutionary rates of the canonical eEF-1α and EFL subtrees are different. This is not to say that EFL and eEF-1α are functionally equivalent: there are, after all, many predicted insertions and FD sites, and many are distributed nonrandomly (Fig. 6). For instance, practically an entire helix predicted to bind actin in eEF-1α (4) is composed of FD sites in EFL. The role of these sites and these processes in EFL is not known at present, but should become clearer when the processes are better characterized at the structural level in eEF-1α. It is also possible that EFL could perform some of the same functions as canonical eEF-1α, but in a slightly different way, as do bacteria. Further biochemical and genetic studies are required to address these functions and to fully understand the pattern of EFL evolution.
Evolutionary History of eEF-1α and EFL. We have shown that a variety of distantly related eukaryotes possess a gene for a derived EFL protein that is expressed and highly represented in EST data. Most of these organisms have no evidence of a canonical eEF-1α, but have close relatives that possess eEF-1α and lack EFL (Figs. 1 and 2). Given the essential nature of eEF-1α and the absence of undue divergence at putative binding sites of known function in translation (Tables 1 and 2), EFL may be capable of performing many of the translation-related functions of eEF-1α (and the possibility that the two proteins are orthologues is not ruled out by the phylogeny). Considering all these characteristics, we propose that EFL has partially or wholly replaced eEF-1α function several times independently. How could the punctate distribution of EFL arise? It is doubtful that EFL genes originated independently by convergence (because they share so many sequence features), which leaves two main alternatives: ancient paralogy or lateral gene transfer. Both are associated with certain expectations, and we can evaluate how well these compare with current observations.
If EFL arose by paralogy, then there must have been at least three gene duplications of ancestral GTPases, yielding four distinct paralogous families. Because EFL is widely distributed among eukaryotes (Fig. 2), the duplication that directly gave rise to EFL would have to have taken place early in eukaryotic evolution and EFL would accordingly be ancestral to most or all eukaryotes. The punctate distribution we see today could only be the result of a high rate of differential loss of EFL or eEF-1α. This model could, in theory, explain the distribution of EFL and eEF-1α, but there are a number of complications. First, the long-term retention of two paralogues is typically due to their adaptation to different functions, so differential loss is complicated by need for some protein to take over the missing functions whenever either of these genes is lost. This is likely why other GTPase families are not distributed in a punctate fashion, which in turn raises the question of why should EFL be different from these other families? We have shown that EFL may be able to perform some eEF-1α functions, but in organisms with eEF-1α some protein would also have to take over whatever function preserved EFL through most of eukaryotic evolution. More importantly, both genes must have coexisted in the ancestors of lineages where some descendents have EFL and others have eEF-1α, such as chromalveolates and plants/green algae. We would expect that both genes should still be found in many of these organisms, but as a rule this is not so and it is certainly not the case in those apicomplexa, ciliates, heterokonts, plants, green algae, red algae, animals, and fungi, where genome data are available. A few more genomes will probably be found to contain both genes, but the overall distribution of EFL and its near-complete absence in genomes with canonical eEF-1α does not currently support the notion that EFL represents and ancient gene family that originated early in eukaryotic evolution.
The alternative possibility, that EFL was distributed by eukaryote-to-eukaryote lateral transfer, fits the current observations better, but is also not without complications. This model of events does not specify the ultimate source of EFL. It may be derived from an eEF-1α that evolved very rapidly in a particular eukaryotic lineage, and, although highly derived, remained a functionally adequate EF. This is an appealing possibility because it suggests a continuous core function of the protein. It is also possible that it is derived from some other subfamily of eukaryotic GTPase, because the phylogeny does not adequately resolve the position of EFL. In either case, if such a gene spread to other eukaryotes by lateral transfer, and its presence encouraged the loss of eEF-1α (e.g., by performing overlapping functions), the observed distribution would result. Eukaryote-to-eukaryote lateral transfer is not well understood, but there are precedents (29–31). This model is also simpler than ancient paralogy because the loss of only one protein (eEF-1α) must be explained, whereas ancient paralogy requires that the loss of eEF-1α and EFL (in different lineages) must be explained. Lateral gene transfer is also consistent with the generally nonoverlapping distribution of the two proteins because it assumes they are similar in function and so redundancy or selection drives the nonoverlapping distribution, whereas paralogues are only maintained over long periods if they differentiate. Indeed, ancient paralogy predicts exactly the opposite of the observed distribution: the proteins coexisted for much of eukaryotic evolution because of differentiated function, so there is no reason to expect them to become mutually exclusive. Overall, both models are possible, but we favor lateral transfer because it is more consistent with the known distribution of proteins.
Lateral transfer assumes few eukaryotes had EFL at first, so why did so many eukaryotes acquire it? There are two equally interesting possibilities. On one hand, if EFL is fixed in a population by chance, then the frequency of transfer must be exceedingly high. The number of successful “takeovers” would be a small fraction of the number of transfers, and EFL transfers probably represent a small fraction of the number of eEF-1α transfers because EFL is comparatively rare. Most transfers of canonical eEF-1α between two eukaryotes would go unnoticed because the phylogeny is poorly resolved and lateral transfer is generally recognized by phylogenetic incongruence. Eukaryote-to-eukaryote transfers are beginning to be recognized to occur at some frequency (29–31), and this interpretation of EFL distribution suggests such transfers may be more frequent than we presently think. On the other hand, if the rate of transfer is not unusually high, then it suggests that EFL is a “supergene” that is very likely to replace eEF-1α once it is introduced into a genome. This conclusion would imply that EFL has some selective advantage over the highly conserved, indispensable eEF-1α protein found in vast majority of eukaryotes. If so, then EFL has made the jump between two selective peaks, further functional studies of EFL will be very important to pin down where such differences are derived. Distinguishing between these two possibilities will require substantially more information both from the distribution of EFL and also its possible functional relationship to eEF-1α.
Supplementary Material
Acknowledgments
We thank B. Lee and T. Borza for providing Prototheca and Scenedesmus data; G. I. McFadden for making a Bigelowiella library available; B. F. Lang for communicating unpublished results of Spizellomyces and Allomyces ESTs; H. Philippe for communicating unpublished results of Monosiga ESTs; T. James and R. Vilgalys for providing fungal data; J. Harper, N. Patron, R. Waller, and A. de Koning for assistance with EST data; J. Harper and R. Waller for critical reading; and J. Leigh, C. Blouin, A. J. Roger, and the Statistical Evolutionary Biology Group (Dalhousie University) for the program puzz2lnf, with assistance with the analyses covares v2.0, and generously allowing use of their computers, respectively. Bigelowiella EST sequencing was supported by Natural Science and Engineering Research Council of Canada Grant 227301-00; Helicosporidium sequencing was supported by Canadian Institutes for Health Research Grant MOP-42517; and EST sequencing from Heterocapsa, Isochrysis, Pavlova, and Acetabularia was supported by the Genome Atlantic/Genome Canada Protist EST Program. Acetabularia and Helicosporidium projects are collaborations with D. Mandoli (University of Washington, Seattle) and B. Drion (University of Florida, Gainesville), respectively. P.J.K. is a Scholar of the Canadian Institute for Advanced Research, Canadian Institutes For Health Research, and the Michael Smith Foundation for Health Research.
Author contributions: P.J.K. designed research and performed research; and P.J.K. and Y.I. analyzed data and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: EF-1α, elongation factor 1α; eEF-1α, eukaryotic EF-1α; aEF-1α, archaebacterial EF-1α; eEF-1αNM, nonmicrosporidian eEF-1α; eEF-1αM, microsporidian eEF-1α; EFL, EF-like; eRF3, eukaryotic release factor 3; aa-tRNAs, aminoacyl-tRNAs; HBS1, heat shock protein 70 subfamily B suppressor 1; ML, maximum likelihood; ASRV, among-sites rate variation; SR, site rates; AU, approximately unbiased; FD, functionally divergent.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AY29485–AY29490).
References
- 1.Keeling, P. J., Fast, N. M. & McFadden, G. I. (1998) J. Mol. Evol. 47, 649-655. [DOI] [PubMed] [Google Scholar]
- 2.Baldauf, S. L., Palmer, J. D. & Doolittle, W. F. (1996) Proc. Natl. Acad. Sci. USA 93, 7749-7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Negrutskii, B. S. & El'skaya, A. V. (1998) Prog. Nucleic Acid Res. Mol. Biol. 60, 47-78. [DOI] [PubMed] [Google Scholar]
- 4.Gaucher, E. A., Das, U. K., Miyamoto, M. M. & Benner, S. A. (2002) Mol. Biol. Evol. 19, 569-573. [DOI] [PubMed] [Google Scholar]
- 5.Gaucher, E. A., Gu, X., Miyamoto, M. M. & Benner, S. A. (2002) Trends Biochem. Sci. 27, 315-321. [DOI] [PubMed] [Google Scholar]
- 6.Gaucher, E. A., Miyamoto, M. M. & Benner, S. A. (2001) Proc. Natl. Acad. Sci. USA 98, 548-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inagaki, Y., Blouin, C., Susko, E. & Roger, A. J. (2003) Nucleic Acids Res. 31, 4227-4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inagaki, Y., Susko, E., Fast, N. M. & Roger, A. J. (2004) Mol. Biol. Evol. 21, 1340-1349. [DOI] [PubMed] [Google Scholar]
- 9.Baldauf, S. L. & Palmer, J. D. (1993) Proc. Natl. Acad. Sci. USA 90, 11558-11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roger, A. J., Sandblom, O., Doolittle, W. F. & Philippe, H. (1999) Mol. Biol. Evol. 16, 218-233. [DOI] [PubMed] [Google Scholar]
- 11.Kamaishi, T., Hashimoto, T., Nakamura, Y., Nakamura, F., Murata, S., Okada, N., Okamoto, K.-I., Shimzu, M. & Hasegawa, M. (1996) J. Mol. Evol. 42, 257-263. [DOI] [PubMed] [Google Scholar]
- 12.Hashimoto, T., Nakamura, Y., Nakamura, F., Shirakura, T., Adachi, J., Goto, N., Okamoto, K. & Hasegawa, M. (1994) Mol. Biol. Evol. 11, 65-71. [DOI] [PubMed] [Google Scholar]
- 13.Baldauf, S. L. & Doolittle, W. F. (1997) Proc. Natl. Acad. Sci. USA 94, 12007-12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwabe, N., Kuma, K.-I., Hasegawa, M., Osawa, S. & Miyata, T. (1989) Proc. Natl. Acad. Sci. USA 86, 9355-9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inagaki, Y., Doolittle, W. F., Baldauf, S. L. & Roger, A. J. (2002) Curr. Biol. 12, 772-776. [DOI] [PubMed] [Google Scholar]
- 16.Ronquist, F. & Huelsenbeck, J. P. (2003) Bioinformatics 19, 1572-1574. [DOI] [PubMed] [Google Scholar]
- 17.Guindon, S. & Gascuel, O. (2003) Syst. Biol. 52, 696-704. [DOI] [PubMed] [Google Scholar]
- 18.Felsenstein, J. (1993) proml (J. Felsenstein, Univ. of Washington, Seattle).
- 19.Shimodaira, H. (2002) Syst. Biol. 51, 492-508. [DOI] [PubMed] [Google Scholar]
- 20.Strimmer, K. & von Haeseler, A. (1996) Mol. Biol. Evol. 13, 964-969. [Google Scholar]
- 21.Shimodaira, H. & Hasegawa, M. (2001) Bioinformatics 17, 1246-1247. [DOI] [PubMed] [Google Scholar]
- 22.Susko, E., Inagaki, Y., Field, C., Holder, M. E. & Roger, A. J. (2002) Mol. Biol. Evol. 19, 1514-1523. [DOI] [PubMed] [Google Scholar]
- 23.Gu, X. (1999) Mol. Biol. Evol. 16, 1664-1674. [DOI] [PubMed] [Google Scholar]
- 24.Blouin, C., Boucher, Y. & Roger, A. J. (2003) Nucleic Acids Res. 31, 790-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keeling, P. J. (2004) Am. J. Bot., in press. [DOI] [PubMed]
- 26.Simpson, A. G. & Roger, A. J. (2002) Curr. Biol. 12, R691-R693. [DOI] [PubMed] [Google Scholar]
- 27.Philippe, H., Snell, E. A., Bapteste, E., Lopez, P., Holland, P. W. & Casane, D. (2004) Mol. Biol. Evol. 21, 1740-1752. [DOI] [PubMed] [Google Scholar]
- 28.Chapman, R. L., Buchheim, M. A., Delwiche, C. F., Friedl, T., Huss, V. A., Karol, K. G., Lewis, L. A., Manhart, J., McCourt, R. M., Olsen, J. L. & Waters, D. A. (1998) in Molecular Systematics of Plants II, eds. Soltis, D. E., Soltis, P. S. & Doyle, J. J. (Kluwer, Boston), pp. 508-540.
- 29.Andersson, J. O., Sjogren, A. M., Davis, L. A., Embley, T. M. & Roger, A. J. (2003) Curr. Biol. 13, 94-104. [DOI] [PubMed] [Google Scholar]
- 30.Archibald, J. M., Rogers, M. B., Toop, M., Ishida, K. & Keeling, P. J. (2003) Proc. Natl. Acad. Sci. USA 100, 7678-7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergthorsson, U., Adams, K. L., Thomason, B. & Palmer, J. D. (2003) Nature 424, 197-201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}