

Abstract
Accurate high-resolution refinement of protein structure models is a formidable challenge because of the delicate balance of forces in the native state, the difficulty in sampling the very large number of alternative tightly packed conformations, and the inaccuracies in current force fields. Indeed, energy-based refinement of comparative models generally leads to degradation rather than improvement in model quality, and, hence, most current comparative modeling procedures omit physically based refinement. However, despite their inaccuracies, current force fields do contain information that is orthogonal to the evolutionary information on which comparative models are based, and, hence, refinement might be able to improve comparative models if the space that is sampled is restricted sufficiently so that false attractors are avoided. Here, we use the principal components of the variation of backbone structures within a homologous family to define a small number of evolutionarily favored sampling directions and show that model quality can be improved by energy-based optimization along these directions.
With the progression of structural genomics initiatives (1–3), comparative modeling has become an increasingly important method for building protein structure models (4, 5). After a suitable structure template is chosen, accurate comparative modeling requires a correct alignment between the target protein sequence and the template sequence, an accurate method for modeling the loops (the insertions and deletions in an alignment) and side chains, and, finally, a method for refining the coordinates derived from the template structure toward those of the true native structure (6–8). In this study, we focus on this last model-refinement step. Improvement of the accuracy of comparative models is very important because accurate comparative models potentially can be used for many applications, such as virtual drug scanning (9), molecular replacement (10), and function prediction (11). Refinement is particularly important when the sequence identity between a target protein and the template protein is <30% (12), because models built by using current methods generally have rms deviations (rmsd) of >1.5 Å (13).
However, high-resolution refinement is as formidable as it is important. This difficulty is due to both the large size of conformational space and the delicate balance of forces in the native state. Indeed, in the recent CASP5 experiment (The 5th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction), most refined structures had larger rmsd to the native structure than the starting template backbone conformation (7). High-resolution refinement is thus a very stringent test of accuracy that perhaps no current force field satisfies.
Progress on this very important but very challenging problem may be facilitated by focusing on more constrained and thus more tractable refinement problems. We were led to thinking about such problems by the observation that a refinement protocol that did not markedly improve de novo structure-prediction models was very much more successful on the more constrained rigid-body protein–protein docking problem (14). The greatly reduced number of backbone degrees of freedom in the protein–protein docking problem significantly reduces the number of false attractors in the free-energy landscape: As illustrated in ref. 14, the docking free-energy landscapes typically are funneled strongly into the native minimum.
The conceptual step forward in this paper is to use evolutionary information to reduce the number of degrees of freedom in the monomeric protein-refinement problem to mimic the situation in the protein–protein docking problem. We accomplish this goal by restricting sampling to the subspace defined by the largest principal components (PCs) of the variation in the structural core of homologous proteins. This strategy greatly enhances the sampling of near-native backbone conformations, and the low-energy models identified by using the Rosetta high-resolution energy function (15, 16) usually have lower rmsd to the native backbones than the starting templates. This restricted refinement problem can provide a testing ground for evaluating and improving potential functions for the unrestricted comparative-modeling refinement problem. More practically, the refinement of structure cores by energy-based sampling along evolutionarily preferred directions can serve as the first step toward improving a model structure built from a template. After a more accurate structure core is obtained, the rest of the structure can be built by using loop modeling and side-chain repacking (6).
Methods
Data Set. A set of protein structure families was derived from the Structural Classification of Proteins (SCOP) ASTRAL95 domain structure database (17, 18). For each structure family, one protein was chosen randomly as the target to be modeled, and information from structures in the family with sequence identity from 10 to 30% and rmsd from 1.0 to 4.0 Å to the target protein was used to caluculate the PCs. Information from more closely related homologues was excluded to mimic the situation in a difficult comparative-modeling scenario. For computational efficiency, only proteins with <150 residues were included in the test set. The 77 structure families in the test set span five SCOP (17) class categories, which are all α proteins, all β proteins, α/β proteins, α + β proteins, and small proteins, to ensure the generality of the reported results. Details on the SCOP families and target proteins used in the tests are provided in Table 1, which is published as supporting information on the PNAS web site.
Protein Structure Data. Each protein with n backbone atoms (N, Cα, C, O, and Cβ) is represented by a vector of length 3 n, which is comprised of the n × (x,y,z) coordinates. Based on the alignment between a model structure and its homologous structures, coordinate displacement vectors (CDVs) are constructed by subtracting the coordinates of the model structure from those of each homologous structure. If residue i (with coordinate xi,yi,zi) in the model structure is aligned with residue j (with coordinate xj,yj,zj) in a homologous structure, the CDV entry for that position is defined by (xi - xj, yi - yj, zi - zj). When there is a gap in the alignment, no variation information can be obtained for that position, and the position is excluded from the displacement vector. Hence, a displacement vector has length 3 na, where na is the number of backbone atoms of the residues at sequence positions that are aligned for all family members. These sequence positions are considered the core region of the structure family.
PC Analysis. The CDVs for a protein family define a high-dimensional space that represents the structural variation within the family. We used PC analysis (19) to identify the major directions of variation in this space. The PCs are orthogonal linear combinations of the CDVs; the first PC accounts for the largest amount of structural variation, and the following PCs account for decreasing amounts of structural variation. To obtain the PCs, we applied singular-value decomposition (20, 21) on the CDVs derived from structurally aligned homologous backbone conformations. Specifically, representing each CDV as a column of the data matrix X, the singular value decomposition of X gives X = ULVT. The columns of matrix U constitute an orthogonal basis for the displacement vector space, and the diagonal matrix L gives the magnitude associated with each basis vector. The PCs of the CDVs then are constructed by multiplying U × L. Note that the PCs are linear combinations of the original CDVs: Because matrix V is orthogonal, UL = XV.
Structure Alignment. To generate the CDVs between protein backbone structures, an alignment of the proteins is needed. mammoth-mult (D. Lupyan, A. Leo-Macias, and A.R.O., unpublished program), a multiple-alignment version of the structural alignment program mammoth (22), is used to align multiple protein backbone structures.
rmsd Calculation. The rmsd of two protein structure backbones is defined as
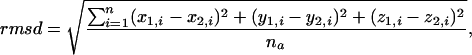 |
[1] |
where na is the number of aligned backbone atoms, and (x1,i, y1,i, z1,i) and (x2,i, y2,i, z2,i) are the ith atom coordinates of proteins 1 and 2, respectively. The rmsd is computed as described in ref. 23.
Sequence Alignment. psi-blast (24) was used to align the sequence to be modeled with a multiple-sequence alignment generated from a multiple-structure alignment of the protein family.
Energy Evaluation. For each backbone conformation, we carried out a combinatorial Monte Carlo optimization (25) of the side-chain conformations in Roland Dunbrack's backbone-dependent rotamer library (26) by using the Rosetta full-atom energy function (the “repacking” process). The energy function is dominated by a Lennard–Jones potential, an implicit solvation model, and an orientation-dependent hydrogen-bonding term; the potential function is specified in detail in the supplementary material to ref. 27. The side-chain torsion angles then are further optimized by continuous-energy minimization by using the Dav idson–Fletcher–Powell (DFP) quasi-Newton method (20).
Simplex and Powell Optimization. The implementations of the Simplex and Powell local optimizations given in Numerical Recipes (20) were used to optimize the Rosetta energy function in the space defined by the PCs of variation in the structural family. The starting structure was selected as described in Test I, II, and III in Results. The variables subjected to the optimization are the amplitudes of the displacement along each of the PCs, and the objective function is the Rosetta energy function. For Simplex optimization, the starting n + 1 points are the starting structure plus n structures generated by shifting the starting structure along one of the PCs by 0.2 times the amplitude of the component. For Powell optimization, the starting point is the starting structure, and the starting directions are the n PC vectors.
Grid Search. The space defined by the three largest PCs of variation in the structural family was sampled by using a grid-searching procedure. Eleven evenly separated points were selected along each of the three directions, which results in 113 = 1331 backbone conformations. The energy of each conformation was determined by side-chain repacking followed by minimization as described above. A second, finer grid search in the neighborhood of the minimum identified in the initial search can be carried out to increase the sampling resolution. To determine the range of values over which sampling is performed, we carried out initial tests on five proteins in which structures were perturbed by up to half the principal component amplitude, and we identified ranges for each of the tests in which the energy minima fell (perturbations of structures by more than half the PC analysis amplitudes can produce large distortions of bond lengths and angles). For test I, a sampling interval of -1/10 to +1/10 of the PC amplitude was found to be sufficient to bracket the global minimum, whereas for test II and III, where the starting structure is further from the native structure, a larger sampling interval from -4/10 to +4/10 of the PC amplitude generally bracketed the minimum.
Eliminating Distortion of Bond Angles and Lengths. The sampled decoys were subjected to a fast minimization (100 standard steps) in the charmm force field (28). This step was solely for eliminating distortions in bond lengths and angles accompanying the displacement of the coordinates when moving along the PCs, while at the same time maintaining the backbone structure almost unchanged. After this minimization, the bond lengths and angles had distributions similar to those in experimentally determined structures.
Refinement Protocol. Starting from a comparative model, the CDVs of all structures in the homologous family relative to the model were extracted, and PC analysis was performed. A grid search then was carried out along the first three PCs. For each conformation, the residue side chains were optimized by repacking, followed by continuous minimization as described above, and the energy was evaluated. The decoy with the lowest energy was selected, and the distortions in backbone bond lengths and angles were eliminated.
Results
Most Structural Variation in a Protein Family Can Be Described by Several PCs. Refinement of protein backbone structure models requires an efficient way to sample the backbone conformation space near the native structure. Here, we explore the use of the space defined by the PCs of the structural variation in naturally occurring protein families for high-resolution refinement.
Fig. 1 shows an example of the variation in the structural core of the insect phospholipase A2 family [a.133.1.1 (SCOP database)] and the variation described by the first three PCs. The amount of the backbone conformational variation described by increasing numbers of PCs is shown in Fig. 2. Although the fraction of the total variation described by a certain number of PCs differs from one structural family to another, most of the variation can be described by the first several PCs. Remarkably, the first PC generally can describe 40–90% of the backbone conformational variation in a structural family, and the first three PCs combined can describe 65–99% of the conformational variation. With the number of family members increasing, there is generally larger variation in a structure family; thus, lesser variation can be explained by the first several PCs. But even in a case where there are 36 structure members, structure family b.1.1.1 (SCOP database), the first three PCs still describe 75% of the variation. This finding suggests that by searching the subspace defined by the first several PCs, we can efficiently sample the majority of backbone conformational variation contained in the homologous structures.
Fig. 1.

Variation in structure family and variation represented by PCs. (a) Superposition of the cores of all eight structures in structure family a.133.1.1. (b–d) Backbone conformations sampled along the direction defined by the first PC (b), the second PC (c), and the third PC (d).
Fig. 2.

Fraction of structural variation explained by PCs. Each line represents a structure family. With additional PCs, more and more variation can be described. The dashed line indicates that the first three PCs can describe at least 65% of the variation observed in each structure family.
The PCs of Conformational Displacement Vectors Define Efficient Searching Directions. The PCs are essentially linear combinations of the variation in different proteins; thus, they contain information on the concerted conformational changes of all backbone residues. Moving protein backbone residues along the directions defined by PCs of backbone conformational variation should be able to search effectively the near-native conformation space. To test this hypothesis, a single structure randomly selected to be the “starting model” was excluded from the multiple structural alignment, and PCs were computed from the remaining family members. The aligned residues in the starting model then were moved in the directions defined by PCs or random directions, and the conformation with the lowest rmsd to the native conformation was recorded. As shown in Fig. 5 (which is published as supporting information on the PNAS web site), with more and more conformation space accessible with an increasing number of components, the sampled model backbone with the lowest rmsd is progressively closer to the native conformation. Sampling along the directions defined by PCs generates models much closer to the native backbone conformation, indicating that using the PC directions is an efficient way to explore near-native backbone conformation space, than does sampling along random directions.
Combining Energy Evaluation and Sampling Along PCs to Improve Model Accuracy. We investigated the extent to which energy calculations can be used to find close-to-native physically plausible models in the set of conformations generated by sampling along the PCs. The Rosetta potential energy function, which is dominated by a 12–6 Lennard–Jones potential, an orientation-dependent hydrogen-bonding term (29), and an implicit solvation model (30), was used to evaluate models sampled along the directions defined by PCs. Three tests were carried out as described below. In all tests, the model selected as the template was excluded from the computation of the PCs.
Three Tests Reflect Three Possible Scenarios in Comparative Modeling. Test I. When the closest structural neighbor to the native structure among all homologous structures is known, it should be used as a backbone template to model the target protein. In reality, the closest structure cannot always be unambiguously chosen a priori, but this test represents a challenge for high-resolution refinement using PCs of natural structure variation, because no better individual backbone structure is present in the homologous set that is used to define the sampling space. To simulate this case, all homologous backbone structures are superimposed on the target native backbone, and the one with the lowest rmsd is used as a template to model the target protein.
Test II. In reality, the structure template used to model a target protein has to be selected according to certain criteria, such as sequence similarity and/or energy evaluation. A comparison between using sequence similarity [both sequence identity- and BLOSUM62 (31) matrix-based similarity score] and the Rosetta potential energy as template-selection criteria revealed that the Rosetta energy function results in a better near-native template on average. Specifically, the templates selected by the lowest Rosetta energy criterion give an average rmsd of 2.00 Å compared with the native structures, whereas the templates selected by the highest sequence ID or the highest BLOSUM62 similarity score criterion give an average rmsd of 2.17 and 2.28 Å, respectively. Therefore, for each test family, the target protein sequence was threaded onto each homologous structure, and the model structure with the lowest Rosetta full-atom energy score was selected as the template.
Test III. The above two tests both use structural alignment between homologous structures and the target native structure to define the mapping of variation in homologous structures to the model structure. In reality, the mapping between the target sequence and homologous sequences has to be determined by an alignment algorithm. This alignment most certainly will introduce errors into the modeling process. To assess the effect of alignment errors, in test III a psi-blast sequence-profile alignment was used for each test family, and the structure template was selected according to the protocol described in test II. This test is not intended to show that the method can recover from bad alignments; rather, it is a way to assess the practical usefulness of the method when alignment errors are present.
Sampling Strategy. As illustrated in Fig. 5, moving along the direction defined by the first PC usually brings about the largest conformational improvement in the backbone, and it is not obvious how many of the remaining PCs should be sampled. Increasing the number of directions will increase the chance of finding native-like conformations, but it also will increase the size of the space that must be sampled and, hence, will make location of the global minimum a much more challenging task. In addition, the increased size of the space will bring in more false attractors in the energy landscape.
We initially sought to refine structures in the space defined by all of the PCs by using both the Simplex and Powell methods (20). As illustrated in Fig. 6, which is published as supporting information on the PNAS web site, optimization of the energy by using the Simplex method often considerably reduced both the energy and the rmsd of the input structures. The more continuous Powell method was less successful, and further examination of the energy landscape showed that it was quite rugged (Fig. 7, which is published as supporting information on the PNAS web site), which is not surprising because for every backbone defined by a particular perturbation along the PCs, a complete side-chain rotamer-packing calculation is carried out, followed by side-chain minimization (see Methods).
Backbone Conformations in the Space Spanned by the Three Largest PCs Are Often Closer to the Native Backbone Conformation than the Starting Template. Because optimization methods such as the Simplex and Powell method are readily trapped in local minima, we experimented with grid sampling, which is much less sensitive to local minima. To make grid sampling feasible, we restricted the search to the space defined by the three largest PCs, which has the further advantage of being enriched in low-rmsd structures, as shown below.
In most of the homologous families, a significant fraction of the sampled backbone conformations have lower rmsd to the native structure than the rmsd between the starting model and the native structure. In comparison, random perturbations of a model backbone structure most often increase the rmsd to the native backbone structure. Fig. 8, which is published as supporting information on the PNAS web site, shows the percentage of decoys with rmsd lower than the starting model for each homologous family in test I, II, and III. For example, in ≈27% of the test sets in test II, >30% of decoys have lower rmsd to native than the starting model; in half of the test sets, >20% of decoys have lower rmsd than the starting model.
The high percentage of sampled decoys with rmsd lower than the starting model can be understood by considering the way that the conformation space is sampled. The directions along which the starting model is perturbed are defined by variation in its structural homologs. If the structural variation contained in the homologous structures is a reasonable representation of the variation in the target native conformation (which of course does not contribute to the PC calculation), then moving a model backbone along the directions defined by the variation can either decrease the rmsd between the model and the native structure when the model backbone is moved “toward” the native backbone or increase the rmsd when the model backbone is moved “away from” the native backbone. When sampled along the directions defined by the first three PCs, both positive and negative sides are explored equally, so that ≈12.5% (1/2 × 1/2 × 1/2) of decoys should have lower rmsd than the starting structure. The instances in Fig. 8 where >12.5% of decoys have lower rmsd than the native structure may be those in which the first PC largely dominates sampling; in the limit in which only this direction is sampled, the frequency would be expected to approach 50% (indeed, there is no percentage >50% in Fig. 8).
Low-rmsd Physically Viable Structures Can Be Selected Based on the Energy. After repacking and minimizing (see Methods) side-chain conformations on the decoy backbone conformations, the Rosetta full-atom energy is evaluated, and the lowest-energy conformation is identified. Fig. 3 shows the difference in rmsd to the native structure between this lowest-energy conformation and the starting model. In all three tests, most of the lowest-energy selected conformations have significantly lower rmsd to the native backbones than do the starting models. Greater improvement throughout the test sets is achieved when the final model is selected from the five lowest-energy decoys from each set (Fig. 9, which is published as supporting information on the PNAS web site). In many, but not all, cases there was a quite strong correlation between rmsd and energy (for example, see Fig. 10, which is published as supporting information on the PNAS web site).
Fig. 3.

Improvement of protein backbone core region by sampling along the directions defined by the first three PCs and selecting low-energy decoys by using the Rosetta energy function. rmsd improvement of the lowest-energy decoys in tests I (a), II (b), and III (c) is shown. The improvement of rmsd is measured by (rmsd of starting model - rmsd of refined model). Positive values indicate improvement of backbone conformations.
The results from test II demonstrate the greatest improvement over all three tests. This result is not surprising, because test II has correct alignments, and the suboptimal starting templates allow larger space for improvement. In the hard case of test I, the starting model is already very close to native, so the improvement is expected to be small. In test III, the improvements are also small, indicating that the method is sensitive to alignment errors. Even so, for most families tested, the improvements are obvious. Note that six families are not tested in test III, because psi-blast could not align these target sequences with their homologous sequence profiles. These six families are still listed in Fig. 3c for comparison purposes and account for six zero-improvement data points. The extent of improvement of the models was similar for the cases in which the similarity between the query sequence and the starting template was between 10% and 20% and the cases in which the similarity was between 20% and 30%.
Fig. 4 shows an example of the improvement of backbone structures after refinement. The structural cores of histidyltRNA synthetase C-terminal domain (domain d1kmma1 from family C.51.1.1) before and after refinement are shown, along with the native conformation. Comparing the refined conformation and the starting model with the native conformation shows the improvement throughout the backbone structure.
Fig. 4.

Example of successful model refinement. Red, model structure; blue, native structure; green, refined structure. The rmsd between the model structure and native structure is 2.36 Å, and the rmsd between the refined structure and native structure is 1.42 Å.
We experimented with a further round of grid-based optimization along the fourth through sixth PCs starting from the minimum found in a previous search along the first three PCs. As shown in Fig. 10, neither the energy nor the rmsd changed significantly in this second optimization step. We also experimented with a second grid search along the first three PCs, starting with the minimum defined in the first grid search after regularization by using charmm (see Methods). We carried out this experiment because it seemed possible that distortions in bond lengths and angles caused by the perturbations along the PCs might limit the extent of perturbation along any one direction and, hence, that further optimization might be possible after regularizing the bond lengths and angles. However, as shown in Fig. 11 (which is published as supporting information on the PNAS web site), the results after this second optimization step again were not significantly different from those after the first step.
To determine whether the decreases in backbone rmsd were associated with improvements in side-chain packing, we compared the accuracy of χ1 recovery in the starting models and the lowest-energy refined decoys. As shown in Fig. 12, which is published as supporting information on the PNAS web site, there was some improvement in χ1 recovery for test I, whereas there was less improvement for tests II and III.
Discussion
By combining energy-based refinement with sampling along evolutionarily observed directions, we partially can overcome the two principal obstacles to energy-based refinement of comparative models: the very large and rugged nature of the landscape being sampled, and the inaccuracy of current force fields. Sampling is greatly facilitated because the PCs represent feasible concerted movements of the chain and, furthermore, represent directions sampled evolutionarily. Problems associated with inaccuracies in the energy function are reduced because the great reduction in the size of the space being sampled eliminates most false attractors.
The improvements of model conformation in test III (with psi-blast alignment) are generally smaller than those in test II (with structural alignment), indicating that the utility of the directions defined by PCs is sensitive to alignment errors. The energy decrease from the starting models to the lowest-energy decoys obtained in the grid search is significantly less with incorrect alignments, and it may be possible to select better alignments by using this criterion. Thus, it may be possible to extend iterative alignment and model evaluation methods (32) to include high-resolution refinement.
Other fields where backbone flexibility must be modeled may profit from the application of the method described here. In flexible backbone protein–protein docking, plausible alternative conformations of the partners may be generated by sampling along the PCs. For generation of amino acid sequence profiles to represent protein families for remote homology detection, backbone sampling along these directions is likely to create more useful and relevant ensembles than random sampling (33, 34). More generally, combining with the evolutionary information contained in families of homologous proteins can increase the limited current utility of physically based refinement methods and provide a stepping stone toward the long-range goal of improving model accuracy by using physically based methods alone.
Acknowledgments
We thank members of the Baker laboratory for helpful discussions, particularly Tanja Kortemme and Dylan Chivian for reading and making valuable comments on the manuscript; Dylan Chivian for helping with the preparation of test data sets and use of the Rosetta comparative modeling method; Philips Bradley for giving stimulating insights into the nature of conformational space search; Chu Wang for helping with side-chain rotamer packing using Rosetta; and John Karanicolas for helping to set up the charmm package. The Rosetta energy function is developed by many scientists in the Baker laboratory. This work is supported by the Howard Hughes Medical Institute and Ministerio de Ciencia y Tecnología Grant BIO2001-3745 (to A.R.O.). Research at Centro Biología Molecular Severo Ochoa is facilitated by an institutional grant from the Ramón Areces Foundation.
Author contributions: B.Q. and A.R.O. designed research; B.Q. performed research; A.R.O. contributed new reagents/analytical tools; B.Q. analyzed data; and B.Q. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: rmsd, rms deviation; CDV, coordinate displacement vectors; PC, principal component.
References
- 1.Sanchez, R., Pieper, U., Melo, F., Eswar, N., Marti-Renom, M. A., Madhusudhan, M. S., Mirkovic, N. & Sali, A. (2000) Nat. Struct. Biol. 7, Suppl., 986-990. [DOI] [PubMed] [Google Scholar]
- 2.Baker, D. & Sali, A. (2001) Science 294, 93-96. [DOI] [PubMed] [Google Scholar]
- 3.Stevens, R. C., Yokoyama, S. & Wilson, I. A. (2001) Science 294, 89-92. [DOI] [PubMed] [Google Scholar]
- 4.Fu, Z., Aronoff-Spencer, E., Backer, J. M. & Gerfen, G. J. (2003) Proc. Natl. Acad. Sci. USA 100, 3275-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa, H. & Toyoshima, C. (2002) Proc. Natl. Acad. Sci. USA 99, 15977-15982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chivian, D., Kim, D. E., Malmstrom, L., Bradley, P., Robertson, T., Murphy, P., Strauss, C. E., Bonneau, R., Rohl, C. A. & Baker, D. (2003) Proteins 53, Suppl. 6, 524-533. [DOI] [PubMed] [Google Scholar]
- 7.Tramontano, A. & Morea, V. (2003) Proteins 53, Suppl. 6, 352-368. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez, R. & Sali, A. (2000) Methods Mol. Biol. 143, 97-129. [DOI] [PubMed] [Google Scholar]
- 9.Lengauer, T., Lemmen, C., Rarey, M. & Zimmermann, M. (2004) Drug. Discov. Today 9, 27-34. [DOI] [PubMed] [Google Scholar]
- 10.Read, R. J. (2001) Acta Crystallogr. D 57, 1373-1382. [DOI] [PubMed] [Google Scholar]
- 11.Skolnick, J., Fetrow, J. S. & Kolinski, A. (2000) Nat. Biotechnol. 18, 283-287. [DOI] [PubMed] [Google Scholar]
- 12.Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F. & Sali, A. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 291-325. [DOI] [PubMed] [Google Scholar]
- 13.Cozzetto, D. & Tramontano, A. (2004) Proteins, in press.
- 14.Gray, J. J., Moughon, S., Wang, C., Schueler-Furman, O., Kuhlman, B., Rohl, C. A. & Baker, D. (2003) J. Mol. Biol. 331, 281-299. [DOI] [PubMed] [Google Scholar]
- 15.Bradley, P., Chivian, D., Meiler, J., Misura, K. M., Rohl, C. A., Schief, W. R., Wedemeyer, W. J., Schueler-Furman, O., Murphy, P., Schonbrun, J., et al. (2003) Proteins 53, Suppl. 6, 457-468. [DOI] [PubMed] [Google Scholar]
- 16.Bonneau, R., Strauss, C. E., Rohl, C. A., Chivian, D., Bradley, P., Malmstrom, L., Robertson, T. & Baker, D. (2002) J. Mol. Biol. 322, 65-78. [DOI] [PubMed] [Google Scholar]
- 17.Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995) J. Mol. Biol. 247, 536-540. [DOI] [PubMed] [Google Scholar]
- 18.Chandonia, J. M., Hon, G., Walker, N. S., Lo Conte, L., Koehl, P., Levitt, M. & Brenner, S. E. (2004) Nucleic Acids Res. 32, D189-D192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez, R. C. & Woods, R. E. (1992) Digital Image Processing (Addison–Wesley, Reading, MA).
- 20.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. (1992) Numerical Recipes in C (Cambridge Univ. Press, Cambridge, U.K.).
- 21.Teodoro, M. L., Phillips, G. N., Jr., & Kavraki, L. E. (2003) J. Comput. Biol. 10, 617-634. [DOI] [PubMed] [Google Scholar]
- 22.Ortiz, A. R., Strauss, C. E. & Olmea, O. (2002) Protein Sci. 11, 2606-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLachlan, A. D. (1979) J. Mol. Biol. 128, 49-79. [DOI] [PubMed] [Google Scholar]
- 24.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhlman, B. & Baker, D. (2000) Proc. Natl. Acad. Sci. USA 97, 10383-10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunbrack, R. L., Jr., & Cohen, F. E. (1997) Protein Sci. 6, 1661-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhlman, B., Dantas, G., Ireton, G. C., Varani, G., Stoddard, B. L. & Baker, D. (2003) Science 302, 1364-1368. [DOI] [PubMed] [Google Scholar]
- 28.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comp. Chem. 4, 187-217. [Google Scholar]
- 29.Kortemme, T., Morozov, A. V. & Baker, D. (2003) J. Mol. Biol. 326, 1239-1259. [DOI] [PubMed] [Google Scholar]
- 30.Lazaridis, T. & Karplus, M. (1999) Proteins 35, 133-152. [DOI] [PubMed] [Google Scholar]
- 31.Henikoff, J. G. & Henikoff, S. (1996) Methods Enzymol. 266, 88-105. [DOI] [PubMed] [Google Scholar]
- 32.John, B. & Sali, A. (2003) Nucleic Acids Res. 31, 3982-3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larson, S. M., England, J. L., Desjarlais, J. R. & Pande, V. S. (2002) Protein Sci. 11, 2804-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larson, S. M., Garg, A., Desjarlais, J. R. & Pande, V. S. (2003) Proteins 51, 390-396. [DOI] [PubMed] [Google Scholar]