

ABSTRACT
WD40 repeat proteins fold into characteristic β-propeller structures and control signaling circuits during cellular adaptation processes within eukaryotes. The RACK1 protein of Saccharomyces cerevisiae, Asc1p, consists exclusively of a single seven-bladed β-propeller that operates from the ribosomal base at the head region of the 40S subunit. Here we show that the R38D K40E ribosomal binding-compromised variant (Asc1DEp) is severely destabilized through mutation of phosphosite T143 to a dephosphorylation-mimicking alanine, probably through proteasomal degradation, leading to asc1− phenotypes. Phosphosite Y250 contributes to resistance to translational inhibitors but does not influence Asc1DEp stability. Beyond its own phosphorylation at T143, Y250, and other sites, Asc1p heavily influences the phosphorylation of as many as 90 proteins at 120 sites. Many of these proteins are regulators of fundamental processes ranging from mRNA translation to protein transport and turnover, cytoskeleton organization, and cellular signaling. Our data expose Asc1p/RACK1 as a key factor in phosphosignaling and manifest it as a control point at the head of the ribosomal 40S subunit itself regulated through posttranslational modification.
KEY WORDS: Asc1/RACK1, Saccharomyces cerevisiae, protein phosphorylation, quantitative proteomics, ribosome, signal transduction network
INTRODUCTION
The Saccharomyces cerevisiae protein Asc1 consists of a single WD40 repeat domain that folds into a characteristic seven-bladed β-propeller (1) serving as a scaffold for protein-protein interactions. Asc1p is involved in cellular signal transduction pathways and is associated with the process of protein biosynthesis through its localization to the head of the 40S subunit of the ribosome. The protein was proposed to function as the Gβ subunit acting together with the Gα GTPase, Gpa2p, within the cyclic AMP/protein kinase A (cAMP/PKA) signal transduction pathway and to physically interact with components of mitogen-activated protein kinase (MAPK) cascades (2, 3). Asc1p is highly conserved in eukaryotes, and its mammalian orthologue RACK1 (receptor for activated protein C kinase 1) has also been described to bind signaling factors, such as kinases and phosphatases, thereby regulating their activity and target specificity (reviewed in reference 4). The RACK1-dependent docking of activated protein kinase C βII (PKCβII) and c-Jun N-terminal kinase (JNK) to the ribosome provides a direct link between signal transduction and mRNA translation (5–7). At the ribosome, PKCβII phosphorylates, for instance, the translation initiation factor 6, thereby supporting ribosomal subunit joining and initiation of translation (5).
Yeast Asc1p has been proposed to be a repressor of translation due to increased translational activity of Asc1p-deficient ribosomes in vitro and in vivo (8–10). Asc1p supports the translation of mRNAs with short open reading frames (11). It further promotes nascent polypeptide-dependent translation arrest, triggered by a sequence of basic amino acids or tandem repeats of rare codons (12, 13), and is required to maintain the reading frame during ribosome stalling caused by inefficiently decoded CGA codon repeats (14). Asc1p/RACK1 affects translation, depending on the 5′ untranslated regions of mRNAs (15) and through its interaction with mRNA-binding proteins, such as Scp160p in S. cerevisiae (16) and ZBP1 in mammalian neuronal cells (17). Mammalian RACK1 also regulates protein expression at a posttranslational level by affecting the degradation of certain proteins, e.g., the α-subunit of the transcription factor hypoxia-inducible factor 1 (HIF1α) (18). The absence of RACK1 leads to lethality at early stages of embryogenesis in animals and plants, whereas Asc1p-deficient yeast cells proliferate well in culture. Yet they also show loss of the dimorphic switch, as evidenced by their inability to grow adhesively on surfaces as haploids or to form filament-like pseudohyphae as diploids (19).
Asc1p/RACK1's ability to contact multiple proteins demands mechanisms that regulate these interactions as needed. Association of mammalian RACK1 with other proteins can be regulated through its phosphorylation (20, 21). Phosphorylation of RACK1 was additionally implicated in the regulation of its stability in Arabidopsis thaliana (22). For S. cerevisiae, Asc1p phosphosites are known from high-throughput phosphoproteome studies (23, 24). We identified additional phosphosites of affinity-captured Strep-tagged Asc1p by using a liquid chromatography-mass spectrometry (LC-MS) approach. Previously known and newly identified phosphosites were systematically replaced by phosphorylation- and dephosphorylation-mimicking amino acids (E and A, respectively), and the resulting mutant strains were phenotypically characterized, revealing the importance of phosphosites T143 and Y250. Phosphorylation/dephosphorylation mimics of Asc1p variants additionally compromised in ribosome binding through an R38D K40E exchange (1) highlighted the significance of phosphosites T143 and Y250 as well as T12, T96, and T99. The role of Asc1p in signal transduction further suggests that it affects the phosphorylation of other proteins. Thus, we quantitatively analyzed the Asc1p-dependent phosphoproteome and identified an unexpectedly large number of regulated proteins (90 proteins). This positions the ribosomal β-propeller at the center line of cellular signaling.
RESULTS
The Asc1 protein becomes phosphorylated at several sites.
Asc1p/RACK1 interacts with protein kinases in both yeast and higher eukaryotes. Phosphorylation of Asc1p has been reported in high-throughput phosphoproteome studies (23, 24) and might provide a mechanism for regulating protein interactions, stability, or subcellular localization. For an in-depth characterization of Asc1p's phosphorylation, we expressed Strep-tagged Asc1p in S. cerevisiae cells. We initially assessed the functionality of the tagged protein by complementation of an Asc1p-depleted strain (Fig. 1A) and subsequently enriched Strep-tagged Asc1p from cell cultures for the further protein analysis by LC-MS. The raw MS data search resulted in the identification of seven singly phosphorylated peptides, two of them with an overlapping sequence due to a cleavage site at K161 that was missed by trypsin (Fig. 1B). Since all identified phosphopeptides contained more than one serine, threonine, and/or tyrosine residue, the phosphoRS algorithm was used to calculate phosphosite probabilities (25, 26). Six amino acid residues of Asc1p, i.e., T12, S120, T143, S166, T168, and Y250, were identified as unambiguous phosphosites, with a site probability of 100%. These data confirmed the already known phosphosites S120, S166, and T168 (23, 24) and led to the identification of three formerly unknown sites, T12, T143, and Y250. Furthermore, we identified a phosphopeptide with a 99.9% highest localization probability for residue T300. For more details on peptide identification and phosphosite localization, see Table S3 and Fig. S1 in the supplemental material. In this study, we focused on the characterization of T12, S120, T143, S166, T168, and Y250 plus T96 and T99. The latter two sites were not detected in our experiments. The positions of these eight phosphosites within the protein bound to the 40S subunit of the ribosome are illustrated in Fig. 1C. T12, T99, and T143 are localized in the βD strands of blades VII, II, and III and thus are at the circumference of the protein. T96, S120, S166, T168, and Y250 are localized at sides of Asc1p that do not directly face the ribosome. T96 lies next to T99, neighboring the ribosomal protein Rps17. S166 is positioned within the loop between the βA and βB strands of blade IV, adjacent to T168, which is the N-terminal amino acid of strand IVβB. None of the phosphosites is localized at the top site of Asc1p that directly contacts the ribosome, revealing that the sites are accessible for kinases when the protein is bound to the ribosome.
FIG 1.

LC-MS-based identification of Asc1p phosphosites. (A) Complementation of asc1− phenotypes with plasmid-borne C- and N-terminally Strep-tagged Asc1p. The Δasc1 strain was transformed with high-copy-number plasmids bearing the ASC1 wild-type gene (pME2624) or the ASC1-Strep (pME2834) or Strep-ASC1 (pME4132) allele under the control of the MET25 promoter. The ASC1 wild-type strain and the Δasc1 strain, both carrying the empty vector (EV; pME2787), served as controls. Cells were tested for their resistance to cell wall stress (Congo red), osmotic stress (NaCl), and inhibition of translation elongation (cycloheximide). The cells were additionally tested for growth at 25°C to assess temperature sensitivity. Haploid adhesive growth was tested on 3-AT. Cell patches are shown before and after washing. Respiratory activity was monitored for single yeast colonies grown on 0.4% glucose by use of the TTC assay. Apart from the reduced growth on cycloheximide, the Strep-tagged Asc1p variants complemented all asc1− phenotypes. (B) Asc1p amino acid sequence with identified phosphopeptides and phosphosites. The amino acid sequence coverage of Asc1p, considering all identified high-confidence peptides, was 97.81% (false discovery rate, ≤0.01). Phosphosites S166 and T168 were identified within a phosphopeptide ranging from residues A162 to K176 and within a second peptide from residues V156 to K176, bearing a missed cleavage site at the C terminus of K161, indicated with a plus sign. Residues R38 and K40, which lead to compromised ribosome association of Asc1p when mutated to D and E, respectively, are shown in red and marked with dots (1, 66). WD40 repeats are indicated as found in the Saccharomyces Genome Database (SGD; SMART domain SM0032). See Table S3 in the supplemental material for details on the identification of phosphopeptides and Fig. S1 for fragmentation spectra. (C) Cartoon view of Asc1p bound to the 40S subunit of the ribosome. The Asc1p β-propeller is depicted in white (loops) and gray (β-strands). Positions of phosphosites that were further analyzed in this study are highlighted in yellow, and the amino acids are depicted as sticks, with carbon atoms shown in yellow, nitrogen atoms in blue, and oxygen atoms in red. For better recognizability, neighboring amino acids within the LC-MS-identified peptides are shown in green. Additionally, residues R38 and K40 are highlighted. For the ribosomal protein Rps3, the C-terminal last 14 amino acids were not structurally resolved. Structure figures were generated with the PyMol Molecular Graphics System software on the basis of the crystal structure of the eukaryotic ribosome (Protein Data Bank [PDB] entry 4V88) (67).
Asc1p phosphosites T143 and Y250 confer resistance to translational inhibitors, whereas T12, T96, T99, and T143 contribute to the stabilization of an R38D K40E amino acid exchange variant of Asc1p.
To analyze how Asc1p is regulated through its phosphorylation, we generated S. cerevisiae Asc1p phosphosite mutant strains. These strains express Asc1p with either glutamate or alanine at one of the phosphosites to mimic the constitutively phosphorylated or dephosphorylated state, respectively. Y250 was mutated to phenylalanine instead of alanine due to the higher similarity between these two amino acids. For phosphosites lying close to each other within the amino acid sequence (T96 and T99 as well as S166 and T168), we additionally generated strains with both sites mutated simultaneously. Furthermore, we constructed strains carrying each phosphosite substitution in combination with two amino acid changes known to unsteady Asc1p's binding to the ribosome: R38D and K40E. An S. cerevisiae asc1 R38D K40E strain (asc1DE) was first described by Coyle and colleagues (1), who showed that the ribosome-repulsive force of amino acids D38 and E40 within this variant results in a shift of the majority of the protein from the ribosomal to the ribosome-free fraction during sucrose gradient centrifugation. Despite the ribosome repulsion of the DE variant, it was recently shown by cross-linking experiments that Asc1DEp still associates with the 40S ribosome in vivo (11). We generated the asc1DE strain as well and further combined the phosphosite exchanges with the DE exchange to investigate whether any of the phosphosites interact synthetically with the DE exchange within Asc1DEp.
All mutated ASC1 alleles were integrated into the yeast genome at the authentic ASC1 locus so that the resulting strains were isogenic to the ASC1 wild-type strain except for the desired codon exchanges. The strains are listed in Table 1, and more detailed information, including complete genotypes and strain designations, is available in the supplemental material. Strains were tested for Asc1p-dependent phenotypes related to respiratory activity, cell wall maintenance, and sensitivity to translation inhibitors. In line with the results of Coyle et al. (1), the asc1DE strain behaved mainly like the ASC1 wild-type strain except for an increased sensitivity to cycloheximide (described below). Furthermore, the protein abundances of the transcription factors Flo8p, Tec1p, and Rap1p, which are heavily affected by the absence of Asc1p (15), remained unchanged in the DE background (Fig. 2A).
TABLE 1.
Overview of Asc1p phosphosite mutant strains and their phenotypesa
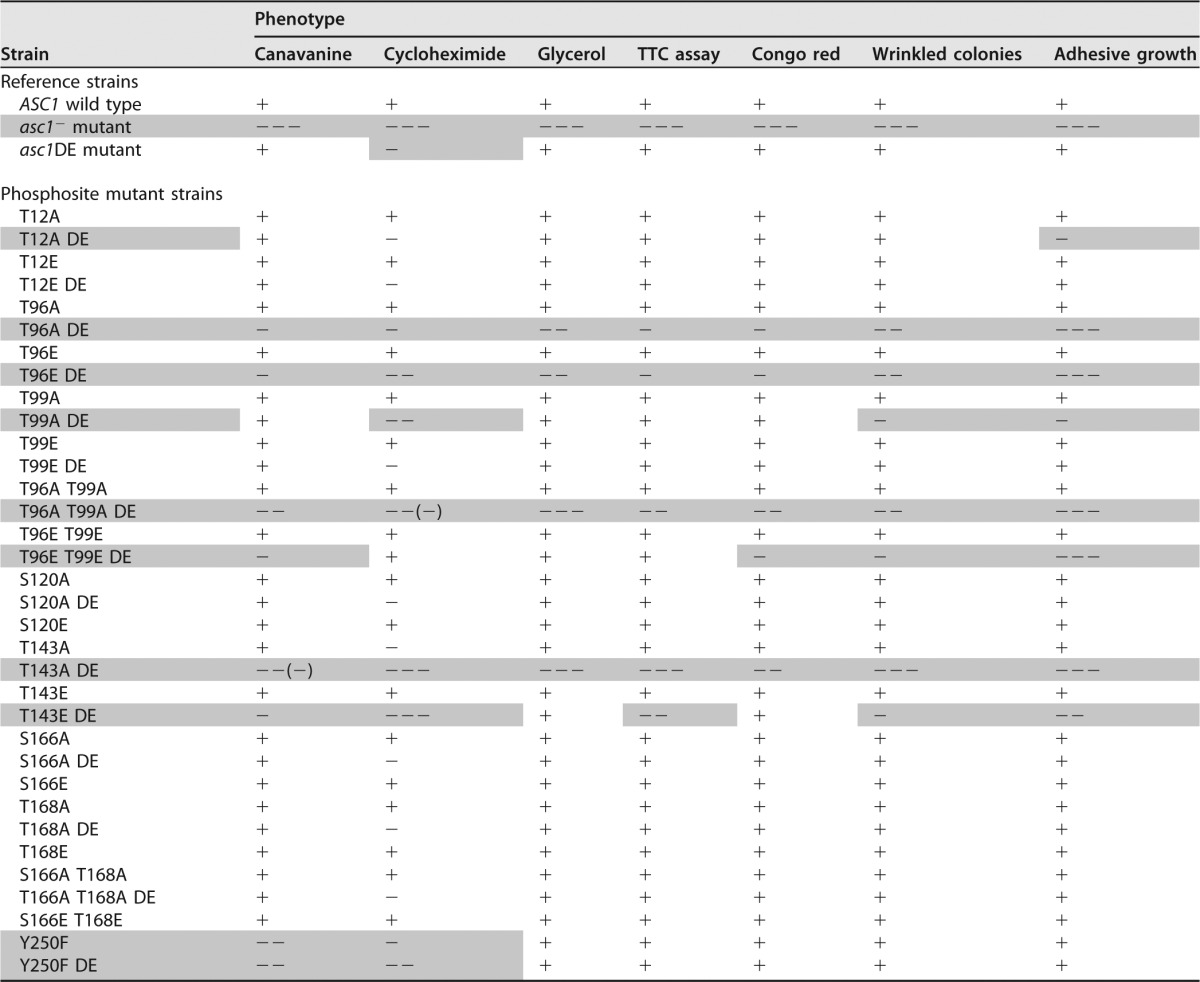
As references, the phenotypes of the ASC1 wild-type strain were defined as “+,” and the phenotypes of the Asc1p-deficient asc1− strain were defined as “−−−.” The phenotypes of the Asc1p phosphosite mutant strains were classified accordingly. Shading highlights strains and their phenotypes that differed significantly from the ASC1 wild-type strain.
FIG 2.


Impacts of the DE and phosphosite mutations on Asc1p-dependent phenotypes. (A) Immunodetection of Flo8p-myc3, Tec1p, and Rap1p within cell extracts derived from ASC1 wild-type, asc1−, and asc1DE cultures. Ponceau red staining of proteins served as a loading control, and part of the lower half of the stained Western blot membrane is depicted. (B) Asc1p dependency of the high-osmolarity response pathway. (B1) Hog1p phosphorylation in ASC1 wild-type, asc1−, and asc1DE cells as detected with a phospho-p38 MAPK-specific antibody. As a loading control, a part of the Ponceau red-stained Western blot membrane, at approximately the level of the Hog1p-P signals, is depicted. (B2) Drop dilution assay to evaluate osmotic stress resistance. (C to F) Phenotypic characterization of Asc1p phosphosite mutant strains. (C) Drop dilution assays. Cell suspensions were spotted on YNB plates with or without cycloheximide or canavanine to assess sensitivity to translation/protein biosynthesis inhibitors. To evaluate cell wall integrity, cells were dropped on YEPD plates with or without Congo red, and to test fermentative activity, cells were spotted onto YEP medium with glycerol instead of glucose. (D) TTC assay to evaluate respiratory activity. (E) Haploid adhesive growth tests on 3-AT. Plates are depicted before and after washing with a constant stream of water. (F) Colony morphology under glucose limitation conditions. Cells were grown on YEP-2% glucose plates (no limitation) and YEP-0.4% glucose plates (glucose limitation).
All phosphosite mutant strains were compared to the ASC1 wild-type strain, the Asc1p-deficient asc1− strain, and the asc1DE strain as references. The results are summarized in Table 1, and the main observations are depicted in Fig. 2C to F. Like the DE exchange variant, dephosphorylation-mimicking amino acid exchanges T143A and Y250F increased the sensitivity to the translation inhibitor cycloheximide. The Y250F mutation also caused sensitivity to the l-arginine analogon canavanine (Fig. 2C). The DE exchange emphasized the importance of phosphosites T143 and Y250: the asc1T143ADE and asc1Y250FDE strains showed synthetic hypersensitivity to cycloheximide (Fig. 2C). They were even phenotypically similar to the asc1− strain. Irrespective of the DE mutation, phenylalanine at position 250 exclusively affected the sensitivity to cycloheximide and canavanine. The asc1T143ADE strain behaved similarly to the asc1− strain in almost all tests. The asc1T143EDE strain, with the phosphorylation mimic glutamate at position 143, partially restored the wild-type phenotypes.
Without the DE exchange, all other phosphosite mutant strains showed wild-type behavior (Fig. 2C to F and data not shown). In combination with the DE exchange, amino acid substitutions at T12, T96, and T99 had the following consequences. The asc1T12ADE strain showed impaired adhesive growth (Fig. 2E). The asc1T96ADE and asc1T96EDE strains were not adhesive and showed almost no wrinkled colony morphology, similar to the asc1− strain. All other phenotypes were only mildly affected (Fig. 2E and F). The T99A mutation combined with the DE exchange affected the same two phenotypes, though less severely. The asc1T96A T99ADE strain behaved like the asc1T96ADE strain and also exhibited slightly increased sensitivity to cycloheximide (Fig. 2C). Interestingly, the sensitivity of the asc1T96EDE strain to cycloheximide and its reduced growth on glycerol were suppressed by the additional T99E exchange (asc1T96E T99EDE strain). For the other tests, the additional T99E mutation also partially suppressed phenotypes caused by the T96E and DE mutations.
Combination of the DE exchange with dephosphorylation mimics at S120, S166, and T168 did not affect any of the tested phenotypes (data not shown). Thus, DE-induced alterations in Asc1p binding to the ribosome caused phenotypes when combined with specific phosphosite mutations. A high-copy-number plasmid bearing the wild-type ASC1 gene complemented all mutations and thus revealed a recessive nature of the described asc1 mutations (Fig. S2).
Dephosphorylation mimics at T12, T96, T99, and T143 but not at Y250 destabilize Asc1DEp.
The protein levels of Asc1p, in general, were unaffected by amino acid exchanges at the phosphosites analyzed in this study (Fig. 3A and data not shown). The cellular abundance of Asc1DEp was rather increased in comparison to that of wild-type Asc1p (Fig. 3A). However, this increased level of Asc1DEp required the integrity of phosphosites T12, T96, T99, and T143 but not Y250. Replacing residue T143 with alanine strongly decreased the abundance of cellular Asc1DEp. This correlates with the phenotypes of this strain that are reminiscent of those of the asc1− strain. The exchange of T143 for a phosphorylation-mimicking glutamate residue partially rescued the low abundance of Asc1T143ADEp.
FIG 3.

Asc1 protein abundance in phosphosite mutant strains. (A to C) Immunodetection of Asc1p within cell extracts derived from ASC1 wild-type, asc1DE, and phosphosite mutant strains and the asc1− strain (negative control). Ponceau red staining of proteins served as a loading control, and a part of the lower half of the stained Western blot membrane is depicted. The graphs provide mean relative intensities for three replicates, with standard deviations indicated by error bars. Fold changes were calculated relative to wild-type signal intensities. All nine values for the asc1DE strain were taken together. Panel C further shows Northern blots for detection of ASC1 mRNA and SNR24 snoRNA U24 levels in T143 phosphosite mutant strains. rRNA served as a loading control.
The combination of the DE exchange with the T96A and T96E mutations also reduced the abundance of the β-propeller to levels far below the wild-type levels. This effect was even enhanced through the simultaneous mutation of the proximal amino acid T99 to alanine in the asc1T96A T99ADE strain. In contrast, the asc1T96E T99EDE strain exhibited a slightly higher Asc1p level, an observation that is also in line with its milder phenotypes. Remarkably, the T96E and T99E exchanges even rescued the sensitivity of the asc1DE strain to cycloheximide.
The asc1Y250FDE strain displayed Asc1p levels as high as those of the asc1DE strain. Thus, the sensitivities of the asc1Y250F strain and the asc1Y250FDE strain to cycloheximide and canavanine do not correlate with a reduced cellular abundance of Asc1Y250FDEp. We tested whether the changes in Asc1p expression were caused by alterations of the ASC1 mRNA level by performing Northern hybridization experiments with a probe against exon 1 of ASC1. The asc1T143ADE strain, which had the strongest reduction of Asc1p abundance, revealed unchanged levels of ASC1 mRNA, providing evidence that the reduced Asc1p levels were caused posttranscriptionally (Fig. 3C).
The T143A exchange does not influence Asc1p's ribosome binding and does not destabilize a D109Y variant.
Binding of Asc1p to the ribosome might contribute to its own stability. To test the impact of phosphorylation site T143 on ribosome binding, the T143A and T143E Asc1p variants, and also the Y250F variant, were tested in ribosome pulldown experiments and compared to wild-type Asc1p and Asc1DEp. Ribosome pulldown assays with Rps2p-GFP confirmed the reduced binding of DE variants but did not show changes for the variants with an amino acid exchange at T143 or Y250 (Fig. 4).
FIG 4.

Ribosome binding of phosphosite mutant Asc1p variants. Asc1p phosphosite mutant strains were transformed with a plasmid bearing GFP-fused RPS2 (pHK697). Rps2p-GFP was pulled from cell extracts by use of GFP trap beads. Protein samples taken before the addition of the beads (input) and the proteins eluted from the GFP trap beads (eluate) were subjected to Western blot experiments. Immunodetection of Asc1p and Rps2p-GFP was performed using Asc1p- and GFP-specific antibodies, respectively. A wild-type strain expressing no GFP-tagged Asc1p was analyzed in parallel to exclude unspecific binding of Asc1p to the beads. Asterisks indicate protein bands that are supposed to be caused by processed Rps2p-GFP or Rps3p.
For the asc1T143ADE strain, additional processed forms of Rps2p-GFP were observed that were also detected to a lesser extent in the asc1Y250F and asc1Y250FDE strains (labeled with asterisks in Fig. 4). These signals could be detected for the asc1− strain as well. Moreover, a processed Rps3p signal that was not present for the ASC1 wild-type strain was detected for the asc1− and asc1T143ADE strains by use of an Rps3p-specific antibody. Accordingly, proteolytic activity that goes beyond Asc1p's own degradation might be organized Asc1p dependently at the ribosome.
We also tested whether the T143A exchange within another known ribosomal binding-compromised variant, namely, Asc1D109Yp (12), leads to phenotypes similar to those caused by Asc1T143ADEp. This was, however, not the case overall (Fig. 5). Only growth on cycloheximide was reduced for the asc1T143AD109Y strain, similar to that of the asc1T143ADE strain. Furthermore, a reduced binding affinity for the ribosome through either R38D K40E or D109Y exchange did not lead per se to the degradation of Asc1p but rather to a stabilization of the protein. Only in combination with the DE exchange, not the D109Y mutation, did the T143A dephosphorylation mimic drastically reduce cellular Asc1p levels. In contrast to D109, K40 is a known site for posttranslational modifications, namely, acetylation and succinylation (27, 28). Thus, the prevention of a posttranslational modification of K40 through mutation to E40 rather than an impaired ribosome association might synthetically affect the impact of the T143A mutation.
FIG 5.

Influence of the D109Y mutation on Asc1p-dependent phenotypes and Asc1p protein levels. The different ASC1 variants were expressed from CEN plasmids under the control of the native ASC1 promoter in the Δasc1 background. The ASC1 wild-type strain and the Δasc1 strain, both transformed with the empty vector (EV), were used as controls. (A) Resistance of the strains to cell wall stress (Congo red), osmotic stress (NaCl), low temperature (25°C), and inhibition of translation elongation (cycloheximide) was tested. (B) Haploid adhesive growth of the mutant strains on 3-AT. (C) Immunodetection of Asc1p within cell extracts obtained from the indicated yeast strains. A part of the Western blot membrane stained with Ponceau red is depicted as a loading control.
Asc1p coordinates the phosphorylation of at least 90 proteins at 120 different sites.
Asc1p's involvement in cellular signal transduction prompted us to investigate Asc1p-sensitive protein phosphorylation beyond its own phosphorylation. Previous studies reported that the absence of Asc1p leads to increased phosphorylation of the MAPKs Kss1p and Slt2p, which control filamentous growth, mating, and cell wall integrity (2, 29). In the present study, we additionally analyzed the phosphorylation of the MAPK of the high-osmolarity response pathway, Hog1p, by using an antibody specific for this protein phosphorylated at residues T174 and/or Y176 by its upstream mitogen-activated protein kinase kinase (MAP2K), Pbs2p. A significant decrease in Hog1p phosphorylation was detected in the asc1− strain (Fig. 2B1). This explains the sensitivity of the asc1− strain to osmotic stress (Fig. 2B2) (30). To gain an unbiased and more comprehensive view of Asc1p-sensitive phosphorylation of other proteins, we performed stable isotope labeling with amino acids in cell culture (SILAC) and LC-MS-based quantitative phosphoproteome analyses to compare the asc1− strain and the ASC1 wild-type strain. In parallel, the total proteome was analyzed to correlate quantitative changes of phosphopeptides to changes in total protein abundance. For efficient incorporation of the isotopically labeled amino acids arginine and lysine into proteins, we generated arginine- and lysine-auxotrophic Δarg4 Δlys1 strains. The workflow for the peptide sample preparation is depicted in Fig. 6A. Detailed descriptions of the LC-MS data evaluation and statistical analysis by use of MaxQuant/Perseus software are provided in Materials and Methods and in Tables S4 (proteome) and S5 (phosphoproteome). The phosphoproteome analysis resulted in the identification of 1,947 distinct phosphopeptides with localization site probabilities of ≥0.5. A total of 1,245 of these sites were detected and quantified in at least two independent replicates of the comparison between the asc1− and ASC1 wild-type strains and were considered for further data analysis.
FIG 6.

Analysis of the Asc1p-dependent phosphoproteome. (A) Peptide sample preparation for phosphoproteome and proteome analyses. S. cerevisiae ASC1 wild-type and asc1− strains auxotrophic for arginine and lysine (Δarg4 Δlys1) were cultivated in the presence of lightly, intermediately (medium), or heavily labeled arginine and lysine as indicated in the figure. Arrows indicate which cultures were pooled and show the number of replicates. In total, nine independent cell pools were obtained and subjected to the subsequent preparation of cell extracts. The protein extracts were split in two and processed separately for proteome and phosphoproteome analyses as described in detail in Materials and Methods. For an extended version of this workflow that comprises additional strains, see Fig. S3 in the supplemental material. (B) Assignment of the Asc1p-dependently regulated phosphoproteins to cellular processes. The 90 proteins were grouped in a nonexclusive manner, meaning that 15 proteins are present in two groups. The numbers of proteins assigned to each group are given on the left and right sides of the graph (see Table S7 for the identities of all proteins in the groups). Each cross represents a single regulated phosphosite and indicates its degree of regulation. (C) Overrepresented motifs for phosphorylated serine and threonine residues. Abundances of motifs for Asc1p-dependently regulated phosphosites were compared to the occurrences of the same motifs for all phosphosites identified in this study. Motifs were searched for sites with localization probabilities of ≥0.75 by using motif-x software (49, 50). Additional motifs identified for the complete proteome are not depicted.
A two-sample t test (P < 0.01) was performed to statistically compare the SILAC ratios for the phosphosites to those for the corresponding proteins. The significantly regulated phosphosites were further filtered based on their degree of regulation. To this end, proteome-corrected log2 SILAC ratios for the phosphopeptides were determined by calculating the differences between the ratios for the phosphopeptides and those for their corresponding proteins. Table 2 lists all phosphosites with proteome-corrected median log2 SILAC ratios of ≤−0.58 or ≥0.58. (A more comprehensive list, containing phosphosites with proteome-corrected median log2 SILAC ratios of <−0.26 or >0.26, is provided in Table S6.) Phosphosites with zero to two quantification values for the corresponding protein were evaluated separately by performing a one-sample t test on their SILAC ratios. In total, 120 sites in 90 proteins were found to be Asc1p-dependently regulated more than 50% in their degree of phosphorylation (log2 SILAC ratios of ≤−0.58 or ≥0.58) (Table 2). Forty-three of these sites were found to be upregulated and 77 downregulated. Twenty-four proteins with more than one Asc1p-dependently regulated phosphosite were identified, and one of these proteins (Acc1p) contained one upregulated as well as one downregulated site.
TABLE 2.
Asc1p-dependently regulated phosphositesa
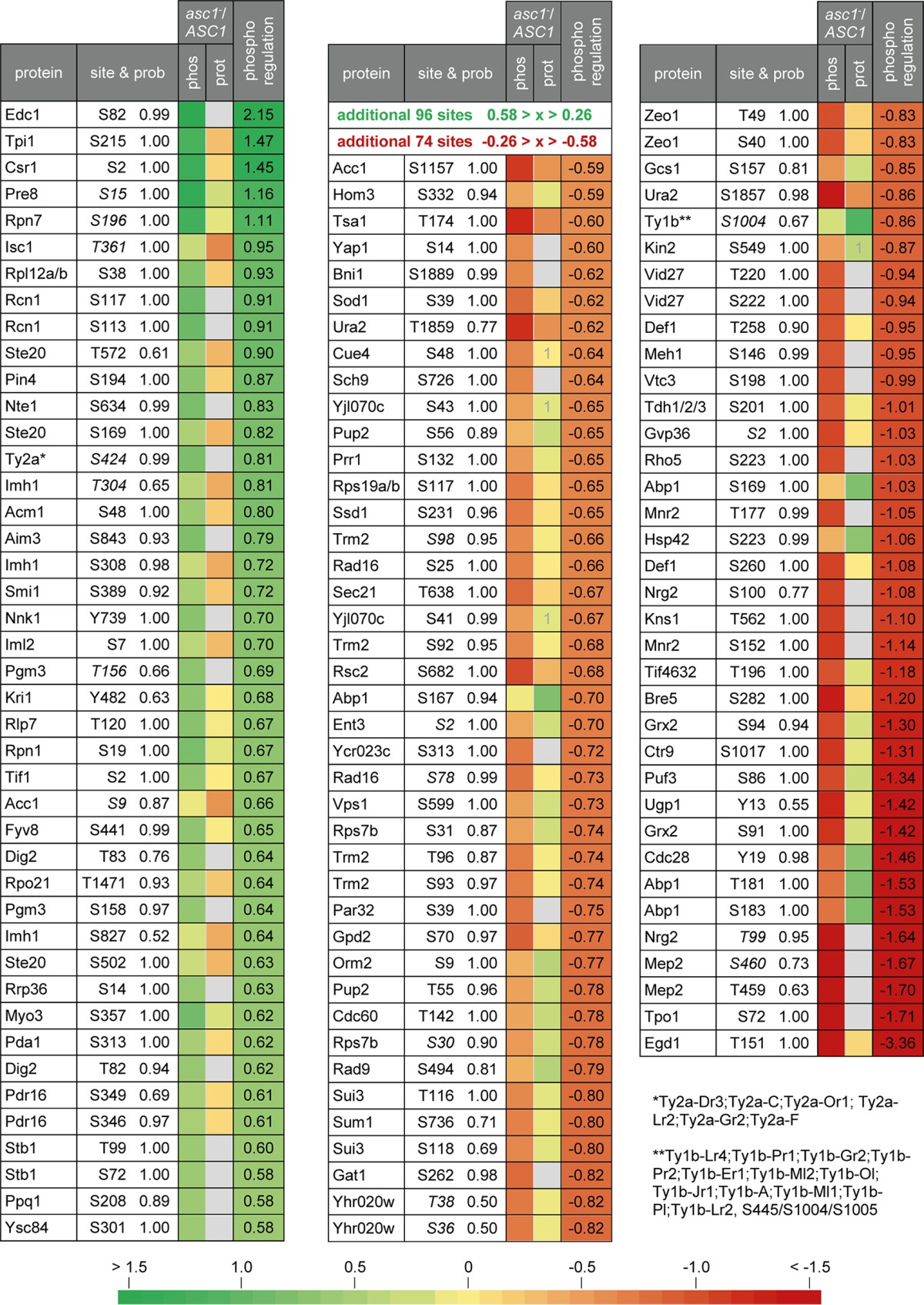
All proteins containing phosphosites with normalized median log2 asc1−/ASC1 SILAC ratios of ≤−0.58 or ≥0.58 are listed together with their regulated sites and the corresponding localization probabilities (site & prob). The colors reflect the median SILAC ratios for the phosphosites (phos), the corresponding proteins (prot), and proteome corrections (log2 ratio for the phosphosite minus log2 ratio for the protein) (phospho regulation). Gray indicates that the protein was not quantified in the whole-proteome analysis. In this case, the phosphoregulation value is identical to the quantification value for the phosphosite. A “1” indicates that only one quantification value was obtained. Phosphosites printed in italics are formerly unknown sites according to PhosphoGRID (74) and the SGD, including data from Swaney et al. (41). See Table S6 in the supplemental material for details and for additional phosphosites with normalized log2 asc1−/ASC1 SILAC ratios of <−0.26 or >0.26.
Asc1p mediates cellular signals for fundamental processes of eukaryotic gene expression.
To evaluate which processes were affected by the Asc1p-dependent phosphorylations, the 90 Asc1p-dependently regulated phosphoproteins were assigned to the biological processes they are involved in (according to gene ontology terms) by making use of the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 (31, 32). From this initial computational analysis, we established seven major groups and assigned all but 10 proteins to these functional categories in a nonexclusive manner (Fig. 6B; Table S7). One of the largest groups contains proteins that are related to mRNA translation. Some are directly involved in this process, such as ribosomal proteins (Rps7b and Rpl12a/bp), mRNA-binding proteins (Puf3p and Ssd1p), and translation factors. Three translation initiation factors comprising the RNA helicase eukaryotic translation initiation factor 4A (eIF4A) (Tif1p) were identified as being regulated. Increased phosphorylation of eIF4A in the absence of Asc1p confirmed an earlier observation from our lab (19). Additionally, proteins indirectly involved in translation, such as the tRNA-modifying enzyme Trm2p, ribosome biogenesis factors, and the leucyl-tRNA-synthetase Cdc60p, were assigned to the translation group.
The Asc1p-dependent phosphorylation network encompasses further processes that contribute to cellular protein homeostasis, including transcription and protein turnover. The absence of Asc1p alters, e.g., the phosphorylation of the oxidative stress-responsive transcription factor Yap1p. The transcription category was additionally expanded for proteins affecting mRNA fate, such as the decapping protein Edc1. The protein turnover group includes three components of the regulatory particle of the 26S proteasome (Rpn1p and Rpn7p) and two components of its catalytic core (Pre8p and Pup2p). Furthermore, an Asc1p-sensitive phosphosite was identified in Bre5p, the deubiquitination cofactor of Ubp3p and thus a regulator of bulk protein degradation during ribophagy and mitophagy (33). The traffic and transport group comprises central structural components of vesicle coats (e.g., Sec21p) and regulators of vesicle-mediated transport (e.g., Gcs1p). Also, transmembrane transporters, such as the polyamine transporter Tpo1p and the ammonium transporter Mep2p, were assigned to this category. Asc1p affects the phosphorylation of several proteins that are related to cytokinesis, cytoskeleton organization, and budding, for instance, the polarisome component and formin Bni1p, actin nucleation-promoting factors (Myo3p and Abp1p), and regulators of actin filament elongation (Aim3p and Abp1p). Another group comprises enzymes of the primary metabolism that are involved, e.g., in amino acid biosynthesis (Hom3p), glycolysis and gluconeogenesis (Tpi1p and Tdh1/2/3p), or pyrimidine biosynthesis (Ura2p).
Finally, the largest group contains proteins implicated in cell signaling. This group stands in close relation to all other groups, since it comprises 7 protein kinases, among them Cdc28p, Sch9p, and Ste20p, and the protein phosphatase Ppq1p, altogether possibly involved in the observed Asc1p-dependent changes in the phosphoproteome. In summary, the presented data reveal an unexpected connectivity of a ribosomal protein to cellular protein phosphorylation.
DISCUSSION
Asc1DEp reveals the functional significance of Asc1p phosphosites.
The systematic phenotypic analysis of Asc1p phosphosite mutant strains showed an increased sensitivity to translation inhibitors caused by dephosphorylation-mimicking T143A and Y250F exchanges. We combined the phosphosite mutations with the amino acid changes R38D and K40E (DE exchange). Both exchanged residues contribute to ribosome binding of Asc1p, and K40 is also known as a natural modification site for acetylation and succinylation (27, 28). The weakened binding of Asc1DEp to the ribosome in vivo was recently questioned in formaldehyde cross-linking experiments (11). Still, the precise positioning of Asc1DEp at the head region of ribosomal 40S should be distorted due to changes in the surface shape and its electrostatic potential (Fig. 7A). Furthermore, acetylation and succinylation of K40 might influence the Asc1p-ribosome interaction. Both modifications change the local basic character of the K40 side chain moiety to either neutral (acetylation) or negative (succinylation). In the crystal structure of the yeast 80S ribosome, the side chain of K40 is buried in a large cavity formed by rRNA. However, it is not involved in direct intermolecular interactions, as potential interacting partners are separated from K40 by distances of more than 6 Å. Modeling of both modifications revealed that the gain in side chain length enables formation of additional stabilizing interactions (Fig. 7B). Acetylated K40 may reach the phosphate moiety of U1340, while succinylated K40 may form interactions with the base of A1410 and C1338. The results of analysis of the asc1T143AD109Y strain imply that the integrity of amino acids R38 and K40 rather than uncompromised ribosome binding is essential for keeping Asc1T143Ap intact.
FIG 7.
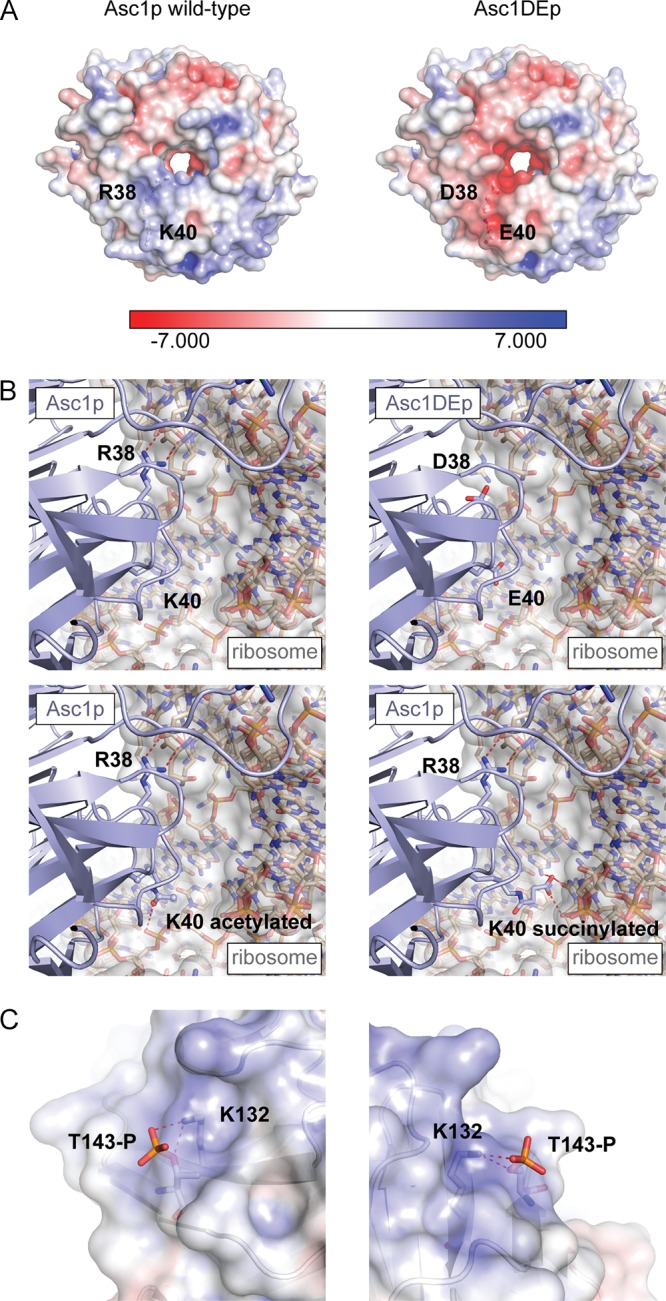
Modeling of Asc1p variants with the DE exchange, K40 modifications, or T143 phosphorylation. (A) Ribosome-binding surface of Asc1p. The electrostatic surface potentials of Asc1p and Asc1DEp are colored from red (−7 kBT/e) to blue (7 kBT/e). The distinct basic patch formed by R38 and K40 (depicted as balls and sticks) is changed in the DE variant to a negatively charged patch that does not form binding interactions with negatively charged phosphate moieties of rRNA. (B) Posttranslational modification of K40. A closeup view of the Asc1p-ribosome binding interface surrounding R38 and K40 is depicted in comparison to models of Asc1DEp and posttranslationally modified Asc1p that can be acetylated or succinylated at K40. Polar interactions of modified K40 residues are marked by dashed lines. (C) Interaction of phosphorylated T143 with K132. Phosphorylation of T143 was modeled and suggested salt bridge formation between the phosphate group and the amino group of the K132 side chain. Figures were generated with the PyMOL Molecular Graphics System software on the basis of PDB files 3FRX (Asc1p) (1) and 4V88 (80S ribosome) (67).
Asc1p's orthologues in other organisms, such as Schizosaccharomyces pombe, Homo sapiens, and Arabidopsis thaliana, become phosphorylated as well. An amino acid sequence alignment of Asc1p and these orthologues revealed a high level of conservation of T12, T143, and Y250 (Fig. 8A). Phosphorylation was also reported for T10 of S. pombe Cpc2p in a high-throughput study (34) and for Y246 and Y248 of the H. sapiens and A. thaliana RACK1 proteins, respectively (35, 36), corresponding to T12 and Y250 of Asc1p. Human RACK1 has been shown to be phosphorylated at Y246 by the Src kinase. Through this modification, the interaction between RACK1 and Src is increased, correlating with an inhibition of the kinase activity (35, 37). Phosphorylation of Y246 is also important for the interaction of RACK1 with the mRNA-binding protein ZBP1, another target of the Src kinase at the ribosome. ZBP1 binds the β-actin mRNA and regulates its local translation together with the Src kinase and RACK1 (17). Our results showed specific sensitivity of the asc1Y250F strain, and especially the asc1Y250FDE strain, to inhibitors of protein biosynthesis, suggesting a relevant role of this phosphosite in translational processes. As shown here for the Y250F exchange within yeast Asc1p, the exchange at the corresponding site within mammalian RACK1 (Y246F) does not affect the association of the protein with ribosomes, implying that the effect of this exchange on cellular processes is not caused by the protein's release from the site of translation (17). Since Y250 is located opposite the ribosome binding site at the bottom of Asc1p, it might be responsible for the binding of other regulatory proteins. To the best of our knowledge, this is the first study showing the in vivo modification of this residue by direct identification of the phosphopeptide through LC-MS. The high conservation of this amino acid residue and its surrounding sequence indicates that its phosphorylation is also highly conserved throughout the eukaryotic domain.
FIG 8.

Phosphosites within Asc1p and its orthologues. (A) Amino acid sequence alignment of Asc1p and its orthologues, with phosphosites. Amino acid residues that are conserved in at least three of the depicted sequences are shaded in black, and those that are similar in at least three sequences are shaded in gray. Phosphosites are indicated above the sequences. For S. cerevisiae Asc1p (S.c. Asc1p), phosphosites T12 (this study), S60 (68), T96 and T99 (69), S108 and S120 (23), T143 (this study), S166 and T168 (24), and Y250 and T300 (this study) are indicated with asterisks. For S. pombe Cpc2p (S.p. Cpc2p), phosphosites T10, S39, Y52, S148, S242, and S255 (34) are labeled with circles. Phosphosites S122, T162, and Y248 of A. thaliana RACK1A (A.t. RACK1A) (T161 of RACK1B and RACK1C) (22, 36) are indicated with minus signs, and phosphosites T50 (70), Y52 (71), S146 (18), Y228 (20), and Y246 (20, 37) of H. sapiens RACK1 (H.s. RACK1) are indicated with plus signs. The seven WD40 repeats are labeled below the sequences. Sequences were retrieved from the Uniprot database, aligned by use of the Clustal Omega program (http://www.uniprot.org/align/), and shaded with the BoxShade tool (http://www.ch.embnet.org/software/BOX_form.html). (B) Phosphorylation in the blade IV region of Asc1p/RACK1. Structures of S. cerevisiae Asc1p (gray), H. sapiens RACK1 (blue), and A. thaliana RACK1A (green) are shown with their respective phosphorylation sites, T143, S146, and S122, highlighted. Phosphosites are depicted as sticks, with carbon atoms shown in yellow, nitrogen atoms in blue, and oxygen atoms in red. Additionally, superpositioning of all three structures is depicted. The Asc1p homodimer is displayed with the two interacting Asc1p molecules in gray and white and with the T143 residues from both proteins highlighted. Structure figures were generated with the PyMOL Molecular Graphics System software on the basis of the PDB files 3FRX (Asc1p) (1), 4AOW (hRACK1) (72), 3DM0 (AtRACK1) (73), and 3RFG (Asc1p dimer) (38).
Amino acid T143 of Asc1p has not been described as a phosphosite in yeast or other organisms so far. However, comparisons of the amino acid sequences and structures of the orthologues show that phosphosites S122 of A. thaliana RACK1A, S146 of human RACK1, and S148 of S. pombe Cpc2p are closely located within the same region of the protein (Fig. 8) (22, 34). For human RACK1, phosphorylation of S146 was proposed to be a prerequisite for the formation of a RACK1 homodimer that mediates degradation of the transcription factor subunit HIF1α (18). So far, there is no evidence of homodimerization of S. cerevisiae Asc1p in vivo, although a crystal structure of a recombinant Asc1p homodimer has been determined (38). The almost identical positions of the yeast protein residue T143 and the human protein residue S146 within the orthologous proteins indicate an involvement of T143 in a putative dimerization process of yeast Asc1p. This idea is supported by the fact that the T143 residues of two Asc1p molecules localize directly at the dimer interface (Fig. 8B) (38). In combination with the DE exchange, the T143A exchange drastically changed the phenotypes and strongly reduced the abundance of the protein, possibly by affecting Asc1p's stability. Secondary structure prediction of Asc1T143Ap by use of the PSIPRED server (39, 40) did not reveal any changes in lengths and positions of the β-strands, implying that mutation of T143 to alanine does not lead to misfolding of the 3βD strand. Phosphorylation of T143 may stabilize the protein's fold through the formation of a salt bridge with K132 (Fig. 7C). K132 is a known ubiquitination site (41), and the phosphorylation status of T143 may directly regulate its ubiquitination to mediate proteasomal degradation of Asc1p. For A. thaliana, phosphorylation of RACK1A at S122 by WNK8 was also proposed to be involved in the regulation of the protein's degradation (22). Remarkably, the known Asc1p-binding protein Bre5p (42), a cofactor of the deubiquitinating isopeptidase Ubp3p and a regulator of bulk protein degradation during ribophagy and mitophagy, was found to be Asc1p-dependently phosphorylated in this study.
The ribosomal scaffold protein Asc1 coordinates fundamental processes of gene expression through phosphoregulation.
Asc1p and its mammalian orthologue RACK1 are emerging as key factors within the network of signaling pathways that adjust the phosphoproteome to cellular needs. Here we report a decreased abundance of the phosphorylated and activated form of the MAPK Hog1p in asc1− cells. A recent study on osteoclastogenesis revealed that RACK1 acts within a signal transduction pathway that leads to phosphorylation of the mammalian orthologue of Hog1p, p38 (43). Reduced RACK1 levels diminished p38 phosphorylation (43), analogous to our observations. Furthermore, p38b of Drosophila melanogaster was shown to bind and phosphorylate RACK1 (44).
Mammalian RACK1 was proposed to localize activated PKCβII and JNK to the ribosome for subsequent phosphorylation of eIF6, eIF4E, and eEF1A2 (5–7). Our study revealed Asc1p-dependently phosphorylated proteins possibly modified at the ribosomes through a similar mechanism. Upon Asc1p deficiency, one of the strongest downregulated phosphosites is S86 of the mRNA-binding protein Puf3p. Recently, Puf3p was shown to become phosphorylated at this and other residues when cells are deprived of glucose. This phosphorylation promotes the translation of bound transcripts that encode mitochondrial proteins (45). In line with this finding, our quantitative proteome analysis revealed reduced abundances of plenty of mitochondrial proteins, including ribosomal proteins and subunits of the transporter complexes of the outer and inner mitochondrial membranes, in the asc1− strain (see Table S8 in the supplemental material). This regulation phenotypically correlates with a reduced respiratory activity of Asc1p-deficient cells and with impaired growth on nonfermentable carbon sources (15; this study).
According to the Saccharomyces Genome Database (SGD), the following five of the Asc1p-dependently regulated phosphoproteins are physical interaction partners of Asc1p: Bre5p, Rlp7p, Rpn1p, Smi1p, and Ste20p. Among others, these factors are putative targets of a direct Asc1p-mediated kinase/phosphatase-substrate interaction according to their physical proximity to Asc1p. The list of physical Asc1p interaction partners includes the MAP4K Ste20p (2), which acts upstream of the MAPKs Kss1p, Fus3p, and Hog1p. We identified Ste20p as having three upregulated phosphosites that may be involved in the regulation of its general kinase activity or target specificity. Ste20p activity was proposed to be inhibited through Asc1p (2). In line with this, we identified increased phosphorylation of the Ste20p target Myo3p at S356 in the absence of Asc1p. This phosphorylation was shown in vitro and is required for Myo3p's function (46). The absence of Asc1p further reduced the phosphorylation of residues Y19 of Cdc28p and S726 of Sch9p, which are known to be involved in the regulation of kinase activities (47, 48). Considering a putative Asc1p-dependent regulation of specific kinases, we searched for overrepresented potential kinase motifs surrounding the regulated phosphosites. For this analysis, we used the motif-x software tool and considered only phosphosites with localization probabilities of ≥0.75 and at least one proteome value (Fig. 6C) (49, 50). Thirty-eight percent of the serine and 53% of the threonine residues were followed by a proline residue at position +1 (Fig. 6C). SP and TP motifs are preferentially recognized by MAPKs and cyclin-dependent kinases (51). It has to be noted that this motif was generally highly abundant—albeit less frequent—among all phosphosites identified here (31% SP and 30% TP).
Ribosomes are the most abundant and energy-consuming biosynthesis machineries in dividing cells, generating the material for all proteinaceous activities. Therefore, they require extensive feedback and feedforward loops to adjust their activity and fate to cellular signals. They thus represent signal control centers on top of their function as mere protein factories. RACK1/Asc1p couples these two functions by channeling phosphorylation to key regulators of affected processes that in turn regulate protein abundances of the involved proteins.
Figure 9 summarizes and emphasizes important aspects of Asc1p-dependent cellular phosphoadaptation, depicting the current and previous observations on Asc1p made in yeast. Through the coordination of a broad phosphorylation network, regulatory Asc1p connects ribosomes with fundamental cellular processes ranging from carbon source utilization to cell wall integrity, transcription, and cell cycle regulation.
FIG 9.

The sphere of influence of Asc1p in a RACK wheel overview. The impacts of Asc1p in the adaptation of the cellular proteome and phosphoproteome are depicted schematically. Asc1p links cellular signal transduction to the ribosome (indicated in gray below the red Asc1p protein; the structure figure was generated with the PyMol Molecular Graphics System software on the basis of the PDB file 4V88), leading to adjustment of the cellular proteome as needed (indicated by red arrows). Simultaneously, Asc1p is important for the phosphorylation status of central components of various processes, named in the outermost ring of the scheme. Phosphorylations are illustrated with a “P” in a star. The inner ring around the central RACK wheel gives examples of proteins containing Asc1p-sensitive phosphorylation sites. The outer ring shows examples of proteins that are Asc1p-dependently regulated at the proteome level. Phosphorylation of Asc1p itself may modulate the protein's integrity as a link between translation and signal transduction. Additional references were used for the Flo11p (19), Kss1p (2), Scp160p (16), and Slt2p (29) data. HSP, heat shock protein; HXTs, hexose transporters.
MATERIALS AND METHODS
Plasmid construction.
A list of plasmids used in this study (see Table S1 in the supplemental material) and a detailed description of their construction can be found in the supplemental material. For expression of N- or C-terminally Strep-tagged Asc1p, the high-copy-number (2μm) plasmid pRS426MET25, carrying a MET25 promoter and a CYC1 terminator, was used as the vector backbone. Plasmids carrying asc1 with codon exchanges were constructed via a two-step PCR strategy. In the first reaction, the codon exchange(s) was introduced within an oligonucleotide bearing the mutated codon(s) in its center, using ASC1-carrying plasmids as templates. In the second PCR, the complete asc1 allele flanked by the respective restriction sites was amplified for cloning into the parent vector.
Yeast strains and growth conditions.
The S. cerevisiae strains used in this study are of the Σ1278b background. Table S2 lists all yeast strains used in this study. A detailed description of their construction can be found in the supplemental material. Strains with codon exchanges within the open reading frame of ASC1 were obtained by transformation of a Δasc1::URA3 strain with the respective asc1 alleles carrying flanking regions identical to those of the ASC1 gene for homologous recombination and selection for resistance against 5-fluoroorotic acid. Arginine- and lysine-auxotrophic strains were generated by successive replacement of the ARG4 and LYS1 genes with the recyclable loxP::URA3::loxP marker cassette, amplified from plasmid pUG72, according to the method of Gueldener et al. (52). An arginine- and lysine-auxotrophic asc1SNR24 strain was generated from the ASC1 wild-type Δarg4::loxP Δlys1::loxP strain as described by Rachfall et al. (15). Generation of an asc1DE strain expressing C-terminally 3×myc-tagged Flo8p was done according to the method of Janke et al. (53). Strains were cultivated in liquid yeast nitrogen base (YNB) medium (1.5 g/liter YNB without amino acids and ammonium sulfate, 5 g/liter ammonium sulfate, 2% glucose) containing the respective supplements or in yeast extract-peptone-dextrose (YEPD) medium (1% yeast extract, 2% peptone, 2% glucose). Solid media contained 2% agar. Experiment-specific growth conditions are described in the respective paragraphs.
Western blot analysis.
Cultures were grown in 50 ml YNB medium to mid-log phase (optical density at 600 nm [OD600] = 0.8) and harvested by centrifugation at 1,650 × g for 4 min at 4°C. Cells were washed with ice-cold breaking buffer (100 mM Tris-HCl, pH 7.5, 200 mM NaCl, 20% glycerol, 5 mM EDTA) and lysed with glass beads in 500 μl breaking buffer containing 0.5% β-mercaptoethanol, 1 tablet of cOmplete protease inhibitor (Sigma-Aldrich) per 50 ml, 1 tablet of PhosSTOP phosphatase inhibitor cocktail (Sigma-Aldrich) per 10 ml, 1 mM NaF, 8 mM β-glycerol phosphate, and 0.5 mM sodium vanadate. Samples were incubated in the presence of 4% SDS at 65°C for 10 min. Samples were centrifuged at 16,200 × g for 15 min at room temperature, and the supernatant was collected as the cell extract. Protein concentrations were determined using the bicinchoninic acid (BCA) reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. The extracts were mixed with 3× loading dye (0.25 M Tris-HCl, pH 6.8, 30% glycerol, 15% β-mercaptoethanol, 7% SDS, 0.3% bromphenol blue) and heated again at 65°C for 10 min before being subjected to SDS-PAGE followed by blotting onto nitrocellulose membranes. The membranes were stained with Ponceau red (0.2% Ponceau S, 3% trichloroacetic acid) for 5 min, washed with water, and photographed using a Fusion-SL-4 instrument (Peqlab Biotechnology GmbH). The membranes were incubated with monoclonal mouse anti-myc (2276; Cell Signaling Inc.), polyclonal goat anti-Rap1p (yN-18; Santa Cruz Biotechnology), polyclonal rabbit anti-Tec1p, polyclonal anti-Asc1p (one provided by Andrew Link and another produced by Davids Biotechnology), polyclonal rabbit anti-Rps3p (provided by Heike Krebber), monoclonal mouse anti-GFP (B-2. sc-9996; Santa-Cruz Biotechnology), or polyclonal phospho-p38 MAPK (Thr180/Tyr182) (9211; Cell Signaling Inc.) antibodies followed by incubation with peroxidase-coupled goat anti-mouse (115-035-003; Dianova), donkey anti-goat (sc-2020; Santa Cruz Biotechnology), or goat anti-rabbit (G21234; MoBiTec) secondary antibodies. Chemiluminescence was detected using a Fusion-SL-4 instrument. Signals were quantified relative to those for Ponceau staining as a loading control, according to the method of Rivero-Gutiérrez et al. (54), using Bio1D software, version 15.01 (Vilber Lourmat GmbH). Western blot experiments for protein expression analysis were done at least in triplicate.
GFP trap assay.
Yeast cells expressing green fluorescent protein (GFP)-tagged Rps2p were cultivated in 150 ml YNB medium to mid-log phase and harvested by centrifugation at 1,650 × g for 4 min at 4°C. GFP trap assays were performed as described by Neumann et al. (55). Cells were washed with phosphate-buffered saline (PBS; 8 mM Na2HPO4, 2 mM NaH2PO4, 150 mM NaCl) and subsequently lysed with glass beads in the presence of 250 μl breaking buffer (PBS, 3 mM KCl, 2.5 mM MgCl2, 0.5% Triton X-100, 1 tablet of cOmplete EDTA-free protease inhibitor [Sigma-Aldrich] per 50 ml, and phosphatase inhibitors as described above). After cell lysis, another 250 μl of breaking buffer was added. Samples were centrifuged at 16,200 × g at 4°C until the lysate appeared clear. Prior to the addition of GFP-Trap_A beads (ChromoTek), a control sample was taken as an input control. GFP trap beads were equilibrated by washing five times with 1 ml washing buffer (PBS, 3 mM KCl, 2.5 mM MgCl2, 0.5% Triton X-100). Between washing steps, beads were collected by centrifugation at 400 × g at 4°C for 2 min. For each sample, 10 μl of GFP trap beads was used. The lysate was incubated for at least 2 h with the GFP trap beads at 4°C with rotation. Subsequently, the beads were washed five times with 1 ml breaking buffer. After the last washing step, 40 μl 2× loading dye was added to the beads, and samples were incubated for 5 min at 95°C for elution of the proteins. GFP trap experiments were performed twice.
Northern hybridization analysis.
Total RNA was isolated from yeast according to the method of Cross and Tinkelenberg (56). Forty micrograms of RNA was mixed with 30 μl sample buffer (66.7% formamide, 9.25% glycerol, 0.18% bromphenol blue, 1.8% ethidium bromide), heated at 65°C for 15 min, and kept on ice for 10 min. RNAs were separated in a 1.4% agarose gel containing 3% formaldehyde and 1% MOPS buffer (23 mM MOPS [morpholinepropanesulfonic acid], 5 mM sodium acetate, 1 mM EDTA, pH 7) in running buffer (6.7% formaldehyde and 1% MOPS) at 70 V. Transfer of RNA to a nylon membrane was performed by capillary blotting overnight. Cross-linking of RNA to the membrane, generation and labeling of the probe, and signal detection were carried out as described by Rachfall et al. (15), using AlkPhos direct labeling reagents (GE Healthcare GmbH), the CDP-Star detection reagent (GE Healthcare GmbH), and a Fusion-SL-4 instrument (Peqlab Biotechnology GmbH) for signal detection. The Northern hybridization experiment was performed with three replicates.
Sample preparation for LC-MS-based analysis of Asc1p phosphorylation.
Strain RH3263 (Δasc1) was transformed with plasmid pME2834 (ASC1-Strep) or pME4135 (Strep-ASC1) and cultivated in 1 to 10 liters of liquid YNB medium to an OD600 of 0.8. Harvested cells were washed with ice-cold breaking buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2). Cells were lysed with glass beads in breaking buffer (approximately 5 times the volume of the cell pellet) supplemented with protease and phosphatase inhibitors as described above. Samples were centrifuged at 16,200 × g for 15 min at 4°C, and the supernatant was applied to Strep-Tactin–Sepharose columns for affinity purification of Strep-tagged Asc1p (Strep-tag starter kit; IBA GmbH). The purification was performed according to the manufacturer's instructions. Eluate fractions were subjected to SDS-PAGE followed by colloidal Coomassie G250 staining of proteins (57) and in-gel digestion with trypsin (58) of the excised protein band containing enriched Strep-tagged Asc1p.
Sample preparation for LC-MS-based proteome and phosphoproteome analyses.
For phosphoproteome and proteome analyses, 40-ml cultures were grown in liquid YNB medium containing 100 mg/liter arginine, 100 mg/liter lysine, 20 mg/liter tryptophan, and 20 mg/liter uracil. The following stable isotopically labeled amino acids were purchased from Silantes GmbH: 13C6-l-arginine HCl, 13C615N4-l-arginine HCl, 4,4,5,5-D4-l-lysine HCl, and 13C6,15N2-l-lysine HCl. Cell extracts were prepared as described in “Western blot analysis,” with the exceptions that cell extracts were subjected to sonication for filter-aided sample preparation (FASP) (59). Cell extract containing 100 μg protein was mixed with loading dye, subjected to SDS-PAGE, and stained with colloidal Coomassie G250 (modified from that described by Kang et al. [57]). Each gel lane was divided into 10 pieces, and proteins were subjected to in-gel digestion according to the method of Shevchenko et al. (58). For phosphoproteome analyses, 1 mg protein from each sample was subjected to chloroform-methanol extraction (60) to reduce the amount of SDS in the samples. The pellets were dissolved in 200 μl 8 M urea (prepared in 0.1 M Tris-HCl, pH 8.5), and proteins were digested in solution with LysC (Wako Pure Chemical Industries) and trypsin (Serva Electrophoresis) according to the FASP protocol, using centrifugal filter units (Merck Millipore). Afterwards, peptides were desalted with C18 cartridges (3M) (59) followed by TiO2 affinity purification to enrich phosphopeptides from the complex peptide mixtures. The protocol for phosphopeptide enrichment was modified from that of Mazanek et al. (61). Desalted peptides from the previous step were dried, reconstituted in 50 μl loading solvent (70% acetonitrile [ACN], 420 mM 1-octanesulfonic acid [Sigma-Aldrich], 50 mg/ml dihydroxybenzoic acid [Fluka, Sigma-Aldrich], 0.1% heptafluorobutyric acid [Sigma-Aldrich], 3% trifluoroacetic acid [TFA]), and applied to equilibrated TiO2 columns (TT2TiO; Glygen Corporation) at an estimated ratio of 400 μg peptides to 1 mg of TiO2 for optimized recovery of phosphopeptides from TiO2, according to the method of Kanshin et al. (62). Equilibration of the columns was done by applying 40 μl of the first washing solution (70% ACN) followed by 40 μl loading solvent. A syringe was used to gently press the liquid through the stationary phase. After loading of the peptide samples, columns were washed twice with 40 μl loading solvent, once with the second washing solution (70% ACN, 125 mM asparagine, 125 mM glutamine, 3% TFA), and twice with 40 μl of the third washing solution (70% ACN, 3% TFA). The second washing solution comprised asparagine and glutamine to displace N/Q-rich peptides on the TiO2 column (62). Peptides were eluted from the column by twice applying 40 μl elution buffer (50 mM ammonium dihydrogen phosphate adjusted to pH 10.5 with ammonium hydroxide). Samples were acidified with 20 μl TFA, dried, reconstituted in sample buffer (2% ACN, 0.1% formic acid), and again desalted using C18 cartridges. For LC-MS analyses, all samples were dissolved in 20 μl sample buffer.
LC-MS analysis.
A liquid chromatograph coupled to an Orbitrap Velos Pro hybrid ion trap-Orbitrap mass spectrometer was employed for protein and phosphopeptide identification and for relative quantification by use of SILAC ratios. Peptides in 1 to 6 μl sample solution were trapped and washed on an Acclaim PepMap 100 precolumn (C18; 100 μm by 2 cm by 3 μm by 100 Å; Thermo Fisher Scientific) at a flow rate of 25 μl/min for 6 min in 100% solvent A (98% water, 2% ACN, 0.07% TFA). Analytical peptide separation by reverse-phase chromatography was performed on an Acclaim PepMap RSLC column (C18; 75 μm by 25 cm or 50 cm by 3 μm by 100 Å; Thermo Fisher Scientific), typically running a gradient from 98% solvent A (water, 0.1% formic acid) and 2% solvent B (80% ACN, 20% water, 0.1% formic acid) to 42% solvent B within 95 min and to 65% solvent B within the next 26 min, at a flow rate of 300 nl/min (both solvents and acids were from Fisher Chemicals). Chromatographically eluting peptides were ionized online by nanoelectrospray ionization (nESI) using a Nanospray Flex ion source (Thermo Fisher Scientific) at 2.4 kV and were continuously transferred into the mass spectrometer. Full scans within the mass range of 300 to 1,850 m/z were taken within the Orbitrap-FT analyzer, at a resolution of 30,000 or 60,000 (SILAC experiments), with parallel data-dependent top 10 MS2 collision-induced dissociation (CID) fragmentation with the LTQ Velos Pro linear ion trap. Phosphopeptide samples were analyzed with CID fragmentation by applying the multistage activation (MSA) method as well as with higher-energy collisional dissociation (HCD) fragmentation, in a separate run. When HCD fragmentation was used, data-dependent top five MS2 fragmentation was performed, and fragment ions were analyzed in the Orbitrap analyzer. For analysis of Asc1p phosphopeptides, targeted data acquisition with parent mass lists was also applied. LC-MS method programming and data acquisition were done with the software XCalibur 2.2 (Thermo Fisher Scientific). For identification of Asc1p-derived phosphopeptides, MS/MS2 data were searched against an S. cerevisiae-specific protein database of 6,110 entries, including common contaminants (Saccharomyces Genome Database [SGD], S288C_ORF_database release, version 2011; Stanford University), by using the Proteome Discoverer software, version 1.4, and phosphosite localization was evaluated using phosphoRS (25, 26). The digestion mode was trypsin, and a maximum of two missed cleavage sites was considered. Carbamidomethylation at cysteine was set as a fixed modification, and oxidation at methionine and phosphorylation at serine, threonine, and tyrosine were considered variable modifications. Mass tolerances of precursors and fragment ions were 10 ppm and 0.6 Da, respectively. False discovery rates were calculated by Proteome Discoverer, using the reverse-decoy mode, and the filter for valid peptide sequence matches was set to 0.01. For quantitative proteome analyses, MS/MS2 data were analyzed with the MaxQuant 1.5.1.0 software with the program's default parameters, using an S. cerevisiae-specific protein database derived from UniProt (Proteome ID UP000002311; 6,721 entries; 2014 download) (63). The digestion mode was trypsin/P, and a maximum of two missed cleavage sites was considered. Carbamidomethylation at cysteine was set as a fixed modification, and acetylation at the N terminus, oxidation at methionine, and phosphorylation at serine, threonine, and tyrosine (only for raw data from MSA and HCD LC-MS runs) were considered variable modifications. Arg6 and Lys4 were defined as medium peptide labels and Arg10 and Lys8 as heavy peptide labels. Mass tolerances of precursors and fragment ions were 4.5 ppm and 0.5 Da, respectively. Match between runs, Fourier transform-based mass spectrometer (FTMS) requantification, and FTMS recalibration were enabled. For protein quantification, the minimum ratio count was 2, and unique plus razor peptides were considered. False discovery rates were calculated by MaxQuant, using the revert-decoy mode, and the filter for valid peptide sequence matches was set to 0.01. MaxQuant output data were further processed using Perseus software 1.5.0.15 (63).
Phenotypic tests.
Yeast cells were grown to mid-log phase and diluted to an OD600 of 0.1. Three consecutive 10-fold dilutions were prepared, and 10 μl or 20 μl of each dilution was dropped on YEP plus 2% glucose or glycerol or on YNB medium, respectively. The media tested were YEP with 2% glycerol, YEPD or YNB with Congo red (125 μg/ml or 225 μg/ml) or NaCl (75 mM), and YNB with canavanine (600 ng/ml) or cycloheximide (0.15 μg/ml). YEPD and YNB plates were used for growth controls. Plates were incubated for 3 to 7 days at 30°C. To test temperature sensitivity, plates were incubated at 25°C for 3 days. To assess the respiratory activity of the strains, a triphenyltetrazolium chloride (TTC) assay (64) was performed as follows. One-hundred-microliter aliquots of the highest dilution were streaked on YNB plates containing 0.4% glucose, and the plates were incubated for 3 days at 30°C. Colonies were then overlaid with liquid 1.2% top agar containing 0.5% TTC and incubated for 20 to 30 min at 30°C. To observe glucose-dependent colony morphology, 100-μl aliquots of the highest dilution of the cell suspensions were streaked on YEP plates containing either 2% glucose or 0.4% glucose to obtain single colonies after 3 days of growth at 30°C. The ability of the yeast strains to grow adhesively was tested by growth on YNB plates containing 10 mM 3-amino-1,2,4-triazole (3-AT) for 3 days at 30°C, followed by washing of the plates under a constant stream of water.
Accession number(s).
The mass spectrometry proteomic data have been deposited with the ProteomeXchange Consortium (65) via the PRIDE partner repository, with the data set identifier PXD003031.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sabrina Sander for her experimental support and Mahdokht Kohansal Nodehi (Department of Neurobiology, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany) for sharing her expertise in phosphopeptide enrichment with us. We thank Heike Krebber for providing plasmid pHK697 and the Rps3p antibody. We thank Andrew Link (Vanderbilt University Medical Center, Nashville, TN) and Hans-Ulrich Mösch (Philipps-Universität, Marburg, Germany) for providing Asc1p and Tec1p antibodies, respectively.
This work was supported by the Göttingen Graduate School for Neurosciences and Molecular Biosciences (DFG Grant GSC 226/2) and by DFG Grant VA352/2-1.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00279-16.
REFERENCES
- 1.Coyle SM, Gilbert WV, Doudna JA. 2009. Direct link between RACK1 function and localization at the ribosome in vivo. Mol Cell Biol 29:1626–1634. doi: 10.1128/MCB.01718-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeller CE, Parnell SC, Dohlman HG. 2007. The RACK1 ortholog Asc1 functions as a G-protein β subunit coupled to glucose responsiveness in yeast. J Biol Chem 282:25168–25176. doi: 10.1074/jbc.M702569200. [DOI] [PubMed] [Google Scholar]
- 3.Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras AC, Nesvizhskii AI, Tyers M. 2010. A global protein kinase and phosphatase interaction network in yeast. Science 328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li JJ, Xie D. 2015. RACK1, a versatile hub in cancer. Oncogene 34:1890–1898. doi: 10.1038/onc.2014.127. [DOI] [PubMed] [Google Scholar]
- 5.Ceci M, Gaviraghi C, Gorrini C, Sala LA, Offenhauser N, Marchisio PC, Biffo S. 2003. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 426:579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 6.Ruan Y, Sun L, Hao Y, Wang L, Xu J, Zhang W, Xie J, Guo L, Zhou L, Yun X, Zhu H, Shen A, Gu J. 2012. Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. J Clin Invest 122:2554–2566. doi: 10.1172/JCI58488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gandin V, Gutierrez GJ, Brill LM, Varsano T, Feng Y, Aza-Blanc P, Au Q, McLaughlan S, Ferreira TA, Alain T, Sonenberg N, Topisirovic I, Ronai ZA. 2013. Degradation of newly synthesized polypeptides by ribosome-associated RACK1/c-Jun N-terminal kinase/eukaryotic elongation factor 1A2 complex. Mol Cell Biol 33:2510–2526. doi: 10.1128/MCB.01362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann B, Mösch HU, Sattlegger E, Barthelmess IB, Hinnebusch A, Braus GH. 1999. The WD protein Cpc2p is required for repression of Gcn4 protein activity in yeast in the absence of amino-acid starvation. Mol Microbiol 31:807–822. doi: 10.1046/j.1365-2958.1999.01219.x. [DOI] [PubMed] [Google Scholar]
- 9.Gerbasi VR, Weaver CM, Hill S, Friedman DB, Link AJ. 2004. Yeast Asc1p and mammalian RACK1 are functionally orthologous core 40S ribosomal proteins that repress gene expression. Mol Cell Biol 24:8276–8287. doi: 10.1128/MCB.24.18.8276-8287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiabudini M, Tais A, Zhang Y, Hayashi S, Wolfle T, Fitzke E, Rospert S. 2014. Release factor eRF3 mediates premature translation termination on polylysine-stalled ribosomes in Saccharomyces cerevisiae. Mol Cell Biol 34:4062–4076. doi: 10.1128/MCB.00799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson MK, Rojas-Duran MF, Gangaramani P, Gilbert WV. 2016. The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs. eLife 5:e11154. doi: 10.7554/eLife.11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuroha K, Akamatsu M, Dimitrova L, Ito T, Kato Y, Shirahige K, Inada T. 2010. Receptor for activated C kinase 1 stimulates nascent polypeptide-dependent translation arrest. EMBO Rep 11:956–961. doi: 10.1038/embor.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuda R, Ikeuchi K, Nomura S, Inada T. 2014. Protein quality control systems associated with no-go and nonstop mRNA surveillance in yeast. Genes Cells 19:1–12. doi: 10.1111/gtc.12106. [DOI] [PubMed] [Google Scholar]
- 14.Wolf AS, Grayhack EJ. 2015. Asc1, homolog of human RACK1, prevents frameshifting in yeast by ribosomes stalled at CGA codon repeats. RNA 21:935–945. doi: 10.1261/rna.049080.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rachfall N, Schmitt K, Bandau S, Smolinski N, Ehrenreich A, Valerius O, Braus GH. 2013. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol Cell Proteomics 12:87–105. doi: 10.1074/mcp.M112.017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baum S, Bittins M, Frey S, Seedorf M. 2004. Asc1p, a WD40-domain containing adaptor protein, is required for the interaction of the RNA-binding protein Scp160p with polysomes. Biochem J 380:823–830. doi: 10.1042/bj20031962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceci M, Welshhans K, Ciotti MT, Brandi R, Parisi C, Paoletti F, Pistillo L, Bassell GJ, Cattaneo A. 2012. RACK1 is a ribosome scaffold protein for β-actin mRNA/ZBP1 complex. PLoS One 7:e35034. doi: 10.1371/journal.pone.0035034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, Liu JO, Semenza GL. 2007. Calcineurin promotes hypoxia-inducible factor 1α expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem 282:37064–37073. doi: 10.1074/jbc.M705015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valerius O, Kleinschmidt M, Rachfall N, Schulze F, López Marín S, Hoppert M, Streckfuss-Bömeke K, Fischer C, Braus GH. 2007. The Saccharomyces homolog of mammalian RACK1, Cpc2/Asc1p, is required for FLO11-dependent adhesive growth and dimorphism. Mol Cell Proteomics 6:1968–1979. doi: 10.1074/mcp.M700184-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Chang BY, Chiang M, Cartwright CA. 2001. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J Biol Chem 276:20346–20356. doi: 10.1074/jbc.M101375200. [DOI] [PubMed] [Google Scholar]
- 21.Kiely PA, O'Gorman D, Luong K, Ron D, O'Connor R. 2006. Insulin-like growth factor I controls a mutually exclusive association of RACK1 with protein phosphatase 2A and β1 integrin to promote cell migration. Mol Cell Biol 26:4041–4051. doi: 10.1128/MCB.01868-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urano D, Czarnecki O, Wang X, Jones AM, Chen JG. 2015. Arabidopsis receptor of activated C kinase1 phosphorylation by WITH NO LYSINE8 KINASE. Plant Physiol 167:507–516. doi: 10.1104/pp.114.247460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smolka MB, Albuquerque CP, Chen SH, Zhou H. 2007. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci U S A 104:10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 26.Olsen JV, Mann M. 2004. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc Natl Acad Sci U S A 101:13417–13422. doi: 10.1073/pnas.0405549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henriksen P, Wagner SA, Weinert BT, Sharma S, Bacinskaja G, Rehman M, Juffer AH, Walther TC, Lisby M, Choudhary C. 2012. Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol Cell Proteomics 11:1510–1522. doi: 10.1074/mcp.M112.017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. 2013. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 4:842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Chasse SA, Flanary P, Parnell SC, Hao N, Cha JY, Siderovski DP, Dohlman HG. 2006. Genome-scale analysis reveals Sst2 as the principal regulator of mating pheromone signaling in the yeast Saccharomyces cerevisiae. Eukaryot Cell 5:330–346. doi: 10.1128/EC.5.2.330-346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melamed D, Bar-Ziv L, Truzman Y, Arava Y. 2010. Asc1 supports cell-wall integrity near bud sites by a Pkc1 independent mechanism. PLoS One 5:e11389. doi: 10.1371/journal.pone.0011389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 32.Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Müller M, Kötter P, Behrendt C, Walter E, Scheckhuber CQ, Entian KD, Reichert AS. 2015. Synthetic quantitative array technology identifies the Ubp3-Bre5 deubiquitinase complex as a negative regulator of mitophagy. Cell Rep 10:1215–1225. doi: 10.1016/j.celrep.2015.01.044. [DOI] [PubMed] [Google Scholar]
- 34.Wilson-Grady JT, Ville J, Gygi SP. 2008. Phosphoproteome analysis of fission yeast. J Proteome Res 7:1088–1097. doi: 10.1021/pr7006335. [DOI] [PubMed] [Google Scholar]
- 35.Chang BY, Conroy KB, Machleder EM, Cartwright CA. 1998. RACK1, a receptor for activated C kinase and a homolog of the β subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol Cell Biol 18:3245–3256. doi: 10.1128/MCB.18.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabila M, Kundu N, Smalls D, Ullah H. 2016. Tyrosine phosphorylation based homo-dimerization of Arabidopsis RACK1A proteins regulates oxidative stress signaling pathways in yeast. Front Plant Sci 7:176. doi: 10.3389/fpls.2016.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang BY, Harte RA, Cartwright CA. 2002. RACK1: a novel substrate for the Src protein-tyrosine kinase. Oncogene 21:7619–7629. doi: 10.1038/sj.onc.1206002. [DOI] [PubMed] [Google Scholar]
- 38.Yatime L, Hein KL, Nilsson J, Nissen P. 2011. Structure of the RACK1 dimer from Saccharomyces cerevisiae. J Mol Biol 411:486–498. doi: 10.1016/j.jmb.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 39.Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. 2013. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res 41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 41.Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J. 2013. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods 10:676–682. doi: 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ossareh-Nazari B, Bonizec M, Cohen M, Dokudovskaya S, Delalande F, Schaeffer C, Van Dorsselaer A, Dargemont C. 2010. Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep 11:548–554. doi: 10.1038/embor.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin J, Lee D, Choi Y, Lee SY. 2015. The scaffold protein RACK1 mediates the RANKL-dependent activation of p38 MAPK in osteoclast precursors. Sci Signal 8:ra54. doi: 10.1126/scisignal.2005867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belozerov VE, Ratkovic S, McNeill H, Hilliker AJ, McDermott JC. 2014. In vivo interaction proteomics reveal a novel p38 mitogen-activated protein kinase/Rack1 pathway regulating proteostasis in Drosophila muscle. Mol Cell Biol 34:474–484. doi: 10.1128/MCB.00824-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee CD, Tu BP. 2015. Glucose-regulated phosphorylation of the PUF protein Puf3 regulates the translational fate of its bound mRNAs and association with RNA granules. Cell Rep 11:1638–1650. doi: 10.1016/j.celrep.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu C, Lytvyn V, Thomas DY, Leberer E. 1997. The phosphorylation site for Ste20p-like protein kinases is essential for the function of myosin-I in yeast. J Biol Chem 272:30623–30626. doi: 10.1074/jbc.272.49.30623. [DOI] [PubMed] [Google Scholar]
- 47.Booher RN, Deshaies RJ, Kirschner MW. 1993. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J 12:3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R. 2007. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell 26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz D, Gygi SP. 2005. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol 23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 50.Chou MF, Schwartz D. 2011. Biological sequence motif discovery using motif-x. Curr Protoc Bioinformatics Chapter 13:13.15–13.24. doi: 10.1002/0471250953.bi1315s35. [DOI] [PubMed] [Google Scholar]
- 51.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma JL, Sheu YJ, Sassi HE, Sopko R, Chan CS, De Virgilio C, Hollingsworth NM, Lim WA, Stern DF, Stillman B, Andrews BJ, Gerstein MB, Snyder M, Turk BE. 2010. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal 3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. 2002. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 54.Rivero-Gutierrez B, Anzola A, Martinez-Augustin O, de Medina FS. 2014. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal Biochem 467:1–3. doi: 10.1016/j.ab.2014.08.027. [DOI] [PubMed] [Google Scholar]
- 55.Neumann B, Wu H, Hackmann A, Krebber H. 2016. Nuclear export of pre-ribosomal subunits requires Dbp5, but not as an RNA-helicase as for mRNA export. PLoS One 11:e0149571. doi: 10.1371/journal.pone.0149571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cross FR, Tinkelenberg AH. 1991. A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell 65:875–883. doi: 10.1016/0092-8674(91)90394-E. [DOI] [PubMed] [Google Scholar]
- 57.Kang D, Gho S, Suh M, Kang C. 2002. Highly sensitive and fast protein detection with Coomassie brilliant blue in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Bull Korean Chem Soc 11:1511–1512. [Google Scholar]
- 58.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 59.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 60.Wessel D, Flügge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 61.Mazanek M, Mituloviae G, Herzog F, Stingl C, Hutchins JR, Peters JM, Mechtler K. 2007. Titanium dioxide as a chemo-affinity solid phase in offline phosphopeptide chromatography prior to HPLC-MS/MS analysis. Nat Protoc 2:1059–1069. doi: 10.1038/nprot.2006.280. [DOI] [PubMed] [Google Scholar]
- 62.Kanshin E, Michnick SW, Thibault P. 2013. Displacement of N/Q-rich peptides on TiO2 beads enhances the depth and coverage of yeast phosphoproteome analyses. J Proteome Res 12:2905–2913. doi: 10.1021/pr400198e. [DOI] [PubMed] [Google Scholar]
- 63.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 64.Ogur M, St John R, Nagai S. 1957. Tetrazolium overlay technique for population studies of respiration deficiency in yeast. Science 125:928–929. doi: 10.1126/science.125.3254.928. [DOI] [PubMed] [Google Scholar]
- 65.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. 2014. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, Frank J. 2004. Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat Struct Mol Biol 11:957–962. doi: 10.1038/nsmb822. [DOI] [PubMed] [Google Scholar]
- 67.Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. 2011. The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 68.Gnad F, de Godoy LM, Cox J, Neuhauser N, Ren S, Olsen JV, Mann M. 2009. High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 9:4642–4652. doi: 10.1002/pmic.200900144. [DOI] [PubMed] [Google Scholar]
- 69.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. 2007. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A 104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Y, Wang Q, Qiu G, Zhou S, Jing Z, Wang J, Wang W, Cao J, Han K, Cheng Q, Shen B, Chen Y, Zhang WJ, Ma Y, Zhang J. 2015. RACK1 promotes autophagy by enhancing the Atg14L-Beclin 1-Vps34-Vps15 complex formation upon phosphorylation by AMPK. Cell Rep 13:1407–1417. doi: 10.1016/j.celrep.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 71.Kiely PA, Baillie GS, Barrett R, Buckley DA, Adams DR, Houslay MD, O'Connor R. 2009. Phosphorylation of RACK1 on tyrosine 52 by c-Abl is required for insulin-like growth factor I-mediated regulation of focal adhesion kinase. J Biol Chem 284:20263–20274. doi: 10.1074/jbc.M109.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruiz Carrillo D, Chandrasekaran R, Nilsson M, Cornvik T, Liew CW, Tan SM, Lescar J. 2012. Structure of human Rack1 protein at a resolution of 2.45 A. Acta Crystallogr Sect F Struct Biol Cryst Commun 68:867–872. doi: 10.1107/S1744309112027480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ullah H, Scappini EL, Moon AF, Williams LV, Armstrong DL, Pedersen LC. 2008. Structure of a signal transduction regulator, RACK1, from Arabidopsis thaliana. Protein Sci 17:1771–1780. doi: 10.1110/ps.035121.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. 2006. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.