

ABSTRACT
The Werner syndrome protein (WRN) suppresses the loss of telomeres replicated by lagging-strand synthesis by a yet to be defined mechanism. Here, we show that whereas either WRN or the Bloom syndrome helicase (BLM) stimulates DNA polymerase δ progression across telomeric G-rich repeats, only WRN promotes sequential strand displacement synthesis and FEN1 cleavage, a critical step in Okazaki fragment maturation, at these sequences. Helicase activity, as well as the conserved winged-helix (WH) motif and the helicase and RNase D C-terminal (HRDC) domain play important but distinct roles in this process. Remarkably, WRN also influences the formation of FEN1 cleavage products during strand displacement on a nontelomeric substrate, suggesting that WRN recruitment and cooperative interaction with FEN1 during lagging-strand synthesis may serve to regulate sequential strand displacement and flap cleavage at other genomic sites. These findings define a biochemical context for the physiological role of WRN in maintaining genetic stability.
KEYWORDS: Werner syndrome, telomeres, DNA replication, lagging-strand synthesis, DNA helicase, Okazaki fragment, aging, lagging strand
INTRODUCTION
The Werner syndrome protein (WRN) is a member of the RecQ family of DNA helicases, whose loss-of-function mutations result in the premature-aging disease Werner syndrome (WS) (1). Cells derived from WS patients undergo premature replicative senescence and display multiple chromosome aberrations, a phenomenon termed variegated translocation mosaicism (2, 3). WRN, like other but not all RecQ helicases, contains molecular domains, including the RecQ C-terminal (RQC) domain with a winged-helix (WH) motif proximal to the helicase carboxyl-terminal region and the helicase and RNase D C-terminal (HRDC) domain, that are thought to control substrate specificity and direct RecQ family members to the appropriate physiological targets through protein-protein interactions and DNA binding (4, 5). WRN, however, is distinct from the other members of this family of helicases in that it possesses an exonuclease domain. WRN unwinds and/or processes a variety of DNA molecules in vitro (6), and both activities are modulated by physical interactions with proteins implicated in various nuclear processes (7–14). WRN interacts with factors involved in DNA replication (9, 10, 13, 15–17) and colocalizes with a subset of replication foci during unperturbed S phase (18). Consistent with these findings, several studies have demonstrated S phase-specific defects in WRN-deficient cells (18–20), suggesting that WRN may play a specialized role during processive DNA synthesis. Although the in vivo substrates of WRN are mostly unknown, it is documented that a subpopulation of WRN localizes at telomeres during S phase (21, 22) and that WRN deficiency results in stochastic loss of telomeres synthesized by lagging-strand synthesis, a process termed sister telomere loss (STL) (21, 23). Although this phenotype is thought to arise from defects that occur during telomere replication, the underlying mechanism remains to be determined. Telomeres are intrinsically difficult to replicate because the repetitive G-rich sequence can form secondary structures, including G quadruplexes (G4), that are barriers for the advancing polymerase (24, 25). It is thought that disruption of these structures by specialized DNA helicases is required to ensure normal progression of the replication fork. Like WRN, the human RecQ family member Bloom syndrome protein (BLM) has also been implicated in telomere replication (26–29), and both proteins establish functional interactions with an overlapping set of proteins that function at the replication fork (6). However, despite great progress in the field, key questions related to the functions and substrate specificities of these helicases during telomere replication are still largely unresolved. In this study, we performed in vitro assays reconstituted with purified human proteins to define the contributions of WRN and BLM to the replication of the telomeric G-rich repeats by lagging-strand synthesis and to identify steps in the process that define the specificity of WRN. We show that WRN, as well as BLM, stimulates progression and strand displacement synthesis by human DNA polymerase δ (hPolδ), the lagging-strand polymerase (30), across telomeric G-rich repeats, which are barriers to hPolδ elongation. Remarkably, only WRN promotes the formation of cleavage products during sequential strand displacement and flap endonuclease 1 (FEN1) cleavage, a critical step in Okazaki fragment maturation (31). Moreover, WRN promotes precise cleavage by FEN1 during strand displacement synthesis on a nontelomeric substrate, suggesting that WRN may also contribute to Okazaki fragment processing during DNA synthesis at other genomic sites.
RESULTS
WRN and BLM stimulate polymerization by DNA polymerase δ across substrates with G-rich telomeric repeats.
To gain mechanistic insight into the role of WRN in telomere lagging-strand synthesis, we employed an in vitro system reconstituted with purified hPolδ, the processive enzyme responsible for the extension of the newly synthesized Okazaki fragment (32, 33). We purified hPolδ from a HeLa cell line stably expressing an epitope-tagged hPolδ p50 subunit (see Fig. S1A and B in the supplemental material) and examined the activity of the purified enzyme on linear oligonucleotide-based templates, which allow high-resolution analysis of products extended by hPolδ, with either a random AT-rich sequence or telomeric G-rich repeats (Fig. 1A). We used a large excess of annealed primer template over polymerase to minimize reinitiation and analyze single-round extension. Incorporation of nucleotides into the radiolabeled products of the extension reactions was visualized and quantified by phosphorimaging and ImageQuant analysis, respectively, and the amount of radioactivity in each extended product was plotted as the band intensity over the length of the template. hPolδ activity generates long, extended products in reactions with the random AT-rich sequence template, whereas elongation is significantly reduced in reactions with the telomeric G-rich-repeat template, even in the presence of the purified processivity factor proliferating cell nuclear antigen (PCNA) (Fig. 1B; see Fig. S2C in the supplemental material). This result is in agreement with previous studies and underscores the problem posed by the telomeric G-rich repeats during hPolδ elongation (34, 35). Next, we determined whether WRN or the RecQ family member BLM helps hPolδ overcome the inhibitory effect posed by the telomeric G-rich repeats. We observed that the addition of WRN or BLM to the reactions with the telomeric G-rich repeats led to a marked shift toward the synthesis of long, extended products, while neither helicase affected polymerase extension on the random AT-rich template (Fig. 1C and D). The synthesis of extended products by hPolδ in the presence of WRN or BLM was further stimulated by PCNA (Fig. 1E), suggesting that cooperation between RecQ helicase and the processivity factor during polymerase extension promotes efficient synthesis through the telomeric G-rich repeats. To ensure that the results were not influenced by possible differences in the efficiencies of initiation of synthesis between the samples, we also examined elongation by hPolδ on templates with telomeric G-rich or C-rich repeats embedded within nontelomeric flanking sequences. Consistent with the previous results, polymerase elongation was strongly inhibited by the telomeric G-rich sequence, and the addition of WRN or BLM resulted in robust synthesis of products that were extended past the nontelomere sequence (NTS) and across the G-rich repeats (see Fig. S2B and D in the supplemental material), confirming that both helicases can relieve the block to DNA synthesis posed by the telomeric G-rich repeats. Identical results were obtained in reactions that were supplemented with an excess of cold-annealed primer template just before the addition of nucleotides (not shown).
FIG 1.

Telomeric (tel) G-rich repeats are barriers for polymerization by hPolδ that are overcome by RecQ helicases. (A) Schematic representation of the templates used in the polymerase extension assays. (B) Purified hPolδ (0.2 nM) was incubated with the indicated 32P-labeled primer template (400 nM) in the absence or presence of purified PCNA (5, 10, or 20 nM) in a 50-μl reaction mixture under standard conditions. The reactions were carried out in the presence of a molar excess of primed template over polymerase, an experimental condition that minimizes polymerase reassociation and allowed us to measure the number of nucleotides incorporated in a single polymerase-DNA binding event. The reactions were terminated and processed as described in Materials and Methods. The products of the reactions were resolved on denaturing gels and visualized by autoradiography and phosphorimager analysis. The signal profiles were generated using ImageJ software and plotted as signal intensity per extended product. Lanes “Primer” contained 32P-labeled primer oligonucleotide only. (C) Purified hPolδ (0.2 nM) was incubated with the indicated radiolabeled primer template in the absence or presence of purified WRN (1, 2, or 4 nM) under standard conditions and processed as described in the legend to panel B. (D) Purified hPolδ (0.2 nM) was incubated with the indicated 32P-labeled primer template in the absence or presence of purified BLM (1, 2, or 4 nM) under the conditions described in the legend to panel B. Arrows (C and D) show fully extended products. (E) Purified hPolδ (0.2 nM) was incubated with the indicated 32P-labeled primer template in the absence (−) or presence (+) of PCNA (5 nM) and either BLM or WRN (1 nM [+] or 2 nM [++]) under the conditions described in the legend to panel B. The data shown are representative of the results of at least three independent experiments.
Stimulation of DNA synthesis across the telomeric G-rich repeat by WRN requires helicase activity and the HDRC domain.
To define the functional and structural domains required to promote polymerization across the telomeric G-rich repeats, we compared hPolδ elongation in the presence of wild-type WRN or mutants with missense mutations that inactivate exonuclease (WRN-D82A) or ATPase/helicase (WRN-K577M) activity (Fig. 2A). This analysis showed that WRN-D82A promoted DNA elongation across the telomeric G-rich sequences, as well as the wild-type protein, whereas WRN-K577M failed to induce any significant elongation past these repeats (Fig. 2B; see Fig. S3 in the supplemental material), demonstrating that only the helicase activity plays a critical role in the process. We noted that WRN-K577M induced nucleolytic processing of the priming oligonucleotide, suggesting that WRN exonuclease activity has harmful effects when active elongation across the telomeric G-rich repeats is inhibited. Consistent with this interpretation, reactions with a WRN nuclease/helicase mutant (WRN-D82A/K577M) lacked any detectable degradation product (Fig. 2B). Next, we asked whether WRN domains implicated in interactions with protein and DNA binding contribute to promoting polymerase elongation across the telomeric G-rich repeats. We generated WRN mutants lacking the winged-helix motif (WRNΔWH) or the helicase and RNase D C-terminal domain (WRNΔHRDC) and tested the purified proteins in the in vitro reconstituted reactions. This analysis demonstrated that the WH motif is not required for promoting hPolδ elongation across the telomeric G-rich repeats (Fig. 2C). On the other hand, elongation across telomeric G-rich repeats was abolished in reactions with WRNΔHRDC (Fig. 2C), indicating that this domain, in coordination with the helicase activity, plays a critical role in promoting hPolδ elongation across the sequence. As was observed in reactions with the helicase mutant WRN, loss of the HRDC domain resulted in nucleolytic processing of the priming oligonucleotide.
FIG 2.
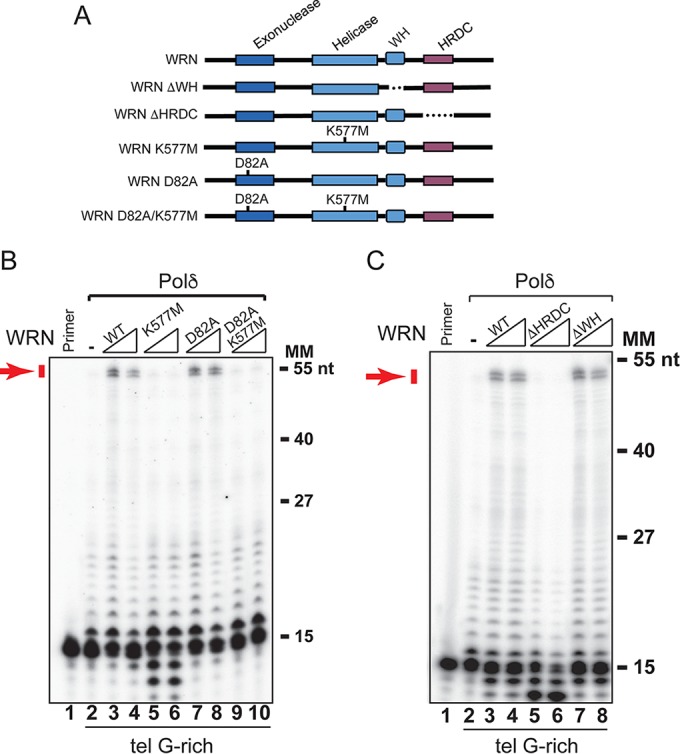
The ATPase/helicase activity and HRDC domains of WRN are required to promote extension through telomeric G-rich repeats by hPolδ. (A) Diagram of WRN domain structure and WRN mutants used in this study. (B) Purified hPolδ (0.2 nM) was incubated with the 32P-labeled primer telomeric G-rich template in the absence or presence of purified wild-type WRN, ATPase/helicase-defective WRN-K577M, exonuclease-defective WRN-D82A, or WRN-D82A/K577M (2 and 4 nM) in a 50-μl reaction mixture under standard conditions. The reactions were terminated and processed as described in Materials and Methods. (C) Purified hPolδ (0.2 nM) was incubated with the 32P-labeled primer telomeric G-rich template in the absence or presence of purified (2 and 4 nM) wild-type WRN, WRN lacking the WH domain (WRNΔWH), and WRN lacking the HRDC domain (WRNΔHRDC) in a 50-μl reaction mixture under standard conditions. The reactions were terminated and processed as described in Materials and Methods. Arrows show fully extended products. Lanes “Primer” contained 32P-labeled primer oligonucleotide only. MM, molecular markers. The data shown are representative of the results of two independent experiments.
WRN facilitates the displacement of a downstream fragment during hPolδ elongation.
During lagging-strand synthesis, Polδ extends a primer to the downstream Okazaki fragment and displaces its 5′ end into a single-stranded flap, which is predominantly removed by the structure-specific FEN1 (36). hPolδ is a poorly processive enzyme, and PCNA is thought to help in displacing the downstream fragment (33, 37). Nevertheless, high-GC content sequences have been shown to inhibit strand displacement synthesis even in the presence of PCNA or the Pif1 helicase (38, 39). We performed strand displacement assays using either the random AT-rich or telomeric G-rich substrates with a 28 downstream (blocking) oligonucleotide positioned 12 or 36 nucleotides (nt) downstream of the primer (Fig. 3A). The 3′ end of the blocking oligonucleotide was conjugated to biotin and bound to streptavidin to prevent nucleolytic attack. For the telomeric G-rich substrates, the 3′ end of the blocking oligonucleotide and the 5′ end of the template were composed of nontelomeric sequences to allow precise annealing. In the absence of PCNA, hPolδ stalls at the junction with the blocking oligonucleotide and is unable to displace the downstream oligonucleotide on either the 12-nt- or 36-nt-gap substrate (Fig. 3B and C). The addition of PCNA results in robust strand displacement synthesis on 12-nt-gap substrates, as well as the 36-nt-gap random AT-rich substrate (Fig. 3A and C). However, we did not observe any strand displacement synthesis on the 36-nt-gap telomeric G-rich substrate (Fig. 3C). We reasoned that structures such as G quadruplexes that can form only on the 36-nt-gap telomeric G-rich substrate may prevent strand displacement by impeding hPolδ/PCNA elongation along the template. We then performed electrophoretic mobility shift assays (EMSA) using antibodies that specifically recognize G4 DNA (40) and observed retarded DNA-antibody complexes indicative of G4 structures on the 36-nt-gap but not the 12-nt-gap substrate (see Fig. S4B in the supplemental material). Next, we tested whether WRN or BLM facilitates strand displacement synthesis on the 36-nt-gap telomeric substrate by aiding polymerase extension through the telomeric G-rich sequences. We observed that the addition of WRN or BLM to the reactions resulted in mild but significant stimulation of strand displacement synthesis by hPolδ, which was further enhanced by the addition of PCNA (Fig. 3D; see Fig. S5 in the supplemental material). These results suggest that WRN- or BLM-mediated resolution of secondary structures formed by the telomeric G-rich repeats in the single-stranded region preceding the downstream fragment promotes hPolδ/PCNA elongation and strand displacement. Interestingly, strand displacement synthesis was stimulated by WRN and BLM on 12-nt-gap substrates, as well as the 36-nt-gap random AT-rich substrate, even in the absence of PCNA (Fig. 3; see Fig. S5 in the supplemental material), suggesting that the function of these helicases in the displacement of a downstream fragment is not restricted to telomeric sequences.
FIG 3.

WRN and BLM promote strand displacement synthesis on gapped substrates. (A) Schematic representation of the substrates used to analyze strand displacement synthesis by hPolδ. Nontelomeric sequences were located at the 5′ and 3′ ends of the telomeric template to allow annealing of primer and blocking oligonucleotides, respectively. Primer and blocking fragments were separated by a 12-nt or 36-nt gap. The 3′ end of the blocking oligonucleotide is conjugated with biotin (B) and bound to streptavidin (ST). (B) Purified hPolδ (0.2, 0.4, or 0.8 nM) was incubated with random AT-rich (lanes 2 to 4) or telomeric G-rich (lanes 9 to 11) substrates with a 12-nt gap between the primer and the blocking oligonucleotide under standard conditions. PCNA (5, 10, or 20 nM) and hPolδ (0.2 nM) were added to the reaction mixtures shown in lanes 5 to 7 and 12 to 14. The reactions were terminated after 10 min, and the products were resolved on a denaturing gel and visualized by autoradiography and phosphorimager analysis. The normalized band intensities of the different-length products were generated using PhosphorImager analysis and ImageJ software and plotted as the signal intensity as a function of band migration. Lanes “Primer” contained 32P-labeled primer oligonucleotide only. (C) Purified hPolδ (0.2 nM) in the absence or presence of PCNA (5, 10, or 20 nM) was incubated with random AT-rich nontelomeric (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates with a 36-nt gap between the primer and the blocking oligonucleotide under standard conditions. The reactions were analyzed as described in the legend to panel B. (D) Purified hPolδ (0.2 nM) in the absence or presence of PCNA (5 nM) (lanes 3, 5 to 6, and 8 to 9), WRN (lanes 4 to 6; 1, 1, and 2 nM, respectively) and BLM (lanes 7 to 9; 1, 1, and 2 nM, respectively) was incubated with a telomeric G-rich substrate with a 36-nt gap between the primer and the blocking oligonucleotide under standard conditions. The reactions were analyzed as described in the legend to panel B. MM, molecular markers. The data shown are representative of the results of at least three independent experiments.
WRN promotes FEN1-directed cleavage during strand displacement synthesis.
FEN1 binds to WRN and BLM (9, 41), and both interactions have been shown to stimulate flap cleavage on preformed flapped substrates (41–43). FEN1 deficiency also results in an STL phenotype that closely resembles that observed in WRN-deficient cells (44). Thus, we tested whether concerted action by WRN or BLM and FEN1 influences the processing of the downstream fragment during strand displacement synthesis. For this purpose, the downstream oligonucleotide in the substrates described above was radiolabeled at the 5′ end to allow detection of FEN1-dependent cleavage products generated during strand displacement synthesis (Fig. 4A). We titrated PCNA, WRN, or BLM in reactions with hPolδ and FEN1 and measured the distribution and signal intensities of cleavage products in reactions with the random AT-rich and telomeric G-rich substrates. Whereas very small amounts of cleavage products were generated in reactions with hPolδ alone, the addition of PCNA to the reactions with the random AT-rich substrates with either a 12-nt or 36-nt gap resulted in a significant increase in the formation of a broad range of cleavage products, ranging in size from 2 to ≥18 nt (Fig. 4B and C). PCNA also promotes the formation of cleavage products on the telomeric G-rich substrate with a 12-nt gap (Fig. 4B). However, no significant amount of cleavage products was observed in the 36-nt-gap telomeric G-rich substrate (Fig. 4C). Remarkably, the addition of WRN to reactions with this substrate resulted in the formation of several cleavage products between 2 and 18 nt in size (Fig. 4D). Moreover, WRN promoted the formation of several minor and one major 5-nt cleavage products on both 12-nt- and 36-nt-gap random AT-rich substrates in the absence or presence of PCNA (Fig. 4D and E; see Fig. S6B in the supplemental material). The finding that WRN stimulated the formation of one major cleavage product on nontelomeric substrates suggests that the functional interaction between WRN and FEN1 during strand displacement influences the choice of cleavage sites on these substrates. In contrast, BLM did not promote FEN1 cleavage on any of the substrates tested in the analysis (Fig. 4F; see Fig. S6C in the supplemental material), underscoring a major functional distinction between WRN and BLM in a step that is critical for lagging-strand synthesis. The formation of cleavage products promoted by WRN on these substrates is strictly dependent on strand displacement synthesis, since it is not observed in the absence of hPolδ or in reactions with WRN mutants (WRN-K577M and WRNΔHRDC) that are unable to promote primer extension by hPolδ (Fig. 5A and B). Significantly, sequential strand displacement and flap cleavage also required the WH motif, which mediates FEN1 binding (Fig. 5C) (9), suggesting that the physical interaction between WRN and FEN1 plays a critical role in coordinating sequential strand displacement and flap cleavage during processive DNA synthesis. To reinforce this conclusion, we performed reactions with a FEN1 protein bearing a point mutation that abrogates WRN binding (FEN1-E359K) (see Fig. S7 in the supplemental material) (45) and showed that cleavage was inhibited in the presence of the mutant FEN1. To confirm that fragment cleavage was mediated by the nuclease activity of FEN1, the reactions were carried out in the presence of FEN1-D181A, a mutant protein that lacks nuclease activity but is still able to bind a flap substrate (Fig. 5C) (46).
FIG 4.

WRN, but not BLM, promotes FEN1 cleavage during strand displacement synthesis. (A) Schematic representation of the substrates used to analyze flap cleavage by FEN1. The 3′ end of the blocking oligonucleotide is conjugated with biotin (B) and bound to streptavidin (ST). Nontelomeric sequences are located at the 5′ and 3′ ends of the telomeric template to allow annealing of the primer and blocking oligonucleotides, respectively. The primer and blocking fragments are separated by a 12-nt or 36-nt gap. The 5′ end of the blocking oligonucleotide is radiolabeled. (B) Purified hPolδ (0.2 nM) and FEN1 (5 nM) were incubated with the 12-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence (lanes 3 to 5 and 8 to 10) of PCNA (5, 10, or 20 nM). The reactions were terminated after 10 min, and the products were resolved on a denaturing gel and visualized by autoradiography. The normalized band intensities of the cleavage products were generated using phosphorimager analysis and ImageJ software and plotted as the signal intensity as a function of band migration. (C) Purified hPolδ (0.2 nM) and FEN1 (5 nM) were incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence (lanes 3 to 5 and 8 to 10) of PCNA (5, 10, or 20 nM). The reactions were analyzed as described in the legend to panel B. (D) Purified hPolδ (0.2 nM) and FEN1 (5 nM) were incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence (lanes 3 to 5 and 8 to 10) of WRN (1, 2, or 4 nM). The reactions were analyzed as described in the legend to panel B. (E) FEN1 cleavage during strand displacement synthesis on the 36-nt-gap random AT-rich nontelomeric (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates was assessed in reactions carried out in the absence (lanes 2 and 7) or presence of hPolδ (0.2 nM) (lanes 3 to 5 and 8 to 10), FEN1 (5 nM) (lanes 2 to 5 and 7 to 10), PCNA (5 nM) (lanes 2, 4 and 5, 7, and 9 and 10), and WRN (1 nM [lanes 2 to 4 and 7 to 9] or 2 nM [lanes 5 and 10]). The reactions were analyzed as described in the legend to panel B. Lanes 2 and 7 show products of polymerase-independent cleavage by FEN1. (F) Purified hPolδ (0.2 nM) and FEN1 (5 nM) were incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence (lanes 3 to 5 and 8 to 10) of BLM (1, 2, or 4 nM). The reactions were analyzed as described in the legend to panel B. The data shown are representative of the results of at least three independent experiments. Lanes Bat and Btel contained radiolabeled random AT-rich or telomeric G-rich blocking oligonucleotides, respectively. MM, molecular markers.
FIG 5.
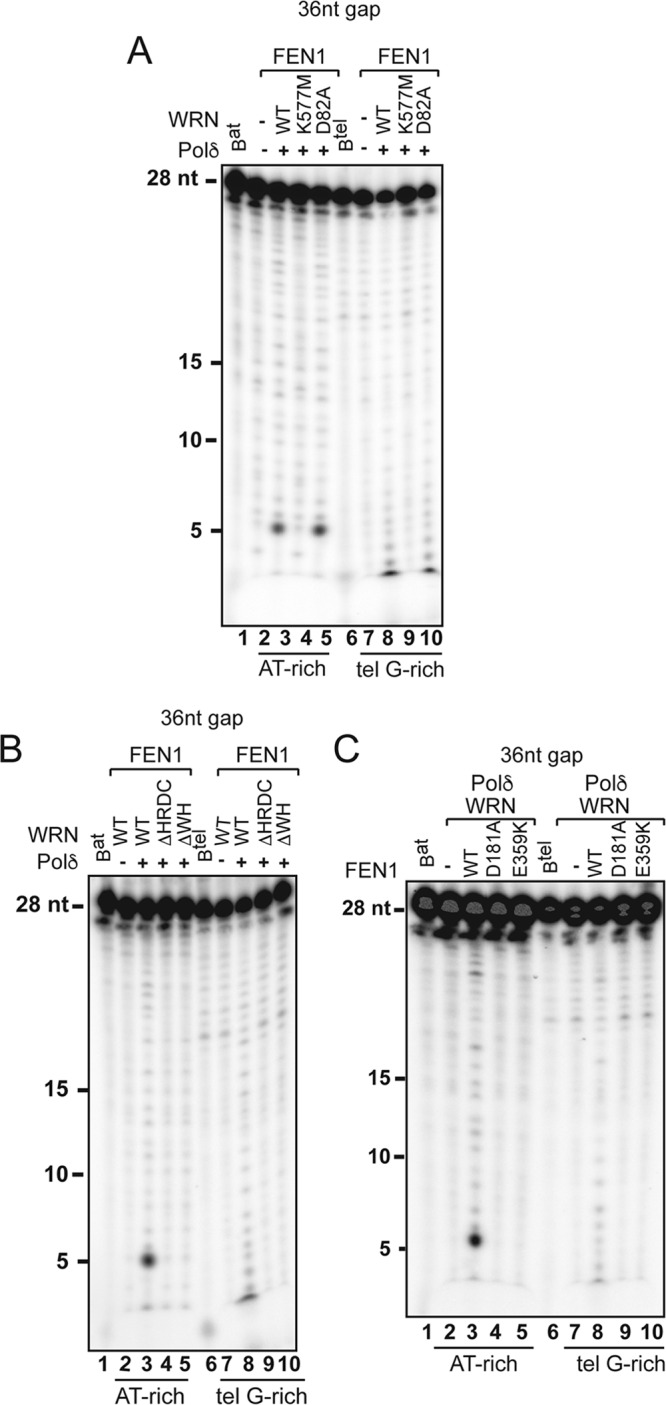
Multiple WRN domains cooperate to promote strand displacement-dependent FEN1 cleavage. (A) FEN1 (5 nM) was incubated with the 36-nt random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence of hPolδ (0.2 nM) (lanes 3 to 5 and 8 to 10) and wild-type WRN (2 nM) (lanes 3 and 8), helicase mutant WRN (K577M) (2 nM) (lanes 4 and 9), or exonuclease mutant WRN (D82A) (2 nM) (lanes 5 and 10). The reactions were terminated after 10 min and analyzed as described in Materials and Methods. (B) FEN1 (5 nM) was incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence of hPolδ (0.2 nM) (lanes 3 to 5 and 8 to 10) and wild-type WRN (2 nM) (lanes 2, 3, 7, and 8), a WRN protein lacking the HRDC domain (WRNΔHRDC) (2 nM) (lanes 4 and 9), or a WRN protein lacking the WH domain (WRNΔWH) (2 nM) (lanes 5 and 10). The reactions were terminated after 10 min and analyzed as described previously. The data shown are representative of the results of two independent experiments. (C) hPolδ (0.2 nM) and WRN (2 nM) were incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence of wild-type FEN1 (5 nM) (lanes 3 and 8), nuclease mutant FEN1-D181A (5 nM) (lanes 4 and 9), or WRN-binding mutant FEN1-E359K (5 nM) (lanes 5 and 10). Lanes Bat and Btel contained 32P-labeled blocking AT-rich and telomeric G-rich oligonucleotides, respectively.
To reconstitute a system that more closely mimics the physiological initiator element of an Okazaki fragment, we carried out experiments with downstream hybrid fragments bearing eight ribonucleotides at the 5′ end. First, we examined strand displacement synthesis and, in line with the results with the DNA fragment, we observed that PCNA alone stimulated strand displacement across the AT-rich fragment but that significant displacement of the telomeric G-rich fragment occurred only in the presence of WRN (Fig. 6B). Next, we performed FEN1 cleavage during strand displacement synthesis assays and showed that WRN promoted the formation of a prominent 8-nt product on the AT-rich substrate, indicating that the major cleavage site is located at the RNA-DNA junction (Fig. 6C). Titration of increasing amounts of WRN into the reaction mixture resulted in a concentration-dependent increase of a second cleavage product of 6 nt. Reactions with the telomeric G-rich substrate yielded several cleavage products, ranging from 5 to 9 nt in size, only in the presence of WRN, with the 5-nt and 9-nt products the most prominent. Importantly, as we demonstrated in reactions with a downstream DNA fragment, FEN1 cleavage of the RNA-DNA hybrid fragment was mediated by the nuclease activity of FEN1 and required physical interaction with WRN (Fig. 6C).
FIG 6.

WRN promotes strand displacement synthesis and FEN1 cleavage on substrates with a downstream RNA-DNA hybrid fragment. (A) Schematic representation of the substrates used to analyze strand displacement synthesis by hPolδ. Nontelomeric sequences are located at the 5′ and 3′ ends of the telomeric template to allow annealing of primer and blocking oligonucleotides, respectively. The primer and blocking fragments are separated by a 36-nt gap. (B) (Left) Purified hPolδ (0.2 nM) was incubated with random AT-rich nontelomeric (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates with a 36-nt gap between the primer and the blocking oligonucleotide in the absence or presence of purified wild-type WRN (1, 2, and 4 nM). (Right) Purified hPolδ (0.2 nM) was incubated with a telomeric G-rich substrate with a 36-nt gap between the primer and the blocking oligonucleotide in the absence or presence of PCNA (5 nM) (lanes 3, 5, 6, 9, 11, and 12), WRN (1, 1, and 2 nM) (lanes 4 to 6 and 10 to 12) under standard conditions. The data shown are representative of the results of at least three independent experiments. (C) (Left) hPolδ (0.2 nM) and FEN1 (5 nM) were incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence of wild-type WRN (1, 2, and 4 nM) (lanes 3 to 5 and 8 to 10). The reactions were terminated after 10 min and visualized by autoradiography and phosphorimager analysis. The signal profiles were generated using ImageJ software and plotted as the signal intensity per extended product. Lanes Bat and Btel (lanes 1 and 6) contained 32P-labeled blocking oligonucleotide only. The data shown are representative of the results of two independent experiments. (Right) hPolδ (0.2 nM) and WRN (2 nM) were incubated with the 36-nt-gap random AT-rich (lanes 2 to 5) or telomeric G-rich (lanes 7 to 10) substrates in the absence (lanes 2 and 7) or presence of wild-type FEN1 (5 nM) (lanes 3 and 8), nuclease mutant FEN1-D181A (5 nM) (lanes 4 and 9), or WRN-binding mutant FEN1-E359K (5 nM) (lanes 5 and 10). Radiolabeled AT-rich or telomeric C-rich oligonucleotides were used to determine the migration of 8-nt fragments. Lanes Bat and Btel (lanes 1 and 6) contained 32P-labeled blocking AT-rich and telomeric G-rich blocking oligonucleotides, respectively. MM, molecular markers.
The WH motif and the HRDC domain influence telomere length homeostasis in vivo.
To determine the roles of the WH motif and HRDC domain in telomere length homeostasis in vivo, we constructed lentiviral vectors for the conditional (doxycycline [dox]-inducible) expression of telomerase and generated telomerase-positive Werner syndrome (WS-teli+dox) cells. This expression system utilizes the third-generation version of the reverse Tet transactivator (Tet-ON) with improved doxycycline sensitivity and activity (47). Ectopic expression of telomerase has been shown to reset telomere length toward normal, to prevent the formation of telomere-induced DNA damage foci (TIFs), and to rescue cell proliferation (48, 49). Next, WS-teli+dox cells were complemented with wild-type or mutant WRN proteins lacking exonuclease or helicase activity, the WH motif, or the HRDC domain, and similar proliferation capacities were observed among the cell lines in the presence of dox (data not shown). We then followed the growth of these cell lines for several weeks after dox removal and observed a significant reduction in the proliferative capacity of the parental cell line (WS-teli+dox) compared to the cell line reconstituted with wild-type WRN (Fig. 7B). WS-teli+dox cells expressing WRN-K577M, WRNΔHRDC, and to a lesser extent WRNΔWH also showed reduced proliferation. In contrast, the proliferation of cells expressing WRN-D82A was indistinguishable from that of WS-teli+dox cells reconstituted with wild-type WRN. To determine whether telomere attrition may contribute to reduced proliferative capacity, we measured telomere lengths before and after doxycycline removal by Southern blotting of the terminal restriction fragment (TRF). These experiments showed that cells expressing telomerase have long telomeres irrespective of the absence or presence of wild-type or mutant WRN proteins (Fig. 7C). After doxycycline removal, the telomeres in all the cell lines were more heterogeneous in length, with WRN-deficient cells or cells expressing WRN-K577M displaying shorter telomeres than cells expressing wild-type or exonuclease mutant WRN (Fig. 7C; see Fig. S9 in the supplemental material). Significantly, deletion of the WH motif or HRDC domain also resulted in pronounced telomere shortening, suggesting that these structural domains, together with helicase activity, contribute to telomere length homeostasis during unperturbed growth.
FIG 7.
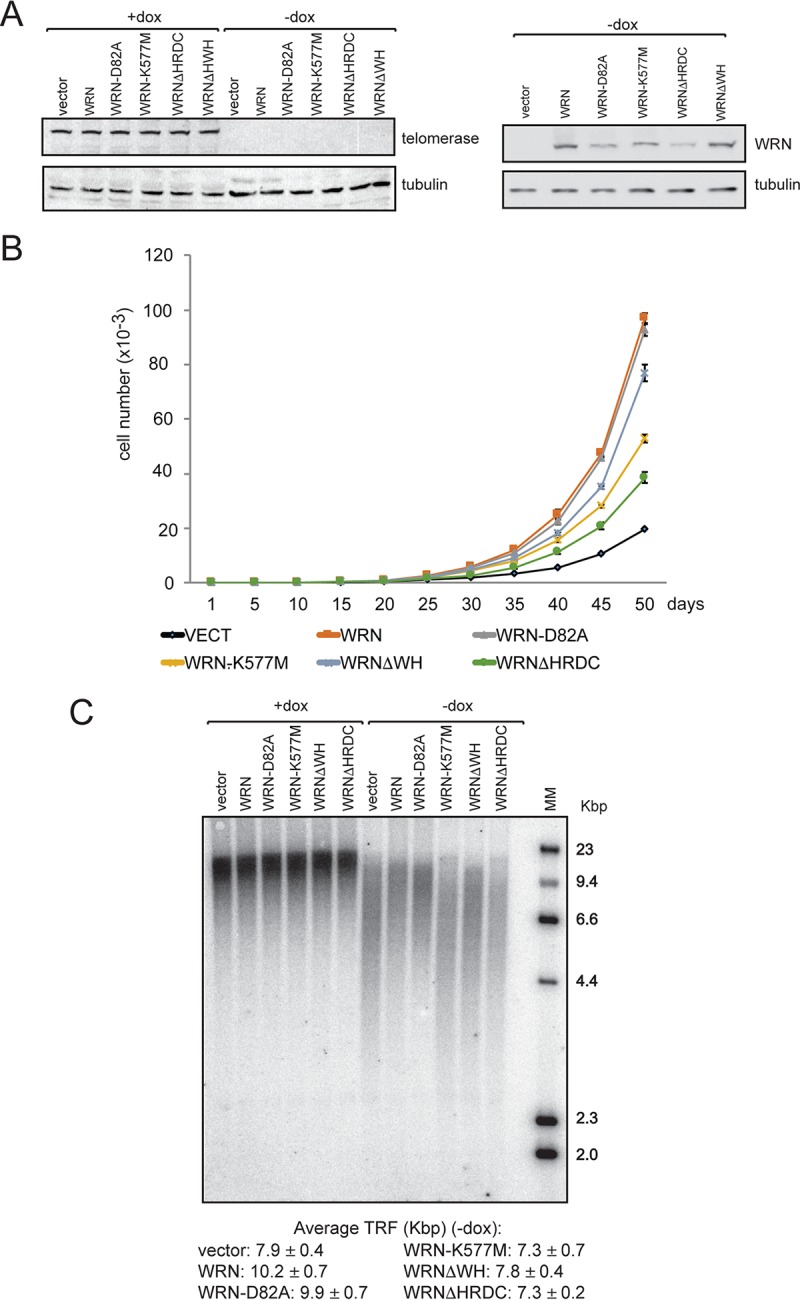
The WH motif and the HRDC domain of WRN contribute to cell proliferation and telomere length homeostasis. (A) Western blot analysis of WS-teli+dox parental cells and cells complemented with wild-type or mutant WRN proteins. +dox, cells grown in medium containing doxycycline; −dox, cells after doxycycline removal. (B) Analysis of proliferation of WS-teli+dox parental cells and cells complemented with wild-type or mutant WRN proteins after doxycycline removal. (C) TRF analysis of WS-teli+dox parental cells and cells complemented with wild-type or mutant WRN proteins before and after doxycycline removal. Average terminal restriction fragment lengths and standard deviations were calculated from the results of three independent experiments, as described in Materials and Methods.
DISCUSSION
WRN is a RecQ helicase that is thought to preserve genome integrity, at least in part by maintaining telomere length homeostasis (50). Our study provides a mechanistic explanation for the loss of telomeres replicated by lagging-strand synthesis in WRN-deficient cells. In agreement with the current model of DNA replication, we show that Polδ performs DNA synthesis and sequential strand displacement and flap cleavage on a random AT-rich substrate in the presence of PCNA, indicating that our experimental system faithfully reconstitutes this process in vitro. However, Polδ processivity and strand displacement are strongly inhibited on telomeric substrates even in the presence of PCNA. The addition of WRN to these reactions stimulates both steps, suggesting that WRN contributes to strand synthesis across the telomeric TTAGGG tandem repeats (see Fig. S10 in the supplemental material). Previous studies have shown that WRN stimulates DNA synthesis by Saccharomyces cerevisiae Polδ, an enzyme that is significantly more processive than its human counterpart (51, 52), on GC-rich templates only in the absence of PCNA and suggested that WRN is not required for general processive DNA synthesis in vivo (16). Our observation that WRN does not influence hPolδ elongation on a nontelomeric template supports this conclusion. However, the finding that WRN promotes robust Polδ extension and FEN1-mediated cleavage during strand displacement at telomeric G-rich repeats strongly suggests that WRN plays a specialized role during replication to ensure proper telomere lagging-strand synthesis. Our analysis indicates that the helicase activity is critically important in this process, possibly aiding the resolution of structural roadblocks on the long single-stranded G-rich priming loop that are thought to form on the lagging strand during replication (53, 54). Telomeric G-rich repeats can fold into noncanonical DNA structures termed G4 (55), and WRN unwinds this type of DNA structure in vitro (56, 57), although a previous report indicated that WRN is not active on telomeric G4 DNA (58). The finding that WRN-K577M fails to promote flap cleavage during strand displacement is consistent with the observation that ectopic expression of this mutant in HeLa cells results in telomere instability and STL (21, 59). Our results demonstrate that the HRDC domain also plays a critical role in strand synthesis across the telomeric G-rich repeats in vitro and contributes to telomere length homeostasis in vivo. The HRDC domain is conserved among many RecQ helicases and is thought to mediate protein interactions and DNA binding (5). Interestingly, a recent study has shown that the HRDC domain of BLM interacts with the single-stranded DNA ahead of G4 DNA and cooperates with helicase activity to unwind these DNA structures (60). Based on this observation, it is likely that the HRDC domain, together with helicase activity, contributes to elongation across telomeric G-rich repeats by making critical physical contacts with the telomeric G-rich strand ahead of the advancing polymerase. Unlike the helicase activity, the exonuclease activity of WRN does not seem to play a prominent role in any of the processes examined in this study. We cannot rule out the possibility that this enzymatic activity contributes to telomere replication in certain situations, such as the removal of mismatched or damaged nucleotides incorporated during processive DNA synthesis, as others have suggested (61).
Our data show that WRN is not unique in promoting Polδ extension and strand displacement synthesis through the telomeric G-rich repeats, since BLM performs as well as WRN in these reactions. These findings indicate some degree of functional overlap between the two helicases, which may explain the stochastic telomere effects observed in WRN-deficient cells. BLM plays a significant role in maintaining genome integrity, and several studies have suggested that it contributes in many ways to telomere stability. BLM binds the telomere binding protein TRF1, and this physical interaction has been implicated in preventing the fragile phenotype during telomere lagging-strand synthesis (28). BLM also resolves late replication intermediates and prevents the formation of ultrafine anaphase bridges (UBFs) at telomeres and common fragile sites (CFSs) (26, 29). More recently, BLM has been shown to stimulate polymerization across telomeric G-rich sequences that are replicated by leading-strand synthesis from origin firing within the telomere (27). In spite of the functional overlaps, our study reveals a significant difference between WRN and BLM in coordinating cleavage of the upstream fragment during strand displacement. Even though both proteins interact with FEN1 and stimulate the cleavage of preformed short flaps (42, 43, 62), we show that only WRN promotes FEN1-mediated cleavage during strand displacement synthesis, clearly pointing to a WRN-specific function. Importantly, FEN1 cleavage is inhibited by a mutation that prevents WRN binding. Given that the sequential strand displacement and cleavage of the upstream fragment by coordinated action of hPolδ and FEN1 are critical steps in Okazaki fragment maturation, our results suggest that the functional interaction between WRN and FEN1 contributes to telomere lagging-strand synthesis. This interpretation is in agreement with data showing that knockdown of FEN1 results in a telomere phenotype (STL) similar to that observed in WRN-deficient cells, and expression of a FEN1 variant lacking the WRN-binding domain does not rescue the telomere loss (44). We suggest that WRN deficiency may result in the synthesis of DNA fragments unsuitable for ligation, thereby preventing the production of a newly synthesized uninterrupted telomere. The partially replicated strand may contain single-strand DNA gaps that are processed into double-strand DNA breaks, leading to the loss of the telomere replicated by lagging-strand synthesis (21). It is also conceivable that unprocessed flaps could bind to the cDNA strand via illegitimate recombination, thereby creating elevated recombination rates between telomeres of sister chromatid and chromosome aberrations or promoting the formation of extrachromosomal telomeric circles, two of the phenotypes that characterize WRN-deficient cells (63, 64). Interestingly, the functional cooperativity in fragment cleavage during strand displacement synthesis between WRN and FEN1 is also observed on nontelomeric substrates, suggesting that WRN has the potential to influence Okazaki fragment metabolism at other loci. In support of this idea, WRN has been shown to colocalize with a subset of DNA replication foci in vivo, and WRN-deficient cells display slow progression or inactivation of a subset of replication forks (18). Although the identities of the genomic sites whose replication is affected by loss of WRN have not been determined, WRN depletion has been shown to affect the expression (breakage) of certain CFSs in vivo (65). Given the mechanistic similarities in gap filling and FEN1 cleavage between Okazaki fragment processing and long-patch base excision repair (36), the functional interaction between WRN and FEN1 may comprise repair processes, as well. However, stimulation of FEN1 cleavage by WRN is more robust on RNA-DNA hybrids than on DNA fragments, suggesting higher specificity for fragments that mimic the natural 5′ ends of Okazaki fragments. Enhanced cleavage in the presence of WRN occurs at the RNA-DNA junction on the AT-rich fragment, while a major cleavage site on the telomeric RNA-DNA hybrid fragment is 1 nucleotide downstream of the RNA-DNA junction. The difference in the positions of the major cleavage sites may reflect differences in hPolδ processivity or the kinetics of strand displacement on substrates with distinct nucleotide compositions. In eukaryotes, the removal of RNA primers from the lagging strand of replicating DNA is thought to occur through two partially overlapping processes that are mediated by RNase H (66, 67), although studies in yeast have also proposed alternative pathways (68). Our data suggest that the recruitment of WRN to replicating DNA may serve as an alternative pathway to stimulate FEN1-directed RNA primer removal, an idea that will be explored in our future investigations. In conclusion, our results provide biochemical evidence for a role of WRN in Okazaki fragment maturation during lagging-strand synthesis that is likely important for maintaining telomere length homeostasis and preventing genome instability, a hallmark of human cancer and other disorders involving tissues that require rapid repair and renewal capacities.
MATERIALS AND METHODS
Cell line and antibodies.
HeLa cells and WS primary fibroblasts (AG00780; nonsense mutation at codon 368 [Arg368Ter]) were purchased from the ATCC and Coriell Biorepository, respectively. The cells were cultured in Dulbecco modified essential medium (DMEM) supplemented with 10% fetal calf serum (FCS) and maintained at 37°C in a humidified incubator at 5% CO2. The plasmid for bacterial expression of the G4 antibody (pSANG10-3F-BG4) was obtained from Addgene; Polδ catalytic subunit antibody (sc-55499), α-tubulin antibody (sc-5286), and FEN1 antibody (sc-13051) were from Santa Cruz Biotechnology; Flag antibody (F3165) was from Sigma-Aldrich; anti-telomerase reverse transcriptase (anti-TERT) antibody (600-401-252) was from Rockland.
Flag-hPolδ2 cell line and purification of human DNA polymerase δ.
To generate a pRRL lentiviral transfer vector (69) for the expression of Flag-hPolδ2 (the P50 subunit of hPolδ) in HeLa cells, the full-length open reading frame of the human Polδ2 gene was amplified from a HeLa cell cDNA library by PCR using the following primers: 5′-TTTTTCATATGTTTTCTGAGCAGGCTGCC-3′ and 5′-TTTTGAATTCTCAGGGGCCCAGCCCCAGGCC-3′. The amplified DNA was digested with NdeI and EcoRI and cloned into the pcDNA3-Flag vector to generate pcDNA3-Flag-hPolδ2. This construct was digested with BamHI and XhoI to isolate a DNA fragment containing the Flag-tagged hPolδ2 cDNA, which was then cloned into the BamHI-XhoI sites of the pRRL vector to generate pRRL-flag-hPolδ2. Sequence analysis of the pRRL-flag-hPolδ2 construct confirmed the identity of the cDNA. The recombinant lentivirus for the expression of Flag-hPolδ2 in HeLa cells was produced by transfection of 293T cells as described previously (63). Expression of Flag-hPolδ2 in HeLa cells was verified by immunoblotting using anti-Flag antibody. For the purification of human Polδ, 100 mg of nuclear extracts prepared from HeLa cells expressing Flag-hPolδ2 was incubated with anti-Flag resin at 4°C for 1 h. After extensive washes, the bound proteins were eluted with elution buffer (1 M KCl, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin). The eluate was dialyzed in buffer containing 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride. An aliquot of the dialyzed material was precipitated with trichloroacetic acid (TCA), resolved by SDS-polyacrylamide gel electrophoresis, and analyzed by silver staining or immunoblotting with antibodies against the P125 subunit of hPolδ and Flag. The concentration of hPolδ was estimated by silver staining using serially diluted bovine serum albumin (BSA) as a standard. hPolδ was aliquoted and snap-frozen in liquid nitrogen. Single-use aliquots were stored at −80°C.
Purification of recombinant factors.
cDNAs for the expression of recombinant Flag-tagged wild-type WRN, WRN-D82A, WRN-K577M, WRN-D82A/K577M, WRNΔWH (Δ956–1064), WRNΔHRDC (Δ1142–1235), and BLM were cloned into baculovirus expression vectors to generate viruses for the infection of Sf9 cells. Forty-eight hours after infection, cells were collected and lysed in lysis buffer (10 mM HEPES, pH 7.5, 100 mM NaCl, 1.5 mM MgCl2, 0.5% Nonidet P-40). Recombinant proteins were purified by affinity chromatography on anti-Flag resin, as described previously (70). cDNAs for FEN1, FEN1-D181A, FEN1-E359K, and PCNA were cloned into bacterial vectors for expressing N-terminal 6×His proteins (pQE32-His-PCNA and pQE31-His-FEN1) and purified from Escherichia coli using His-Select HF nickel affinity gel (Sigma-Aldrich), as described in the manufacturer's manual. Protein concentrations were measured by the Bio-Rad protein assay using BSA as the standard.
hPolδ extension assay.
Oligonucleotides were purchased from Integrated DNA Technologies and purified using high-performance liquid chromatography (HPLC). The oligonucleotide primers were 5′-end labeled with [γ-32P]ATP (3,000 Ci/mmol; MP Biomedicals) using T4 polynucleotide kinase (New England BioLabs). Unincorporated [γ-32P]ATP was removed by fractionation on a ProbeQuant G-50 microcolumn. The labeled primer was mixed with an equivalent amount of unlabeled complementary template DNA in buffer containing 20 mM Tris-HCl, pH 7.5, 10 mM KCl, 5 mM MgCl2. The mixture was boiled for 5 min at 100°C, and the denatured oligomers were allowed to anneal by slowly cooling to room temperature. Primer extension synthesis was carried out in extension buffer (40 mM Tris, pH 7.5, 20 mM KCl, 5 mM MgCl2, 5 mM dithiothreitol, 0.1 mg/ml bovine serum albumin, and 0.2 mM [each] dATP, dGTP, dCTP, and dTTP) containing 400 nM 32P-labeled primer template substrate and hPolδ in the absence or presence of increasing amounts of purified wild-type or mutant WRN, BLM, or PCNA in a final volume of 50 μl. We used a >20-fold excess of PCNA over polymerase in all the reactions. Reaction mixtures were incubated at 37°C for 10 min, unless otherwise noted, and the reactions were terminated by addition of 10 μl of 5% SDS buffer. The reaction mixtures were incubated with 10 μg proteinase K at 45°C for 30 min and phenol-chloroform extracted, and DNA was precipitated with ethanol. The DNA pellet was solubilized in 15 μl of Tris-EDTA (TE) buffer, and then 2 μl of 95% formamide denaturing loading buffer was added to each reaction mixture. Samples were boiled for 5 min and electrophoresed on 10% polyacrylamide-urea gels. The gels were dried, and the radiolabeled products were visualized by autoradiography, quantitated with a phosphorimager (FLA-7000; Fujifilm), and analyzed using the open source ImageJ software (NIH). A representative experiment with at least three replicates is shown in each figure.
Electrophoretic mobility shift assays.
BG4 antibodies were expressed in E. coli BL21(DE3) with the pSANG10-3F-BG4 vector, and then the BG4 antibodies were purified by affinity chromatography on anti-Flag resin as described previously (40). 32P-labeled 55-nt primer template or 12-nt- or 36-nt-gap G-rich templates (500 nM) were incubated with 200 ng, 400 ng, or 800 ng BG4 antibodies in 15 μl of extension buffer (40 mM Tris, pH 7.5, 20 mM KCl, 5 mM MgCl2, 5 mM dithiothreitol, 0.1 mg/ml bovine serum albumin) or EMSA buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM KCl, 2 mM MgCl2, 1 mM EDTA, and 5% glycerol) at 25°C for 30 min. The samples were resolved by electrophoresis at 10 V/cm through a 4.5% polyacrylamide gel at 8°C. The gels were dried on Whatman 3MM paper and subjected to autoradiography and phosphorimager analysis (FLA-7000; Fujifilm).
Strand displacement assay.
The oligonucleotide primer was 5′ end labeled and purified as described above. The labeled primer was mixed with an equivalent amount of unlabeled complementary template DNA and the blocking oligonucleotide in buffer containing 20 mM Tris, pH 7.5, 10 mM KCl, 5 mM MgCl2. The radiolabeled substrates (400 nM) were incubated with 1.0 μg of streptavidin at room temperature for 20 min to block the 3′ biotinylated end of the downstream oligonucleotide. Strand displacement reactions were carried out in a final volume of 50 μl for 10 min at 37°C and analyzed as described above.
Sequential strand displacement synthesis and FEN1 cleavage assay.
The radiolabeled substrates (400 nM) were incubated with 1.0 μg of streptavidin at room temperature for 20 min to block the 3′ biotinylated end of the downstream oligonucleotide. In each reaction, the substrate was incubated with hPolδ and FEN1 in the absence or presence of different amounts of purified WRN (wild type or mutants), BLM, and PCNA in a final volume of 50 μl. After 10 min at 37°C, the samples were processed as described for the primer extension assays.
Protein-protein interaction assay.
Purified Flag-WRN or BSA (5 μg) was absorbed on anti-Flag beads and then incubated with 200 μl of purified His-FEN1 or His-FEN1-E359K (5 μg). After extensive washing, bound proteins were released by boiling the beads at 100°C for 3 min and analyzed by Western blotting with FEN1 and WRN antibodies.
Inducible expression of TERT in WS cells.
To generate lentiviral vectors for the conditional expression of TERT, TERT cDNA was inserted into the pENTT-MCS vector to produce pENTT-TERT vectors, and then pSLIK-TERT-Neo was generated by in vitro recombination between pENTT-TERT and pSLIK-Neo using the Gateway LR Clonase enzyme mix kit (Invitrogen, Carlsbad, CA). Recombinant lentiviruses were produced as previously described (63) and used to infect primary WS fibroblasts. Transduced cells were selected in DMEM supplemented with 400 μg/ml Geneticin (G418) for 7 days. Doxycycline (1 μg/ml) was added to the media to induce telomerase protein (TERT) expression.
Expression of wild-type and mutant WRN proteins in WS cells.
Flag-tagged wild-type WRN, exonuclease mutant WRN(E82A), helicase mutant WRN(K577M), WRNΔWH, and WRNΔHRDC cDNAs were cloned into pRRLsin.hCMV-Puro vectors as described previously (63). WS cells/TERT (plus dox) were transduced with lentiviruses encoding wild-type and mutant forms of WRN and then subjected to puromycin selection. Cell extracts were analyzed for expression of telomerase, WRN, and α-tubulin by Western blotting.
Telomere restriction fragment analysis.
Flag-tagged wild-type or mutant WRN proteins were expressed in WS cells/TERT (without dox), and then the cells were collected. Genomic DNA was isolated by standard protocols, and equal amounts of DNA were digested with HinfI and RsaI. Two micrograms of DNA was separated on 0.8% agarose gels, transferred to a Hybond membrane (GE Healthcare Life Sciences), and hybridized with a radiolabeled (CCCTAA)4 probe. The blots were analyzed with a phosphorimager (FLA-700; Fujifilm), and telomeric repeat signals were quantitated using the open source ImageJ software (NIH). The median telomere restriction fragment length was estimated by comparing the median of the distribution of telomere values within each lane with the migration of DNA size standards. Average TRF lengths and standard deviations were calculated from the results of three independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Lieber for FEN1 cDNA and members of the Comai laboratory for suggestions and valuable discussions.
This work was supported by a grant from the National Institutes of Health (R01AG034156 to L.C.) and was conducted in a facility constructed with support from Research Facilities Improvement Program grants C06 RR014514-01, C06 RR10600-01, and C06 CA62528 from the National Center for Research Resources, National Institutes of Health.
B.L. designed, performed, and analyzed all the experiments described and contributed to the preparation of the manuscript. S.R. contributed to the design of the study and the interpretation of data. L.C. conceived and coordinated the study and wrote the paper, together with B.L. We all analyzed the results and approved the final version of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00560-16.
REFERENCES
- 1.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. 1996. Positional cloning of the Werner's syndrome gene. Science 272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 2.Salk D. 1982. Werner's syndrome: a review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Hum Genet 62:1–5. doi: 10.1007/BF00295598. [DOI] [PubMed] [Google Scholar]
- 3.Scappaticci S, Cerimele D, Fraccaro M. 1982. Clonal structural chromosomal rearrangements in primary fibroblast cultures and in lymphocytes of patients with Werner's syndrome. Hum Genet 62:16–24. doi: 10.1007/BF00295599. [DOI] [PubMed] [Google Scholar]
- 4.Bennett RJ, Keck JL. 2004. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol 39:79–97. doi: 10.1080/10409230490460756. [DOI] [PubMed] [Google Scholar]
- 5.Kitano K. 2014. Structural mechanisms of human RecQ helicases WRN and BLM. Front Genet 5:366. doi: 10.3389/fgene2014.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croteau DL, Popuri V, Opresko PL, Bohr VA. 2014. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem 83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. 2000. Ku complex interacts with and stimulates the Werner protein. Genes Dev 14:907–912. [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, Comai L. 2000. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J Biol Chem 275:28349–28352. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- 9.Brosh RM Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. 2001. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J 20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doherty KM, Sommers JA, Gray MD, Lee JW, von Kobbe C, Thoma NH, Kureekattil RP, Kenny MK, Brosh RM Jr. 2005. Physical and functional mapping of the replication protein A interaction domain of the Werner and Bloom syndrome helicases. J Biol Chem 280:29494–29505. doi: 10.1074/jbc.M500653200. [DOI] [PubMed] [Google Scholar]
- 11.Li B, Comai L. 2001. Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-Ku70/80 complex. J Biol Chem 276:9896–9902. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- 12.Li B, Comai L. 2002. Displacement of DNA-PKcs from DNA ends by the Werner syndrome protein. Nucleic Acids Res 30:3653–3661. doi: 10.1093/nar/gkf488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Lopez AM, Jackson DA, Nehlin JO, Iborra F, Warren AV, Cox LS. 2003. Characterisation of the interaction between WRN, the helicase/exonuclease defective in progeroid Werner's syndrome, and an essential replication factor, PCNA. Mech Ageing Dev 124:167–174. doi: 10.1016/S0047-6374(02)00131-8. [DOI] [PubMed] [Google Scholar]
- 14.von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng WH, Bohr VA. 2003. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol 23:8601–8613. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szekely AM, Chen YH, Zhang C, Oshima J, Weissman SM. 2000. Werner protein recruits DNA polymerase delta to the nucleolus. Proc Natl Acad Sci U S A 97:11365–11370. doi: 10.1073/pnas.97.21.11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamath-Loeb AS, Johansson E, Burgers PM, Loeb LA. 2000. Functional interaction between the Werner syndrome protein and DNA polymerase delta. Proc Natl Acad Sci U S A 97:4603–4608. doi: 10.1073/pnas.97.9.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebel M, Leder P. 1998. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci U S A 95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez-Lopez AM, Jackson DA, Iborra F, Cox LS. 2002. Asymmetry of DNA replication fork progression in Werner's syndrome. Aging Cell 1:30–39. doi: 10.1046/j.1474-9728.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara Y, Higashikawa T, Tatsumi M. 1977. A retarded rate of DNA replication and normal level of DNA repair in Werner's syndrome fibroblasts in culture. J Cell Physiol 92:365–374. doi: 10.1002/jcp.1040920305. [DOI] [PubMed] [Google Scholar]
- 20.Poot M, Hoehn H, Runger TM, Martin GM. 1992. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res 202:267–273. doi: 10.1016/0014-4827(92)90074-I. [DOI] [PubMed] [Google Scholar]
- 21.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. 2004. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 22.Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA. 2004. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell 14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Cheung HH, Liu X, Canterel-Thouennon L, Li L, Edmonson C, Rennert OM. 2014. Telomerase protects Werner syndrome lineage-specific stem cells from premature aging. Stem Cell Rep 2:534–546. doi: 10.1016/j.stemcr.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilson E, Geli V. 2007. How telomeres are replicated. Nat Rev Mol Cell Biol 8:825–838. doi: 10.1038/nrm2259. [DOI] [PubMed] [Google Scholar]
- 25.Pfeiffer V, Lingner J. 2013. Replication of telomeres and the regulation of telomerase. Cold Spring Harb Perspect Biol 5:a010405. doi: 10.1101/cshperspect.a010405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schawalder J, Paric E, Neff NF. 2003. Telomere and ribosomal DNA repeats are chromosomal targets of the Bloom syndrome DNA helicase. BMC Cell Biol 4:15. doi: 10.1186/1471-2121-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. 2015. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol 210:191–208. doi: 10.1083/jcb.201410061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann M, Kibe T, Kabir S, de Lange T. 2014. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev 28:2477–2491. doi: 10.1101/gad.251611.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barefield C, Karlseder J. 2012. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res 40:7358–7367. doi: 10.1093/nar/gks407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunkel TA, Burgers PM. 2008. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol 18:521–527. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balakrishnan L, Bambara RA. 2013. Okazaki fragment metabolism. Cold Spring Harb Perspect Biol 5:a010173. doi: 10.1101/cshperspect.a010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balakrishnan L, Bambara RA. 2011. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. J Biol Chem 286:6865–6870. doi: 10.1074/jbc.R110.209502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgers PM. 2009. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem 284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards DN, Machwe A, Wang Z, Orren DK. 2014. Intramolecular telomeric G-quadruplexes dramatically inhibit DNA synthesis by replicative and translesion polymerases, revealing their potential to lead to genetic change. PLoS One 9:e80664. doi: 10.1371/journal.pone.0080664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lormand JD, Buncher N, Murphy CT, Kaur P, Lee MY, Burgers P, Wang H, Kunkel TA, Opresko PL. 2013. DNA polymerase delta stalls on telomeric lagging strand templates independently from G-quadruplex formation. Nucleic Acids Res 41:10323–10333. doi: 10.1093/nar/gkt813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balakrishnan L, Bambara RA. 2013. Flap endonuclease 1. Annu Rev Biochem 82:119–138. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Podust VN, Chang LS, Ott R, Dianov GL, Fanning E. 2002. Reconstitution of human DNA polymerase delta using recombinant baculoviruses: the p12 subunit potentiates DNA polymerizing activity of the four-subunit enzyme. J Biol Chem 277:3894–3901. doi: 10.1074/jbc.M109684200. [DOI] [PubMed] [Google Scholar]
- 38.Pike JE, Henry RA, Burgers PM, Campbell JL, Bambara RA. 2010. An alternative pathway for Okazaki fragment processing: resolution of fold-back flaps by Pif1 helicase. J Biol Chem 285:41712–41723. doi: 10.1074/jbc.M110.146894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossi ML, Bambara RA. 2006. Reconstituted Okazaki fragment processing indicates two pathways of primer removal. J Biol Chem 281:26051–26061. doi: 10.1074/jbc.M604805200. [DOI] [PubMed] [Google Scholar]
- 40.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. 2013. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S, Sommers JA, Wu L, Bohr VA, Hickson ID, Brosh RM Jr. 2004. Stimulation of flap endonuclease-1 by the Bloom's syndrome protein. J Biol Chem 279:9847–9856. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- 42.Brosh RM Jr, Driscoll HC, Dianov GL, Sommers JA. 2002. Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry 41:12204–12216. doi: 10.1021/bi026031j. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Bambara RA. 2005. Human Bloom protein stimulates flap endonuclease 1 activity by resolving DNA secondary structure. J Biol Chem 280:5391–5399. doi: 10.1074/jbc.M412359200. [DOI] [PubMed] [Google Scholar]
- 44.Saharia A, Guittat L, Crocker S, Lim A, Steffen M, Kulkarni S, Stewart SA. 2008. Flap endonuclease 1 contributes to telomere stability. Curr Biol 18:496–500. doi: 10.1016/j.cub.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung L, Onyango D, Guo Z, Jia P, Dai H, Liu S, Zhou M, Lin W, Pang I, Li H, Yuan YC, Huang Q, Zheng L, Lopes J, Nicolas A, Chai W, Raz D, Reckamp KL, Shen B. 2015. The FEN1 E359K germline mutation disrupts the FEN1-WRN interaction and FEN1 GEN activity, causing aneuploidy-associated cancers. Oncogene 34:902–911. doi: 10.1038/onc.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen B, Nolan JP, Sklar LA, Park MS. 1996. Essential amino acids for substrate binding and catalysis of human flap endonuclease 1. J Biol Chem 271:9173–9176. doi: 10.1074/jbc.271.16.9173. [DOI] [PubMed] [Google Scholar]
- 47.Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI, Rebres R, Roach T, Seaman W, Simon MI, Fraser ID. 2006. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc Natl Acad Sci U S A 103:13759–13764. doi: 10.1073/pnas.0606179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J. 2007. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci U S A 104:2205–2210. doi: 10.1073/pnas.0609410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hisama FM, Chen YH, Meyn MS, Oshima J, Weissman SM. 2000. WRN or telomerase constructs reverse 4-nitroquinoline 1-oxide sensitivity in transformed Werner syndrome fibroblasts. Cancer Res 60:2372–2376. [PubMed] [Google Scholar]
- 50.Oshima J, Sidorova JM, Monnat RJ Jr. 2016. Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev 15:S1568–1637(16)30026-5. doi: 10.1016/j.arr.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu Z, Perumal SK, Yue H, Benkovic SJ. 2012. The human lagging strand DNA polymerase delta holoenzyme is distributive. J Biol Chem 287:38442–38448. doi: 10.1074/jbc.M112.404319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langston LD, O'Donnell M. 2008. DNA polymerase delta is highly processive with proliferating cell nuclear antigen and undergoes collision release upon completing DNA. J Biol Chem 283:29522–29531. doi: 10.1074/jbc.M804488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamdan SM, Loparo JJ, Takahashi M, Richardson CC, van Oijen AM. 2009. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 457:336–339. doi: 10.1038/nature07512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinha NK, Morris CF, Alberts BM. 1980. Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J Biol Chem 255:4290–4293. [PubMed] [Google Scholar]
- 55.Phan AT, Mergny JL. 2002. Human telomeric DNA: G-quadruplex, i-motif and Watson-Crick double helix. Nucleic Acids Res 30:4618–4625. doi: 10.1093/nar/gkf597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mohaghegh P, Karow JK, Brosh RM Jr, Bohr VA, Hickson ID. 2001. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res 29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamath-Loeb AS, Loeb LA, Johansson E, Burgers PM, Fry M. 2001. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J Biol Chem 276:16439–16446. doi: 10.1074/jbc.M100253200. [DOI] [PubMed] [Google Scholar]
- 58.Fry M, Loeb LA. 1999. Human Werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J Biol Chem 274:12797–12802. doi: 10.1074/jbc.274.18.12797. [DOI] [PubMed] [Google Scholar]
- 59.Bai Y, Murnane JP. 2003. Telomere instability in a human tumor cell line expressing a dominant-negative WRN protein. Hum Genet 113:337–347. doi: 10.1007/s00439-003-0972-y. [DOI] [PubMed] [Google Scholar]
- 60.Chatterjee S, Zagelbaum J, Savitsky P, Sturzenegger A, Huttner D, Janscak P, Hickson ID, Gileadi O, Rothenberg E. 2014. Mechanistic insight into the interaction of BLM helicase with intra-strand G-quadruplex structures. Nat Commun 5:5556. doi: 10.1038/ncomms6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamath-Loeb AS, Shen JC, Schmitt MW, Loeb LA. 2012. The Werner syndrome exonuclease facilitates DNA degradation and high fidelity DNA polymerization by human DNA polymerase delta. J Biol Chem 287:12480–12490. doi: 10.1074/jbc.M111.332577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma S, Sommers JA, Gary RK, Friedrich-Heineken E, Hubscher U, Brosh RM Jr. 2005. The interaction site of flap endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res 33:6769–6781. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li B, Jog SP, Reddy S, Comai L. 2008. WRN controls formation of extrachromosomal telomeric circles and is required for TRF2DeltaB-mediated telomere shortening. Mol Cell Biol 28:1892–1904. doi: 10.1128/MCB.01364-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laud PR, Multani AS, Bailey SM, Wu L, Ma J, Kingsley C, Lebel M, Pathak S, DePinho RA, Chang S. 2005. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev 19:2560–2570. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pirzio LM, Pichierri P, Bignami M, Franchitto A. 2008. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol 180:305–314. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kao HI, Bambara RA. 2003. The protein components and mechanism of eukaryotic Okazaki fragment maturation. Crit Rev Biochem Mol Biol 38:433–452. doi: 10.1080/10409230390259382. [DOI] [PubMed] [Google Scholar]
- 67.Zheng L, Shen B. 2011. Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol 3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. 1999. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol Cell Biol 19:8361–8371. doi: 10.1128/MCB.19.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J Virol 72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li B, Reddy S, Comai L. 2009. Sequence-specific processing of telomeric 3′ overhangs by the Werner syndrome protein exonuclease activity. Aging 1:289–302. doi: 10.18632/aging.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.