

Abstract
Observational studies have shown that elevated systolic blood pressure (SBP) is associated with future onset of type 2 diabetes, but whether this association is causal is not known. We applied the Mendelian randomization framework to evaluate the causal hypothesis that elevated SBP increases risk for type 2 diabetes. We used 28 genetic variants associated with SBP and evaluated their impact on type 2 diabetes using a European-centric meta-analysis comprising 37,293 case and 125,686 control subjects. We found that elevation of SBP levels by 1 mmHg due to our genetic score was associated with a 2% increase in risk of type 2 diabetes (odds ratio 1.02, 95% CI 1.01–1.03, P = 9.05 × 10−5). To limit confounding, we constructed a second score based on 13 variants exclusively associated with SBP and found a similar increase in type 2 diabetes risk per 1 mmHg of genetic elevation in SBP (odds ratio 1.02, 95% CI 1.01–1.03, P = 1.48 × 10−3). Sensitivity analyses using multiple, alternative causal inference measures and simulation studies demonstrated consistent association, suggesting robustness of our primary observation. In line with previous reports from observational studies, we found that genetically elevated SBP was associated with increased risk for type 2 diabetes. Further work will be required to elucidate the biological mechanism and translational implications.
Introduction
Metabolic syndrome is defined by a collection of risk factors that strongly predict the onset of type 2 diabetes later in life (1). A component of this syndrome—hypertension—is decidedly associated with type 2 diabetes (2). Beyond association with this clinical classification, observational studies have also demonstrated that, measured as a continuous trait, systolic blood pressure (SBP) is associated with type 2 diabetes, with a 1-mmHg increase associated with a 1%–4% increase in type 2 diabetes risk (3–6). It would desirable for mechanistic and therapeutic reasons to better understand the relationship—causal or correlation—between these factors (7,8).
Investigating a causal relationship between blood pressure and type 2 diabetes is a difficult problem. Observational studies are limited in their ability to investigate causality due to confounding factors such as obesity and use of medications and due to issues of reverse causality. The randomized clinical trial can assess causality, and such studies have been used to demonstrate the therapeutic benefit of blood pressure–lowering medication on cardiovascular events (9–11). Because elevated blood pressure is causally related to cardiovascular disease, a well-controlled, ethical placebo-based randomized intervention study for the effects of antihypertension medication on populations with type 2 diabetes is inconceivable. Previous clinical trials designed to assay the diabetic properties of antihypertension medications follow an “add-on” regimen of these medications in the control arm. Thus, in well-designed studies, “untreated” groups are simply treated less often with a specific therapeutic (12,13). Although powered to study drug efficacy, such trials are weakly powered to evaluate causality between blood pressure and type 2 diabetes. Blood pressure is a complex trait with multiple etiological underpinnings (e.g., insulin resistance, obesity, inflammation, etc.). It may be that some, all, or none of these underlying factors directly relate to type 2 diabetes etiology. Thus, investigating specific, physiological pathways that putatively elevate blood pressure and type 2 diabetes risk is another obstacle to progress.
Recent advances in our genetic understanding of blood pressure provide a new avenue to characterize this relationship: the approach of Mendelian randomization (MR). MR is a form of instrumental variable analysis whereby selected genetic variants related to a specific exposure of interest are used to statistically evaluate a causal hypothesis between the exposure and an outcome (14,15). Because genotypes assort randomly during the process of meiosis and genotypes precede phenotype, MR addresses the issue of reverse causality. Confounding can be addressed in part by selecting genetic factors that are exclusively associated with SBP (16–18). Multiple variants can be combined into a genetic risk score (GRS) and subsequently used for hypothesis testing to provide a causal effect estimate of genetically elevated SBP on risk to type 2 diabetes. As it is difficult to guarantee that each variant used in a GRS is a statistically valid instrument, it is also desirable to quantify and control potential confounding bias that may impact the inference. Toward this end, Egger regression and a novel weighted-median effect estimator have been proposed as sensitivity analyses for causal effects estimated from traditional MR (19,20).
Here, we present evidence that genetically elevated SBP increases the risk of type 2 diabetes. To perform our test, we meta-analyzed case and control cohorts to summarize the association with type 2 diabetes, evaluated a set of genetic variants reproducibly associated with SBP, and used multiple MR inference techniques robust to assumptions of bias in the analysis.
Research Design and Methods
Selection of Genetic Variants for the SBP Genotype Risk Score
We developed two instruments for MR analysis. First, we selected 26 single nucleotide polymorphisms (SNPs) with established association from a recent meta-analysis of SBP, spanning 69,395 individuals from a three-stage validation experiment using association data from up to 133,661 additional individuals (16). We added three variants (rs13359291, rs1563788, and rs2014912) reported in a recent genome-wide association study (GWAS) of blood pressure–related phenotypes in up to 320,251 individuals, and an additional SNP (rs12946454) drawn from a combined analysis of Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium results with an additional study of 34,433 subjects (17,18). Two SNPs were eliminated due to associations with potential confounding factors (BMI, coronary heart disease, and type 1 diabetes), resulting in 28 SNPs for our “expanded” genetic instrument. Our “conservative” instrument comprised 13 SNPs used by Yin and Voight (21), which were selected based on criteria to minimize linkage disequilibrium (LD) among SNPs and to reduce potential confounding from cardiometabolic phenotypes (Supplementary Table 1). All SNPs used were genome-wide significantly associated with SBP and were not in strong LD with one another (CEU r2 < 0.05). Where possible, we obtained effect sizes for SNPs from the winner’s curse–adjusted estimated effects (see Supplementary Table A.2 in Ehret et al. [16]). We scrutinized rs2521501, which fell into a region of established association with T2D (22,23). However, tags for this association (rs12899811 and rs8042680) were not in strong LD with rs2521501 (CEU r2 < 0.01), suggesting our selected SBP-associated SNP is independent of these tags. Moreover, several of our casual effect estimators are designed to be robust against the potential invalidity of specific instruments, so we opted to retain this SNP in our primary analysis. We note that a sensitivity analysis that excluded rs2521501 did not alter our central findings (Supplementary Tables 2A–C).
Obtaining Summary Association Data for Type 2 Diabetes
We combined type 2 diabetes association data obtained from the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium, which included stage 1 GWAS meta-analysis and stage 2 Metabochip genotyping. This comprised up to 34,840 case and 114,981 control subjects (24). To these data, we incorporated summary association data from three additional cohorts (PennCATH, MedStar, and the FINRISK study), totaling an additional 2,453 case and 10,705 control subjects (25–27). Odds ratios (ORs) with SEs for each SNP were generated using inverse-variance weighted (IVW) fixed-effects meta-analysis. All data sets were based on summary-level (not individual-level) data. Our causal inference tests assumed that the samples used to estimate the genetic effects for SBP and type 2 diabetes are nonoverlapping. As we used the effect estimates for SBP obtained in the replication (not discovery), the sample overlap was slight (∼4%–5%).
Causal Effect Estimate via Genotype Risk Score and Heterogeneity Analysis
We estimated a causal effect from our SNP collections using the GRS method, as previously described (28). Briefly, under the standard MR assumptions and provided that each SNP is not strongly linked to one another, a causal effect (α̂) and error [SE(α̂)] between SBP and type 2 diabetes can be estimated by (28):
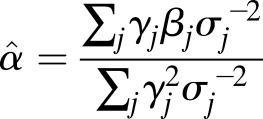 |
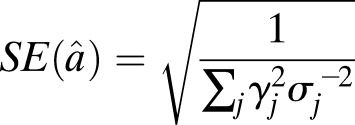 |
where for all j SNPs, βj represents the estimated natural log odds effect of the jth SNP on type 2 diabetes, σj represents the standard error on the log odds effect of the jth SNP on type 2 diabetes, and γj represents the effect of the SNP on SBP (i.e., the weight) in millimeters of mercury. Assuming 17.87 mmHg per SD of SBP, we estimated ∼1% and ∼0.5% variance explained in SBP levels and F-statistics of 11.6 and 11.9 for the n = 28 and n = 13 variant scores, respectively (28). Sample size used for the F-statistic calculations was estimated as the median across SNPs, calculated for each given the allele frequencies, OR, and SE of the type 2 diabetes association (Supplementary Tables 3 and 4). We also computed a causal estimate using an unweighted GRS (i.e., weights set to one for all variants). As for the weighted scores, the type 2 diabetes log-odds was polarized to the SBP-increasing allele. The Cochran Q test, implemented in the gtx package (v0.0.8) in R (v3.3.0), was used to assess heterogeneity for each risk score (Supplementary Table 2B). Estimates for the effect of SBP on type 2 diabetes for individual SNPs was calculated using Eqs. 1 and 2. The number of SNPs reporting a positive association between SBP and type 2 diabetes was totaled, and statistical significance was calculated using the binom.test function in R (v3.2.1).
Causal and Bias Estimation Using Egger Regression and Weighted-Median Estimation
Egger regression analyses were performed in R (v3.3.0, code available at https://github.com/raikens1/T2D_MR), and details on the specific approach are described elsewhere (20). Briefly, Egger regression is analogous to the causal effect approach that uses IVW regression on the summary estimates of the effect of the biomarker versus the effect on the outcome. In traditional IVW, the intercept term is fixed at zero, which provides an estimate of causal effects that is asymptotically equivalent to the GRS method (20). Egger regression instead estimates an intercept term in the regression. A significant, nonzero intercept implies directional bias among the selected genetic instruments, and the resulting effect estimate is a valid causal effect in the context of estimated directional bias. Significance tests for both approaches were performed using the Student t test distribution, and CIs for the estimated causal effects were obtained via bootstrapping. We also used a simulation approach to explore the sensitivity of Egger regression for detecting different bias levels in our data. By constructing simulated data sets (n = 1,000) with a preset effect of SBP on type 2 diabetes, we noted the behavior of Egger regression estimates of SBP effect and average bias under different patterns of pleiotropic bias (see Supplementary Data and sample code).
A weighted-median method for assessing causal effect is unbiased asymptotically (as sample size increases) and requires at least 50% of the weight for the score to derive from valid instruments (19). Weighted-median estimation was carried out in R (v3.3.0) using sample code previously made available (19). SE and 95% CI were estimated by bootstrapping.
Exploring Potential for Bias Due to Adiposity Adjustments via Simulation
To explore potential sources of bias due to adjustments in the primary GWAS analysis (16), we used the MR predictor tool (29) to generate sets of simulated phenotypic data for 150,000 individuals under a model whereby SBP and BMI increase type 2 diabetes risk. To construct our simulations, estimates of type 2 diabetes prevalence (30), association of SBP with BMI (31), and BMI-associated type 2 diabetes risk (32) were drawn from the literature. We then used PLINK (v1.07) (33) to generate linear SBP association estimates for each of the 13 SNPs in our conservative instrument set (1,000 simulations). To quantify the deviation under different models, for each SNP we measured the deviation between the true SBP effect and the estimate for each simulation, averaged over all 13 loci (see Supplementary Data, sample code is available at https://github.com/raikens1/T2D_MR). Differences in the distributions were evaluated for significance using a two-sided t test in R (v3.3.0).
Results
Genetically Elevated SBP Associates With Type 2 Diabetes
We calculated a causal effect estimate with the GRS method, using an expanded instrument with the greatest number of SBP associations (n = 28 SNPs) and our European meta-analysis association for type 2 diabetes (see research design and methods). We found that a 1-mmHg genetic elevation of SBP was associated with a ∼2% increase in type 2 diabetes risk (OR 1.018, 95% CI 1.009–1.028, P = 9.1 × 10−5) (Fig. 1A, Table 1, and Supplementary Table 3). Next, we applied a more conservative genetic instrument comprising SNPs exclusively associated with SBP (n = 13) to evaluate a causal effect for type 2 diabetes. Consistent with the above, we found that a 1-mmHg genetic increase in SBP associated with a ∼2% increase in type 2 diabetes risk (OR 1.021, 95% CI 1.008–1.033, P = 1.5 × 10−3) (Fig. 1B, Table 1, and Supplementary Table 4). Our causal genetic estimates are consistent with observational reports suggesting a 1%–4% increase in hazard of type 2 diabetes for a 1-mmHg elevation in SBP (3–6).
Figure 1.

Summary of association between genetic variants and type 2 diabetes with causal effect estimated via GRS. Genetic variant associations from the expanded instrument of 28 SBP-associated variants (A) and the conservative instrument of 13 SNPs (B). ORs and 95% CI are shown in units per 1-mmHg risk increase. P values were calculated using a χ2 distribution.
Table 1.
Summary statistics for genetic instruments used for causal inference analysis for SBP to type 2 diabetes
| Expanded set (n = 28) | Conservative set (n = 13) | |||||
|---|---|---|---|---|---|---|
| OR* | 95% CI | P | OR* | 95% CI | P | |
| Cumulative GRS†‡ |
1.018 |
1.009–1.028 |
9.1 × 10−5 |
1.021 |
1.008–1.033 |
1.5 × 10−3 |
| Unweighted GRS†‡ |
1.008 |
1.003–1.013 |
2.0 × 10−3 |
1.008 |
1.001–1.015 |
0.018 |
| Weighted-median GRS§ |
1.018 |
1.006–1.033 |
< 0.05 |
1.020 |
1.003–1.039 |
< 0.05 |
| Egger regression (causal estimate)§|| |
1.043 |
1.006–1.064 |
5.5 × 10−3 |
1.052 |
1.004–1.086 |
0.018 |
| Egger regression (bias estimate) |
−0.014 |
— |
0.08 |
−0.018 |
— |
0.10 |
| Heterogeneity (Cochran Q) | — | — | 0.2 | — | — | 0.59 |
*ORs are given in units of fold increase in type 2 diabetes risk per unit increase in mmHg, except the unweighted score, which is given in unit increase per allele.
†95% CI from normal distribution.
‡P value from χ2 test.
§95% CI calculated by bootstrapping.
||P value from Student t test.
Type 2 Diabetes Risk Estimates From Individual SNPs and Risk Score Heterogeneity
To determine if an individual SNP used in our score drove this result, we calculated effect estimates for all variants used in our risk scores on type 2 diabetes risk. A total of 19 out of 28 SNPs in our expanded set reported a positive, consistent association between the allele increasing SBP and type 2 diabetes, and 9 out of 13 SNPs for the conservative score (binomial test P = 0.09 and P = 0.27 for the expanded and conservative sets, respectively) (Fig. 1 and Supplementary Tables 2B, 3, and 4). While 3 of 28 SNPs in the expanded set and 2 of 13 SNPs in the conservative set were nominally associated with type 2 diabetes (P < 0.05), this was not unusual (binomial test P = 0.16 and P = 0.14, respectively). A formal test for heterogeneity using Cochran Q did not detect evidence for heterogeneity (P = 0.2 and P = 0.59 for the expanded and conservative scores, respectively) (Table 1 and Supplementary Table 2B). These results suggest that the effects estimated from our GRSs derived above are not driven by outlier genetic variants with strong effects on type 2 diabetes risk (Fig. 1 and Supplementary Fig. 1).
Sensitivity Analysis for SBP Genetic Instruments Using Alternative Causal Inference Methods
We next performed an Egger regression analysis using the expanded and conservative SBP genetic variant sets described above. This approach provides a point estimate and test of directional bias that might result due to pleiotropy with causal factors that may confound our interpretation of causality, in addition to a causal estimate in the context of estimated bias (20). Although less well powered than the GRS method, this method provides an appropriate sensitivity analysis to help interpret results (19): if our result was completely explained by directional bias due to a confounding (but unmeasured) factor, this effect could be discovered and our results reinterpreted in this light.
After applying the Egger regression approach, we did not detect bias for either genetic score (P = 0.08 and P = 0.10 for the expanded and conservative SNP sets, respectively) (Table 1 and Supplementary Table 2C). This itself may not be surprising, as empirically it has been shown that this test is modestly powered to detect bias when present (Supplementary Data and Supplementary Tables 5 and 6) (20). We noted that the direction of estimated bias was negative in both cases, suggesting that the average effect of putative bias present across the selected genetic variants results in an underestimation of the causal effect on type 2 diabetes risk (Supplementary Data and Supplementary Fig. 2). We next generated a causal effect estimate using Egger regression for each of our expanded and conservative SNP sets. We found that a 1-mmHg genetic increase in SBP was associated with a 4%–5% increase in type 2 diabetes risk for both the expanded (OR 1.04, bootstrap 95% CI 1.006–1.064, P = 5.5 × 10−3) and conservative SNP sets (OR 1.05, bootstrap 95% CI 1.004–1.086, P = 1.8 × 10−2) (Table 1 and Supplementary Table 2C). Simulations we performed to understand the impact of negative bias on the analysis (Supplementary Data) suggest that the effect estimates reported by Egger regression are unbiased on average, although the variance in estimation is higher than when no bias is at work (Supplementary Fig. 3). This is consistent with previous simulation studies for Egger regression (20).
We next obtained a weighted-median estimate for the causal effect. This method provides a point estimate analogous to the GRS that is unbiased asymptotically and only requires 50% or more of the weight for the score to derive from valid instruments, a less stringent requirement compared with the GRS score method (19). For both expanded and conservative SNP sets, the weighted-median estimated for 1-mmHg genetic elevation of SBP on type 2 diabetes risk agreed with estimates from our previous analyses (OR 1.02 and P < 0.05 for both) (Table 1 and Supplementary Table 2A). These data suggest that 1) our result cannot be fully explained by an unknown confounding factor contributing positive, directional bias to our causal estimate, and 2) if bias is present, the estimated causal effect of SBP on type 2 diabetes may be greater than reported by our GRS.
Estimate of Causal Effects Are Robust to Blood Pressure Effect Estimates
To investigate if our inference was robust to the choice of weights used in our GRS analysis, we estimated a causal effect using an unweighted GRS for both the expanded and conservative SNP sets. This score can be interpreted as the average increase in odds of type 2 diabetes per SBP-increasing allele and should be robust to concerns on the proper selection of weights used above (i.e., sample overlap between genetic association studies), albeit with a reduction in statistical power (29). We found that type 2 diabetes risk was increased by ∼1% per SBP-increasing allele for both the conservative (OR 1.008, 95% CI 1.001–1.015, P = 1.8 × 10−2) and expanded (OR 1.008, 95% CI 1.003–1.013, P = 2.0 × 10−3) risk score (Table 1 and Supplementary Table 2A). Using simulations, we also evaluated if adjustment for adiposity and/or exclusion of type 2 diabetes case subjects contributing to the SBP association scan could contribute bias to the SBP effects used in our risk scores, but we did not observe substantial effects (Supplementary Data and Supplementary Figs. 4–6). These results suggest that our findings are somewhat robust to the choice of SBP effects used in our risk scores.
Discussion
Causal inference is one of the most challenging problems in medicine and biology. Here, we report that a theoretic 1-mmHg genetic increase in SBP translated into 2% increased risk of type 2 diabetes, consistent with evidence obtained from observational studies of these traits. We also performed sensitivity analyses to demonstrate the robustness of our estimates to the chosen method or sample exclusion filters (16), employed MR analyses sensitive to directional pleiotropic bias, incomplete validity of genetic variants used in our score, and weights used in the score. Our report follows an initial screen that reported an initially positive association (21). Compared with that study, we increased the number of samples contributing to the type 2 diabetes association, expanded the list of SBP-associated genetic variants used for testing, and performed additional sensitivity analyses using three MR methods. A recent study reported a MR analysis between SBP and type 2 diabetes in ∼96,000 subjects (5). However, this study reported an effect opposite of observational epidemiology, with limited interpretation power owing to the genetic variants used (n = 5, compared with the n = 13 or 28 we used) and sample size (2,859 incident cases, compared with 37,293 subjects with type 2 diabetes in this report).
Our results do not indicate a mechanism, and speculation on that score should be interpreted cautiously. One possibility is that SBP directly increases risk to type 2 diabetes. One suggested model is that elevated blood pressure manifests as vasoconstriction, impairing blood flow and glucose disposal in peripheral tissues (e.g., skeletal muscles or adipose) and subsequently causes type 2 diabetes (34). This model could explain, in part, why therapeutic perturbations of the renin or angiotensin systems could result in putative protection against type 2 diabetes (35). In support of this model, previous studies have shown that use of antihypertension drugs in type 2 diabetes mouse models lower blood pressure, body weight, and plasma glucose levels and improve insulin sensitivity and resistance, along with additional, favorable metabolic parameters (36,37).
The more likely possibility is that our genetic score for SBP is a close proxy for another etiological factor. One potential candidate risk factor is insulin resistance. The long-standing view is that insulin resistance is the first “step” in a metabolic cascade (7,8) that ultimately results in hyperinsulinemia and elevated blood pressure through several mechanisms, including enhanced sodium absorption in distal nephrons, sodium retention, vascular defects, or others (38). But if our score captured insulin resistance by proxy, we would expect to observe an association with other insulin resistance traits, as previously demonstrated with fasting insulin levels (39). However, the conservative risk score we used was not associated with triglycerides, HDL cholesterol, or fasting insulin levels, all hallmarks of insulin resistance phenotypes orthogonal to obesity. A second candidate could be inflammation. Inflammation and biomarkers associated with inflammation are associated with hypertension (40) and type 2 diabetes (41,42); a causal relationship here could also explain our result. However, previous MR studies for a causal relationship here have yielded inconsistent results (43). Still, we cannot rule out that our genetic score implicates a previously unknown inflammatory pathway common to both conditions.
It is important to consider other sources of bias that may contribute to our observation. Because our estimates of SBP associations used in our GRS are taken from a large meta-analysis, conditioning on adiposity or type 2 diabetes could result in associations that could confound our interpretation of causality. This effect is referred to as collider bias, which is the induction of false associations between two or more variables due to conditioning on a common cause (Supplementary Data and Supplementary Fig. 6) (44). There are two possibilities to consider in which collider bias would result in inaccurate estimates of SBP association for a given variant: 1) if elevated SBP increased BMI, then conditioning on BMI will cause a BMI-elevating variant to falsely associate with SBP, and 2) if elevated SBP causes type 2 diabetes, excluding subjects with type 2 diabetes would cause type 2 diabetes–causing variants to falsely associate with SBP. However, each is addressable. It is understood that elevated BMI raises blood pressure, but there is little evidence to suggest that the reverse is true (31,45). This indicates that the first case is unlikely. The second case could be of issue if a substantial proportion of the cohorts used to identify SBP-associated variants systematically excluded diabetes case subjects. However, upon closer investigation of cohorts participating in the SBP association study, results there were based heavily on prospective cohort studies without a disease restriction, with <5% of the total sample sizes for the GWAS meta-analyses drawn from control cohorts for type 2 diabetes (16). Thus, each SNP in our genetic instrument is quite close to the population-level estimate of SBP effect rather than a false association to type 2 diabetes induced by potential collider bias. Our simulation analyses also support this supposition.
Two additional limitations of our study involve the participants in the type 2 diabetes meta-analysis studies. The first involves antihypertension medication use: if patients with diabetes were more likely to be using antihypertension medications relative to control subjects, and if antihypertension therapeutics actually increased the risk for type 2 diabetes, then we might falsely observe an association between SBP and type 2 diabetes. In particular, there is increasing evidence that the use of β-blockers and thiazide diuretics is associated with increased risk of type 2 diabetes (46,47). A simulation analysis suggested that this type of effect could result in bias to our GRS, but would also be detectable as positive bias through Egger regression and could be accounted for in our causal inference (Supplementary Table 7). In the real data, we observed that Egger regression did not detect the presence of bias, and the magnitude of that bias was negative rather than positive bias (Table 1), the opposite of what would be expected from this type of effect. A second issue that could arise is due to ascertainment of coronary heart disease (CHD) cases among subjects with type 2 diabetes. Because patients with type 2 diabetes are at increased risk for CHD and because SBP is causal for CHD, our observed association could reflect an excess number of residual and/or undiagnosed participants with CHD in patients with type 2 diabetes. In this scenario, it would follow that a GRS combining variants reproducibly associated with CHD would also associate with type 2 diabetes (48), which we do not observe (n = 41 variants, GRS P = 0.16, data not shown). It is apparent that prospective cohort studies would certainly be of benefit in further clarifying these potential issues, and our findings certainly motivate such analyses in the future.
Ultimately, the translational benefits based on these results are not obvious. This is due to the complexity that different blood pressure–lowering medications may ultimately have on type 2 diabetes risk, through on- or off-target effects (46,47). However, our findings here do make a specific prediction: genetic pathways for SBP either intrinsically or through existing and/or as-yet-undescribed mechanisms relate to type 2 diabetes. This prediction adds increasing weight to both identify and characterize the causal genes underlying blood pressure associations as a strategy toward novel therapeutics for both hypertension and type 2 diabetes.
Supplementary Material
Article Information
Acknowledgments. The authors are indebted to Tim Frayling (University of Exeter, Exeter, U.K.) and three anonymous reviews for helpful comments on the manuscript.
Funding. R.C.A. is grateful for support from the Institute of Translational Medicine and Therapeutics summer research fellowship and the Summer Undergraduate Internship Program at the Perelman School of Medicine, University of Pennsylvania. B.F.V. was supported by grants from the American Heart Association (13SDG14330006) and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (R01DK101478). V.S. was supported by the Finnish Foundation for Cardiovascular Research.
The funding bodies had no role in the design of the study, results, or decision to publish.
Author Contributions. R.C.A. and B.F.V. conceived and designed the study, acquired the data, drafted the tables and figures, and wrote the first draft of the manuscript. R.C.A., W.Z., and B.F.V. analyzed the data. R.C.A., W.Z., D.S., M.P.R., S.E.E., E.T., V.S., and B.F.V. edited the manuscript. M.P.R., S.E.E., E.T., and V.S. provided additional data. B.F.V. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db16-0868/-/DC1.
References
- 1.Wilson PWF, D’Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 2005;112:3066–3072 [DOI] [PubMed] [Google Scholar]
- 2.Wei GS, Coady SA, Goff DC Jr, et al. . Blood pressure and the risk of developing diabetes in African Americans and whites: ARIC, CARDIA, and the Framingham Heart study. Diabetes Care 2011;34:873–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NR; Anglo-Scandinavian Cardiac Outcomes Trial Investigators . Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial--Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008;31:982–988 [DOI] [PubMed] [Google Scholar]
- 4.Lindholm LH, Ibsen H, Borch-Johnsen K, et al.; LIFE study group . Risk of new-onset diabetes in the Losartan Intervention For Endpoint reduction in hypertension study. J Hypertens 2002;20:1879–1886 [DOI] [PubMed] [Google Scholar]
- 5.Emdin CA, Anderson SG, Woodward M, Rahimi K. Usual blood pressure and risk of new-onset diabetes: evidence from 4.1 million adults and a meta-analysis of prospective studies. J Am Coll Cardiol 2015;66:1552–1562 [DOI] [PMC free article] [PubMed]
- 6.Marott SCW, Nordestgaard BG, Tybjærg-Hansen A, Benn M. Components of the metabolic syndrome and risk of type 2 diabetes. J Clin Endocrinol Metab 2016;101:3212–3221 [DOI] [PubMed] [Google Scholar]
- 7.DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991;14:173–194 [DOI] [PubMed] [Google Scholar]
- 8.Ferrannini E, Buzzigoli G, Bonadonna R, et al. . Insulin resistance in essential hypertension. N Engl J Med 1987;317:350–357 [DOI] [PubMed] [Google Scholar]
- 9.Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration . Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000;356:1955–1964 [DOI] [PubMed] [Google Scholar]
- 10.Karnes JH, Cooper-DeHoff RM. Antihypertensive medications: benefits of blood pressure lowering and hazards of metabolic effects. Expert Rev Cardiovasc Ther 2009;7:689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins R, Peto R, MacMahon S, et al. Blood pressure, stroke, and coronary heart disease. Part 2, short-term reductions in blood pressure: overview of randomised drug trials in their epidemiological context. Lancet 1990;335:827–838 [DOI] [PubMed]
- 12.ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group, The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002;288:2981–2997 [DOI] [PubMed] [Google Scholar]
- 13.Julius S, Kjeldsen SE, Weber M, et al.; VALUE trial group . Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet 2004;363:2022–2031 [DOI] [PubMed] [Google Scholar]
- 14.Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med 2008;5:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernán MA, Robins JM. Instruments for causal inference: an epidemiologist’s dream? Epidemiology 2006;17:360–372 [DOI] [PubMed] [Google Scholar]
- 16.Ehret GB, Munroe PB, Rice KM, et al.; International Consortium for Blood Pressure Genome-Wide Association Studies; CARDIoGRAM consortium; CKDGen Consortium; KidneyGen Consortium; EchoGen consortium; CHARGE-HF consortium . Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011;478:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, Kelly TN, Saleheen D, Chambers JC et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet 2015;47:1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newton-Cheh C, Johnson T, Gateva V, et al.; Wellcome Trust Case Control Consortium . Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 2009;41:666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin P, Voight BF. MeRP: a high-throughput pipeline for Mendelian randomization analysis. Bioinformatics 2015;31:957–959 [DOI] [PubMed] [Google Scholar]
- 22.Voight BF, Scott LJ, Steinthorsdottir V, et al.; MAGIC investigators; GIANT Consortium . Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010;42:579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahajan A, Go MJ, Zhang W, et al.; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium . Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 2014;46:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris AP, Voight BF, Teslovich TM, et al.; Wellcome Trust Case Control Consortium; Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium . Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vartiainen E, Jousilahti P, Alfthan G, Sundvall J, Pietinen P, Puska P. Cardiovascular risk factor changes in Finland, 1972-1997. Int J Epidemiol 2000;29:49–56 [DOI] [PubMed] [Google Scholar]
- 26.Grant SFA, Thorleifsson G, Reynisdottir I, et al. . Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 2006;38:320–323 [DOI] [PubMed] [Google Scholar]
- 27.Kathiresan S, Voight BF, Purcell S, et al.; Myocardial Infarction Genetics Consortium. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 2009;41:334–341 [DOI] [PMC free article] [PubMed]
- 28.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 2016;35:1880–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voight BFMR. MR_predictor: a simulation engine for Mendelian randomization studies. Bioinformatics 2014;30:3432–3434 [DOI] [PubMed] [Google Scholar]
- 30.Cowie CC, Rust KF, Byrd-Holt DD, et al. . Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health and Nutrition Examination Survey 1999-2002. Diabetes Care 2006;29:1263–1268 [DOI] [PubMed] [Google Scholar]
- 31.Dudina A, Cooney MT, Bacquer DD, et al.; SCORE investigators . Relationships between body mass index, cardiovascular mortality, and risk factors: a report from the SCORE investigators. Eur J Cardiovasc Prev Rehabil 2011;18:731–742 [DOI] [PubMed] [Google Scholar]
- 32.Sheikh MA, Lund E, Braaten T. The predictive effect of body mass index on type 2 diabetes in the Norwegian Women and Cancer study. Lipids Health Dis 2014;13:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell S, Neale B, Todd-Brown K, et al. . PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blackburn DF, Wilson TW. Antihypertensive medications and blood sugar: theories and implications. Can J Cardiol 2006;22:229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 2001;37:1053–1059 [DOI] [PubMed] [Google Scholar]
- 36.Frantz EDC, Crespo-Mascarenhas C, Barreto-Vianna ARC, Aguila MB, Mandarim-de-Lacerda CA. Renin-angiotensin system blockers protect pancreatic islets against diet-induced obesity and insulin resistance in mice. PLoS One 2013;8:e67192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwai M, Kanno H, Tomono Y, et al. . Direct renin inhibition improved insulin resistance and adipose tissue dysfunction in type 2 diabetic KK-A(y) mice. J Hypertens 2010;28:1471–1481 [DOI] [PubMed] [Google Scholar]
- 38.Oktay AA, Akturk HK, Jahangir E. Diabetes mellitus and hypertension: a dual threat. Curr Opin Cardiol 2016;31:402–409 [DOI] [PubMed] [Google Scholar]
- 39.Yaghootkar H, Scott RA, White CC, et al. . Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 2014;63:4369–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-α) and essential hypertension. J Hum Hypertens 2005;19:149–154 [DOI] [PubMed] [Google Scholar]
- 41.Duncan BB, Schmidt MI, Pankow JS, et al.; Atherosclerosis Risk in Communities Study. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes 2003;52:1799–1805 [DOI] [PubMed]
- 42.Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 2004;25:4–7 [DOI] [PubMed] [Google Scholar]
- 43.Brunner EJ, Kivimäki M, Witte DR, et al. Inflammation, insulin resistance, and diabetes—Mendelian randomization using CRP haplotypes points upstream. PLoS Med 2008;5:e155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole SR, Platt RW, Schisterman EF, et al. . Illustrating bias due to conditioning on a collider. Int J Epidemiol 2010;39:417–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dua S, Bhuker M, Sharma P, Dhall M, Kapoor S. Body mass index relates to blood pressure among adults. N Am J Med Sci 2014;6:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam SKH, Owen A. Incident diabetes in clinical trials of antihypertensive drugs. Lancet 2007;369:1513–1514; author reply 1514–1515 [DOI] [PubMed] [Google Scholar]
- 47.Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007;369:201–207 [DOI] [PubMed] [Google Scholar]
- 48.Nikpay M, Goel A, Won H-H, et al.; CARDIoGRAMplusC4D Consortium . A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.