

SUMMARY
Targeted mass spectrometry assays for protein quantitation monitor peptide surrogates, which are easily multiplexed to target many peptides in a single assay. However, these assays have generally not taken advantage of sample multiplexing which allows up to 10 analyses to occur in parallel. We present a two-dimensional multiplexing workflow that utilizes synthetic peptides for each protein to prompt the simultaneous quantification of >100 peptides from up to 10 mixed sample conditions. We demonstrate that targeted analysis of unfractionated lysates (2-hr) accurately reproduces the quantification of fractionated lysates (72-hr analysis), while obviating the need for peptide detection prior to quantification. We targeted 131 peptides corresponding to 69 proteins across all 60 National Cancer Institute cell lines in biological triplicate, analyzing 180 samples in only 48 hours (the equivalent of 16 min/sample). These data further elucidated a correlation between the expression of key proteins and their cellular response to drug treatment.
Keywords: proteomics, targeted proteomics, multiplexing, isobaric tag, high-throughput
Graphical abstract
INTRODUCTION
In proteomics, two fundamental types of multiplexing are available to increase throughput. During a single run, many peptides can be targeted for quantification (peptide multiplexing) (Picotti and Aebersold, 2012). Additionally, higher-order multiplexing is possible at the sample level. This is most commonly achieved via isobaric labeling strategies (Ross et al., 2004; Thompson et al., 2003). For example, tandem mass tags (TMT) are chemically-reactive reagents that impart isotope-based differences to each sample (Thompson et al., 2003). After mixing, a single analytical run contains the information for up to 10 different samples. However, sample multiplexing with isobaric reagents comes with a significant caveat. The quantitative accuracy can be severely distorted by co-isolated and co-fragmented peptides (Ting et al., 2011; Wenger et al., 2011). Nevertheless, quantitative accuracy is restored when reporter ion quantification occurs in a dedicated MS3 fragmentation event using a technique called synchronous precursor selection (SPS-MS3) to remove interference. This is in contrast to how reporter ions are normally recorded—directly from a combined event which includes both quantification and identification (MS2 scan) (Erickson et al., 2015; McAlister et al., 2014; Ting et al., 2011).
Targeted mass spectrometry (MS)-based analyses have become vital for multiplexing the measurement of many peptides as biomarkers or predictive endpoints (Jaffe et al., 2013), such as those collected during clinical trials (Domon and Gallien, 2015; Gillette and Carr, 2013). Recent advances have focused on increasing the level of peptide multiplexing within the same run or improving assay performance (Escher et al., 2012; Kennedy et al., 2014; Burgess et al., 2014; Gallien et al., 2015). In addition, enterprise-grade software supports the facile development of targeted methods (MacLean et al., 2010). Sample multiplexing, on the other hand, has only rarely been employed in targeted assays. Coon and colleagues (Potts et al., 2016) demonstrated a clever approach to sample multiplexing that relied on metabolic labeling with neutron encoding (NeuCode). Three groups have demonstrated sample multiplexing using isobaric labels (Curran et al., 2015; Everley et al., 2013; Savitski et al., 2010). Importantly, each group’s method was only applied to affinity-enriched subproteomes of reduced complexity. For example, Curran et. al., proposed a method termed MARQUIS (Curran et al., 2015) that utilized both IMAC and phosphotyrosine enrichment prior to analysis of iTRAQ-labeled phosphopeptides by MRM methods. Savitski and coworkers reported an iTRAQ-based targeted assay to measure kinase levels after isolation via kinobeads (Savitski et al., 2010). Thus, isobaric tagging-based multiplexing in targeted assays has not been demonstrated with unfractionated mixtures. With the evolution of MS instrumentation capable of sensitive MS3 analysis of reporter ions (Huguet et al., 2015), we hypothesized that 10-plex sample multiplexing with accurate, targeted quantification might finally be possible directly from proteolyzed cell lysates.
DESIGN
Overview of a two-dimensional targeted proteomics approach
Figure 1 shows our 2D multiplexing method for targeted proteomics (TOMAHAQ—triggered-by-offset, multiplexed, accurate-mass, high-resolution, and absolute quantification). Most targeted proteomics methods utilize complex retention-time scheduling when multiplexing many peptide targets per assay. As an alternative to retention time scheduling, spiked-in internal standard (IS) peptides have been used to enable robust sampling of target peptides independent of chromatography conditions (Gallien et al., 2015; Yan et al., 2011). TOMAHAQ utilizes a similar strategy, where synthetic trigger peptides are monitored during analysis and identified in real time to prompt quantitation of multiplexed targets present at a known offset (Fig. 1a). Labeling the trigger peptide with an alternative form of TMT (termed TMT0) results in co-elution with the corresponding target peptide but at an offset mass (Fig. 1b). Ideally, the trigger peptide is present at sufficient abundance such that it is reproducibly detected in survey scans, enabling sampling of the target peptides even when the multiplexed precursor ion is not detected (Fig. 1b). MS2 analysis of target peptides enables the selection of interference-free b- or y-type fragment ions as precursors (synchronous precursor selection, SPS) for an MS3 spectrum with reporter ion quantification (Fig. 1c–d).
Figure 1. The 10-plex TOMAHAQ (Triggered by offset, multiplexed, accurate-mass, high-resolution, absolute quantification) workflow.
(a) Spiked-in internal standards are mixed with up to ten multiplexed endogenous proteomes and combined before analysis. (b) This results in two precursor ion clusters that have identical elution profiles. Synthetic trigger and multiplexed target peptides are separated by a known mass offset. The presence of synthetic trigger peptides is used to prompt selection (gray bar) and quantitative analysis of target peptides at all precursor abundances, obviating the need for target detection in the MS1. (c) MS2 analysis of target peptides is used to select b- and y-type fragment ions for synchronous precursor selection (SPS, gray bars). (d) Targeting interference-free fragment ions yields accurate quantitation in an MS3 scan, even if multiple interfering peptides are co-isolated.
To date, isobaric labels have rarely been used in targeted proteomics experiments and only on highly simplified or enriched subproteomes. We sought to compare the methods, MS2-only (Curran et al., 2015; Savitski et al., 2010), MS3 (Everley et al., 2013) and pre-selected SPS ions (this work) to evaluate accuracy and precision of measurements from complex unfractionated whole proteome digests.
RESULTS
Constructing a two-proteome model of interference
Targeted mass spectrometry methods, SRM (selected reaction monitoring) and PRM (parallel reaction monitoring), have demonstrated limits of quantification near 100 amol for human peptides spiked into a yeast background (Peterson et al., 2012). We surmised TOMAHAQ could be used to quantify a similarly low amount of peptide, but in a multiplexed experiment. To demonstrate the advantages of TOMAHAQ over existing multiplexed analysis techniques, we compared each method’s ability to measure an 8-fold change at both 400 and 100 amol on column. The model for this comparison used thirteen synthetic human peptides mixed at a known ratio into a background of yeast peptides (Fig. 2). To enable sampling of targeted peptides independent of precursor ion detection, a portion of each human peptide was labeled with TMT0 and spiked into yeast at 500 fmol on-column (Fig. 2b). A separate portion of each human peptide was labeled with two channels of TMT10 reagent, mixed at an 8:1 ratio, and diluted into a background of yeast peptides (labeled at a 1:1 ratio) such that the lowest target channel represented either 400 or 100 amol on-column (Fig. 2c). For each sample, reporter ion quantification was prompted by the TMT0 peptide, but three different quantification methods were evaluated: MS2-only (no SPS ion selection), standard SPS-MS3 (SPS ions are selected via their abundance), or TOMAHAQ analysis (SPS ions are preselected and purity checked on-line) (Fig. 2d).
Figure 2. Comparison of method accuracy for three reporter-ion quantification techniques (MS2-only, standard MS3, and TOMAHAQ) using a combined interference model and dilutions.
(a) Thirteen TMT0-labeled synthetic human peptides (500 fmol) served as the triggering event that was monitored throughout the run and used to prompt analysis of multiplexed targets. A separate portion of each peptide was labeled with two channels of the TMT10 reagent in an 8:1 ratio. Two samples were created, each with 500 fmol trigger peptide and a decreasing amount of multiplexed peptides as measured by the on-column amount of the lowest channel (i.e., Sample 1: 400 amol, and Sample 2: 100 amol). The yeast background comprised 250 ng of yeast labeled with the same isobaric labels as the multiplexed target ions, but in equimolar amounts. (b) The 400 and 100 amol samples were then separately spiked into whole yeast lysate to create two interference model samples. (c) For all samples the trigger peptides were monitored and identified in real-time, prompting quantitative analysis of the target peptides. (d) Each sample was quantified using either MS2-only, standard MS3, or TOMAHAQ analysis. MS2 analysis fragments all isolated precursor species, including interfering ions (red bars) resulting in a compressed ratio. MS3 analysis removes interference by selecting the top 10 peaks (synchronous precursor selection—SPS) from an MS2 spectrum to perform SPS-MS3. However, for low abundance targets, many of the largest peaks in an MS2 spectrum are interfering ions. TOMAHAQ analysis targets pure fragment ions in an MS2 spectrum, resulting in accurate quantification even at the lowest molar amount on column. (e) Comparison of the results for three quantification methods. The black line represents the median (expected value = 8) and the box represents the inner quartile range demonstrating the precision of each method. Each point represents the fold change for each peptide (n=13). (f) Comparison of replicates (n=3) for the 100 amol dilution analyzed with all three quantification methods. Bars represent mean fold change (± 1 s.d.) for each peptide across technical replicates. See also Fig. S1, S2
Assessing low attomole accurate quantification of target peptides
As described previously (Savitski et al., 2011; Ting et al., 2011; Wenger et al., 2011), the MS2-only method returned highly inaccurate quantification due to the isolation and fragmentation of all precursors, even though isolation was achieved via the smallest allowed setting of 0.4 m/z. The median ratios for the 400 and 100 amol samples were 1.08 and 1.01 fold, respectively, as compared to the true ratio of 8 fold (Fig. 2e). The SPS-MS3 method has been shown to remove nearly all precursor interference in large-scale experiments (Isasa et al., 2015; McAlister et al., 2014), but in targeted experiments a greater portion of fragment ions belong to interfering ions—each representing a potential source of ratio compression. For SPS-MS3 analysis, the median fold changes were 6.95 and 1.13 for the 400 and 100 amol dilutions, respectively. This demonstrates that interference in targeted experiments is highly dependent on precursor abundance and selection of the correct SPS ions for MS3 analysis.
Online filtering improves quantitative accuracy
To avoid ratio compression, TOMAHAQ successively applies three filters during the selection of SPS-MS3 ions. First, only peaks corresponding to fragment ions of the target peptide (± 10 ppm) are considered. Second, the relative abundances of remaining fragment ions are compared to a library spectrum and only those matching the expected relative abundances are retained. Third, the SPS ion isolation purity (i.e., percentage of signal belonging to the fragment ion) is calculated for each remaining candidate, and only pure ions are included as precursors in the SPS-MS3 scan (Fig. S1). Although the online filters provide selection of higher purity SPS-ions, the overwhelming proportion of interference is alleviated by targeting specific SPS-MS3 ions (Fig. S1h). Using these filters TOMAHAQ returns median fold changes of 7.60 and 6.93 for the 400 and 100 amol samples, respectively (Fig. 2e). To determine the reproducibility of the multiplexed targeted measurements, the lowest dilution, 100 amol, was analyzed in triplicate by each method (Fig. 2f). When analyzed by TOMAHAQ, 12 of 13 peptides exhibited both a relative error (percent difference between measured and expected ratio) and a coefficient of variation (CV) below 20%. Conversely, only one MS3 and no MS2 measurements met this criterion. These data clearly show that in unfractionated digests, pre-selecting MS3 precursor ions provides a substantial improvement in both accuracy and precision.
TOMAHAQ enables absolute quantification of endogenous peptides
SRM (selected reaction monitoring) and PRM (parallel reaction monitoring) are often used to determine the absolute molar amounts of a peptide in a sample. This is accomplished by spiking a peptide standard into the sample at a known molar amount and using the ratio of target to standard peptide to determine absolute abundance (Gerber et al., 2003). Similarly, within a TOMAHAQ assay, a standard peptide of a known molar amount can be labeled with the isobaric label used to quantify endogenous samples (e.g., TMT10 for Fig. 2) and used for absolute quantification. Using a standard peptide labeled with TMT10 and spiked into the interference model described in Fig. 2, we were able to determine the absolute molar amounts of peptides within the interference model (Fig. S2a). The absolute amounts for each peptide were typically within 20% of the correct value (Fig. S2b–c) with nearly all variable measurements (>20% error) occurring for peptides at the 100 amol level, signifying that we are approaching the limit of quantification for a subset of peptides (Fig. S2b–c).
Comparing quantitative reproducibility of a fully fractionated and unfractionated proteome
Several studies have demonstrated deep quantitative analyses of protein expression levels using 10-plex sample multiplexing and the SPS-MS3 method (Chick et al., 2016; Christoforou et al., 2016; Egan et al., 2015). To reduce sample complexity in large-scale multiplexed proteome analyses, peptides are fractionated prior to LC-MS/MS analysis. Offline peptide fractionation improves the depth of proteome quantification, but also increases the time required for analysis. We surmised that without any fractionation, TOMAHAQ could accurately reproduce the quantification gathered in a large-scale proteome analysis, drastically reducing the time needed to quantify a set of target proteins.
We compared the results from a multiplexed analysis of eight cancer cell lines, labeled with TMT10 reagents, which were either deeply fractionated (24 fractions) or left unfractionated (Fig. 3). For large-scale discovery, 400 μg of lysate was fractionated, then fractions were analyzed via 3-hr gradients using the SPS-MS3 approach (72 hrs of total analysis time), resulting in nearly full proteome quantification (10,214 quantified proteins across all 8 samples). For TOMAHAQ, a set of 131 trigger peptides, corresponding to 69 cancer-related proteins (See Table S1 for a literature reference), were labeled with TMT0, spiked into the unfractionated sample, and 8 μg of lysate was analyzed in a single 2-hr experiment. Finally, a 2-hr unfractionated, discovery analysis was also performed. Leveraging the offset triggering resulted in the identification of 95% (124/131) of targeted peptides; however, 22 peptides did not meet our signal-to-noise threshold for quantitation. After filtering, TOMAHQ quantified 102 peptides from 61 proteins in all 8 samples. When comparing TOMAHAQ to a discovery approach with the same chromatographic conditions, TOMAHAQ uniquely quantified 83 peptides. We next compared quantification at both the peptide and protein level between the discovery and TOMAHAQ methods (Fig. 3a).
Figure 3. TOMAHAQ analysis has precision and sensitivity comparable to a fractioned proteome-wide experiment.
(a) Eight cancer cell lines were digested, labeled with TMT, mixed and split into two samples. One was separated via basic pH reverse-phase fractionation into 24 fractions (requiring 72 hr of analysis), and the remaining sample was left unfractionated (used for 2 hr TOMAHAQ analysis). Peptides and proteins quantified in both methods were compared for quantitative accuracy and precision. An example correlation for Superoxide dismutase (SOD1) is displayed. (b) Pearson correlation was calculated for all peptides identified in both methods (median = 0.97, mean = 0.85). Rarely, peptide measurements derived from the full proteome dataset showed reduced correlation values likely due to interference. Utilizing protein-level quantification (combined peptides) resulted in improved correlations (median = 0.99, mean = 0.94). (c) The correlation for all protein measurements from both methods was assessed to be 0.95. Notable proteins exhibiting dynamic protein abundance in the eight cell lines are highlighted. See also Fig. S3, Table S2
For the 80 peptides measured by the full proteome and TOMAHAQ analyses, expression levels across the eight cancer cell lines were directly compared and typically demonstrated remarkable peptide-to-peptide correlations (median=0.97, mean=0.85; Fig. 3b) despite the significant reduction in lysate and analysis time consumed by the TOMAHAQ approach. Upon inspection, most cases of poor peptide-to-peptide correlation were caused by erroneous measurements in the full proteome analysis due to co-isolated interference (see Fig. S3 for an example). TOMAHAQ avoids SPS ion interference by only isolating and fragmenting peaks corresponding to interference-free fragments of the target peptide. These additional filters reduced the incidence of inaccurate peptide measurements and resulted in high correlations for protein measurements between both methods (median=0.99, mean=0.94). These results highlight TOMAHAQ’s ability to fully reproduce the quantitative accuracy of deeply fractionated samples in a single unfractionated analysis. Remarkably, the correlation of all 60 proteins between the full proteome and TOMAHAQ was determined to be 0.95 (Fig. 3c). This includes a number of proteins that exhibited dramatic differences in their expression. Two proteins, ERBB2 (SK-OV-3) and CTNNB1 (SW-620), exhibited near complete changes in protein expression between cell lines, consistent with previously reported data (Gholami et al., 2013; Lattrich et al., 2008). Other notable proteins, including BRAF, ATM, NFKB1, and EGFR, showed consistent, yet dynamic expression among the eight cell lines.
Targeted analysis of 60 cancer cell lines in biological triplicate
The National Cancer Institute’s collection of 60 cancer cell lines (NCI-60) was introduced in 1990 and is composed of cell lines derived from nine cancerous tissues (Shoemaker, 2006). Due to the longevity of the panel, a vast repository of multi-omics characterizations is available, including a curated database of cell line response to tens of thousands of drugs and compounds (Holbeck et al., 2010). We utilized TOMAHAQ to assay expression of 69 protein targets across the entire NCI-60 panel in biological triplicate. Leveraging the ten-fold gain in throughput provided by TOMAHAQ, all 180 samples were analyzed in just 2 days (Fig. 4a). In total, TOMAHAQ quantified 68 proteins in at least one cell line, and 54 proteins in all 60 cell lines in multiple biological replicates (≥2 bio. reps., Fig. S4). Quantitative reproducibility was assessed by determining the coefficient of variation (CV) for each protein within each cell line. When combined, the mean and median CVs for all proteins were less than 6% (Fig. 4b). Hierarchical clustering of all 180 cell lines revealed a strong association between the biological replicates, as well as three primary clusters (Fig. 4c). A distant cluster (Fig. 4c, cluster 1), consisting of cell lines SK-OV-3 (ovarian) and H226 (non-small cell lung) was explored for significant dynamic protein expression. A Benjamini-Hochberg corrected t-test was performed for each protein within the two cell lines in cluster 1 and all the other 58 cell lines. Proteins that exhibited significant (FDR < 0.01) expression dynamics between cluster 1 and all other cell lines are displayed in Figure 4d. Key protein classes widely implicated in cancer progression differed significantly in their expression including DNA mismatch repair / binding, metabolic processes, kinases and membrane trafficking. Several proteins exhibited dramatic expression departures from their mean expression (fold change)—PML (3.0 fold up), SHMT1 (2.9 fold down), PCNA (2.2 fold down), and GLS (2.0 fold up).
Figure 4. Application of TOMAHAQ analysis to the NCI-60 cancer cell line panel.
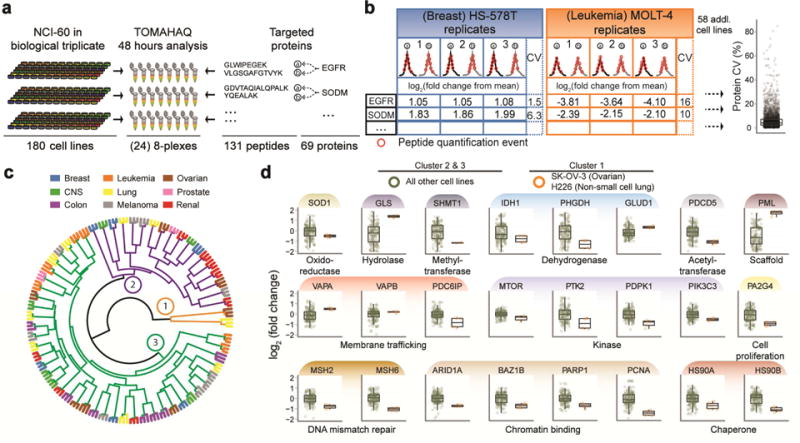
(a) Lysates of the NCI-60 panel in biological triplicate were prepared for TOMAHAQ analysis (24, 8-plexes using 131 triggering peptides). Each 8-plex was analyzed for 2 hr, and the complete dataset was collected in 48 hr. (b) Protein abundance (fold change from mean, log2) was determined by summing the reporter ion intensity for each corresponding peptide, which was generally composed of multiple quantification events across the peptide elution. The coefficient of variation (%CV) for each protein, within each tissue, was calculated and assessed for reproducibility among biological replicates (median = 5.02, mean = 6.16). (c) Hierarchical clustering of the complete quantitative proteomic dataset exhibited robust replicate clustering and varied clustering among cell line origins. Three primary clusters are highlighted (1 = orange, 2 = purple, 3= green). (d) Example box plots of significant expression differences between cluster 1 from panel c and the remaining lines in clusters 2 and 3. See also Fig. S4, Table S3.
Previous proteomic analyses of the NCI-60 have resulted in large datasets that illustrate key protein expression differences among cell lines (Federici et al., 2013; Gholami et al., 2013; Park et al., 2010). Unfortunately, the stochastic nature of shotgun proteomics left many proteins undetected. Among the 54 proteins, we characterized six proteins that were previously unmeasured across the NCI-60. Notably, BAZ1B and PTK2 exhibited tissue specific regulation (Fig. 5a). PTK2, a tyrosine-kinase involved in a number cellular roles including cell migration and adhesion, exhibited a marked reduction in protein expression within the six leukemia cell lines. We hypothesize that PTK2’s role in regulating cellular adhesion may correlate with non-adherent nature of leukemia cells. We observed that CTNB1, which also regulates cellular adhesion, exhibits a nearly identical expression profile within the six leukemia cell lines assayed. Furthermore, down-regulation of PTK2 was found to be correlated with reduced migration of acute myeloid cells and increased survival of acute myeloid leukemia patients (Recher et al., 2004). BAZ1B, also a tyrosine-kinase, showed nearly 2-fold lower expression relative to the mean for six of the seven ovarian cell lines. Remarkably, BAZ1B expression was shown to correlate with drug response sensitivity and is highlighted below.
Figure 5. TOMAHAQ identifies correlations between protein expression and drug sensitivity.
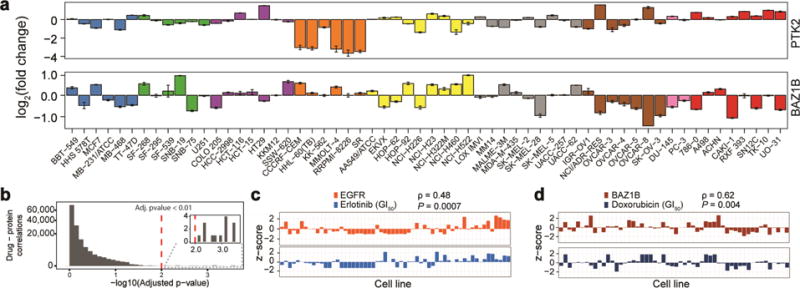
(a) Example proteins quantified via TOMAHAQ, but not detected in any of the deep proteome studies (mean ± sd, n=3). (b) Histogram of correlations comparing cell line specific protein expression and the NCI-60 developmental therapeutics program (DTP)(Holbeck et al., 2010). Fourteen significant drug – protein correlations were identified (inset). (c) Example of correlation (Spearman, ρ = 0.48, adj. p-value = 0.0007) between EGFR expression and Erlotinib treatment. (d) Example of correlation (Spearman, ρ = 0.62, adj. p-value = 0.004) between the protein BAZ1B and the chemotherapy agent doxorubicin (dox). See also Fig S4.
Protein expression of BAZ1B correlates with drug response sensitivity in the NCI-60
The NCI-60 has been thoroughly assayed for response to thousands of drug compounds (Holbeck et al., 2010), providing a vast resource to compare against the protein expression levels measured by TOMAHAQ. We selected a subset of drugs that have been FDA approved (2,535 drug treatment experiments) and correlated z-transformed growth inhibition (GI50) to the expression of proteins measured by TOMAHAQ. We observed 14 significant correlations (spearman, adj. p-value < 0.01, Fig. 5b). Four significant correlations were observed between EGFR expression and Erlotinib, a tyrosine-kinase inhibitor (Fig. 5c). Additionally, we observed significant correlations between GLS / ARID1A and Erlotinib. Finally, the tyrosine-kinase, BAZ1B, which functions in DNA damage through histone phosphorylation was found to have a significant correlation (ρ=0.62, adj. p-value = 0.004) with Doxorubicin (dox) treatment (Fig. 5d). Dox, a DNA damaging agent, is a widely utilized chemotherapeutic for a variety of solid tumors and blood based cancers (Tacar et al., 2013). The strong correlation observed between BAZ1B and dox highlights the potential application of TOMAHAQ for rapid and sensitive drug screening analysis. As shown here, the discovery of protein abundance-drug sensitivity correlations for FDA approved drugs, indicates that this dataset may also be useful for understanding how publicly unavailable and yet-to-be-developed drugs perform among the varying cancer cell lines. We have therefore provided a web resource for viewing and downloading these data for future studies (https://gygi.med.harvard.edu/publications/tomahaq). For example, output from the website for the transcription factor Stat3 displays a large difference in Stat3 between the two prostate cancer cell lines DU145 and PC3, despite both being androgen-independent. This expression difference is consistent with the differing response during a Stat3 DNA-binding activity assay (Mora et al., 2002) and with sensitivity to Stat3 pathway inhibition (Chesnokov et al., 2009) between these two cell lines.
DISCUSSION
Sample multiplexing in targeted assays can increase throughput by as much as 10 fold while providing exceptional quantitative accuracy. Yet, this is likely only a starting point. As next generation isobaric reagents are developed (Braun et al., 2015), multiplexing capabilities of 16 or 24 or even higher reagent sets are possible. These would be directly amenable to the current TOMAHAQ strategy only using longer ion injection times. Finally, since retention time scheduling is not used, we are also far from reaching an upper limit on the level of analyte multiplexing. For the 131 peptides monitored here, depending on local sample complexity, we permitted the instrument to collect up to eight consecutive MS3 spectra, although one was sufficient for quantification. We commonly met that mark (Fig. 4b—red circles), which suggests that in the future TOMAHAQ assays could target many hundreds of peptides in the same assay. The ability to simultaneously and accurately quantify hundreds of peptides across tens of samples highlights the potential of the TOMAHAQ assay. High-throughput assays for drug-development and biomarker validation (Rifai et al., 2006), which were once logistically daunting, might now be addressed via sample multiplexing.
Limitations
The TOMAHAQ approach achieves highly accurate and precise quantification by employing the additional gas-phase purification of a high-resolution MS3 quantitative scan. Currently, only two models of mass spectrometer provide the correct architecture of mass analyzers to permit this mode of operation. Although instrumentation costs can be high, the ability to multiplex the samples dramatically improves the throughput of the instrumentation and can ultimately offset the significant capital costs.
Finally, additional consideration is necessary when performing absolute quantification with TOMAHAQ. For MS3 absolute quantitation, careful consideration of internal standard concentration is necessary to prevent dynamic range issues. If performing MS1 level absolute quantitation, via the trigger peptide, the range of internal standard concentrations is wider, however low abundance targets may not be comparable. Additional MS1 quantitative scans could overcome this limitation.
EXPERIMENTAL PROCEDURES
Standard Peptide Preparation
Peptides (Table S1) were synthesized by Cell Signaling Technologies (Danvers, MA) at a scale of 5 micromole each. Peptides were dissolved in 100 μL DMSO and 1 mg was brought up in 1 mL of 1% formic acid in water. The peptides were then purified using a 50 mg capacity SepPak cartridge (Waters, Milford, MA). Upon reconstitution in HPLC grade H2O, peptide concentration was measured by BCA assay (Pierce, Rockford, IL). 2 μg of each peptide was combined and the pH was adjusted to 8.5 in 200mM EPPS. HPLC grade acetonitrile was added to a final concentration of 10% (V/V). TMT0 or TMT10 reagents (Pierce, Rockford, IL) were then added at a ratio of 2:1 (TMT:Peptide) by mass. The reaction proceeded at room temperature for 1 hr before quenching with a final volume of 0.5% hydroxylamine (Sigma, St. Louis, MO) in 200mM EPPS pH 8.5. The peptides were then acidified and diluted to a final volume of acetonitrile ≤ 5% and purified using a 50 mg SepPak. The peptides were then reconstituted in 1% formic acid in water and ready for MS analysis.
Whole Proteome Sample Preparation
Yeast Sample Preparation
Yeast was grown to an O.D. of 0.8 in a 125mL flask overnight in YPD media at 37 °C. The cells were then washed with cold PBS 3× and lysed in a bead beater in 1 mL of 1% SDS, 100 mM NaCl in 50 mM Tris pH 8.5 with protease inhibitors added. The supernatant was then transferred to a 1.5 mL tube and proteins were reduced in 5 mM DTT for 1 hr at 37 °C. After the sample cooled to room temperature the samples were alkylated using 15 mM of iodoacetamide in the dark. Protein concentration was then assessed using the BCA Assay (Pierce). The protein was then purified using chloroform methanol precipitation and the pellet was washed 3× with cold HPLC grade methanol. The pellet was dissolved in 8M urea in 20 mM EPPS pH 8.5 and diluted to 4M urea using 20 mM EPPS pH 8.5 and digested overnight at room temperature using LysC (Wako) at an enzyme:substrate ratio of 1:75. The samples were then diluted to 1.6M urea using 20mM EPPS pH 8.5 and digested with trypsin (Promega, Madison, WI) for 6 hours at 37 °C using the same 1:75 ratio as before. HPLC grade acetonitrile was then added to a final volume of 10% and TMT was added at a ratio of 2.5:1 and allowed to react as before. The TMT labelled samples were then combined and purified via SepPak (Fig. 2, S3).
NCI-60 Sample Preparation
Three replicate, non-viable, pellets of the NCI-60 cell line panel were obtained from the Developmental Therapeutics Program (DTP) of the NCI. The pellets were lysed via syringe lysis with 12 pumps using a 21 gauge needle and 8 pumps using a 25 gauge needle. The same lysis buffer and subsequent procedures used for yeast were repeated for the 180 human cancer cell pellets. The digested peptides were labelled with the inner 8 channels (127n to 130c) of the 10-plex reagent leaving 126 channel open for any potential TMT0 contamination and 131 open for the bridge channel. The bridge sample consisted of equal amounts of each of the 60 different cell lines.
Liquid Chromatography
A homemade analytical column of 100 μm id was packed with 0.5 cm of C4 5μm beads (Sepax Technologies, Baltimore, MD) and 30 cm of 1.8 μm C18 material (Sepax) and heated to 60 °C. The mobile phases were 3% ACN, 0.1% FA in water (A) and 0.1% FA in ACN (B). The interference sample utilized a 6–30%B gradient while the NCI-60 samples utilized a 8–27%B gradient. All samples were analyzed over a 2 hr gradient. For the interference sample 0.250 μg of yeast protein and 500 fmol of the TMT0 labelled trigger peptides was loaded on column. The NCI-60 samples contained 8 μg on column with 1 ng of trigger peptide. See Data File S1 for a detailed protocol and buffer recipes.
Mass Spectrometry (MS) Analysis
All MS analyses were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo, San Jose, CA).
Standard Peptide “Priming Run”
A mix of all TMT10 labeled standard peptides was first analyzed alone to determine elution order, most intense charge state of each peptide, pre-select synchronous precursor selection (SPS) ions, and calculate relative fragment ion intensities. MS analysis utilized a targeted mass list comprising the neutral mass (M) and charge range (2–5) for each targeted peptide. Precursor m/z values (± 10 ppm, including isotopes) were dynamically excluded for 5 sec if they were chosen 2 times within a 12 sec period. An intensity threshold of 5e5 was used for precursor selection. Candidate precursors were isolated using the quadrupole (isolation window = 0.4), activated with CID in the ion trap (NCE = 35), and analyzed at high-resolution and high-mass accuracy in the Orbitrap (resolution = 15,000 and AGC = 5e4). The SPS ions chosen for targeted analysis had to meet the following criteria: 1) the product ion intensity was at least 15% of the base peak within the fragment spectrum 2) the product ion was not a b1 or b2 / y1 or y2 ion, and 3) the product ion was not within the region between −19 and +7 of the precursor m/z.
TOMAHAQ MS Analysis
The TOMAHAQ workflow includes a series of decisions enabled by the instrument software (Fig S5a, grey triangles) to prompt the collection of a quantitative SPS-MS3 scan. Additional modifications written in the instrument language (Lua) increase the robustness of the method and accuracy of the quantitation (Fig. S5a, red triangles). These modifications include: elution order scheduling of precursors, online identification of trigger peptides, SPS ion filtering, scaling of MS3 injection times and MS3 specific repeat count exclusion. An example scan sequence for a peptide (Fig. S5b) demonstrates the order and relationship of the scan events. The entire method could be made available via an application programming interface (API). An API-enabled method for a different targeted assay is already available (Gallien et al., 2015).
For all experiments, a target list containing TMT0 labeled trigger peptide m/z, z, and scan event index was loaded into the instrument method editor to guide MS/MS analysis of precursors detected (±10 ppm) in MS1 survey scans. For NCI-60 experiments, minimum and maximum elution order (EO) indexes (Bailey et al., 2014) were placed in the fields reversed for the start and stop retention time values. These EO indexes were used so that only ~17 trigger peptides were considered at any given point in the experiment.
The quadrupole was used to isolate a region +50 of the highest and −50 of the lowest trigger peptide m/z, subsequently Orbitrap MS1 survey scans (resolution 60,000 and AGC = 3e5) were used to analyze intact precursors and guide selection of trigger peptides based on their mass (±10 ppm) and charge such that an MS1 scan was obtained every 5 sec. Trigger peptide precursors were only selected if their intensity was greater than 1e5.
Trigger peptides were isolated using the quadrupole (isolation window = 0.4), activated by CID (NCE=34), and analyzed in the Orbitrap (resolution 15,000, AGC = 1e4, max injection time = 35 ms). Trigger peptide MS2 scans were analyzed and identified in real time, as described previously (Bailey et al., 2012), to prompt MS2 analysis of multiplexed target peptides. Briefly, each target peptide along with its corresponding fragment ions were loaded into memory of the instrument computer. The fragment ions of each trigger peptides within ±15 ppm of the precursor were matched to the spectrum (± 10 ppm) and considered to be identified in real-time by the presence of ≥6 matching peaks. A positive identification led to the isolation and fragmentation of target peptides.
Target peptides were isolated by the quadrupole (isolation window = 0.4) at an offset from the identified trigger peptide. Isolation offsets were determined based on the number of isobaric labels on the peptide and precursor charge state (e.g., 5.01045 Da for z = 2 and two TMT tags). Target peptides were activated using CID (NCE = 35) and analyzed in the Orbitrap (resolution 60,000, AGC = 5e4, max injection time = 900 ms). Target peptide MS2 scans were analyzed in order to select ions for synchronous precursor selection (SPS).
For SPS ion selection, only peaks corresponding to target peptide b- or y-type ions (±10 ppm) were considered for selection. Next, matched fragment ions were compared to a library spectrum to ensure that fragment ratios were within ±50% of the expected value for more than ½ of the comparisons. Lastly, each targeted fragment ion was assessed for purity by determining the percentage of total ion current (TIC) that belongs to a fragment ion in an SPS isolation notch. The SPS purity threshold was 0.9 and 0.85 for the interference for NCI-60 analysis, respectively. To determine MS3 injection time, the predicted signal-to-noise (SN) of fragment ions that passed these filters were determined based on the intensity and m/z of the peak. Based on the predicted SN, the target peptide MS2 injection time was scaled in order to obtain ~1,000 SN in the subsequent MS3 scan. This injection time was used for the SPS-MS3 scan. After SPS ion selection, the target peptide fragment ions were activated by HCD (NCE = 55) and the reporter ions were analyzed in the Orbitrap (resolution = 60,000, max injection time = 2,500 ms).
TOMAHAQ can be implemented through the standard method editor (see https://gygi.med.harvard.edu/publications/tomahaq for detailed method construction) and enables: product ion triggering, offset isolation and the targeting of SPS ions based on expected rank order.
Data Processing
Raw data was searched using the Sequest algorithm with 10 ppm and 0.02 Da precursor and fragment ion tolerances respectively. The database contained the full forward and reverse sequence for each target protein. Peptides were filtered using linear discriminate analysis (Huttlin et al., 2010). Since many of the Target ID MS2 spectra were either too complex or too low abundant to yield an ID, the target reporter ion quantification was mapped back to the Trigger peptide identification. An in-house software suite was used for processing peptide identification and quantification information (Erickson et al., 2015). The isotope correction values provided by the TMT manufacturer were adjusted due to the 0.4 m/z isolation widths used here. Peptides were considered quantified if they had sum reporter ion SN ≥ 200 for the NCI-60 analyses.
For the NCI-60 analysis, each of the twenty-four 8-plexes were individually analyzed in a non-targeted approach to provide a sufficient dataset to assess the mixing errors for each cell line. The resulting mixing correction factors were then independently applied to the corresponding twenty-four TOMAHAQ datasets. Furthermore, a sample consisting of a mixture of each of the 60 cell lines from the NCI-60 was spiked into the 10th TMT channel (“bridge channel”) of each of the twenty-four 8-plexes at an equal concentration. Following the application of the mixing correction factors, each of the 24 TOMAHAQ datasets were further normalized to the bridge channel which accounted for deviations in the injection concentration. The final dataset was filtered to proteins found in all 60 cell lines and in at least two of the three biological replicates. When present, incomplete replicate data was imputed as the mean of the two existing replicates.
Supplementary Material
Acknowledgments
We thank members of the Gygi laboratory for helpful discussions. We thank Derek Bailey and Shannon Eliuk at Thermo Fisher for instrumentation assistance. Funding was partially provided by NIH grants DK098285 (J.A.P) and GM67945 (S.P.G.). R.A.E and A.R.E were supported by NIH grants P50-GM107618 and U54- HL127365. M.W was supported by the Charles A. King Trust and NIH grant R01GM103785.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
B.K.E, C.M.R, R.A.E, and S.P.G designed research; B.K.E, R.A.E, and A.R.E prepared samples; J.K. designed and synthesized peptides. J.A.P. and C.R.B aided with experimentation; B.K.E, C.M.R, R.A.E performed research and analyzed data; Targeting peptides based on the TMT0 reagent was first discussed by G.C.M. and M.W. with S.P.G; B.K.E, C.M.R, R.A.E, and S.P.G wrote the paper.
References
- Bailey DJ, McDevitt MT, Westphall MS, Pagliarini DJ, Coon JJ. Intelligent Data Acquisition Blends Targeted and Discovery Methods. J Proteome Res. 2014;13:2152–2161. doi: 10.1021/pr401278j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DJ, Rose CM, McAlister GC, Brumbaugh J, Yu P, Wenger CD, Westphall MS, Thomson JA, Coon JJ. Instant spectral assignment for advanced decision tree-driven mass spectrometry. Proc Natl Acad Sci U S A. 2012;109:8411–8416. doi: 10.1073/pnas.1205292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun CR, Bird GH, Wühr M, Erickson BK, Rad R, Walensky LD, Gygi SP, Haas W. Generation of Multiple Reporter Ions from a Single Isobaric Reagent Increases Multiplexing Capacity for Quantitative Proteomics. Anal Chem. 2015;87:9855–9863. doi: 10.1021/acs.analchem.5b02307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess MW, Keshishian H, Mani DR, Gillette MA, Carr SA. Simplified and Efficient Quantification of Low-abundance Proteins at Very High Multiplex via Targeted Mass Spectrometry. Mol Cell Proteomics. 2014;13:1137–1149. doi: 10.1074/mcp.M113.034660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokov V, Sun C, Itakura K. Glucosamine suppresses proliferation of human prostate carcinoma DU145 cells through inhibition of STAT3 signaling. Cancer Cell Int. 2009;9:25. doi: 10.1186/1475-2867-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick J, Munger S, Simecek P, Huttlin E, Choi K, Gatti D, Raghupathy N, Svenson K, Churchill G, Gygi S. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016 doi: 10.1038/nature18270. Ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou A, Mulvey CM, Breckels LM, Geladaki A, Hurrell T, Hayward PC, Naake T, Gatto L, Viner R, Arias AM, Lilley KS. A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun. 2016;7:9992. doi: 10.1038/ncomms9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran TG, Zhang Y, Ma DJ, Sarkaria JN, White FM. MARQUIS: A multiplex method for absolute quantification of peptides and posttranslational modifications. Nat Commun. 2015;6:5924. doi: 10.1038/ncomms6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, Gallien S. Recent advances in targeted proteomics for clinical applications. PROTEOMICS – Clin Appl. 2015;9:423–431. doi: 10.1002/prca.201400136. [DOI] [PubMed] [Google Scholar]
- Egan ES, Jiang RHY, Moechtar MA, Barteneva NS, Weekes MP, Nobre LV, Gygi SP, Paulo JA, Frantzreb C, Tani Y, Takahashi J, Watanabe S, Goldberg J, Paul AS, Brugnara C, Root DE, Wiegand RC, Doench JG, Duraisingh MT. A forward genetic screen identifies erythrocyte CD55 as essential for Plasmodium falciparum invasion. Science. 2015;348:711–714. doi: 10.1126/science.aaa3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson BK, Jedrychowski MP, McAlister GC, Everley RA, Kunz R, Gygi SP. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal Chem. 2015;87:1241–1249. doi: 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. PROTEOMICS. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everley RA, Kunz RC, McAllister FE, Gygi SP. Increasing Throughput in Targeted Proteomics Assays: 54-Plex Quantitation in a Single Mass Spectrometry Run. Anal Chem. 2013;85:5340–5346. doi: 10.1021/ac400845e. [DOI] [PubMed] [Google Scholar]
- Federici G, Gao X, Slawek J, Arodz T, Shitaye A, Wulfkuhle JD, De Maria R, Liotta LA, Petricoin EF. Systems Analysis of the NCI-60 Cancer Cell Lines by Alignment of Protein Pathway Activation Modules with “-OMIC” Data Fields and Therapeutic Response Signatures. Mol Cancer Res. 2013;11:676–685. doi: 10.1158/1541-7786.MCR-12-0690. [DOI] [PubMed] [Google Scholar]
- Gallien S, Kim SY, Domon B. Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM) Mol Cell Proteomics. 2015;14:1630–1644. doi: 10.1074/mcp.O114.043968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami AM, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M, Kuster B. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration–Approved Anticancer Agents in the NCI60 Panel of Human Tumor Cell Lines. Mol Cancer Ther. 2010;9:1451–1460. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet R, Blank M, Sharma S, Soltero N, Zabrouskov V. Low attomole limit of quantification on a Q-q-OT-IT hybrid mass spectrometer. 63rd Proc Am Soc Mass Spectrom 2015 [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isasa M, Rose CM, Elsasser S, Navarrete-Perea J, Paulo JA, Finley DJ, Gygi SP. Multiplexed, Proteome-Wide Protein Expression Profiling: Yeast Deubiquitylating Enzyme Knockout Strains. J Proteome Res. 2015;14:5306–5317. doi: 10.1021/acs.jproteome.5b00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HC, Taylor JE, Hu M, Englund NP, Yan F, Wang Z, Robert McDonald E, III, Wei L, Ma J, Easton J, Yu Z, deBeaumount R, Gibaja V, Venkatesan K, Schlegel R, Sellers WR, Keen N, Liu J, Caponigro G, Barretina J, Cooke VG, Mullighan C, Carr SA, Downing JR, Garraway LA, Stegmeier F. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45:1386–1391. doi: 10.1038/ng.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JJ, Abbatiello SE, Kim K, Yan P, Whiteaker JR, Lin C, Kim JS, Zhang Y, Wang X, Ivey RG, Zhao L, Min H, Lee Y, Yu MH, Yang EG, Lee C, Wang P, Rodriguez H, Kim Y, Carr SA, Paulovich AG. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods. 2014;11:149–155. doi: 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattrich C, Juhasz-Boess I, Ortmann O, Treeck O. Detection of an elevated HER2 expression in MCF-7 breast cancer cells overexpressing estrogen receptor β1. Oncol Rep. 2008 doi: 10.3892/or.19.3.811. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, et al. Constitutive activation of STAT3 in human prostate tumors and cell lines direct inhibition of STAT3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62:6659–6666. [PubMed] [Google Scholar]
- Park ES, Rabinovsky R, Carey M, Hennessy BT, Agarwal R, Liu W, Ju Z, Deng W, Lu Y, Woo HG, Kim SB, Cheong JH, Garraway LA, Weinstein JN, Mills GB, Lee JS, Davies MA. Integrative Analysis of Proteomic Signatures, Mutations, and Drug Responsiveness in the NCI 60 Cancer Cell Line Set. Mol Cancer Ther. 2010;9:257–267. doi: 10.1158/1535-7163.MCT-09-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol Cell Proteomics. 2012;11:1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Potts GK, Voigt EA, Bailey DJ, Rose CM, Westphall MS, Hebert AS, Yin J, Coon JJ. Neucode Labels for Multiplexed, Absolute Protein Quantification. Anal Chem. 2016;88:3295–3303. doi: 10.1021/acs.analchem.5b04773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recher C, Ysebaert L, Beyne-Rauzy O, Mas VMD, Ruidavets JB, Cariven P, Demur C, Payrastre B, Laurent G, Racaud-Sultan C. Expression of Focal Adhesion Kinase in Acute Myeloid Leukemia Is Associated with Enhanced Blast Migration, Increased Cellularity, and Poor Prognosis. Cancer Res. 2004;64:3191–3197. doi: 10.1158/0008-5472.CAN-03-3005. [DOI] [PubMed] [Google Scholar]
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Savitski MM, Fischer F, Mathieson T, Sweetman G, Lang M, Bantscheff M. Targeted data acquisition for improved reproducibility and robustness of proteomic mass spectrometry assays. J Am Soc Mass Spectrom. 2010;21:1668–1679. doi: 10.1016/j.jasms.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Savitski MM, Sweetman G, Askenazi M, Marto JA, Lang M, Zinn N, Bantscheff M. Delayed Fragmentation and Optimized Isolation Width Settings for Improvement of Protein Identification and Accuracy of Isobaric Mass Tag Quantification on Orbitrap-Type Mass Spectrometers. Anal Chem. 2011;83:8959–8967. doi: 10.1021/ac201760x. [DOI] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric labeling-based multiplexed quantitative proteomics. Nat Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger CD, Lee MV, Hebert AS, McAlister GC, Phanstiel DH, Westphall MS, Coon JJ. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods. 2011;8:933–935. doi: 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Luo J, Robinson M, Eng J, Aebersold R, Ranish J. Index-ion Triggered MS2 Ion Quantification: A Novel Proteomics Approach for Reproducible Detection and Quantification of Targeted Proteins in Complex Mixtures. Mol Cell Proteomics. 2011;10:M110.005611. doi: 10.1074/mcp.M110.005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.