

Abstract
A 60-nucleotide region (S1) downstream of the transcription start site of the cauliflower mosaic virus 35S RNA can enhance gene expression. By using transient expression assays with plant protoplasts, this activity was shown to be at least partially due to the effect of transcriptional enhancers within this region. We identify sequence motifs with enhancer function, which are normally masked by the powerful upstream enhancers of the 35S promoter. A repeated CT-rich motif is involved both in enhancer function and in interaction with plant nuclear proteins. The S1 region can also enhance expression from heterologous promoters.
Without doubt, the most comprehensively studied plant promoter is the 35S promoter of cauliflower mosaic virus (CaMV). Exhaustive investigation has catalogued its behavior in many plant species and in many expression systems (reviewed in reference 32). The promoter architecture has been extensively characterized, and the combinatorial nature of its complement of enhancers is fairly well understood. Despite this, the full intricacy of the CaMV 35S promoter remains to be unraveled. In this report, we present evidence uncovering a hitherto overlooked feature of this promoter.
Previous work in our laboratory reported enhancement of expression by inclusion of the first 60 nucleotides (nt) of the 35S RNA leader sequence (stimulatory region 1 [S1]). At that time, we ascribed this solely to an effect on translation (20). More recently, we characterized an element in the equivalent position in the leader of the pregenomic RNA of rice tungro bacilliform virus (RTBV) that also enhances the expression of reporter genes in protoplast expression systems. A DNA-based element within this region was found to contribute to promoter activity and to be the target for the binding of several proteins from rice nuclear extracts (12, 28). These findings led us to reassess the CaMV leader S1 region, asking whether, in this case also, there could be elements in the transcribed region that contribute to promoter activity.
Here we describe the effect of the S1 sequence on reporter gene expression in protoplast transient expression systems. We find that repeats of a CT-rich motif positively affect promoter activity, both from their usual location within the leader and when translocated to a position upstream of the transcription start site. Plant nuclear proteins bind specifically to these motifs.
MATERIALS AND METHODS
Plasmids.
Plasmids p35S-GUS (carrying the wild-type 35S promoter [−270 to −1 relative to the transcription start site] from strain CM4-184) and pMTF-GUS (carrying a methylation target-free derivative of 35S [MTF]), both with the wild-type S1 leader between the promoter and the β-glucuronidase (GUS) reporter gene, were constructed as described by Hohn et al. (31). Deletion of the 5′ part of the MTF up to position −90 (BamHI-EcoRV deletion) yielded the truncated version p−90MTF-GUS.
Replacement of the promoter fragment BamHI-BglII (−270 to −46) in pMTF-GUS with the −402-to-−130 region of the potato gst-1 promoter (26, 47) created pgst-MTF-GUS.
Mutations in the S1 leader were constructed by replacing the KpnI-NcoI S1 leader fragment of either p35S-GUS or p−90MTF-GUS with synthetic oligonucleotides carrying the desired mutations (sequences are given in Fig. 5A and B). The same strategy was used to clone leaderless constructs (nL) by using a short synthetic oligonucleotide with the sequence GTACCACCAC.
FIG. 5.
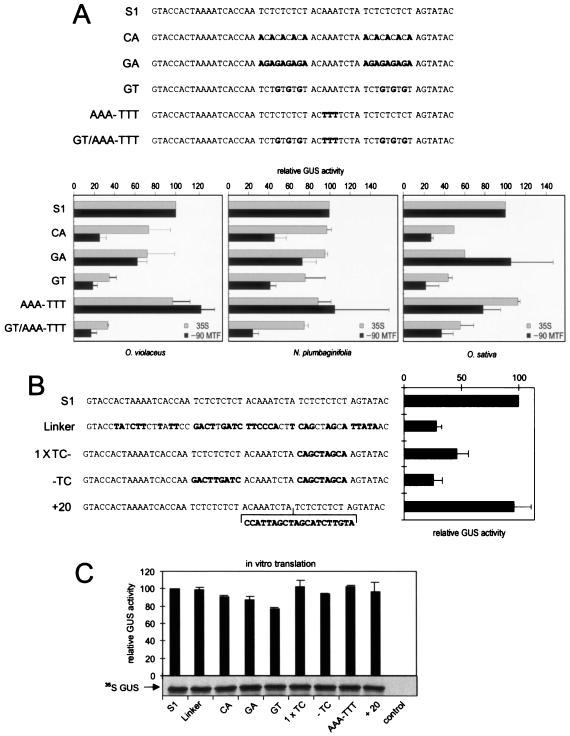
Mutations in the CT-rich motifs compromise expression in vivo. (A) Sequences of the wild-type S1 region and mutated derivatives, and graphs showing GUS activities in transfected plant protoplasts with the different leader sequences linked to either the 35S or the −90MTF promoter. (B) Like panel A, except that activities shown are with the −90MTF promoter and O. violaceus protoplasts only. (C) In vitro translation of mRNAs corresponding to the constructs shown in panels A and B. (Top) Graph showing GUS activity measured in wheat germ extracts after in vitro translation of GUS mRNAs bearing the leaders indicated. (Bottom) Representative autoradiograph showing sodium dodecyl sulfate-polyacrylamide gel electrophoretic analysis of in vitro translation reactions in the presence of [35S]methionine.
Synthetic oligonucleotides spanning the S1 leader region flanked by XbaI sites were inserted into the unique polylinker-derived XbaI site in p−90MTF-nL-GUS exactly adjacent to the 5′ end of the promoter to create plasmids S1-, S1rev-, and 2×S1rev−90MTF-nL-GUS (see Fig. 2).
FIG. 2.
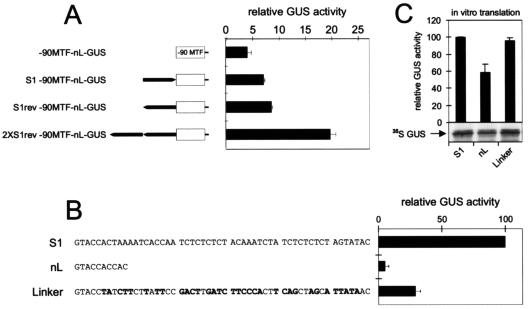
S1 functions as a position- and orientation-independent enhancer of gene expression. (A) The 60-nt S1 region (black arrow) was translocated to upstream of the −90MTF promoter in both orientations (S1- and S1rev−90MTF-nL-GUS) in a GUS expression construct. In 2×S1rev−90MTF-nL-GUS, 2 copies of S1 are present upstream of the promoter in reverse orientation. After transient expression in O. violaceus protoplasts, GUS activities arising from these constructs were compared to those of −90MTF-S1-GUS (100%) and a leaderless derivative, −90MTF-nL-GUS. (B) GUS activities measured in O. violaceus protoplasts transfected with −90MTF-GUS constructs with the leader sequences indicated. (C) In vitro translation of mRNAs corresponding to the constructs shown in panel B. (Top) Graph showing GUS activity measured in WGE after in vitro translation of GUS mRNAs bearing the leaders indicated. (Bottom) Representative autoradiograph showing sodium dodecyl sulfate-polyacrylamide gel electrophoretic analysis of in vitro translation reactions in the presence of [35S]methionine.
Protoplast transfection and detection of reporter gene activities.
Conditions for growth of suspension cultures of Orychophragmus violaceus, preparation of protoplasts, and transformation by electroporation have been described previously (19). Preparation and polyethylene glycol (PEG)-mediated transfection of Nicotiana plumbaginifolia protoplasts were performed as described by Goodall et al. (23). Conditions for growth of suspension cultures of the Oryza sativa line Oc and preparation of protoplasts have been described previously (11). Plasmid DNA was introduced into rice protoplasts by PEG-mediated transfection as for N. plumbaginifolia except that PEG 4000 instead of PEG 6000 was used. Routinely, 5 to 10 μg of plasmid DNA was used per transformation, and an internal control plasmid expressing chloramphenicol acetyltransferase (CAT) was cotransformed in all experiments.
GUS activities in protoplast extracts prepared after overnight incubation were determined as described previously (21). CAT protein levels were determined by using a CAT-ELISA kit (Roche Molecular Biochemicals) according to the manufacturer's instructions. GUS activities were calculated relative to the CAT internal control in all cases. Activities cited are averages of at least three independent transformations, unless otherwise stated.
RNA analysis.
Total RNA was isolated from protoplasts 6 h after transfection and subjected to RNase protection analysis according to published protocols (23). A radioactively labeled probe for S1 or mutant constructs was synthesized by in vitro transcription (in the presence of [α-32P]UTP) using SP6 RNA polymerase from a plasmid constructed by inserting the NcoI-SnaBI fragment from the 5′ end of the GUS gene into the NcoI-EcoRV polylinker sites of pGEM5 linearized at the NcoI site. An antisense probe for the internal control (CAT) was derived from a pGEM1 plasmid carrying the PvuII-EcoRI fragment from near the 5′ end of the CAT gene cloned into HincII-EcoRI polylinker sites. This plasmid was then linearized with HindIII and transcribed with T7 RNA polymerase. Protected fragments were resolved on 6% polyacrylamide denaturing gels and visualized by autoradiography or by PhosphorImager analysis (Molecular Dynamics). Fragments corresponding to GUS and CAT mRNAs were identified based on their sizes. The different protected fragments were quantified by PhosphorImager analysis, and the relative abundance of transcripts produced from each tested mutant was calculated relative to the internal control. Variations were within 10% of the mean.
In vitro translation.
To generate plasmids driven by the T7 promoter, the KpnI-EcoRI cassette containing the leader, the GUS gene, and the poly(A) site of the appropriate clones was cloned into the T7 expression vector pBluescript SK(−) (Stratagene).
T7-directed transcripts were transcribed in the presence of the cap analogue m7GpppG and translated to wheat germ extract (WGE) as described by Ryabova and Hohn (54). Globin RNA and WGE were from Roche Molecular Biochemicals.
Preparation of nuclear extracts.
Crude nuclear extracts were prepared from cell suspension cultures of O. violaceus or O. sativa line Oc as follows. About 4 × 108 suspension cells or protoplasts were disrupted in 30 ml of homogenization buffer (20 mM morpholineethanesulfonic acid [MES] [pH 6.0], 5 mM EDTA, 0.15 mM spermine, 0.5 mM spermidine, 10 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, 1 μg of leupeptin/ml, 1 μg of antipain/ml) with about 10 strokes in a Dounce homogenizer. The slurry was filtered sequentially through 60-, 40-, and 10-μm nylon mesh filters. The nuclei were pelleted by centrifugation at 1,200 × g for 5 min and resuspended in 50 ml of homogenization buffer. The solution was filtered again through 10-μm nylon mesh, and the nuclei were collected by centrifugation at 1,500 × g for 5 min and taken up in 20 ml of lysis buffer (20 mM HEPES [pH 7.5], 50 mM KCl, 2 mM MgCl2, 1 mM EDTA, 10% [vol/vol] glycerol, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 μg of leupeptin/ml, 1 μg of antipain/ml). The nuclei were broken by a few strokes in a Dounce homogenizer and diluted to an optical density at 260 nm of 10. Saturated ammonium sulfate (one-ninth of the volume) was added and shaken to mix, and the solution was left to stand for 30 min. After centrifugation at 100,000 × g for 90 min, 0.33 g of ammonium sulfate/ml was added to the supernatant. After gentle shaking for 15 min to dissolve the ammonium sulfate completely, the solution was left to stand for 30 min and then centrifuged for 30 min at 20,000 × g. The pellet was resuspended in 1 ml of dialysis buffer (same as lysis buffer but without leupeptin and antipain) and dialyzed for 4 h against 200-ml batches of dialysis buffer (buffer exchanged hourly). The dialyzed solution was spun for 10 min at 10,000 × g, and the supernatant was collected and stored in aliquots at −80°C. All of these steps were performed at 4°C.
Crude nuclear extracts from O. sativa plant seedlings were prepared according to the work of He et al. (29).
Gel retardation assays.
The DNA probe for gel shift analysis was produced by annealing two synthetic oligonucleotides corresponding to the first 60 nt of the 5′ end of the CaMV leader sequence flanked by KpnI and NcoI sites. Note that, in the case of the S1 probe, the GTA present at position 53-55 (which derives from a Bst11071 cloning site present in the expression constructs) is replaced with the wild-type sequence TAA. The annealed products were digested with NcoI and labeled with 32P. Competitor DNA fragments were prepared by directly annealing two synthetic complementary oligonucleotides.
Binding reactions contained crude nuclear extracts (3 μl) and 5,000 cpm of labeled DNA (about 0.05 to 0.1 pmol) in a final volume of 15 μl of buffer containing 10 mM HEPES (pH 7.6), 8 mM MgCl2, 1 mM dithiothreitol, 4 mM spermidine, and 5% (vol/vol) glycerol. Ten micrograms of poly(dI-dC) · poly(dI-dC) (Pharmacia) was included as a nonspecific competitor. For the competition assays, variable amounts of competitor as indicated in the figure legends were included. Binding reactions were begun by addition of nuclear extract to the buffer. This reaction mixture was preincubated for 10 min at room temperature and incubated for a further 20 min at room temperature after addition of the probe and the competitors. Samples were then loaded onto a 5% native polyacrylamide gel (polyacrylamide to bisacrylamide, 40:1) in 1× Tris-borate-EDTA buffer. Following electrophoresis at 30 mA for 2 to 3 h at 4°C, gels were dried and autoradiographed.
RESULTS
The S1 leader enhances reporter gene expression.
Previous results from our laboratory had shown that the first 60 nt of the 35S RNA could enhance gene expression; this region is referred to as S1 (20). To investigate the precise effect of the S1 leader sequence on gene expression, a series of constructs expressing the GUS reporter gene in the presence and absence of S1 was constructed. We tested the effect of the S1 leader under the control of an almost full-length (from −270 to −1) 35S promoter. Because the effect of elements downstream of the transcription start site may be masked by the strong upstream enhancers of the 35S promoter, we also wished to test a weaker promoter. We had previously developed a weakened, methylation-target-free (MTF) derivative of 35S, as part of a study designed to examine the effects of coding region methylation in the absence of promoter methylation (31). In the MTF promoter, all CG methylation targets in the 35S promoter have been mutated. In the present study, we used an MTF derivative truncated to position −90 (−90MTF; (Fig. 1A), which retains only about 5% of wild-type 35S activity. We also tested the effect of the S1 leader on expression from a heterologous promoter: in gst-MTF-GUS, the elicitor response and enhancer elements of the potato gst-1 promoter (26, 47) are fused to the −90MTF promoter (Fig. 1A). The activity of the gst-MTF promoter is only slightly less than that of the 35S promoter (Fig. 1B inset). A pair of constructs differing only in the presence or absence of the S1 region was used for each promoter.
FIG. 1.
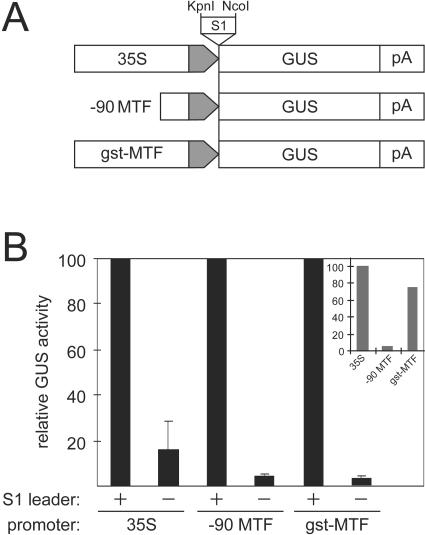
The S1 leader region stimulates reporter gene expression in plant protoplasts. (A) Three different promoters were used to direct expression of the GUS reporter gene: the wild-type 35S promoter (strain CM4-184, −270 to −1); −90MTF, a weakened, methylation target-free derivative of 35S (31), truncated to −90; and gst-MTF, the elicitor response and enhancer elements of the potato gst-1 promoter (26, 47) fused to the core 35S promoter. The promoters are represented as large open arrows, with the shaded end of each arrow representing the common “core” promoter region (−46 to −1). The S1 leader sequence was inserted as indicated, on a KpnI-NcoI fragment. The GUS reporter gene and 35S polyadenylation region (pA) are represented as boxes. (B) Relative GUS activities in O. violaceus protoplasts of the constructs depicted in panel A with (+) or without (−) the S1 leader. Activity in the presence of S1 was set to 100% for each promoter. Inset shows the levels of GUS expression from the three promoters (in the presence of S1) relative to each other.
GUS expression from these plasmids was measured after transient expression in O. violaceus protoplasts. The effect of the S1 sequence on GUS expression was clear (Fig. 1B): in all cases, a dramatic enhancement of GUS expression was observed in the presence of the S1 leader, particularly with the weak −90MTF promoter and the gst-MTF promoter. Results with the complete 35S promoter, although still clearly showing an effect of S1 on expression, were more variable, with pronounced seasonal variations in the results that did not occur with the other promoters. For this and other reasons (see below), the −90MTF promoter was used for further characterization of the S1 effect.
S1 sequences can enhance reporter gene expression from an upstream position.
The results shown in Fig. 1 gave the first hint that the S1 sequences might be affecting transcriptional as well as posttranscriptional processes. Since the S1 region is transcribed and is thus present on the resulting mRNA, an obvious potential explanation for its stimulatory effects is that it acts as an enhancer of translation. Indeed, the S1 sequence is known to be relatively unstructured, a feature known to facilitate translation of a downstream open reading frame. However, if this were the only effect of the S1 region, the extent of stimulation would be expected to be independent of the promoter used, but this is not what was observed. Different promoters were affected to different extents by deletion of the S1 region. This led us to investigate whether S1 contains enhancer-like promoter elements.
The 60-nt S1 leader sequence was transferred from its downstream position to a location upstream of the weakened −90MTF promoter (S1−90MTF-nL-GUS, S1rev−90MTF-nL-GUS). Compared to that of the leaderless construct (−90MTF-nL-GUS), reporter gene expression was roughly twofold higher regardless of the orientation of the S1 sequence (Fig. 2). A duplicated S1 in the upstream position (2×S1rev−90MTF-nL-GUS) stimulated expression more than a single copy. These results indicate that S1 contains a DNA-based enhancer element(s).
The drop in the expression level upon deletion of the 5′ untranslated region can be partially ascribed to reduced translatability due to the short leader remaining (20 bp in contrast to 63 bp in S1). By replacing the S1 leader with a random sequence (Linker) of the same length and with a similar secondary structure (with a free energy of −1.7 compared to −1.2 for S1), we were able to distinguish translational effects deriving from shortening of the leader from sequence-specific effects influencing promoter strength by comparing results from in vivo expression in protoplasts (Fig. 2B) with results from in vitro translation assays carried out in WGE (Fig. 2C). Replacing the S1 leader region with the Linker did not cause any changes in translatability (Fig. 2C), although there was a striking drop in GUS expression in vivo (Fig. 2B). In contrast, deletion of S1 (nL) negatively influenced the translational process. RNA protection assays performed with protoplasts transiently transfected with various leader mutants also demonstrated a clear correlation between reporter gene expression and mRNA amount (data not shown). This latter correlation, together with the results shown in Fig. 1A, which clearly show differing GUS expression levels from identical mRNAs, lead us to consider it unlikely that changes in mRNA stability and half-life contribute to the differences in expression seen in Fig. 2B.
Nuclear proteins bind to CT stretches within S1.
DNA gel mobility shift experiments were performed to search for binding of potential transcription factors to the S1 element. Radioactively labeled double-stranded S1 DNA was used as a probe in experiments with nuclear extracts from protoplasts of O. violaceus and O. sativa. Two complexes were observed with all extract preparations tested (see Fig. 3A, complexes I and II, for O. sativa nuclear extracts; results with O. violaceus extracts were similar [data not shown]). Both shifts are specific: they can be competed by unlabeled S1 but not by unrelated DNA fragments from either the promoter or parts of an unrelated reporter gene (CAT) (Fig. 3A). A third complex (complex III) appeared with some protoplast extract preparations tested, but the corresponding protein factor(s) is either more abundant or more stable in extracts prepared from seedlings (see below).
FIG. 3.
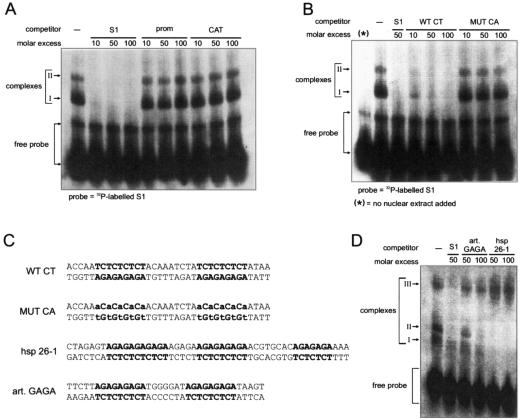
CT motifs within S1 interact with nuclear proteins. (A and B) Gel mobility shift assays. The labeled DNA probe corresponds to the first 60 bp of the 5′ end of the CaMV RNA leader sequence (S1). Nuclear extracts used for the gels shown were prepared from O. sativa protoplasts. The positions of free probe and shifted complexes I and II are indicated. Fragments introduced as competitors are given above the relevant gel lanes, at the molar excess indicated: −, no competitor; S1, unlabeled probe fragment; prom, a CaMV 35S promoter fragment from −70 to −2; CAT, a 120-bp fragment from the beginning of the CAT gene; WT CT and MUT CA, 36-bp DNA fragments whose sequences are given in panel C. (C) Sequences of double-stranded DNA oligonucleotides used as competitors in the experiments for which results are shown in panels B and D. CT and GAGA motifs are boldfaced; mutated nucleotides are lowercased. The hsp-26-1 GAGA motif sequence is from the work of Lu et al. (45). (D) Gel mobility shift assay using labeled S1 as a probe, with the competitors indicated. Complex III is an additional, higher-molecular-weight complex observed in extracts of O. sativa seedlings.
The most eye-catching motifs within S1 are the two stretches of multiple CT repeats (Fig. 3C). Similar sequence motifs (albeit in reverse orientation) are known to be the interaction targets for transcription factors in other systems, e.g., the GAGA elements found in Drosophila heat shock genes (61). To investigate whether these motifs represent protein binding sites, a double-stranded 36-bp DNA fragment covering the two CT repeat sequences (WT CT) was used as a competitor in the binding assays. This fragment could compete both complexes almost as well as the full-length wild-type S1 region (Fig. 3B), while a mutated derivative, where each T residue was replaced with an A (MUT CA), could not, indicating that one or both of the two CT motifs are involved in the interaction with nuclear proteins.
A competitor fragment corresponding to part of the hsp26-1 gene sequence containing the GAGA element (45) was able to compete with S1 for binding of these proteins (Fig. 3D). However, an artificial GAGA fragment competed much less efficiently, suggesting an influence either of the number of GAGA motifs (hsp26-1 contains three, while S1 and artificial GAGA have only two each) or of context and flanking sequences on protein binding.
A nuclear extract from O. sativa seedlings was used for the latter experiment. In this extract, an additional, higher-molecular-weight complex was also observed. Addition of competitors revealed that this complex (Fig. 3D, complex III) is specific for S1 but is not significantly competed by the hsp26-1 GAGA element.
Competition gel shift assays were then performed to further characterize the binding sites and the types of complexes formed between the S1 region and nuclear proteins. An oligonucleotide in which the CT motifs were mutated to GA motifs (MUT GA) competed only weakly with wild-type S1 sequences for the formation of DNA-protein complexes, confirming the importance of the flanking sequences for protein binding. Interestingly, the MUT GA oligonucleotide appears to compete slightly with S1 for the formation of complex I but hardly at all for complex II, suggesting that there might be an additional protein(s) involved in the formation of the latter that can stabilize the proteins that bind to CT motifs.
Two additional competitor variants were tested. In MUT GT, every C in the CT motifs of the oligonucleotide was changed to G (Fig. 4A); in MUT CA (partial), only the three internal T residues of each motif were changed to A. Both these oligonucleotides competed as well as wild-type S1 for complex II while losing completely the ability to compete complex I (Fig. 4B). Again, this is consistent with a requirement for another protein component in complex II that recognizes either the sequence between the two CT stretches or sites overlapping the CT motifs. Different mutations were introduced in the context of the full-length S1 sequence and used as labeled probes in gel shift assays. The MUT GT mutation supported the formation of complexes II and III but not that of complex I (Fig. 4C). The latter cannot be formed on this sequence, because the CT motifs are lacking. However, we interpret complex II as being composed of an additional protein interacting with the sequence between the CT stretches on the probe and recruiting the CT-binding factor(s) through protein-protein interactions. Consistent with this interpretation, mutation of this region resulted in loss of complex II formation (Fig. 4A, MUT AAA-TTT; Fig. 4C, S1 AAA-TTT).
FIG. 4.
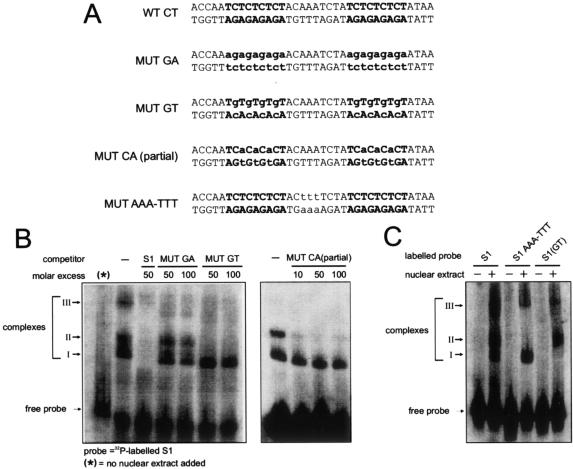
Additional proteins may stabilize complexes. (A) Sequences of double-stranded DNA oligonucleotides used as competitors in the experiments for which results are shown in panel B. CT motifs are boldfaced; mutated nucleotides are lowercased. (B) Gel mobility shift assays using labeled S1 as a probe, with the competitors indicated. Nuclear extract was from O. sativa seedlings. (C) Gel mobility shift assays using labeled probes as indicated. S1 AAA-TTT carries the mutation shown in MUT AAA-TTT in panel A in the context of the entire S1 sequence; likewise, the S1(GT) probe carries the MUT GT mutation.
The formation of complex III can be competed by all of the S1 mutants (Fig. 4B), suggesting that the protein binding site is located outside the two CT motifs, although the weak competition observed in some cases suggests that there may be some overlap in the binding sites involved.
As evidence supporting this suggestion, when hsp26-1 DNA was used as a competitor, the formation of complex III was stimulated while complexes I and II were completely competed, suggesting competition for overlapping binding sites (see Fig. 3D). Further studies of complex III should be carried out with seedling extracts, where the abundance of the corresponding proteins is apparently much greater.
CT-rich motifs in the S1 region enhance expression in vivo.
To investigate the importance of the CT motifs in vivo, S1 mutants were created in which the CT motifs were changed to CA motifs. In the three protoplast types tested and in the context of the weak −90MTF promoter, this led to a substantial decrease in expression (Fig. 5A, mutant CA). As expected, the decrease was only minor in the context of the full-strength wild-type 35S promoter with dicot protoplasts, but still substantial with rice protoplasts. When the CT motifs were mutated to GA motifs to restore GAGA-like elements, activity in the context of weak upstream promoter was partially recovered in O. violaceus and N. plumbaginifolia protoplasts and completely restored in rice protoplasts (Fig. 5A, mutant GA). Mutant GT (Fig. 5A), in which CT motifs were replaced with GT motifs, also led to a drastic decrease in activity. These results correlate well with the results of the gel shift assays, i.e., mutants unable to compete to form complex I are also unable to direct full-strength expression. Consistently, the AAA-TTT mutant, which in gel shift assays still allowed formation of complex I, had no detrimental effect on expression in vivo.
We next tested the effect of removing only one of the CT stretches, or altering the distance between the two motifs. Mutation of one CT motif to an unrelated sequence reduced reporter gene activity by roughly half, with a further reduction if both CT motifs were changed. Increasing the distance between the two CTmotifs did not affect expression (Fig. 5B).
Since all of these mutations are downstream of the transcription start site, they might potentially exert their effects at a posttranscriptional level. To check if any of these mutations affected translation, a series of mRNAs corresponding to the S1 leader (wild type or mutated) plus the GUS coding region was prepared by in vitro transcription. Production of GUS protein from these RNAs was monitored after in vitro translation in WGE, both by direct visualization of the translation product and by quantification of GUS activity in the extract. The results demonstrated that the mutations introduced into the leader sequence had only slight effects on translatability (Fig. 5C).
DISCUSSION
Specific sequences downstream of the transcription initiation site of the CaMV 35S promoter (S1 region) play a role in transcriptional enhancement. Several lines of evidence support this contention: the stimulatory effect of S1 is most pronounced in connection with a weak promoter; S1 can enhance expression in a position- and orientation-independent manner; increasing the number of S1 copies inserted upstream of the initiation site further enhances expression; nuclear proteins specifically interact with S1 sequences, and mutations that disrupt protein binding also affect reporter gene expression levels.
To reveal these effects of the S1 region, it was necessary to use the relatively weak −90MTF promoter. A promoter can be weak due to reduced TFIID stability on the promoter (27, 38, 52). Many activators or enhancers influence promoter strength by facilitating TFIID association with the cognate promoter and/or stabilization of the preinitiation complex. In a strong promoter such as the wild-type 35S promoter, the redundant and combinatorial effects of the powerful upstream enhancers make it likely that the subtle effects of S1 sequences are normally overshadowed.
Sequences involved in transcriptional activation can function in a distance- and orientation-independent manner (5, 10), and the S1 leader sequence fits these criteria (Fig. 2). Consistent with the synergism of enhancer elements (13, 33, 43, 51), a copy number-dependent increase in reporter gene expression could be demonstrated.
A growing number of regulatory elements located downstream of transcription start sites have been reported, many of which affect gene expression on the transcriptional level. Two types of such elements can be distinguished. The first (designated DPE, for downstream promoter element) is located close to the transcription start site and interacts directly with subunits of the transcription factor TFIID to maximize the stability of the TFIID-DNA complex, thereby contributing to promoter strength. DPEs, apparently essential for full promoter activity, are associated either with TATA-less promoters (1, 8, 37, 49, 62) or with promoters endowed with a weak TATA box context and an Inr consensus (36, 41). The second type of element influences promoter activity more indirectly; it is the target of sequence-specific nuclear DNA binding proteins that function either as activators or repressors or by altering DNA conformation and even chromatin structure. Such proteins are often involved in temporal and spatial regulation of gene expression (14-16, 30, 53).
The involvement of CT-rich sequences (inverted: GAGA) in gene regulation has been reported for many years. (CT)/(GA) repeats were first described in Drosophila heat shock protein and histone gene promoters (22, 45) and were later assigned to diverse groups of genes, including inducible, homeotic, and housekeeping genes (24). In addition to Drosophila, such elements have been shown to play an important role in transcriptional regulation in many other organisms, including vertebrates (4, 35, 59), plants (2, 6, 7, 15, 16, 55), and viruses (28), suggesting the involvement of a universally valid mechanism of gene regulation.
GAGA elements have been most thoroughly examined in Drosophila melanogaster, where they are the binding targets of “GAGA transcription factor” (GAF) and the Pipsqueak (Psq) protein (56). A single trinucleotide sequence (GAG) represents a sufficient binding site for GAF (61), in contrast to Psq, which needs a longer GAGA element (39). The two proteins can bind directly to each other, thus establishing a protein complex bound to chromosomal target sites in vivo (56). Biochemical studies indicate that GAF activates RNA polymerase II transcription by counteracting chromatin repression (18). At the hsp70 and hsp26 promoters, GAF directs the formation of an open chromatin structure by nucleosome positioning that allows the association of additional factors necessary for transcriptional competence and creation of nucleosome-free DNase I-hypersensitive sites (44, 60). It appears that the chromatin remodeling complex NURF plays a key role in this process by altering the nucleosome structure in an ATP-dependent manner to allow GAGA factor binding (57). Recent studies on GAF showed that its binding on DNA can positively contribute to the recruitment of TFIID, confirming the notion that it is a widely used transcription factor (40).
GAGA-binding proteins (GBP) have also been identified in plants. Originally detected in soybeans, several GBP homologues have been found to date in several dicot plants and in the monocot rice (O. sativa). In soybeans, GBP binds to the promoter of the heme and chlorophyll synthesis gene Gsa1, which contains a GAGA element, and is implicated in the control of that gene (55).
Several lines of evidence support our speculation that the protein(s) forming complex I on the S1 leader region of CaMV belongs to the class of GBP found in plants. Like soybean GBP, our protein(s) seems to bind the GAGA motif in a very sequence-specific manner, and binding correlates with enhanced promoter activity. Deletion of one of the two CT motifs in the S1 leader region reduced activity almost as much as deletion of both motifs, suggesting a synergistic effect mediated by the bound proteins. On the other hand, insertion of a random 20-bp fragment between the CT motifs had no effect (Fig. 5B). Similar characteristics have been reported for Drosophila GAF, which was shown to oligomerize into higher-order complexes that could then bind to multiple sites spaced at variable distances to synergistically activate transcription (17, 34). The notion that our CT-binding protein has features similar to those of Drosophila GAF was further supported by the observation that an hsp26 gene fragment containing a known GAGA element could efficiently compete with S1 for binding of this protein(s) (Fig. 3C).
The second complex (complex II) seems also to be formed by a protein(s) that binds in a sequence-specific manner to the region located between the two CT motifs (CAAAT), although adjacent sequences overlapping the CT motifs also seem to be involved (Fig. 3B and 4B and C). In contrast to the CT motifs, no functional influence on transcriptional efficiency could be detected by mutagenesis (Fig. 5A), precluding any speculation on biological relevance.
In addition to the CT motif interacting with the GAGA-binding factor, a search of plant databases for potential transcription factor binding sites (by using the Patch program, version 1.0-public, and the TRANSFAC 6.0-Public database; Biobase GmbH, Wolfenbüttel, Germany [http://www.gene-regulation.com/pub/databases.html#transfac]) using the S1 sequence revealed a CAAAT motif specifically bound by the maize high-mobility-group (HMG) protein (25). HMG proteins are relatively abundant chromatin-associated nonhistone nucleoproteins that are able to bend DNA, thus facilitating the binding of various transcription factors to their cognate DNA sequences (9, 42). Sequence-specific binding of HMG proteins involves not only the core binding sequence but also 5′ and 3′ flanking sequences (58), which would be consistent with our observations.
Interestingly, several high-affinity binding sites for HMG1 have been identified in the 5′ long terminal repeat of human immunodeficiency virus type 1. One of these coincides with a transcriptional activator binding site located downstream of the transcriptional start site in the 5′ untranslated region. It was therefore suggested that HMG1 might play a fundamental role in the expression of human immunodeficiency virus type 1 by determining the nature of transcription factor-promoter interactions (30). A similar case was found in plants at the phytochrome A (PHYA) gene promoter, where HMG-I/Y was found to stimulate the sequence-specific binding of the transcriptional activator GT-2 to the promoter (46).
Like NURF, which is required for efficient binding of GAF to GAGA motifs, the protein(s) forming complex II may function to modify DNA or chromatin structure to facilitate the binding of GAF-like factors to their cognate DNA sequences. The reason why no transcriptional effect is observed upon mutation of the binding site for complex II may lie in the experimental setup. Functional tests were performed by transiently transfecting DNA into protoplasts, followed by determination of reporter gene expression after 12 h of incubation. In contrast to the CaMV minichromosome, which has been shown to exist as a chromatin-like nucleoprotein complex with nucleosome subunits in the nuclei of infected leaves (48, 50), the transfected DNA is originally neither arranged in nucleosomes nor properly packed in chromatin. In this situation, the GAGA-binding protein could bind by itself without the help of the complex II-forming protein, which would, however, be absolutely required under natural conditions. Alternatively, the protein may simply be inactive in protoplasts.
Whether the positive effect of the S1 leader is restricted to specific cellular or environmental conditions in the context of the viral life cycle remains an open question. Taking into account the modular structure of the complete 35S promoter and the demonstrated tissue specificity of the different domains 5′ of the TATA box (3), it is possible that the cis elements in the S1 region are responsible for guaranteeing a minimal basal activity of the promoter under every possible circumstance. This notion is supported by the occurrence of CT motifs in plant housekeeping genes, such as those encoding ribosomal proteins L12 and L13, suggesting involvement of these motifs in a universally valid mechanism of gene regulation (6). Transcription from the genomic promoter is of critical importance to a pararetrovirus, not only for production of viral proteins but also for viral replication. The use of a common, and thus abundant, transcription factor to ensure a minimal level of promoter activity could reflect a fundamental survival strategy in these viruses, as evidenced by the finding of a similar mechanism in the plant pararetrovirus RTBV (28).
Acknowledgments
We gratefully acknowledge the expertise of Matthias Müller in plant tissue culture and the preparation of protoplasts. The rice Oc suspension culture was provided by K. Shimamoto (Graduate School of Biological Sciences, Nara Institute of Science and Technology, Ikoma, Nara, Japan). We are grateful to Diana Dominguez for help in setting up the gel-shift experiments.
H.M.R. was supported by a grant from the Swiss National Research Foundation.
REFERENCES
- 1.Abrescia, C., E. De Gregorio, M. Frontini, R. Mantovani, and P. Di Nocera. 2002. A novel intragenic sequence enhances initiator-dependent transcription in human embryonic kidney 293 cells. J. Biol. Chem. 277:19594-19599. [DOI] [PubMed] [Google Scholar]
- 2.Bao, X., B. S. Shorrosh, and J. B. Ohlrogge. 1997. Isolation and characterization of an Arabidopsis biotin carboxylase gene and its promoter. Plant Mol. Biol. 35:539-550. [DOI] [PubMed] [Google Scholar]
- 3.Benfey, P., L. Ren, and N. Chua. 1990. Combinatorial and synergistic properties of CaMV 35S enhancer subdomains. EMBO J. 9:1685-1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bevilacqua, A., M. T. Fiorenza, and F. Mangia. 2000. A developmentally regulated GAGA box-binding factor and Sp1 are required for transcription of the hsp70.1 gene at the onset of mouse zygotic genome activation. Development 127:1541-1551. [DOI] [PubMed] [Google Scholar]
- 5.Blackwood, E. M., and J. T. Kadonaga. 1998. Going the distance: a current view of enhancer action. Science 281:60-63. [DOI] [PubMed] [Google Scholar]
- 6.Bolle, C., R. G. Herrmann, and R. Oelmüller. 1996. Different sequences for 5′-untranslated leaders of nuclear genes for plastid proteins affect the expression of the β-glucuronidase gene. Plant Mol. Biol. 32:861-868. [DOI] [PubMed] [Google Scholar]
- 7.Bolle, C., S. Sopory, T. Lübberstedt, R. G. Herrmann, and R. Oelmüller. 1994. Segments encoding 5′-untranslated leaders of genes for thylakoid proteins contain cis-elements essential for transcription. Plant J. 6:513-523. [DOI] [PubMed] [Google Scholar]
- 8.Burke, T. W., and J. T. Kadonaga. 1997. The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of Drosophila. Genes Dev. 11:3020-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustin, M. 1999. Regulation of DNA-dependent activities by the functional motif of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19:5237-5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler, J. E. F., and J. T. Kadonaga. 2001. Enhancer-promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev. 15:2515-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, G., M. Müller, I. Potrykus, T. Hohn, and J. Fütterer. 1994. Rice tungro bacilliform virus: transcription and translation in protoplasts. Virology 204:91-100. [DOI] [PubMed] [Google Scholar]
- 12.Chen, G., H. M. Rothnie, X. He, T. Hohn, and J. Fütterer. 1996. Efficient transcription from the rice tungro bacilliform virus promoter requires elements downstream of the transcription start site. J. Virol. 70:8411-8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Courey, A. J., and S. Jia. 2001. Transcriptional repression: the long and the short of it. Genes Dev. 15:2786-2796. [DOI] [PubMed] [Google Scholar]
- 14.Curie, C., M. Axelos, C. Bardet, R. Atanassova, N. Chaubet, and B. Lescure. 1993. Modular organization and developmental activity of an Arabidopsis thaliana EF-1α gene promoter. Mol. Gen. Genet. 238:428-436. [DOI] [PubMed] [Google Scholar]
- 15.de Boer, G.-J., C. Testerink, G. Pielage, H. J. J. Nijkamp, and A. R. Stuitje. 1999. Sequences surrounding the transcription initiation site of the Arabidopsis enoyl-acyl carrier protein reductase gene control seed expression in transgenic tobacco. Plant Mol. Biol. 39:1197-1207. [DOI] [PubMed] [Google Scholar]
- 16.Enjuto, M., V. Lumbreras, C. Marin, and A. Boronat. 1995. Expression of the Arabidopsis HMG2 gene, encoding 3-hydroxy-3-methylglutaryl coenzyme A reductase, is restricted to meristematic and floral tissues. Plant Cell 7:517-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Espinas, M. L., E. Jimenez-Garcia, A. Vaquero, S. Canudas, J. Bernues, and F. Azorin. 1999. The N-terminal POZ domain of GAGA mediates the formation of oligomers that bind DNA with high affinity and specificity. J. Biol. Chem. 274:16461-16469. [DOI] [PubMed] [Google Scholar]
- 18.Farkas, G., B. A. Leibovitch, and S. C. R. Elgin. 2000. Chromatin organization and transcriptional control of gene expression in Drosophila. Gene 253:117-136. [DOI] [PubMed] [Google Scholar]
- 19.Fütterer, J., K. Gordon, P. Pfeiffer, H. Sanfacon, B. Pisan, J.-M. Bonneville, and T. Hohn. 1989. Differential inhibition of downstream gene expression by the cauliflower mosaic virus 35S RNA leader. Virus Genes 3:45-55. [DOI] [PubMed] [Google Scholar]
- 20.Fütterer, J., K. Gordon, H. Sanfacon, J.-M. Bonneville, and T. Hohn. 1990. Positive and negative control of translation by the leader sequence of cauliflower mosaic virus pregenomic 35S RNA. EMBO J. 9:1697-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fütterer, J., and T. Hohn. 1991. Translation of a polycistronic mRNA in the presence and absence of the cauliflower mosaic virus transactivator protein. EMBO J. 10:3887-3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilmour, D. S., G. H. Thomas, and S. C. R. Elgin. 1989. Drosophila nuclear proteins bind to regions of alternating C and T residues in gene promoters. Science 245:1487-1490. [DOI] [PubMed] [Google Scholar]
- 23.Goodall, G. J., K. Wiebauer, and W. Filipowicz. 1990. Analysis of pre-mRNA processing in transfected plant protoplasts. Methods Enzymol. 181:148-161. [DOI] [PubMed] [Google Scholar]
- 24.Granok, H., B. A. Leibovitch, C. D. Shaffer, and S. C. R. Elgin. 1995. Gaga over GAGA factor. Curr. Biol. 5:238-241. [DOI] [PubMed] [Google Scholar]
- 25.Grasser, K. D., U.-G. Maier, M. M. Haass, and G. Feix. 1990. Maize high mobility group proteins bind to CCAAT and TATA boxes of a zein gene promoter. J. Biol. Chem. 265:4185-4188. [PubMed] [Google Scholar]
- 26.Hahn, K., and G. Strittmatter. 1994. Pathogen-defence gene prp1-1 from potato encodes an auxin-responsive glutathione S-transferase. Eur. J. Biochem. 226:619-626. [DOI] [PubMed] [Google Scholar]
- 27.Hampsey, M. 1998. Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol. Mol. Biol. Rev. 62:465-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He, X., J. Fütterer, and T. Hohn. 2002. Contribution of downstream promoter elements to transcriptional regulation of the rice tungro bacilliform virus promoter. Nucleic Acids Res. 30:497-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He, X., T. Hohn, and J. Fütterer. 2000. Transcriptional activation of the rice tungro bacilliform virus gene is critically dependent on an activator element located immediately upstream of the TATA box. J. Biol. Chem. 275:11799-11808. [DOI] [PubMed] [Google Scholar]
- 30.Henderson, A., M. Bunce, N. Siddon, R. Reeves, and D. J. Tremethick. 2000. High-mobility-group protein I can modulate binding of transcription factors to the U5 region of the human immunodeficiency virus type 1 proviral promoter. J. Virol. 74:10523-10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hohn, T., S. Corsten, S. Rieke, M. Müller, and H. M. Rothnie. 1996. Methylation of coding region alone inhibits gene expression in plant protoplasts. Proc. Natl. Acad. Sci. USA 93:8334-8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hull, R., S. N. Covey, and P. Dale. 2002. Genetically modified plants and the 35S promoter: assessing the risk and enhancing the debate. Microb. Ecol. Health Dis. 12:1-5. [Google Scholar]
- 33.Iniguez-Lluhi, J. A., and D. Pearce. 2000. A common motif within the negative regulatory regions of multiple factors inhibits their transcriptional synergy. Mol. Cell. Biol. 20:6040-6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsani, K. R., M. A. N. Hajibagheri, and C. P. Verrijzer. 1999. Co-operative DNA binding by GAGA transcription factor requires the conserved BTB/POZ domain and reorganizes promoter topology. EMBO J. 18:698-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenyon, J. R., and I. W. Craig. 1999. Analysis of the 5′ regulatory region of the human Norrie's disease gene: evidence that a non-translated CT dinucleotide repeat in exon one has a role in controlling expression. Gene 227:181-188. [DOI] [PubMed] [Google Scholar]
- 36.Knutson, A., E. Castano, T. Oelgeschlager, R. G. Roeder, and G. Westin. 2000. Downstream promoter sequences facilitate the formation of a specific transcription factor IID-promoter complex topology required for efficient transcription from the megalin/low density lipoprotein receptor-related protein 2 promoter. J. Biol. Chem. 275:14190-14197. [DOI] [PubMed] [Google Scholar]
- 37.Kutach, A. K., and J. T. Kadonaga. 2000. The downstream promoter element DPE appears to be as widely used as the TATA box in Drosophila core promoters. Mol. Cell. Biol. 20:4754-4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, T. I., and R. A. Young. 2000. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 34:77-137. [DOI] [PubMed] [Google Scholar]
- 39.Lehmann, M., T. Siegmund, K.-G. Lintermann, and G. Korge. 1998. The Pipsqueak protein of Drosophila melanogaster binds to GAGA sequences through a novel DNA-binding domain. J. Biol. Chem. 273:28504-28509. [DOI] [PubMed] [Google Scholar]
- 40.Leibovich, B. A., Q. Lu, L. R. Benjamin, Y. Liu, D. S. Gilmour, and S. C. R. Elgin. 2002. GAGA factor and the TFIID complex collaborate in generating an open chromatin structure at the Drosophila melanogaster hsp26 promoter. Mol. Cell. Biol. 22:6148-6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis, B. A., T.-K. Kim, and S. H. Orkin. 2000. A downstream element in the human beta-globin promoter: evidence of extended sequence-specific transcription factor IID contacts. Proc. Natl. Acad. Sci. USA 97:7172-7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lichota, J., and K. D. Grasser. 2001. Differential chromatin association and nucleosome binding of the maize HMGA, HMGB, and SSRP1 proteins. Biochem. J. 40:7860-7867. [DOI] [PubMed] [Google Scholar]
- 43.Lin, Y.-S., M. Carey, M. Ptashne, and M. R. Green. 1990. How different eukaryotic transcriptional activators can cooperate promiscuously. Nature 345:359-361. [DOI] [PubMed] [Google Scholar]
- 44.Lu, Q., L. L. Wallrath, and S. C. R. Elgin. 1995. The role of a positioned nucleosome at the Drosophila melanogaster hsp26 promoter. EMBO J. 14:4738-4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu, Q., L. L. Wallrath, H. Granok, and S. C. R. Elgin. 1993. (CT)n(GA)n repeats and heat shock elements have distinct roles in chromatin structure and transcriptional activation of the Drosophila hsp26 gene. Mol. Cell. Biol. 13:2802-2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Garcia, J. F., and P. H. Quail. 1999. The HMG-I/Y protein PF1 stimulates binding of the transcriptional activator GT-2 to the PHYA gene promoter. Plant J. 18:173-183. [DOI] [PubMed] [Google Scholar]
- 47.Martini, N., M. Egen, I. Rüntz, and G. Strittmatter. 1993. Promoter sequences of a potato pathogenesis-related gene mediate transcriptional activation selectively upon fungal infection. Mol. Gen. Genet. 236:179-186. [DOI] [PubMed] [Google Scholar]
- 48.Menissier, J., G. de Murcia, G. Lebeurier, and L. Hirth. 1983. Electron microscopic studies of the different topological forms of the cauliflower mosaic virus DNA: knotted encapsidated DNA and nuclear minichromosome. EMBO J. 2:1067-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minchiotti, G., C. Contursi, and P. Di Nocera. 1997. Multiple downstream promoter modules regulate the transcription of the Drosophila melanogaster I, Doc and F elements. J. Mol. Biol. 267:37-46. [DOI] [PubMed] [Google Scholar]
- 50.Olszewski, N., G. Hagen, and T. J. Guilfoyle. 1982. A transcriptionally active, covalently closed minichromosome of cauliflower mosaic virus DNA isolated from infected turnip leaves. Cell 29:395-402. [DOI] [PubMed] [Google Scholar]
- 51.Ondek, B., A. Shepard, and W. Herr. 1987. Discrete elements within the SV40 enhancer region display different cell-specific enhancer activities. EMBO J. 6:1017-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pugh, B. F. 2000. Control of gene expression through regulation of the TATA-binding protein. Gene 255:1-14. [DOI] [PubMed] [Google Scholar]
- 53.Rouster, J., J. van Mechelen, and V. Cameron-Mills. 1998. The untranslated leader sequence of the barley lipoxygenase 1 (Lox1) gene confers embryo-specific expression. Plant J. 15:435-440. [DOI] [PubMed] [Google Scholar]
- 54.Ryabova, L. A., and T. Hohn. 2000. Ribosome shunting in the cauliflower mosaic virus 35S RNA leader is a special case of reinitiation of translation functioning in plant and animal systems. Genes Dev. 14:817-829. [PMC free article] [PubMed] [Google Scholar]
- 55.Sangwan, I., and M. R. O'Brian. 2002. Identification of a soybean protein that interacts with GAGA element dinucleotide repeat DNA. Plant Physiol. 129:1788-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwendemann, A., and M. Lehmann. 2002. Pipsqueak and GAGA factor act in concert as partners at homeotic and many other loci. Proc. Natl. Acad. Sci. USA 99:12883-12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsukiyama, T., and C. Wu. 1995. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 83:1011-1020. [DOI] [PubMed] [Google Scholar]
- 58.van Beest, M., D. Dooijes, M. van De Wetering, S. Kjaerulff, A. Bonvin, O. Nielsen, and H. Clevers. 2000. Sequence-specific high mobility group box factors recognize 10-12-base pair minor groove motifs. J. Biol. Chem. 275:27266-27273. [DOI] [PubMed] [Google Scholar]
- 59.Volpi, S., C. Rabada-Diehl, N. Cawley, and G. Aguilera. 2002. Transcriptional regulation of the pituitary vasopressin V1b receptor involves a GAGA-binding protein. J. Biol. Chem. 31:27829-27838. [DOI] [PubMed] [Google Scholar]
- 60.Wilkins, R. C., and J. T. Lis. 1997. Dynamics of potentiation and activation: GAGA factor and its role in heat shock gene regulation. Nucleic Acids Res. 25:3963-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilkins, R. C., and J. T. Lis. 1998. GAGA factor binding to DNA via a single trinucleotide sequence element. Nucleic Acids Res. 26:2672-2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou, T., and C.-M. Chiang. 2001. The intronless and TATA-less human TAFII55 gene contains a functional initiator and a downstream promoter element. J. Biol. Chem. 276:25503-25511. [DOI] [PubMed] [Google Scholar]