

Abstract
Gammaherpesviruses can establish lifelong latent infections in lymphoid cells of their hosts despite active antiviral immunity. Identification of the immune mechanisms which regulate gammaherpesvirus latent infection is therefore essential for understanding how gammaherpesviruses persist for the lifetime of their host. Recently, an individual with chronic active Epstein-Barr virus infection was found to have mutations in perforin, and studies using murine gammaherpesvirus 68 (γHV68) as a small-animal model for gammaherpesvirus infection have similarly revealed a critical role for perforin in regulating latent infection. These results suggest involvement of the perforin/granzyme granule exocytosis pathway in immune regulation of gammaherpesvirus latent infection. In this study, we examined γHV68 infection of knockout mice to identify specific molecules within the perforin/granzyme pathway which are essential for regulating gammaherpesvirus latent infection. We show that granzymes A and B and the granzyme B substrate, caspase 3, are important for regulating γHV68 latent infection. Interestingly, we show for the first time that orphan granzymes encoded in the granzyme B gene cluster are also critical for regulating viral infection. The requirement for specific granzymes differs for early versus late forms of latent infection. These data indicate that different granzymes play important and distinct roles in regulating latent gammaherpesvirus infection.
Gammaherpesviruses are characterized by their association with tumor induction and by their ability to establish lifelong latent infection in lymphoid cells. Human gammaherpesviruses, such as Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus, can cause a number of malignancies and lymphoproliferative disorders. These include Burkitt's lymphoma, Hodgkin's disease, and nasopharyngeal carcinoma for EBV (36, 54) and Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma for Kaposi's sarcoma-associated herpesvirus (43). Disease caused by gammaherpesviruses during chronic infection is associated with latent infection and/or reactivation from latency and is more common in immunocompromised individuals. However, the molecular basis of immune regulation of gammaherpesvirus chronic infection is incompletely understood due, in part, to the strict species specificity of the human viruses. We and others have therefore studied murine gammaherpesvirus 68 (γHV68) as a small-animal model for defining mechanisms of immune control of chronic gammaherpesvirus infection in vivo.
γHV68 is a natural murine pathogen (6, 7) which readily infects inbred and outbred mice and has extensive genome homology to the human gammaherpesviruses (18, 76). γHV68 acute infection in mice is characterized by lytic viral replication in multiple organs (8, 53, 69), which is typically cleared by 15 days postinfection (dpi) (9, 69, 80). Acute infection is succeeded by a latent infection in B cells, macrophages, and dendritic cells (20, 70, 82). The early form of γHV68 latent infection is characterized by efficient viral reactivation ex vivo and by high-level expression of the M2 latency gene, whereas the late form of latency has less efficient viral reactivation and lower levels of M2 expression (11, 21, 27, 74, 81). These data suggest variances in viral gene expression between early and late forms of viral latency and demonstrate that γHV68 transitions from an early form of latency which reactivates efficiently to a more stable form of latency at later times postinfection.
Studies examining host immune responses to γHV68 infection have demonstrated that CD8+ T cells are important for clearance of γHV68 acute infection (19, 68, 80) and that CD8+ T cells specific for viral lytic antigens can significantly decrease viral titers following vaccination with those antigens (37, 67). Moreover, CD8+ T-cell-deficient mice exhibit defects in regulating γHV68 chronic infection, including delayed clearance of persistent replication and increased frequencies of cells which reactivate virus ex vivo and which harbor latent viral genome (72). The relative contributions of specific immune effector mechanisms to host regulation of γHV68 latent infection can differ between tissues (72), suggesting site-dependent mechanisms for controlling viral infection. The observed differences between early and late forms of latency additionally suggest the possibility that different immune mechanisms regulate early versus late forms of latency, although this has not previously been demonstrated. The host immune response regulates latency by limiting the number of cells harboring viral genome and by modulating the efficiency with which latently infected cells reactivate virus when explanted (72). How this regulation is achieved in vivo is not clear, although it may include controlling different phases of infection (for example, establishment or maintenance of latency or reactivation from latency) and altering various aspects of infection, such as cell tropism and viral gene expression.
A primary mechanism by which cytotoxic T lymphocytes (CTLs) kill target cells is through the perforin/granzyme granule exocytosis pathway. Observations that perforin-deficient (perforin−/−) mice have impaired regulation of γHV68 latency and reactivation (72) suggest that the granule exocytosis pathway is important in regulating γHV68 latent infection. Cytotoxicity via the granule exocytosis pathway involves targeted secretion of CTL granule contents, which include the pore-forming protein, perforin, and a family of serine proteases known as granzymes (Gzms). Perforin is essential for cytotoxicity through the granule exocytosis pathway (30, 78) and is responsible for proper trafficking of granzymes (57, 60), which are required for induction of target cell apoptosis (26, 62-64). Eleven murine granzymes have been identified—granzymes A and K are encoded on chromosome 13 and granzyme M is encoded on chromosome 10, while granzymes B, C, D, E, F, G, L, and N are in a multigene cluster on chromosome 14 (reviewed in reference 25). Of these, granzymes A and B have been most extensively studied; the remaining granzymes have unknown functions and are collectively known as “orphan” granzymes.
In vitro cytotoxicity assays using immune effector cells from GzmA- or GzmB-deficient mice indicate that GzmB, but not GzmA, is required for rapid induction of target cell apoptosis (17, 26, 63), although GzmA can compensate for GzmB deficiency with delayed kinetics (62). Moreover, effector cells deficient in both granzymes A and B are as defective as perforin−/− cells in mediating target cell cytotoxicity (62, 64). The original GzmB-deficient mice, which were on a mixed B6/129 background, were later found to also have downregulated expression of the downstream orphan granzymes C, D, and F due to a neighborhood effect caused by retention of a PGK-Neo cassette in the GzmB gene (49). These mice are therefore more accurately described as GzmB-cluster−/− mice. Subsequent generation of the same mutation on a pure 129/SvJ background revealed restriction of the neighborhood effect to granzymes C and F. Removal of the PGK-Neo cassette eliminated this effect, resulting in mice deficient only in GzmB (GzmB−/−). Recent comparisons of CTLs from GzmB-cluster−/− and GzmB−/− mice revealed a more severe defect in cytotoxicity in GzmB-cluster−/− effector cells compared to GzmB−/− cells (P. A. Revell, W. Grossman, D. A. Thomas, R. Behl, X. Cao, Z. Lu, and T. J. Ley, unpublished data), and GzmC has been shown to induce target cell death in vitro (29). Together, these studies suggest that orphan granzymes encoded in the GzmB gene cluster have a role in cytotoxicity.
The granule exocytosis pathway has been shown to be important for regulation of infection with a number of viruses (reviewed in reference 57). Perforin mediates resistance to acute infection with the murine poxvirus ectromelia, the alphaherpesvirus herpes simplex virus, and the betaherpesvirus murine cytomegalovirus (MCMV) (15, 23, 45, 55). Mutations in perforin were recently described in an individual with chronic active EBV infection (33), suggesting the importance of perforin in human gammaherpesvirus infection. Additionally, EBV-specific CD4+ and CD8+ CTLs can utilize the perforin/granzyme pathway to kill EBV-infected cell lines (35, 83). Although studies with γHV68 indicate that perforin is not essential for regulating acute infection (73), perforin−/− mice exhibit increased frequencies of cells which reactivate virus ex vivo and which harbor viral genome at early and late time points during γHV68 latent infection (72). Thus, perforin is an important regulator of both the early and late forms of γHV68 latency. Less is known about the function of granzymes during viral infection. Granzymes have been shown to be important during acute infection with ectromelia (44, 46), herpes simplex virus (48), and MCMV (55). However, the role of granzymes in regulating chronic viral infection is unknown, and the specific contributions of orphan granzymes to antiviral immunity have not been previously examined.
In this study, we utilized mice deficient for specific granzymes to identify those which are important for immune regulation of γHV68 latent infection in vivo. We show that granzymes A and B regulate γHV68 latent infection at an early time point, although their roles appear to be functionally redundant. We further demonstrate that orphan granzymes are critical for regulating both early and late forms of γHV68 latent infection. This represents the first demonstration of a role for orphan granzymes in control of viral infection in vivo. In addition, caspase 3, which can be activated by GzmB, is involved in regulating γHV68 latent infection at an early time point. Thus, immune regulation of γHV68 latent infection involves multiple components of the perforin/granzyme pathway, including granzymes A and B, orphan granzymes, and the GzmB substrate caspase 3.
MATERIALS AND METHODS
Animals.
Mice were housed and bred in a specific-pathogen-free environment at Washington University School of Medicine in accordance with all federal and university policies. Granzyme-deficient (GzmA−/−, GzmB−/−, GzmAXB−/−, GzmB-cluster−/−, and GzmAXB-cluster−/−) mice on the 129/SvJ background were genotyped by Southern blotting (26, 61). Caspase 3−/− mice on the C57BL/6J (B6) background were kindly provided by Donald Nicholson (Merck Frosst) and were genotyped by PCR (65). B6 and 129/SvJ (129X1/SvJ) mice were purchased from Jackson Laboratories. Mutant mice were infected at 8 to 12 weeks of age and compared to age- and sex-matched wild-type mice.
Infections and tissue harvests.
γHV68 clone WUMS (ATCC VR1465) was passaged and the titer was determined by plaque assay on NIH 3T12 cells (80). Mice were infected intraperitoneally with 106 PFU of γHV68 in 0.5 ml of Dulbecco's modified Eagle's medium containing 10% fetal calf serum. For determination of acute viral titers, mice were sacrificed and half of the spleen and half a lobe of liver were harvested and mechanically disrupted using 1-mm silica beads, and titers were determined by plaque assay (10, 75). For limiting dilution analysis of latency, reactivation, and persistent replication, mice were sacrificed and peritoneal cells were harvested and pooled from three to five mice per experimental group (75, 80, 81).
Limiting dilution assay and PCR for latency, reactivation, and persistent replication.
Assays to determine the frequency of cells which reactivated virus ex vivo and which harbored viral genome were performed as previously described (75, 80, 81). Briefly, the frequency of cells which reactivated virus ex vivo was determined by plating serial twofold dilutions of cell samples onto permissive mouse embryonic fibroblast monolayers and scoring for cytopathic effect caused by reactivated virus after 21 days. Persistent lytic viral replication during chronic infection of mice was assessed by quantitating preformed infectious virus in mechanically disrupted cell samples. Mechanical disruption kills >99% of cells but has at most a twofold effect on viral titer (81), thus enabling experimental distinction between reactivation from latency, which requires live cells, and persistent replication. We did not detect any persistent lytic replication in either granzyme-deficient or wild-type mice in these experiments (data not shown). The frequency of cells harboring viral genome was determined by subjecting serial threefold dilutions of cell samples to nested PCR sensitive to a single copy of the γHV68 gene 50. Positive control reaction mixtures containing 10, 1, and 0.1 copies of a plasmid containing gene 50 scored positive in 99, 58, and 9% of all reactions, respectively (data not shown). No positive signal was observed in negative control reactions containing no plasmid (data not shown).
Statistical analysis.
For acute viral titers, data represent the mean ± standard error of the mean from at least two independent experiments, and statistical significance was determined using the Mann-Whitney test. For the frequency of cells which reactivated virus ex vivo and which harbored viral genome, data represent the mean ± the standard error of the mean from at least three independent experiments and were analyzed by nonlinear regression (sigmoidal dose curve with nonvariable slope) using GraphPad Prism (GraphPad, San Diego, Calif.). The frequency of cells which reactivated virus ex vivo and which harbored viral genome were determined based on a Poisson distribution by calculating the cell density at which 63.2% of the wells scored positive for reactivation or viral genome, respectively. Similar results were obtained when linear regression analysis was performed on data points within the linear ranges of the assay by using L-Calc (StemCell Technologies, Vancouver, Canada). Results from different experimental groups were compared using a paired t test to determine statistical significance.
RESULTS
Granzymes A and B have functionally redundant roles in regulating γHV68 early latency but are not required for regulating late latency.
Since perforin is a critical regulator of γHV68 latency and reactivation (72), we tested the effects of GzmA or GzmB deficiency on both early and late forms of γHV68 latency at 16 or 42 dpi, respectively. We found that GzmA deficiency had no effect on frequencies of cells which reactivated virus ex vivo or which harbored viral genome at either early (Fig. 1A) or late (Fig. 1B) times postinfection. In GzmB−/− mice, we observed a twofold increase in the frequency of cells which reactivated virus ex vivo compared to that in wild-type mice at the early time point. Although this difference was statistically significant (P = 0.002), there was no increase in frequency of cells harboring viral genome in GzmB−/− mice at this time point (Fig. 1A), nor was there a significant effect of GzmB deficiency on latent infection at the late time point (Fig. 1B). In contrast, GzmAXB−/− mice, which are deficient in both granzymes A and B, had an eightfold increase in the frequency of cells which reactivated virus ex vivo compared to wild-type mice (P = 0.001) at the early time point (Fig. 1A). These mice also exhibited a small but statistically significant (P = 0.03) twofold increase in the frequency of cells harboring viral genome relative to that in wild-type mice (Fig. 1A). The frequency of cells which reactivated virus ex vivo in GzmAXB−/− mice was eightfold higher than in GzmA−/− mice (P = 0.0004) and threefold higher than in GzmB−/− mice (P = 0.002), suggesting that both granzymes A and B are involved in regulating early γHV68 latent infection but that their roles are functionally redundant. At the late time point, however, simultaneous deficiencies in granzymes A and B had no significant effects on γHV68 latency or reactivation (Fig. 1B). Thus, granzymes A and B have functionally redundant roles in regulating the early form of γHV68 latent infection, but are not essential for regulation of late latency. That a combined deficiency in granzymes A and B had no effect on late latency was surprising, since perforin is important for regulating latency and reactivation at the late time point (72).
FIG. 1.
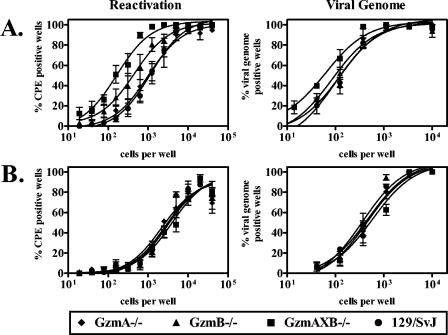
Granzymes A and B have functionally redundant roles in regulating γHV68 early latent infection. Peritoneal cells from infected GzmA−/−, GzmB−/−, GzmAXB−/−, and 129/SvJ mice were analyzed for the frequency of cells which reactivated virus ex vivo and which harbored viral genome at 16 (A) or 42 (B) dpi. No preformed infectious virus was detected in mechanically disrupted samples (data not shown). Number of independent experiments: GzmA−/−, three; GzmB−/−, four; GzmAXB−/−, four; 129/SvJ, seven.
Orphan granzymes in the GzmB locus regulate both early and late forms of γHV68 latent infection.
To test whether granzymes other than GzmA and GzmB regulate γHV68 latent infection, we examined GzmB-cluster−/− mice, which are deficient in expression of GzmB as well as orphan granzymes C and F encoded downstream of GzmB (49). At the early time point, GzmB-cluster−/− mice had a 10-fold increase in the frequency of cells which reactivated virus ex vivo (P = 0.001) and a 3-fold increase in the frequency of cells harboring viral genome (P = 0.03), compared to that in wild-type mice (Fig. 2A). Moreover, GzmB-cluster−/− mice had a fourfold increase in the frequency of cells which reactivated virus ex vivo (P = 0.002) as well as a threefold increase in the frequency of cells harboring viral genome (P = 0.03), compared to that in GzmB−/− mice (Fig. 2A). The increased defect observed in GzmB-cluster−/− mice relative to that in GzmB−/− mice indicates a role for orphan granzymes encoded in the GzmB locus in regulating γHV68 latent infection. We confirmed this finding using GzmAXB-cluster−/− mice, which are deficient in GzmA in addition to the granzymes absent in GzmB-cluster−/− mice. We found that GzmAXB-cluster−/− mice had a 13-fold increase in the frequency of cells which reactivated virus ex vivo (P = 0.001) and a 7-fold increase in the frequency of cells harboring viral genome (P = 0.04), compared to that in wild-type mice (Fig. 2B). Although the frequency of cells which reactivated virus ex vivo in GzmAXB-cluster−/− mice was only slightly higher than that in GzmAXB−/− mice (P = 0.02), GzmAXB-cluster−/− mice exhibited a threefold increase in frequency of cells harboring viral genome (P = 0.03) relative to that in GzmAXB−/− mice (Fig. 2B).
FIG. 2.
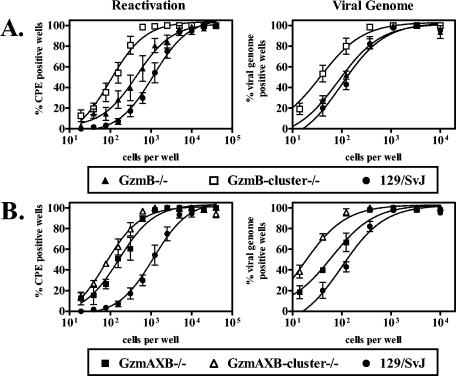
Orphan granzymes in the GzmB locus regulated γHV68 latent infection at the early time point. Peritoneal cells from infected mice were analyzed for the frequency of cells which reactivated virus ex vivo and which harbored viral genome at 16 dpi. (A) Comparison of GzmB-cluster−/−, GzmB−/−, and 129/SvJ mice. (B) Comparison of GzmAXB-cluster−/−, GzmAXB−/−, and 129/SvJ mice. No preformed infectious virus was detected in mechanically disrupted samples (data not shown). Number of independent experiments: GzmB-cluster−/−, five; GzmB−/−, four; GzmAXB-cluster−/−, five; GzmAXB−/−, four; 129/SvJ, seven.
At the late time point, GzmB-cluster−/− mice had a fourfold increase in the frequency of cells which reactivated virus ex vivo compared to that in either wild-type (P = 0.0005) or GzmAXB−/− (P = 0.0008) mice (Fig. 3A). GzmBcluster−/− mice also had a small, but statistically significant, increase in the frequency of cells harboring viral genome compared to wild-type (P = 0.05) or GzmAXB−/− (P = 0.02) mice (Fig. 3B). Similarly, GzmAXB-cluster−/− mice had a fivefold increase in the frequency of cells which reactivated virus ex vivo compared to that in either wild-type (P = 0.0001) or GzmAXB−/− (P = 0.0002) mice (Fig. 3A) and had a threefold increase in the frequency of cells harboring viral genome compared to wild-type (P = 0.04) or GzmAXB−/− (P = 0.02) mice (Fig. 3B). Thus, orphan granzymes encoded in the GzmB locus regulate γHV68 latent infection at early times postinfection and also at late times postinfection when granzymes A and B are not essential for regulation.
FIG. 3.
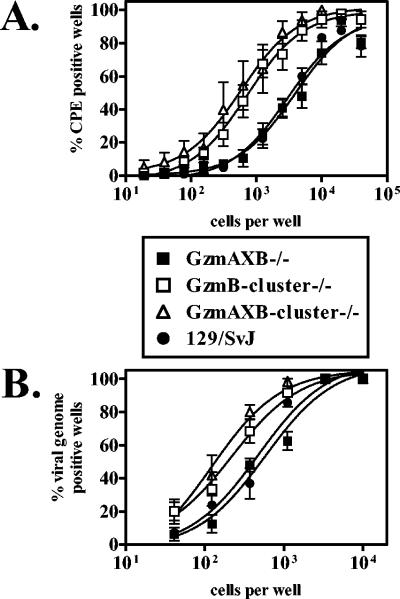
Orphan granzymes, but not granzymes A and B, regulated γHV68 latent infection at the late time point. Peritoneal cells from infected GzmAXB−/−, GzmB-cluster−/−, GzmAXB-cluster−/−, and 129/SvJ mice were analyzed at 42 dpi for the frequency of cells which reactivated virus ex vivo (A) or cells which harbored viral genome (B). No preformed infectious virus was detected in mechanically disrupted samples (data not shown). Number of independent experiments: GzmB-cluster−/−, five; GzmAXB-cluster−/−, four; GzmAXB−/−, four; 129/SvJ, eight.
Impaired regulation of viral latent infection in granzyme-deficient mice is not due to uncontrolled lytic replication during acute infection.
CD8+ T cells, which utilize the perforin/granzyme granule exocytosis pathway, have a critical role in regulating γHV68 acute infection (19, 68, 80). Therefore, the defects in regulating γHV68 latent infection observed in mice lacking multiple granzymes may be due to uncontrolled viral lytic replication during the acute phase of infection. Although the observations that perforin is dispensable for regulation of acute γHV68 infection (73) and that granzyme-deficient mice do not have detectable persistent replication at either early or late time points (data not shown) argue against this possibility, we compared acute viral titers in GzmAXB−/−, GzmB-cluster−/−, and GzmAXB-cluster−/− mice to those in wild-type mice. We found no significant increases in acute viral titers from liver (data not shown) or spleen of granzyme-deficient mice relative to those in wild-type mice at either 4 (Fig. 4A) or 9 (Fig. 4B) dpi. We also did not observe significant increases in acute viral titers in liver or spleen of GzmA−/− and GzmB−/− mice compared to those in wild-type mice at these time points (data not shown). Thus, there was no correlation between increased viral titers during acute infection and dysregulated latent infection in granzyme-deficient mice. We therefore conclude that impaired regulation of γHV68 latent infection in GzmAXB−/−, GzmB-cluster−/−, and GzmAXB-cluster−/− mice is not due to uncontrolled viral replication during acute infection of these mice.
FIG. 4.
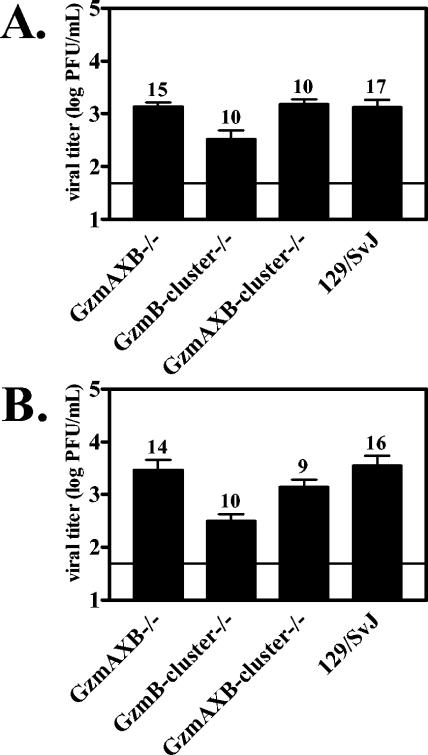
Impaired regulation of latent infection in granzyme-deficient mice is not due to uncontrolled lytic replication during acute infection. Titers in spleens from infected GzmAXB−/−, GzmB-cluster−/−, GzmAXB-cluster−/−, and 129/SvJ mice were determined by plaque assay at 4 (A) or 9 (B) dpi. The limit of detection is indicated by the horizontal line. The number of mice analyzed is given above each bar.
Caspase 3-dependent processes regulate γHV68 latent infection at the early time point.
Utilization of the perforin/granzyme pathway by immune effector cells results in granzyme-mediated induction of apoptosis in target cells. Caspase 3 is an effector caspase which lies downstream of multiple cellular apoptotic pathways (24) and can be activated by GzmB (12, 39, 52). We therefore tested whether caspase 3 is required for regulation of γHV68 latent infection. At the early time point, caspase 3−/− mice had a fivefold increase in frequency of cells which reactivated cells ex vivo (P = 0.006) and an eightfold increase in frequency of cells harboring viral genome (P = 0.01), compared to responses in wild-type mice (Fig. 5A). However, we did not observe any increase in γHV68 latency or reactivation in caspase 3−/− mice at the late time point (Fig. 5B). Thus, caspase 3 is important for regulating γHV68 latency and reactivation at an early time point during latent infection.
FIG. 5.
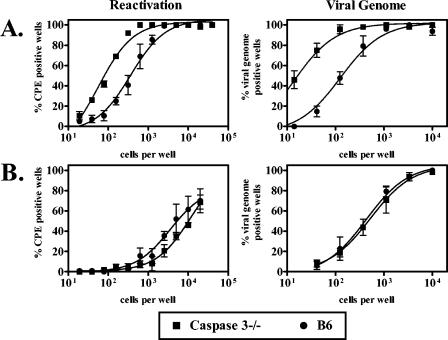
Caspase 3-dependent processes regulated γHV68 chronic infection at the early time point. Peritoneal cells from infected caspase 3−/− and B6 mice were analyzed for the frequency of cells which reactivated virus ex vivo and which harbored viral genome at 16 (A) or 42 (B) dpi. No preformed infectious virus was detected in mechanically disrupted samples (data not shown). Number of independent experiments: caspase 3−/−, four; B6, four.
DISCUSSION
In this study, we examined the role of various components of the perforin/granzyme granule exocytosis pathway in regulating γHV68 latent infection. As perforin is a critical regulator of γHV68 latent infection (72), we focused on the function of granzymes to elucidate the relative contributions of specific granzymes to the host antiviral response. We found that granzymes A and B are important for regulating γHV68 latent infection at an early time point. The observation that regulation of latent infection was more impaired in GzmAXB−/− mice than in either GzmA−/− or GzmB−/− mice suggests that the roles of granzymes A and B are functionally redundant. Additionally, we found that orphan granzymes encoded in the GzmB locus are important for regulating early γHV68 latent infection, particularly the frequency of cells harboring viral genome. Interestingly, these orphan granzymes also contributed to regulation of γHV68 latent infection at a late time point, when simultaneous deficiencies in granzymes A and B had no effect on γHV68 latency and reactivation. Finally, we examined the role of a GzmB substrate, the effector caspase 3, in regulating γHV68 infection and found an important role for caspase 3 at an early time point during latent infection. The results from these studies are summarized in Table 1.
TABLE 1.
Summary of frequencies of cells which reactivated virus ex vivo and which harbored viral genome
| Mouse strain | Frequencya
|
|||
|---|---|---|---|---|
| Early (16 dpi)
|
Late (42 dpi)
|
|||
| Reactivating cells | Genome-positive cells | Reactivating cells | Genome-positive cells | |
| 129/SvJ | 1/2,086 | 1/207 | 1/6,194 | 1/695 |
| GzmA−/− | 1/2,090 | 1/219 | 1/5,036 | 1/720 |
| GzmB−/− | 1/868* | 1/166 | 1/5,884 | 1/572 |
| GzmAXB−/− | 1/280* | 1/100* | 1/7,405 | 1/835 |
| GzmB-cluster−/− | 1/201* | 1/67* | 1/1,777* | 1/406 |
| GzmAXB-cluster−/− | 1/153* | 1/30* | 1/1,263* | 1/233* |
| B6 | 1/621 | 1/227 | 1/9,051 | 1/709 |
| Caspase 3−/− | 1/125* | 1/30* | 1/14,250 | 1/1,121 |
*, statistically significant increase in frequency compared to response in wild-type mice.
Regulation of gammaherpesvirus infection by granzymes A and B.
Our examination of the role of granzymes in regulating γHV68 infection focused initially on granzymes A and B because they are the most extensively characterized granzymes and the only two for which specific knockout mice are available. Our finding that GzmA is not essential for regulating γHV68 latent infection is consistent with observations that GzmA is not required for inducing cytotoxicity in vitro (17). The modest effect observed in GzmB−/− mice likely reflects the in vitro observations that while GzmB is required for rapid induction of apoptosis (26, 63), its absence can be compensated for by the activity of GzmA (62). As expected based on in vitro studies, simultaneous deficiencies in granzymes A and B resulted in a significant defect in regulating γHV68 latent infection at an early time point, which mirrors the defect observed in perforin−/− mice (72). This suggests that the roles for granzymes A and B in regulating the early form of γHV68 latency are functionally redundant. However, the abilities of GzmA and GzmB to compensate for loss of the other is not symmetrical and, as granzymes A and B target different cellular substrates, they likely induce cell death through distinct pathways (57, 62). At the late time point, however, GzmAXB−/− mice were indistinguishable from wild-type mice, which was unexpected because perforin−/− mice fail to control viral latent infection at both the early and late time points (72).
As perforin−/− and GzmAXB−/− mice are on different genetic backgrounds, strain differences may account for this difference. We have observed slight differences between B6 and 129/SvJ mice in our assays: at the early time point, peritoneal cells from 129/SvJ mice reactivate virus ex vivo at a threefold-lower frequency compared to cells from B6 mice, although the frequency of cells harboring viral genome is equivalent between the two strains. This appears specific to the 129/SvJ substrain, since 129/SvEv mice do not differ from B6 mice in these assays (32). However, we did not observe any significant differences in frequencies of cells which reactivated virus ex vivo or which harbored viral genome between B6 and 129/SvJ mice at the late time point, when perforin−/− and GzmAXB−/− mice have disparate phenotypes. Thus, it is unclear whether the observed strain differences at the early time point might account for the results at the late time point. An alternate explanation which we chose to investigate is that perforin-mediated regulation of viral infection employs other granzymes in addition to GzmA and GzmB.
Involvement of orphan granzymes in control of gammaherpesvirus infection.
The potential for involvement of orphan granzymes in regulating γHV68 latent infection was interesting because little is currently known about their functions (25). Using mice with severely reduced expression of orphan granzymes C and F, we found that these orphan granzymes are involved in regulating γHV68 latency and reactivation. Moreover, we found that regulation of γHV68 latent infection at the late time point requires only orphan granzymes, since GzmAXB−/− mice have no apparent defects relative to wild-type mice at that time. We are unable to determine which specific orphan granzymes are responsible for this activity, since mice deficient for individual orphan granzymes are not yet available. However, the observation that orphan granzymes, but not caspase 3, are required for regulating γHV68 latent infection at the late time point suggests that the orphan granzymes involved induce cell death independently of caspase 3. Since GzmC-induced apoptosis proceeds through a caspase-independent pathway and exhibits kinetics and potency similar to those of GzmB (29), it is a promising candidate for further investigation.
Our studies did not address which effector cell population is responsible for the observed activity of orphan granzymes in regulating γHV68 latent infection. The granule exocytosis pathway is utilized by multiple effector cell types, including CTLs, natural killer (NK) cells, and lymphokine-activated killer cells. Orphan granzymes have been shown to be highly expressed in NK and lymphokine-activated killer cells (25, 49), but whether these cells regulate herpesvirus latent infection is unknown. Since activated CD8+ T cells and CTL cell lines can also express orphan granzymes (25) and CD8+ T cells are critical for regulating γHV68 latency and reactivation (72), it is likely that the observed role of orphan granzymes is through employment by CTLs.
The observation that γHV68 latent infection is regulated by different granzymes at early versus late time points postinfection may reflect qualitative differences between virus-specific immune effector cells at the two time points. Expression of different combinations of perforin and granzyme genes has been observed for CD8+ T-cell populations as well as individual CD8+ T cells (16, 22, 34, 50), which is consistent with the possibility of differential granzyme usage by different CTL cells or populations. It is also possible that chronic viral infection can trigger differential expression or usage of granzymes in immune effector cells, as different methods of activating T cells have been reported to result in different granzyme expression patterns (22, 50). However, the biological consequences of differential granzyme expression by CD8+ T-cell populations have not been evaluated in the context of an antiviral immune response.
Alternately, different requirements for specific granzymes between early and late times in viral latent infection may be due to differences in the target cells. It has been observed that CTL-mediated cytotoxicity induces varying types of DNA damage and exhibits different requirements for granzymes A and B, depending on the target cell used (47, 58). This suggests that factors intrinsic to target cells can modulate the effects of CTL-induced cell death. Since current understanding of granzyme activity indicates that the substrates of different granzymes are largely nonoverlapping (57), differential expression of granzyme substrates by target cells could potentially result in different susceptibility of target cells to cytotoxicity mediated by various granzymes. γHV68 has been shown to establish early and late forms of latency which differ in efficiency of reactivation ex vivo (81) and in expression of a viral latency transcript (74). Although the effects of CTL-mediated cytotoxicity on these two latently infected cell populations have not been compared, it is possible that the early form of γHV68 latency is susceptible to induction of cytotoxicity by multiple granzymes, whereas the form which predominates at the later time point is resistant to cytotoxicity mediated by granzymes A and B.
Interestingly, inhibition of GzmB has been reported for several viral proteins, including poxvirus CrmA (51), adenovirus L4-100K assembly protein (1), herpes simplex virus glycoprotein J (28), and the EBV viral Bcl-2 homolog BHRF1 (13). Like EBV, γHV68 expresses a viral Bcl-2 homolog, M11, which inhibits apoptosis in vitro (4, 56, 79), is expressed during latent infection (38, 40, 77), and is required for efficient reactivation from latency ex vivo and for persistent replication in gamma interferon-deficient mice (21). But whether M11 can inhibit granzyme-mediated cell death and whether it is preferentially expressed during late latency are unknown. Nonetheless, the observations that multiple viruses inhibit GzmB activity and that orphan granzymes can regulate viral infection suggest that expression of multiple granzymes in mice serves as a fail-safe mechanism for antiviral immunity.
Regulation of gammaherpesvirus latent infection by caspase 3.
We also observed differential regulation of γHV68 latent infection at early versus late time points postinfection in our studies using caspase 3−/− mice. Caspase 3 deficiency resulted in significantly impaired regulation of γHV68 latency and reactivation at the early time point but not the late time point, as was observed with GzmAXB−/− mice. This confirms that different cellular factors are required for host regulation of early versus late forms of γHV68 latent infection. Although caspase 3 can be cleaved and activated by GzmB (12, 39, 52), granzymes A, B, and C can all induce apoptosis in a caspase-independent manner (2, 5, 29, 59, 71). Moreover, caspase 3 mediates death through other cellular apoptotic pathways, including the death receptor and cellular stress pathways (reviewed in reference 24). It is therefore unclear whether similarities between the phenotypes of caspase 3−/− and GzmAXB−/− mice necessarily indicate that caspase 3 and granzymes function within the same pathway to regulate γHV68 latent infection. It is also possible that caspase 3 functions in controlling γHV68 acute as well as latent infections. Our data demonstrate the importance of caspase 3 in regulating γHV68 latency and reactivation, and they underscore the importance of host apoptotic processes in regulating gammaherpesvirus latent infection.
Function of the granule exocytosis pathway during gammaherpesvirus infection.
The studies presented here, as well as previous studies using perforin−/− mice (72), clearly demonstrate a critical role for the granule exocytosis pathway in host regulation of gammaherpesvirus latent infection. However, the mechanisms by which this pathway regulates gammaherpesvirus latent infection are not clear. For example, mice deficient in components of this pathway may fail to regulate the maintenance of, and reactivation from, an established latent infection. Alternately, defects in granule exocytosis may enable establishment of latency in different cell types or employment of different latency gene expression programs which result in the observed phenotypes ex vivo. While we have not observed substantial differences between granzyme-deficient and wild-type mice in acute viral titers, we cannot rule out the possibility that changes in acute infection, for example in cell tropism, due to granzyme deficiency contribute directly to the differences in latency and reactivation observed during latent infection.
Since the granule exocytosis pathway is important for regulating lymphocyte homeostasis (reviewed in reference 14), it is also possible that perforin- or granzyme-deficient mice fail to control gammaherpesvirus latent infection due to defective lymphocyte development and regulation. In humans, defects in perforin (66), trafficking of granule cytotoxic enzymes (3), or exocytosis of cytotoxic granules (42) are associated with development of hemophagocytic syndrome (HPS). HPS is believed to be triggered by viral infection, particularly with herpesviruses, including EBV and CMV (14), but whether development of HPS in humans results in dysregulation of an existing herpesvirus latent infection is unknown. While perforin−/− mice do not spontaneously develop HPS, infection with lymphocytic choriomeningitis virus can result in uncontrolled expansion of virus-specific T cells, leading to immune-mediated pathology resembling that of human disease (31, 41). Induction of hemophagocytosis by viral infection of granzyme-deficient mice has not been examined, nor is it known whether all murine granzymes are involved in regulation of lymphocyte homeostasis via the granule exocytosis pathway.
In summary, the defects in regulation of gammaherpesvirus chronic infection observed in granzyme-deficient mice may be due to a combination of factors, including impaired T-cell-mediated regulation of viral infection, altered establishment of viral latency, and/or dysregulated lymphocyte homeostasis. The identification of specific granzymes which regulated early or late forms of gammaherpesvirus latent infection in this study will facilitate further examination of how the perforin/granzyme pathway contributes to host antiviral defense and of the functions of orphan granzymes in the host immune response in vivo.
Acknowledgments
H.W.V. was supported by National Institutes of Health (NIH) grant R01 CA74730. T.J.L. was supported by NIH grant DK49786. J.L. and P.A.R. were supported by NIH training grant 5-T32-AI07163-26.
We thank Donald Nicholson for generously providing us with caspase 3-deficient mice and Darren Kreamalmeyer for expert assistance in the generation and breeding of knockout mice. We thank Feng Gao for assistance with statistical analyses of our data. We thank members of the labs of H.W.V. and T.J.L. for helpful suggestions and comments throughout the course of this study.
REFERENCES
- 1.Andrade, F., H. G. Bull, N. A. Thornberry, G. W. Ketner, L. A. Casciola-Rosen, and A. Rosen. 2001. Adenovirus L4-100K assembly protein is a granzyme B substrate that potently inhibits granzyme B-mediated cell death. Immunity 14:751-761. [DOI] [PubMed] [Google Scholar]
- 2.Anel, A., S. Gamen, M. A. Alava, A. M. Schmitt-Verhulst, A. Pineiro, and J. Naval. 1997. Inhibition of CPP32-like proteases prevents granzyme B- and Fas-, but not granzyme A-based cytotoxicity exerted by CTL clones. J. Immunol. 158:1999-2006. [PubMed] [Google Scholar]
- 3.Baetz, K., S. Isaaz, and G. M. Griffiths. 1995. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J. Immunol. 154:6122-6131. [PubMed] [Google Scholar]
- 4.Bellows, D. S., B. N. Chau, P. Lee, Y. Lazebnik, W. H. Burns, and J. M. Hardwick. 2000. Antiapoptotic herpesvirus bcl-2 homologs escape caspase-mediated conversion to proapoptotic proteins. J. Virol. 74:5024-5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beresford, P. J., Z. Xia, A. H. Greenberg, and J. Lieberman. 1999. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 10:585-594. [DOI] [PubMed] [Google Scholar]
- 6.Blasdell, K., C. McCracken, A. Morris, A. A. Nash, M. Begon, M. Bennett, and J. P. Stewart. 2003. The wood mouse is a natural host for Murid herpesvirus 4. J. Gen. Virol. 84:111-113. [DOI] [PubMed] [Google Scholar]
- 7.Blaskovic, D., M. Stancekova, J. Svobodova, and J. Mistrikova. 1980. Isolation of five strains of herpesviruses from two species of free living small rodents. Acta Virol. 24:468. [PubMed] [Google Scholar]
- 8.Blaskovic, D., D. Stanekova, and J. Rajcani. 1984. Experimental pathogenesis of murine herpesvirus in newborn mice. Acta Virol. 28:225-231. [PubMed] [Google Scholar]
- 9.Cardin, R. D., J. W. Brooks, S. R. Sarawar, and P. C. Doherty. 1996. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 184:863-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clambey, E. T., H. W. Virgin, and S. H. Speck. 2000. Disruption of the murine gammaherpesvirus 68 M1 open reading frame leads to enhanced reactivation from latency. J. Virol. 74:1973-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clambey, E. T., H. W. Virgin, and S. H. Speck. 2002. Characterization of a spontaneous 9.5-kilobase-deletion mutant of murine gammaherpesvirus 68 reveals tissue-specific genetic requirements for latency. J. Virol. 76:6532-6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darmon, A. J., D. W. Nicholson, and R. C. Bleackley. 1995. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377:446-448. [DOI] [PubMed] [Google Scholar]
- 13.Davis, J. E., V. R. Sutton, M. J. Smyth, and J. A. Trapani. 2000. Dependence of granzyme B-mediated cell death on a pathway regulated by Bcl-2 or its viral homolog, BHRF1. Cell Death Differ. 7:973-983. [DOI] [PubMed] [Google Scholar]
- 14.de Saint, B. G., and A. Fischer. 2001. The role of cytotoxicity in lymphocyte homeostasis. Curr. Opin. Immunol. 13:549-554. [DOI] [PubMed] [Google Scholar]
- 15.Dix, R. D., E. R. Podack, and S. W. Cousins. 2003. Loss of the perforin cytotoxic pathway predisposes mice to experimental cytomegalovirus retinitis. J. Virol. 77:3402-3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ebnet, K., J. Chluba-de Tapia, U. Hurtenbach, M. D. Kramer, and M. M. Simon. 1991. In vivo primed mouse T cells selectively express T cell-specific serine proteinase-1 and the proteinase-like molecules granzyme B and C. Int. Immunol. 3:9-19. [DOI] [PubMed] [Google Scholar]
- 17.Ebnet, K., M. Hausmann, F. Lehmann-grube, A. Mullbacher, M. Kopf, M. Lamers, and M. M. Simon. 1995. Granzyme A-deficient mice retain potent cell-mediated cytotoxicity. EMBO J. 14:4230-4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Efstathiou, S., Y. M. Ho, S. Hall, C. J. Styles, S. D. Scott, and U. A. Gompels. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J. Gen. Virol. 71:1365-1372. [DOI] [PubMed] [Google Scholar]
- 19.Ehtisham, S., N. P. Sunil-Chandra, and A. A. Nash. 1993. Pathogenesis of murine gammaherpesvirus infection in mice deficient in CD4 and CD8 T cells. J. Virol. 67:5247-5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flano, E., S. M. Husain, J. T. Sample, D. L. Woodland, and M. A. Blackman. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074-1081. [DOI] [PubMed] [Google Scholar]
- 21.Gangappa, S., L. F. Van Dyk, T. J. Jewett, S. H. Speck, and H. W. Virgin. 2002. Identification of the in vivo role of a viral bcl-2. J. Exp. Med. 195:931-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Sanz, J. A., H. R. MacDonald, D. E. Jenne, J. Tschopp, and M. Nabholz. 1990. Cell specificity of granzyme gene expression. J. Immunol. 145:3111-3118. [PubMed] [Google Scholar]
- 23.Ghiasi, H., S. Cai, G. Perng, A. B. Nesburn, and S. L. Wechsler. 1999. Perforin pathway is essential for protection of mice against lethal ocular HSV-1 challenge but not corneal scarring. Virus Res. 65:97-101. [DOI] [PubMed] [Google Scholar]
- 24.Green, D. R. 1998. Apoptotic pathways: the roads to ruin. Cell 94:695-698. [DOI] [PubMed] [Google Scholar]
- 25.Grossman, W. J., P. A. Revell, Z. H. Lu, H. Johnson, A. J. Bredemeyer, and T. J. Ley. 2003. The orphan granzymes of humans and mice. Curr. Opin. Immunol. 15:544-552. [DOI] [PubMed] [Google Scholar]
- 26.Heusel, J. W., R. L. Wesselschmidt, S. Shresta, J. H. Russel, and T. J. Ley. 1994. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 76:977-987. [DOI] [PubMed] [Google Scholar]
- 27.Jacoby, M. A., H. W. Virgin, I. V., and S. H. Speck. 2002. Disruption of the M2 gene of murine gammaherpesvirus 68 alters splenic latency following intranasal, but not intraperitoneal, inoculation. J. Virol. 76:1790-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jerome, K. R., Z. Chen, R. Lang, M. R. Torres, J. Hofmeister, S. Smith, R. Fox, C. J. Froelich, and L. Corey. 2001. HSV and glycoprotein J inhibit caspase activation and apoptosis induced by granzyme B or Fas. J. Immunol. 167:3928-3935. [DOI] [PubMed] [Google Scholar]
- 29.Johnson, H., L. Scorrano, S. J. Korsmeyer, and T. J. Ley. 2003. Cell death induced by granzyme C. Blood 101:3093-3101. [DOI] [PubMed] [Google Scholar]
- 30.Kagi, D., B. Lederman, K. Burki, P. Seiler, B. Odermatt, K. J. Olsen, E. R. Podack, R. M. Zinkernagel, and H. Hengartner. 1994. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369:31-37. [DOI] [PubMed] [Google Scholar]
- 31.Kagi, D., B. Odermatt, and T. W. Mak. 1999. Homeostatic regulation of CD8+ T cells by perforin. Eur. J. Immunol. 29:3262-3272. [DOI] [PubMed] [Google Scholar]
- 32.Kapadia, S. B., B. Levine, S. H. Speck, and H. W. Virgin. 2002. Critical role of complement and viral evasion of complement in acute, persistent, and latent gamma-herpesvirus infection. Immunity 17:143-155. [DOI] [PubMed] [Google Scholar]
- 33.Katano, H., M. A. Ali, A. C. Patera, M. Catalfamo, E. S. Jaffe, H. Kimura, J. K. Dale, S. E. Straus, and J. I. Cohen. 2004. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood 103:1244-1252. [DOI] [PubMed] [Google Scholar]
- 34.Kelso, A., E. O. Costelloe, B. J. Johnson, P. Groves, K. Buttigieg, and D. R. Fitzpatrick. 2002. The genes for perforin, granzymes A-C and IFN-gamma are differentially expressed in single CD8+ T cells during primary activation. Int. Immunol. 14:605-613. [DOI] [PubMed] [Google Scholar]
- 35.Khanolkar, A., H. Yagita, and M. J. Cannon. 2001. Preferential utilization of the perforin/granzyme pathway for lysis of Epstein-Barr virus-transformed lymphoblastoid cells by virus-specific CD4+ T cells. Virology 287:79-88. [DOI] [PubMed] [Google Scholar]
- 36.Kieff, E., and A. B. Rickinson. 2001. Epstein-Barr virus and its replication, p. 2511-2573. In D. M. Knipe and P. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 37.Liu, L., E. J. Usherwood, M. A. Blackman, and D. L. Woodland. 1999. T-cell vaccination alters the course of murine herpesvirus 68 infection and the establishment of viral latency in mice. J. Virol. 73:9849-9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques, S., S. Efstathiou, K. G. Smith, M. Haury, and J. P. Simas. 2003. Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J. Virol. 77:7308-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin, S. J., G. P. Amarante-Mendes, L. Shi, T. H. Chuang, C. A. Casiano, G. A. O'Brien, P. Fitzgerald, E. M. Tan, G. M. Bokoch, A. H. Greenberg, and D. R. Green. 1996. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO J. 15:2407-2416. [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Guzman, D., T. Rickabaugh, T. T. Wu, H. Brown, S. Cole, M. J. Song, L. Tong, and R. Sun. 2003. Transcription program of murine gammaherpesvirus 68. J. Virol. 77:10488-10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matloubian, M., M. Suresh, A. Glass, M. Galvan, K. Chow, J. K. Whitmire, C. M. Walsh, W. R. Clark, and R. Ahmed. 1999. A role for perforin in downregulating T-cell responses during chronic viral infection. J. Virol. 73:2527-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menasche, G., E. Pastural, J. Feldmann, S. Certain, F. Ersoy, S. Dupuis, N. Wulffraat, D. Bianchi, A. Fischer, F. Le Deist, and B. G. de Saint. 2000. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 25:173-176. [DOI] [PubMed] [Google Scholar]
- 43.Moore, P. S., and Y. Chang. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57:609-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullbacher, A., K. Ebnet, R. V. Blanden, R. T. Hla, T. Stehle, C. Museteanu, and M. M. Simon. 1996. Granzyme A is critical for recovery of mice from infection with the natural cytopathic viral pathogen, ectromelia. Proc. Natl. Acad. Sci. USA 93:5783-5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mullbacher, A., R. T. Hla, C. Museteanu, and M. M. Simon. 1999. Perforin is essential for control of ectromelia virus but not related poxviruses in mice. J. Virol. 73:1665-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullbacher, A., P. Waring, H. R. Tha, T. Tran, S. Chin, T. Stehle, C. Museteanu, and M. M. Simon. 1999. Granzymes are the essential downstream effector molecules for the control of primary virus infections by cytolytic leukocytes. Proc. Natl. Acad. Sci. USA 96:13950-13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pardo, J., S. Balkow, A. Anel, and M. M. Simon. 2002. The differential contribution of granzyme A and granzyme B in cytotoxic T lymphocyte-mediated apoptosis is determined by the quality of target cells. Eur. J. Immunol. 32:1980-1985. [DOI] [PubMed] [Google Scholar]
- 48.Pereira, R. A., M. M. Simon, and A. Simmons. 2000. Granzyme A, a noncytolytic component of CD8+ cell granules, restricts the spread of herpes simplex virus in the peripheral nervous systems of experimentally infected mice. J. Virol. 74:1029-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pham, C. T., D. M. Macivor, B. A. Hug, J. W. Heusel, and T. J. Ley. 1996. Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. USA 93:13090-13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prendergast, J. A., C. D. Helgason, and R. C. Bleackley. 1992. Quantitative polymerase chain reaction analysis of cytotoxic cell proteinase gene transcripts in T cells. Pattern of expression is dependent on the nature of the stimulus. J. Biol. Chem. 267:5090-5095. [PubMed] [Google Scholar]
- 51.Quan, L. T., A. Caputo, R. C. Bleackley, D. J. Pickup, and G. S. Salvesen. 1995. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 270:10377-10379. [DOI] [PubMed] [Google Scholar]
- 52.Quan, L. T., M. Tewari, K. O'Rourke, V. Dixit, S. J. Snipas, G. G. Poirier, C. Ray, D. J. Pickup, and G. S. Salvesen. 1996. Proteolytic activation of the cell death protease Yama/CPP32 by granzyme B. Proc. Natl. Acad. Sci. USA 93:1972-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rajcani, J., D. Blaskovic, J. Svobodova, F. Ciampor, D. Huckova, and D. Stanekova. 1985. Pathogenesis of acute and persistent murine herpesvirus infection in mice. Acta Virol. 29:51-60. [PubMed] [Google Scholar]
- 54.Rickinson, A. B. and E. Kieff. 2001. Epstein-Barr virus, p. 2575-2627. In D. M. Knipe and P. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 55.Riera, L., M. Gariglio, G. Valente, A. Mullbacher, C. Museteanu, S. Landolfo, and M. M. Simon. 2000. Murine cytomegalovirus replication in salivary glands is controlled by both perforin and granzymes during acute infection. Eur. J. Immunol. 30:1350-1355. [DOI] [PubMed] [Google Scholar]
- 56.Roy, D. J., B. C. Ebrahimi, B. M. Dutia, A. A. Nash, and J. P. Stewart. 2000. Murine gammaherpesvirus M11 gene product inhibits apoptosis and is expressed during virus persistence. Arch. Virol. 145:2411-2420. [DOI] [PubMed] [Google Scholar]
- 57.Russell, J. H., and T. J. Ley. 2002. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 20:323-370. [DOI] [PubMed] [Google Scholar]
- 58.Sellins, K. S., and J. J. Cohen. 1991. Cytotoxic T lymphocytes induce different types of DNA damage in target cells of different origins. J. Immunol. 147:795-803. [PubMed] [Google Scholar]
- 59.Sharif-Askari, E., A. Alam, E. Rheaume, P. J. Beresford, C. Scotto, K. Sharma, D. Lee, W. E. DeWolf, M. E. Nuttall, J. Lieberman, and R. P. Sekaly. 2001. Direct cleavage of the human DNA fragmentation factor-45 by granzyme B induces caspase-activated DNase release and DNA fragmentation. EMBO J. 20:3101-3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi, L., S. Mai, S. Israels, K. Browne, J. A. Trapani, and A. H. Greenberg. 1997. Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J. Exp. Med. 185:855-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shresta, S., P. Goda, R. Wesselschmidt, and T. J. Ley. 1997. Residual cytotoxicity and granzyme K expression in granzyme A-deficient cytotoxic lymphocytes. J. Biol. Chem. 272:20236-20244. [DOI] [PubMed] [Google Scholar]
- 62.Shresta, S., T. A. Graubert, D. A. Thomas, S. Z. Raptis, and T. J. Ley. 1999. Granzyme A initiates an alternative pathway for granule-mediated apoptosis. Immunity 10:595-605. [DOI] [PubMed] [Google Scholar]
- 63.Shresta, S., D. M. Macivor, J. W. Heusel, J. H. Russell, and T. J. Ley. 1995. Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc. Natl. Acad. Sci. USA 92:5679-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simon, M. M., M. Hausmann, T. Tran, K. Ebnet, J. Tschopp, R. ThaHla, and A. Mullbacher. 1997. In vitro- and ex vivo-derived cytolytic leukocytes from granzyme A × B double knockout mice are defective in granule-mediated apoptosis but not lysis of target cells. J. Exp. Med. 186:1781-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simpson, M. T., J. G. MacLaurin, D. Xu, K. L. Ferguson, J. L. Vanderluit, M. A. Davoli, S. Roy, D. W. Nicholson, G. S. Robertson, D. S. Park, and R. S. Slack. 2001. Caspase 3 deficiency rescues peripheral nervous system defect in retinoblastoma nullizygous mice. J. Neurosci. 21:7089-7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stepp, S. E., R. Dufourcq-Lagelouse, F. Le Deist, S. Bhawan, S. Certain, P. A. Mathew, J. I. Henter, M. Bennett, A. Fischer, B. G. de Saint, and V. Kumar. 1999. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 286:1957-1959. [DOI] [PubMed] [Google Scholar]
- 67.Stevenson, P. G., G. T. Belz, M. R. Castrucci, J. D. Altman, and P. C. Doherty. 1999. A gamma-herpesvirus sneaks through a CD8+ T cell response primed to a lytic-phase epitope. Proc. Natl. Acad. Sci. USA 96:9281-9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stevenson, P. G., R. D. Cardin, J. P. Christensen, and P. C. Doherty. 1999. Immunological control of a murine gammaherpesvirus independent of CD8+ T cells. J. Gen. Virol. 80:477-483. [DOI] [PubMed] [Google Scholar]
- 69.Sunil-Chandra, N. P., S. Efstathiou, J. Arno, and A. A. Nash. 1992. Virological and pathological features of mice infected with murine gammaherpesvirus 68. J. Gen. Virol. 73:2347-2356. [DOI] [PubMed] [Google Scholar]
- 70.Sunil-Chandra, N. P., S. Efstathiou, and A. A. Nash. 1992. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73:3275-3279. [DOI] [PubMed] [Google Scholar]
- 71.Thomas, D. A., C. Du, M. Xu, X. Wang, and T. J. Ley. 2000. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 12:621-632. [DOI] [PubMed] [Google Scholar]
- 72.Tibbetts, S. A., L. Van Dyk, S. H. Speck, and H. W. Virgin. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J. Virol. 76:7125-7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Usherwood, E. J., J. W. Brooks, S. R. Sarawar, R. D. Cardin, W. D. Young, D. J. Allen, P. C. Doherty, and A. A. Nash. 1997. Immunological control of murine gammaherpesvirus infection is independent of perforin. J. Gen. Virol. 78:2025-2030. [DOI] [PubMed] [Google Scholar]
- 74.Usherwood, E. J., D. J. Roy, K. Ward, S. L. Surman, B. M. Dutia, M. A. Blackman, J. P. Stewart, and D. L. Woodland. 2000. Control of gammaherpesvirus latency by latent antigen-specific CD8+ T cells. J. Exp. Med. 192:943-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Dyk, L. F., H. W. Virgin, and S. H. Speck. 2000. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J. Virol. 74:7451-7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Virgin, H. W., P. Latreille, P. Wamsley, K. Hallsworth, K. E. Weck, A. J. Dal Canto, and S. H. Speck. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Virgin, H. W., R. M. Presti, X.-Y. Li, C. Liu, and S. H. Speck. 1999. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J. Virol. 73:2321-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walsh, C. M., M. Matloubian, C.-C. Liu, R. Ueda, C. G. Kurahara, J. L. Christensen, M. T. F. Huang, J. D. Young, R. Ahmed, and W. R. Clark. 1994. Immune function in mice lacking the perforin gene. Proc. Natl. Acad. Sci. USA 91:10854-10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang, G. H., T. L. Garvey, and J. I. Cohen. 1999. The murine gammaherpesvirus-68 M11 protein inhibits Fas- and TNF-induced apoptosis. J. Gen. Virol. 80:2737-2740. [DOI] [PubMed] [Google Scholar]
- 80.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. W. Virgin. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weck, K. E., S. S. Kim, H. W. Virgin, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weck, K. E., S. S. Kim, H. W. Virgin, and S. H. Speck. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73:3273-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoshimi, A., I. Tsuge, H. Namizaki, Y. Hoshino, H. Kimura, Y. Takahashi, N. Watanabe, K. Kuzushima, and S. Kojima. 2002. Epstein-Barr virus-specific T-cell cytotoxicity is mediated through the perforin pathway in patients with lymphoproliferative disorders after allogeneic bone marrow transplantation. Br. J. Haematol. 116:710-715. [DOI] [PubMed] [Google Scholar]