

Abstract
This study evaluated and compared delivery of the tumor necrosis factor alpha receptor (TNFR)-immunoglobulin G1 (IgG1) Fc fusion (TNFR:Fc) gene to the lung by single and repeat administrations of multiple pseudotyped adeno-associated virus (AAV) vectors as a means for achieving systemic distribution of the soluble TNFR:Fc protein. A single endotracheal administration of AAV[2/5]cytomegalovirus (CMV)-TNFR:Fc vector (containing the AAV2 inverted terminal repeats and AAV5 capsid) to the rat lung resulted in long-term, high levels of serum TNFR:Fc protein that gradually declined over a period of 8 months. Endotracheal delivery of AAV[2/1]CMV-TNFR:Fc resulted in serum TNFR:Fc protein levels that were detectable for at least 4 months but were 10-fold lower than that of the AAV[2/5] vector. In contrast, secretion of the TNFR:Fc protein following pulmonary delivery of AAV[2/2]CMV-TNFR:Fc vector was very inefficient, and the protein was detected in the blood only when an airway epithelial cell-specific promoter, CC10, was substituted for the CMV enhancer/promoter to control transgene expression. In the context of AAV[2/5], the CC10 promoter was as efficient as CMV enhancer/promoter in generating similar levels of systemic TNFR:Fc protein, suggesting that this protein is secreted primarily from the airway epithelium. In mice, comparable long-term secretion of TNFR:Fc protein was demonstrated after AAV[2/2] and AAV[2/5] delivery, although the kinetics of transduction appeared to be different. All pseudotyped AAV vectors elicited serum anti-AAV capsid-neutralizing antibody responses, but these did not prevent lung transduction and efficient secretion of TNFR:Fc protein to the circulation following readministration with AAV[2/5]. These results highlight the potential utility of AAV vectors containing serotype 5 capsid to deliver and redeliver genes of secreted proteins to the lung to achieve long-term systemic protein expression.
Delivery of therapeutic proteins to the systemic circulation using gene therapy has the potential to improve the efficacy, duration, convenience, and cost effectiveness of chronic disease treatment by replacing the frequent and often invasive administrations of recombinant proteins with infrequent delivery of the corresponding therapeutic genes. For rheumatoid arthritis, long-lasting systemic production of a therapeutic protein such as the anti-tumor necrosis factor alpha (anti-TNF-α) inhibitor etanercept (TNF-α receptor-immunoglobulin G1 [IgG1] Fc fusion [TNFR:Fc]), by noninvasive administration of gene therapy vectors, may improve the pharmacokinetics of this relatively short-lived protein.
Recombinant adeno-associated virus (AAV) vector is one of the most promising delivery vehicles under development for gene therapy which has been shown to efficiently direct long-term transgene expression in multiple tissues, including skeletal muscle, liver, brain, and retina, with little or no inflammation or cellular immune response (1, 8, 26, 30, 35, 41, 42, 49, 53). Although most of the earlier studies were carried out with vectors based on AAV serotype 2 (AAV2), recent experiments with vectors derived from alternate primate AAV serotypes, of which at least eight have been isolated to date (11, 12, 20, 34, 39, 50), have demonstrated marked increase in transduction efficiency of various target tissues due to distinct mechanisms of tropism, uptake, and trafficking (5, 16, 28, 47, 48, 52). In vitro and in vivo studies with AAV5 have demonstrated improved binding and transduction of murine lung airway epithelia compared with that of AAV2 (52). Within the brain, while vectors derived from AAV2 efficiently transduce neurons in close proximity to the site of administration, AAV5-based vectors are capable of diffusion within the mouse striatum well beyond the injection site and have demonstrated efficient gene transfer to the ependymal cells of the ventricles and the cerebral hemisphere (14). AAV1 and AAV7 vectors appear to be the most efficient gene transfer vehicles in skeletal muscle transduction compared with AAV2, -3, -4, and -5 vectors (10, 20, 50), whereas AAV vectors containing the type 5 and 8 capsids have been shown to direct high levels of transgene expression in liver (20, 32, 40).
Since the tissue tropism of each viral serotype is determined by the interaction of the capsid with a distinct receptor and/or coreceptor(s) on target cells, pseudotyped vectors can be efficiently produced by cross-packaging of an AAV2 vector genome into the capsids of other serotypes (15, 37, 38, 45, 46, 48).
The lung can potentially be employed as a metabolic factory for the ectopic production and secretion of therapeutic proteins into the circulation because it has one of the most expansive epithelium-blood capillary networks and is easily accessible for noninvasive gene delivery. In addition, the lung offers a large number of target cells with adequate access to the blood. In the context of recombinant AAV, successful in vivo airway gene transfer with AAV vectors, carrying reporter genes and therapeutic genes including the cystic fibrosis transmembrane regulator transgene, has been demonstrated in multiple species, including rabbits (6, 23), mice (24), nonhuman primates (2, 7, 13, 17), and human cystic fibrosis patients (3, 18, 33). In addition, the lung appears to be an ideal target organ in terms of safety parameters, given the lack of observable AAV vector DNA biodistribution to gonads and given its decreased likelihood of generating immune responses compared to muscle or liver following the delivery of AAV vectors (13, 17, 51).
The concept of using the lung for the ectopic production of secreted proteins has recently been employed by Auricchio and colleages, who demonstrated that intranasal delivery of therapeutic genes, including erythropoietin and factor IX, to mice by using pseudotyped AAV vectors led to long-term and apparently functional secretion of the corresponding proteins into the systemic circulation (4).
In this report, we evaluated and compared delivery of the TNFR:Fc fusion gene to the lung by single and repeat administrations of pseudotyped AAV vectors of alternate serotypes as a means to achieve systemic distribution of a TNFR:Fc fusion protein. Here, we show that in rats, pseudotyped AAV5 (AAV[2/5]) vector is more efficient than AAV[2/1] or AAV[2/2]. However, in mice AAV[2/2] appears to be as efficient as AAV[2/5] in mediating long-term secretion of TNFR:Fc protein from the lung to the systemic circulation, albeit at different initial kinetics of transduction. In addition, we examined the types of lung cells transduced by AAV vectors pseudotyped with type 2 or 5 capsid and provide evidence to suggest that airway epithelium is the primary source of TNFR:Fc protein secretion to the blood. Finally, AAV[2/5] could be readministered to animals that were previously treated with either the same or an alternate pseudotyped vector and efficiently mediate secretion of TNFR:Fc protein to the circulation.
MATERIALS AND METHODS
Cells.
Human 293 (22) and HeLa (21) cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (BioWhittaker; Cambrex, Walkersville, Md.) supplemented with 100 U of penicillin G/ml, 100 μg of streptomycin/ml, 4.5 mg of glucose/ml, 4 mM l-glutamine, and 10% heat-inactivated (55°C; 30 min) fetal bovine serum (HyClone, Logan, Utah) in a humidified atmosphere of air with 10% CO2 at 37°C.
Plasmids.
Plasmid pAAVCMV-EGFP contains the enhanced green fluorescent protein (EGFP) gene under the control of the human cytomegalovirus (CMV) major immediate-early gene enhancer/promoter region, a chimeric human β-globin splice donor-human Ig splice acceptor site, and a bovine growth hormone (BGH) poly(A) signal. The EGFP transcriptional unit is flanked at each end by the AAV2 145-bp inverted terminal repeat sequence, derived from pAV2 (27, 43). The structure of plasmid pAAVCMV-hFIX is essentially the same as that of pAAVCMV-EGFP, except that human factor IX (hFIX) cDNA was substituted for the EGFP sequences. Construction of plasmid pAAVCMV-TNFR:Fc was previously described (9). Plasmid pAAVCC10-TNFR:Fc was constructed as follows. A 326-bp DNA fragment, containing the rat CC10 promoter (44), was amplified by PCR from rat lung genomic DNA with an upstream primer sequence, 5′-CTCGAGCAATTGCTTCCCGGAACCT-3′, and a downstream primer, 5′-GCGGCCGCATGTGTGGGTATGCGTGTGGC-3′, containing unique XhoI and NotI restriction sites at the 5′ and 3′ ends, respectively. The 326-bp PCR product was cloned into plasmid pCR2.1-TOPO TA (Invitrogen, Carlsbad, Calif.) and propagated in Escherichia coli TOP10 cells. Plasmid pAAVCMV-TNFR:Fc was digested with XhoI and NotI to remove the CMV immediate-early enhancer/promoter. The 326-bp CC10 promoter was liberated from plasmid pCR2.1CC10 via XhoI/NotI digestion and ligated into the XhoI/NotI sites in plasmid pAAVCMV-TNFR:Fc. The trans-packaging plasmids pBSHSPR2C1 and pBSHSPR2C5 were constructed as follows. Genomic DNA was extracted from AAV1 (American Type Culture Collection, Manassas, Va.), and the cap encoding sequence was amplified by PCR with Pfx polymerase (Invitrogen). The AAV2 cap gene was excised from the AAV2 helper plasmid pBSHSPRC2.3 and replaced with the amplified AAV1 cap sequence with an SwaI restriction site in the rep-cap intergenic junction and a BsrGI site engineered just upstream of the AAV2 poly(A) signal. The resulting trans-packaging construct, pBSHSPR2C1, contains the AAV2 rep gene under the control of a minimal eukaryotic promoter and the AAV1 cap open reading frame positioned between the AAV2 rep-cap intergenic junction and the AAV2 poly(A) signal. The trans-packaging construct, pBSHSPR2C5, containing the AAV2 rep and AAV5 cap genes, was assembled similarly except that the template for AAV5 cap was the plasmid pACK2/5 (25), and the insertion sites in pBSHSPRC2.3 were SwaI/XbaI. The plasmid pAd Helper 4.1 expresses the E2a, E4-orf6, and VA genes of adenovirus type 5 (Ad5).
Vector production and purification.
Recombinant AAV vectors were produced by a standard calcium phosphate transfection method in adherent human 293 cells, using the Ad helper, trans-packaging, and AAV vector plasmids. Briefly, cells were seeded in 225-cm2 T-flasks and grown to subconfluency in complete DMEM (BioWhittaker). Plasmids pAd helper 4.1, pBS-HSP-RC2.3 (or pBS-HSP-R2C5 or pBS-HSP-R2C1), and AAV vector plasmid (i.e., pAAVCMV-TNFR:Fc) were added (2:2:1 molar ratio) to a 300 mM CaCl2 solution. To form a calcium phosphate precipitate, the plasmid mixture was added to a 2× HBS buffer (280 mM NaCl, 1.5 mM Na2PO4, 5 mM HEPES [pH 7.1]), incubated for 30 s, and then added directly onto the 293 cell monolayer. The cells were incubated for 6 to 8 h at 37°C, after which the medium was aspirated and replenished with fresh complete but serum-free DMEM. Three days posttransfection, the cells were lysed by the addition of deoxycholate (Fisher, Houston, Tex.) to a final concentration of 0.5%, releasing all cellular and nuclear contents into the solution. The cellular DNA was digested by the addition of Benzonase (EM Sciences, Gibbstown, N.J.) at a concentration of 10 U/ml for 1 h at 37°C. Tween 20 (Fisher) was added to a final concentration of 1% with a further incubation for 60 min at 37°C, followed by the addition of NaCl to a final concentration of 1 M, before clarification through depth filters (Millipore, Bedford, Mass.). Clarified lysates were concentrated 15- to 20-fold by tangential flow filtration with Pellicon 2 minicassettes (110-kDa molecular mass cut off; Fisher) and were exchanged against formulation buffer (20 mM Tris [pH 8.0], 0.2 M NaCl, 2 mM MgCl2, and 2% glycerol). The lysates were diluted through a 0.45-μm-pore-size Pall suporcap filter unit (Gelman; Fisher) and stored at −80°C. The harvested lysates were purified by ion-exchange chromatography essentially as previously described (29, 54). Vector-containing elutes were formulated in a buffer containing 20 mM Tris (pH 8.0), 0.2 M NaCl, 2 mM MgCl2, and 2% glycerol and aseptically filtered. Final vector preparations were >95% pure, with low endotoxin levels (<0.5 endotoxin units/ml). The purified vectors were nonaggregated, as determined by dynamic laser light-scattering analysis with a DynaPro-99 instrument (Protein Solutions, High Wycombe, United Kingdom). Vector titers were determined by real-time PCR with an Applied Biosystems Prism 7900 sequence detector (Perkin-Elmer, Foster City, Calif.) and were between 5 × 1012 and 20 × 1012 DNase-resistent particles (DRP)/ml. Vector infectivity was assessed in a 50% tissue culture infective dose assay with the HeLa-based B50 cell line (19). The average particle-to-infectivity ratio of AAV[2/1], AAV[2/2], and AAV[2/5] was 191, 138, and 881, respectively. Average vector productivity for AAV[2/2], AAV[2/1], and AAV[2/5] was 8 × 104, 105, and 3 × 105 DRP/cell, respectively.
Administration of pseudotyped AAV vectors to animals.
Female Lewis rats, 5 to 7 weeks old, were purchased from Charles River Laboratories (Wilmington, Mass.). Male Rag1−/− mice, 6 to 8 weeks old, were purchased from Jackson Laboratories (Bar Harbor, Maine). Animals were maintained and treated in accordance with the Institutional Animal Care and Use Committee of Targeted Genetics Corporation and the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were allowed to acclimate 7 days before experimentation and were fed water and chow ad libitum. Animals were anesthetized with isoflurane (5% with O2 for induction and 3% for maintenance) (Abbott Laboratories, North Chicago, Ill.). For endotracheal administrations, a dissecting microscope light was placed at the ventral neck to allow visualization of the tracheal opening, with the mouth opened and the tongue extended to visualize the larynx. A catheter stylet (Cathlon; Johnson & Johnson, Arlington, Tex.) was advanced when the vocal chords were relaxed and the tracheal opening was visible. The stylet was then withdrawn and a syringe containing either vehicle or AAV vector (100 and 300 μl for mice and rats, respectively) was attached to the catheter. Following vector administration, a bolus of air (approximately 100 to 200 μl) was pushed through to clear the catheter of any remaining vector solution. Animals were placed in a recovery cage and monitored for signs of respiratory distress. For intramuscular administration, rats were anesthetized as described above. The quadriceps was visualized, the injection site was sterilized, and the rats were injected intramuscularly with vector with a 0.5-ml insulin syringe and a 28.5-gauge needle. Each animal was injected with a total volume of 150 μl of vector, with each hind limb receiving 75 μl. After the procedure, the animals were placed in a recovery cage and observed for normal ambulation.
Immunohistochemical analysis of EGFP expression.
EGFP expression in the rat lung was assessed 42 days following endotracheal administration of either AAV[2/2]CMV-EGFP or AAV[2/5]CMV-EGFP vector (at a dose of 1012 DRP). Briefly, rats were humanely sacrificed, and lungs were perfused and fixed overnight with 10% neutral-buffered-formalin (Sigma, St. Louis, Mo.). Tissues were then processed and embedded in paraffin. Sections of 0.5-μm tissue were cut, placed on poly-l-lysine-coated slides, and processed for immunohistochemical analysis of EGFP protein as follows. The sections were deparaffinized and rehydrated in a graded series of ethanol washes. Slides underwent antigen retrieval in citrate buffer (pH 6.0) in a steamer for 30 min prior to being blocked in the peroxidase block of the Rabbit/Peroxidase EnVision+ System (DAKO, Carpinteria, Calif.). The slides were washed three times with phosphate-buffered saline (PBS) and incubated for 1 h at room temperature with either anti-EGFP antibody (1:2,000 dilution; Molecular Probes, Eugene, Oreg.) or the isotype control rabbit IgG antibody whole-molecule antibody (1:20,000 dilution; Jackson ImmunoResearch, West Grove, Pa.) Both anti-EGFP and isotype control antibodies were at a concentration of 2 μg/ml. After being washed with PBS, the slides were incubated for 30 min at room temperature with labeled polymer (DAKO) according to the manufacturer's directions. The peroxidase-catalyzed product was visualized with 3,3′-diaminobenzidine (DAKO). Unless otherwise noted, the slides were rinsed between steps three times for 2 min each in PBS (pH 7.6). After light counterstaining with methylene blue (Sigma), the slides were dehydrated in sequential concentrations of ethanol and xylene, and the coverslips were mounted with Permount (Fisher).
Anti-AAV capsid-neutralizing antibody assay.
Anti-AAV2 or AAV1 capsid-neutralizing antibody titers were determined by analyzing the ability of serum from either AAV[2/2]CMV-TNFR:Fc or AAV[2/5]CMV-TNFR:Fc vector-treated animals to inhibit the transduction of reporter vector AAV[2/2]CMV-EGFP or AAV[2/1]CMV-EGFP, respectively, in Ad5-infected human 293 cells. Cells were seeded in a 96-well plate and grown overnight until they reached 70 to 80% confluency. To inactivate the complement, serum samples were incubated for 30 min at 56°C. Twofold serial dilutions of serum were incubated with AAVCMV-EGFP (5 ×103 DRP/cell) of the corresponding serotype for 1 h at 37°C. The cells were infected with Ad5 (multiplicity of infection = 2.5) and the serum-AAVCMV-EGFP mixtures were added to the cells. Cells were incubated for 48 h, and the expression of EGFP was analyzed by FluoroImaging (Cytofluor 4000; PerSeptive Biosystems, Framingham, Mass.). The neutralizing antibody titer was calculated as the highest dilution of serum at which there was 50% fluorescence. Anti-AAV5 capsid-neutralizing antibody titers were determined as described above, except that HeLa cells were substituted for the 293 cells and AAV[2/5]CMV-EGFP vector was used in the assay.
RT-PCR analysis of TNFR:Fc mRNA. (i) RNA isolation.
Rat lung tissues were ground to a fine powder in liquid nitrogen with a 6750 Freezer/Mill (Spex CertiPrep, Metuchen, N.J.). Samples were kept frozen (−70°C) until aliquoted for RNA isolation. Total RNA was isolated with the RNeasy Fibrous Tissue Kit (QIAGEN, Valencia, Calif.) according to the manufacturer's protocol. This included the use of the optional on-column DNase step. After isolation, RNA was quantified with a standard spectrophotometer at A260 absorbance.
(ii) cDNA synthesis.
For first-strand cDNA synthesis, the sample size per single replicate was 650 ng of RNA per 20 μl of reaction mixture (56.65 ng/μl) containing 0.1 volume 10× buffer (Invitrogen), 2.5 mM MgCl2, 0.5 mM deoxynucleotide triphosphates, 10 mM dithiothreitol, 10 U of RNase OUT, 50 U of Superscript II RNase H− reverse transcriptase (Invitrogen), and 0.5 μM primerXBrt(T) (5′-UUGGCAGAGGGCGACUGUCATUTTUTTTTTUTTTTTUTT-3′). Each sample was incubated with or without an internal spike control (synthetic TNFR:Fc RNA) to evaluate cDNA and PCR efficiencies as well as to monitor for gross inhibition in test samples. The reverse transcription (RT)-PCR was carried out at 1 cycle in a thermal cycler (GeneAmp PCR 9700; Applied Biosystems, Foster City, Calif.) for 50 min at 42°C. Following first-strand synthesis, 1 U of RNase H (Invitrogen) was added to degrade a portion of RNA from the RNA-DNA hybrid. To synthesize second-strand cDNA from the first-strand cDNA template, 0.5 μM of the forward primer 2557U17 (5′-GCCAGCCATCTGTTGTT-3′), 2 U of DNA polymerase, 2.5 mM MgCl2, and 0.2 μl of 10× buffer (Invitrogen) were added to each reaction mixture. The second-strand synthesis reaction was carried out at 1 cycle for 40 min at 42°C. After second-strand synthesis, 43 μl of TE buffer (10 mM Tris, 1 mM EDTA, [pH. 8]) (Ambion, Austin, Tex.) was added to bring the reaction volume up to 65 μl.
(iii) Quantitative RT-PCR.
All PCRs were carried out in a 50-μl volume with 10 μl of the cDNA. The master mixture consisted of 0.3 μM forward primer BGH (5′-GACCCTGGAAGGTGCCACT-3′), 0.3 μM reverse primer Xpcr.rev (5′-GCACG AGGGCGACTGTCAT-3′), 0.1 μM primer BGH.mgb (5′-(FAM)CCACTGTCCTTTCCTAAT-3′) (where FAM is 6-carboxyfluoroscein), and a 0.5 volume of a real-time 2× PCR buffer (Applied Biosystems). As an endogenous control, the assay included PCR primers and probe from a Taqman Rodent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control reagent kit (Applied Biosystems), targeting the rat GAPDH housekeeping gene, to qualitatively assess samples for amplifiable RNA. The PCR was carried out with a real-time thermal cycler (Sequence Detection System model 7700; Applied Biosystems) according to the following program: 10 min at 50°C, 10 min at 95°C, 50 cycles of 15 s at 95°C, and 1 min at 60°C. The quantitation of the test samples was determined relative to the synthetic TNFR:Fc RNA standard curve.
Protein analyses.
Serum samples were assayed for the presence of rat TNFR:Fc fusion protein with an enzyme-linked immunosorbent assay (ELISA). Briefly, 96-well plates were coated with a goat anti-murine TNF receptor II antibody (R&D Systems, Minneapolis, Minn.) overnight at 4°C. Plates were washed three times with PBS supplemented with 0.05% Tween 20 (Fisher) and blocked with 1% bovine serum albumin (Sigma) for 3 h at room temperature on an orbital shaker. After being washed, the plates were incubated with the serum samples for 1 h at room temperature on a shaker plate. Following sample incubation, the wells were washed and incubated with a biotin-conjugated mouse anti-rat IgG1 antibody (PharMingen/BD Biosciences, San Jose, Calif.) for 1 h, washed, and then incubated with streptavidin-horseradish peroxidase (Zymed, San Francisco, Calif.) for 30 min at room temperature. The presence of rat TNFR:Fc protein was detected with the TMB colormetric substrate (Pierce, Rockford, Ill.) and stopped with 1 M sulfuric acid (Sigma). Quantitation of the amount of rat TNFR:Fc protein was based on optical density values at 450 nm (SpectrMax 250 plate reader; Molecular Devices, Sunnyvale, Calif.) compared with a standard curve of purified rat TNFR:Fc protein. The limit of detection of the assay was 2.19 ng/ml. The levels of human FIX in mouse plasma were determined by ELISA. Mouse blood was obtained in heparinized capillary tubes and was subjected to centrifugation at 800 × g for 10 min. Plasma was removed and placed at −80°C until analyzed. Ninety-six-well immunoplates (Nunc, Rochester, N.Y.) were coated with affinity-purified sheep anti-hFIX IgG (KPL, Inc., Gaithersburg, Md.) overnight at 4°C. Plates were washed three times with PBS containing 0.05% Tween 20 (Fisher) and blocked with 1% bovine serum albumin (Sigma) for 3 to 4 h at room temperature. After being washed, the plates were incubated with the serum samples for 1 to 2 h at room temperature on a shaker plate. Following sample incubation, the wells were washed four times with PBS-Tween 20 and incubated with peroxidase-labeled affinity-purified sheep anti-hFIX antibody (KPL, Inc.) for 2 h, washed four times with PBS-Tween, incubated with the colorimetric substrate ABTS (2,2′-azino-di[3-ethylbenzthiazoline-6-sulfonate]) (Zymed) for 15 min, and then quenched with 1% sodium dodecyl sulfate. Quantitation of hFIX protein was based on optical density values at 450 nm (SpectrMax 250 plate reader; Molecular Devices) compared to a standard curve of purified recombinant hFIX protein (BeneFIX; Wyeth/Genetics Institute, Cambridge, Mass.). The limit of detection of the assay was 20 ng/ml.
Statistical analysis.
Data were pooled by experimental group and the means ± the standard error of the mean (SEM) were calculated with the Microsoft Excel graphing and statistical program. The Student t test was conducted to determine the presence of any significant differences between two groups. P values of less than 0.05 were considered statistically significant.
RESULTS
Single pulmonary delivery of AAV[2/2]CMV-TNFR:Fc or AAV[2/5]CMV-TNFR:Fc vectors to rat lung.
We evaluated and compared the serum levels and duration of TNFR:Fc protein expression following a single endotracheal administration of either AAV[2/2]TNFR:Fc or AAV[2/5]TNFR:Fc vector. Vehicle or pseudotyped vectors at doses of 1010, 1011, or 1012 DRP were administered to rats, and the serum levels of TNFR:Fc protein were monitored over time by ELISA. As expected, there were no detectable circulating levels of TNFR:Fc protein in the sera of vehicle-treated animals. Additionally, rats treated with the AAV[2/2]CMV-TNFR:Fc vector had no detectable levels of circulating TNFR:Fc protein over a period of 9 weeks postvector administration. In contrast, treatment with the AAV[2/5]CMV-TNFR:Fc vector resulted in a dose- and time-dependent serum expression of TNFR:Fc protein. Maximal mean serum TNFR:Fc protein levels of 7.83 ± 0.83, 33.60 ± 11, and 370 ± 39 ng/ml were observed at day 40 postadministration at vector doses of 1010, 1011, and 1012 DRP, respectively (Fig. 1a). A gradual decline in serum TNFR:Fc protein expression was observed over a period of 9 months, although significant levels (mean, 48 ± 2.2 ng/ml) were still detectable in the sera of rats treated at the highest vector dose 252 days postadministration (Fig. 1a). Both AAV[2/2]CMV-TNFR:Fc and AAV[2/5]CMV-TNFR:Fc vectors elicited high titers of circulating anti-AAV 2 and anti-AAV5 capsid-neutralizing antibodies, respectively. There was no cross-neutralization between the two serotypes (Fig. 2a and b).
FIG. 1.

Serum TNFR:Fc protein levels after a single endotracheal administration of pseudotyped AAV vectors to female Lewis rats. (a) Administration of either vehicle or 1010, 1011, or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector. (b) Administration of either vehicle or 1011 or 1012 DRP of AAV[2/1]CMV-TNFR:Fc vector. The serum levels of soluble TNFR:Fc protein were monitored by ELISA at the time points indicated. The data are expressed as the means ± SEM of five animals tested.
FIG. 2.
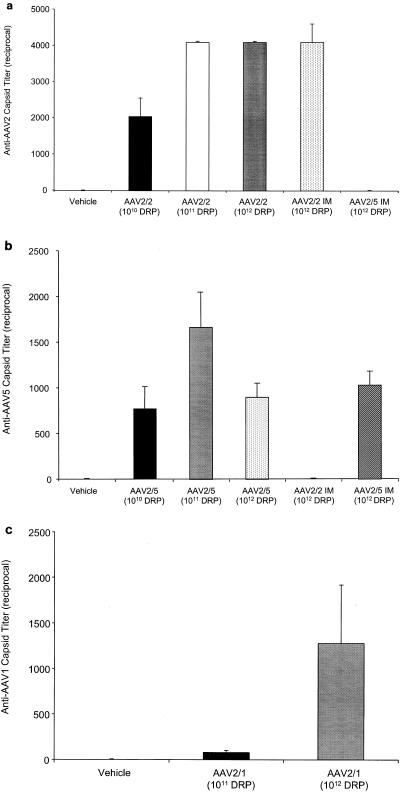
Serum-neutralizing antibody levels elicited against AAV capsid in rats. Sera were analyzed for neutralizing activity against AAV2, AAV5, or AAV1 as described in Materials and Methods. The reciprocal dilution of the neutralizing antibody titer is plotted. Each bar represents the mean ± SEM of five experiments. Each serum sample was tested in triplicate. Sera were titrated in sequential twofold dilutions. (a) Anti-AAV2 capsid titer, (b) Anti-AAV5 capsid titer, (c) Anti-AAV1 capsid titer.
Cell types transduced by AAV[2/2] or AAV[2/5] vectors in rat lung.
To better understand the differences between AAV[2/2] and AAV[2/5] in rat lung transduction, we examined and compared the type of transduced cells and the overall expression levels of EGFP in rat lung following endotracheal administration of AAV[2/2]CMV-EGFP or AAV[2/5]CMV-EGFP vectors by immunohistochemistry. The results in Fig. 3 show that both pseudotyped vectors transduced a variety of cells with a comparable distribution pattern, including airway epithelium in trachea, bronchi, and bronchiole; cells in the alveoli; vascular endothelium; bronchus-associated lymph tissues; and muscle cells of large blood vessels. However, there was significantly greater staining intensity of EGFP in sections of AAV[2/5] than in sections from AAV[2/2]-treated animals (Fig. 3A and C). This result indicates that although the spectrum of cells transduced by AAV[2/2] and AAV[2/5] are the same, the overall transduction efficiency of lung cells by AAV[2/5] is superior.
FIG. 3.
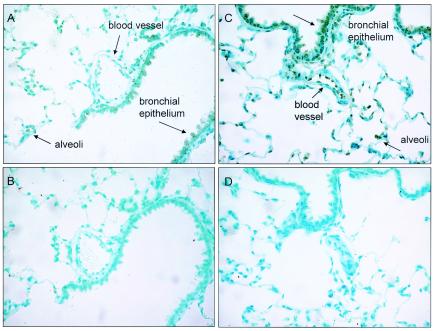
Immunohistochemical analysis of EGFP expression in 0.5-μm sections of rat lung, 42 days after AAV[2/2]CMV-EGFP or AAV[2/5]CMV-EGFP vector-mediated endotracheal administration. Positive cells are present in alveolar and airway epithelia as well as om vascular endothelia when AAV[2/2] and AAV[2/5] were given, although they are significantly more intense in the AAV[2/5]-treated lung. (A and B) AAV[2/2]CMV-EGFP vector-treated lung sections reacted with rabbit anti-EGFP IgG or the isotype control antibody, respectively. (C and D) AAV[2/5]CMV-EGFP vector-treated lung sections reacted with rabbit anti-EGFP IgG or the isotype control antibody, respectively.
Secretion of TNFR:Fc protein by airway epithelial cells.
To examine the contribution of AAV[2/2]- or AAV[2/5]-transduced airway epithelial cells to TNFR:Fc protein secretion to the systemic circulation, the rat CC10 gene promoter, which primarily drives gene expression in these cells, was substituted for the CMV enhancer/promoter (44). The results in Fig. 4 show that initially there was a more rapid onset in transduction of the airway epithelium following endotracheal administration of AAV[2/5]CC10-TNFR:Fc vector than in transduction of lung tissue by AAV[2/5]CMV-TNFR:Fc vector. However, in both vector-treated groups, the transduction resulted in comparable peak serum TNFR:Fc levels of approximately 400 ng/ml at 41 days postvector administration. These levels declined to nearly 200 ng/ml over a period of 85 days, consistent with an airway epithelial cell half-life of about 3 months. Furthermore, for both treatment groups, the decline in the level of expression was comparable with respect to the serum levels and the kinetics and duration of TNFR:Fc protein expression, as both treatment groups displayed a slow and gradual decline in serum TNFR:Fc protein to a low but detectable level of 80 to 100 ng/ml over a period of 7 months. Pulmonary delivery of AAV[2/2]CC10-TNFR:Fc vector resulted in increased transduction of the airway epithelium compared with animals treated with AAV[2/2]CMV-TNFR:Fc vector, but the levels of serum TNFR:Fc protein were at least 10-fold lower than in samples from either AAV[2/5]CMV-TNFR:Fc or AAV[2/5]CC10-TNFR:Fc vector-treated rats (Fig. 4). Consistent with this data, comparison of TNFR:Fc mRNA levels by quantitative RT-PCR analysis, demonstrated 16-fold more TNFR:Fc molecules per cell in the lungs of AAV[2/5]CC10-TNFR:Fc vector-treated animals than in lungs of AAV[2/2]CC10-TNFR:Fc vector-treated animals (mean, 32 and 2 TNFR:Fc mRNA molecules/cell, respectively) (Fig. 7b).
FIG. 4.
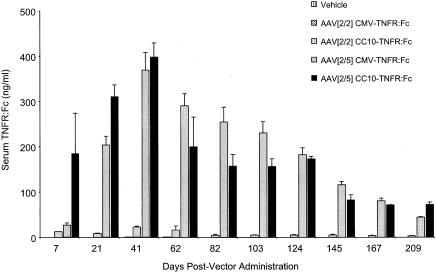
Serum TNFR:Fc levels after a single endotracheal administration of either vehicle or 1012 DRP of AAV[2/2]CMV-TNFR:Fc, AAV[2/5]CMV-TNFR:Fc, AAV[2/2]CC10-TNFR:Fc, or AAV[2/5]CC10-TNFR:Fc vector to female Lewis rats. The serum levels of soluble TNFR:Fc protein were monitored by ELISA at the time points indicated. The data are expressed as the means ± SEM of five animals tested.
FIG. 7.
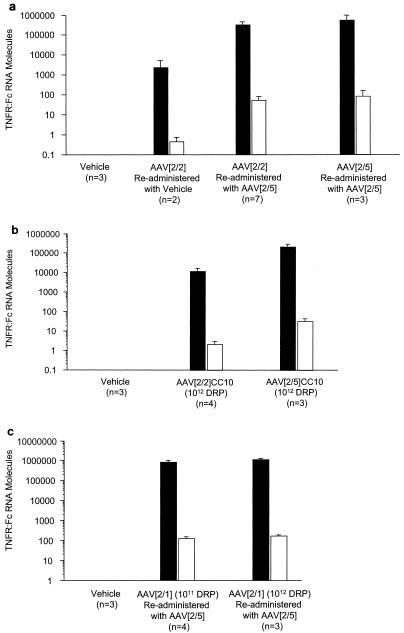
Quantitative real-time RT-PCR analysis of TNFR:Fc mRNA in rat lung. (a) Vehicle- or AAV[2/2]CMV-TNFR:Fc vector-treated animals, given additional doses of either vehicle or two more doses of AAV[2/5]CMV-TNFR:Fc vector, were sacrificed after 475 days. Animals treated twice with AAV[2/5]CMV-TNFR:Fc vector were sacrificed after 497 days. (b) Vehicle-, AAV[2/2]CC10-TNFR:Fc vector-, or AAV[2/5]CC10-TNFR:Fc vector-treated animals were sacrificed after 209 days. (c) Vehicle- or AAV[2/1]CMV-TNFR:Fc vector-treated animals, retreated with either vehicle or with 1011 or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector, were sacrificed after 355 days. Total RNA was isolated from whole lung tissue, and the amount of TNFR:Fc mRNA was determined by quantitative RT-PCR as described in Materials and Methods. The number of animals in each group is shown. The data represent the means, and the error bars indicate standard deviations. Black bars, TNFR:Fc mRNA molecules per 100 ng of total RNA. Open bars, TNFR:Fc mRNA molecules per cell.
Pulmonary delivery of AAV[2/1]CMV-TNFR:Fc vector to rat lung.
Similar to the kinetics of AAV[2/5] vector transduction in the rat lung, endotracheal delivery of AAV[2/1]CMV-TNFR:Fc vector at a dose of either 1011 or 1012 DRP also resulted in peak expression approximately 6 weeks postadministration of the vector. However, the levels of circulating TNFR:Fc protein (mean, 3.7 ± 0.6 and 50 ± 5 ng/ml at 1011 and 1012 DRP, respectively) were seven- and ninefold lower than those observed in rats treated with similar doses of AAV[2/5]CMV-TNFR:Fc vector (Fig. 1b). Following peak expression, the levels of serum TNFR:Fc protein gradually declined more than threefold over a period of 3 to 4 months. Additionally, analysis of serum from AAV[2/1]CMV-TNFR:Fc-treated animals 104 days postadministration of the vector revealed moderate titers of anti-AAV1 capsid-neutralizing antibodies (Fig. 2c).
Comparison of AAV[2/2]- and AAV[2/5]-mediated secretion of soluble proteins following pulmonary administration to immunodeficient mice.
To test whether the differences between AAV[2/2] and AAV[2/5] in rat lung transduction were also reproducible in other rodent species, we administered 6 × 1011 DRP of either AAV[2/2]CMV-TNFR:Fc or AAV[2/5]CMV-TNFR:Fc vector to the lungs of Rag-1−/− mice and compared the levels of serum TNFR:Fc protein over time. There was a gradual increase in serum TNFR:Fc protein expression in both AAV[2/2] and AAV[2/5] vector-treated animals over a period of 12 weeks. However, AAV[2/5]-treated animals displayed increased systemic TNFR:Fc protein expression with faster kinetics during the first 5 weeks postvector delivery, reaching a mean of 107 ± 56 ng/ml compared with 31 ± 24 ng/ml of TNFR:Fc protein in AAV[2/2]-treated mice (Fig. 5a). Interestingly, at 12 weeks postvector administration (the latest time point tested), the serum levels of TNFR:Fc protein in AAV[2/2]- and AAV[2/5]-treated mice were comparable (mean, 111 ± 81 and 147 ± 82 ng/ml, respectively).
FIG. 5.
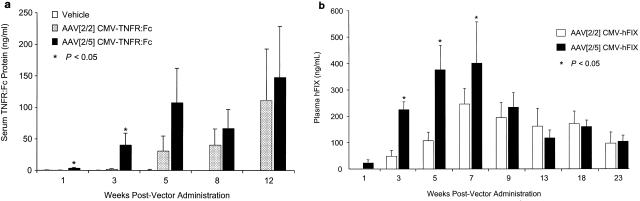
(a) Serum TNFR:Fc levels after a single endotracheal administration of either vehicle or 6 × 1011 DRP of AAV[2/2]CMV-TNFR:Fc or AAV[2/5]CMV-TNFR:Fc vector to male Rag-1−/− mice. The serum levels of TNFR:Fc protein were monitored by ELISA at the time points indicated. (b) Plasma levels of recombinant hFIX protein after a single endotracheal administration of either 6 × 1011 DRP of AAV[2/2]CMV-hFIX or AAV[2/5]CMV-hFIX vector to male Rag-1−/− mice. The plasma levels of hFIX protein were monitored, at the time points indicated, by ELISA. The values shown represent the means ± SEM of five animals tested. Asterisks denote values that differ statistically at P < 0.05 compared with serum TNFR:Fc protein levels (a) or plasma hFIX protein levels (b) in AAV[2/2]CMV-TNFR:Fc-treated animals.
To test if these results reflected differences in animal species rather than the nature of the transgene product, we also compared the plasma levels of hFIX following endotracheal administration of either AAV[2/2]CMV-hFIX (6 × 1011 DRP) or AAV[2/5]CMV-hFIX (6 × 1011 DRP) to Rag-1−/− mice. Consistent with the previous data of TNFR:Fc protein expression in Rag-1−/− mice, there was a gradual increase in plasma hFIX protein over a period of 7 weeks in both treatment groups, but AAV[2/5]-treated mice displayed an earlier onset and more robust expression of hFIX protein than AAV[2/2]-treated animals (Fig. 5b). At 9 weeks after vector administration, the levels of hFIX protein in the plasma of AAV[2/2]- and AAV[2/5]-treated animals were comparable (180 ± 28 and 220 ± 30 ng/ml, respectively). In both vector treatment groups, expression of plasma hFIX protein remained comparable and sustained but slowly declined almost twofold over a period of 23 weeks (Fig. 5b). Thus, it appears that there is a fundamental difference between rats and mice with respect to AAV2 transduction of the lung. In rats, AAV[2/2] transduces lung cell types similar to AAV[2/5] albeit at very low efficiency, while in mice AAV[2/2] and AAV[2/5] achieve similar efficiencies of transduction but with different kinetics.
Readministration of AAV[2/5]CMV-TNFR:Fc vector to rat lung.
Since delivery of the AAV[2/5]CMV-TNFR:Fc vector to the lung elicited serum anti-AAV5-neutralizing antibody response and since lung transduction significantly declined after 8 months, we wanted to examine the effect of existing serum anti-AAV type 5 capsid-neutralizing antibodies on the levels of circulating TNFR:Fc protein following readministration with AAV[2/5]CMV-TNFR:Fc vector. To this end, rats inistially treated with the highest dose (1012 DRP) of AAV[2/5]CMV-TNFR:Fc vector (Fig. 1a) were administered a second dose (1012 DRP) of the same vector 253 days after the first. The results in Fig. 6a show that following a second dose of AAV[2/5]CMV-TNFR:Fc vector, the levels of serum TNFR:Fc protein gradually increased over a period of 6 months (mean, 295 ± 9.5 ng/ml by day 455), but then rapidly declined more than 10-fold over a period of 6 weeks (mean, 26 ± 1.5 ng/ml by day 497). Quantitative analyses of TNFR:Fc mRNA from whole lungs of three animals receiving a second dose of AAV[2/5] showed 76 TNFR:Fc mRNA molecules per cell (Fig. 7a). These results are consistent with the data reported for mice by Auricchio and colleagues (4), indicating not only that readministration of AAV[2/5] vector to the lung in the presence of preexisting serum-neutralizing antibodies is feasible but also that protein levels were comparable to those in naïve animals, potentially allowing for repeated treatment of systemic chronic diseases such as rheumatoid arthritis, psoriasis, or hemophilia.
FIG. 6.
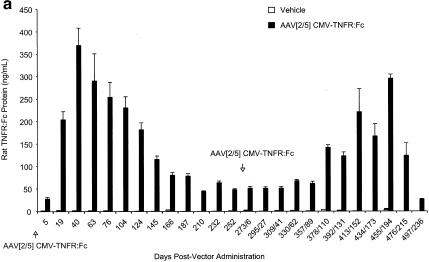
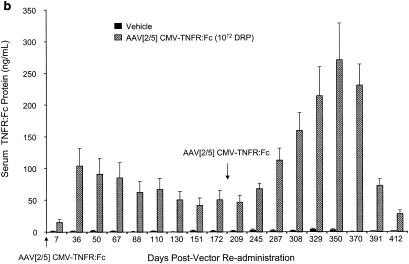
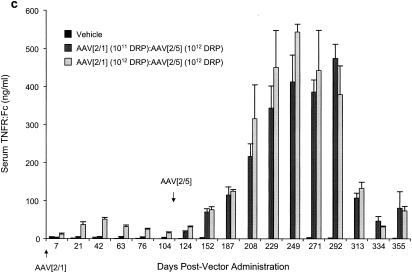
Serum TNFR:Fc levels following repeat endotracheal administrations of pseudotyped AAV vectors to female Lewis rats. (a) Administration of either vehicle or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector. First and second administrations were carried out on day 1 and day 252, respectively. (b) Two endotracheal administrations of either vehicle or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector to rats, previously treated with AAV[2/2]CMV-TNFR:Fc vector. The serum levels of TNFR:Fc protein after treatment with either vehicle or AAV[2/2]CMV-TNFR:Fc vector were below the limits of detection (2.19 ng/ml) and are not shown. Nine weeks after treatment with AAV[2/2], animals were given either vehicle or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector (day 1 on graph). After 180 days, animals were given another dose of either vehicle or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector endotracheally. (c) Readministration of AAV[2/5]CMV-TNFR:Fc vector to AAV[2/1]CMV-TNFR:Fc-vector treated rats. On day 1, animals were administered with either vehicle or 1011 or 1012 DRP of AAV[2/1]CMV-TNFR:Fc vector. On day 105, the vehicle-treated group was administered with vehicle, while the two groups that were treated with AAV[2/1]CMV-TNFR:Fc vector received a dose of 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector. The serum levels of soluble TNFR:Fc protein were monitored by ELISA at the time points indicated. The values shown represent the means ± SEM for five animals tested
Pulmonary readministration of AAV[2/5]CMV-TNFR:Fc vector to AAV[2/2]CMV-TNFR:Fc vector-treated rats.
Although pulmonary delivery of AAV[2/2]CMV-TNFR:Fc did not result in detectable serum TNFR:Fc protein, the treated animals elicited anti-AAV2 capsid-neutralizing antibody response. To test the effect of these preexisting neutralizing antibodies on lung transduction following a repeat delivery of an AAV[2/5]CMV-TNFR:Fc vector, animals that were previously given AAV[2/2] were given either vehicle or 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector. The results in Fig. 6b show that while there was little or no serum TNFR:Fc protein following the delivery of vehicle, animals that were given AAV[2/5]CMV-TNFR:Fc vector expressed the TNFR:Fc protein with kinetics similar to those of animals given AAV[2/5]CMV-TNFR:Fc vector for the first time (Fig. 1a). However, the peak serum levels of TNFR:Fc protein in the animals that were previously treated with AAV[2/2] were almost fourfold lower than those receiving AAV[2/5]CMV-TNFR:Fc as their first treatment (mean, 105 ± 27 ng/ml and 370 ± 39 ng/ml, respectively) (Fig. 6b versus Fig. 1a). It is feasible that preexisting serum anti AAV2 capsid antibodies may have an inhibitory effect on AAV[2/5] vector transduction of the lung. However, when a second dose of 1012 DRP of AAV[2/5]CMV-TNFR:Fc vector was given to these animals after 180 days, serum TNFR:Fc protein expression increased to levels similar to those in naïve animals that were treated with AAV[2/5]CMV-TNFR:Fc vector for the first time (271 ± 57 and 370 ± 39 ng/ml, respectively) (Fig. 6b and 1a). Consistent with the data shown in Fig. 1a, there was a decline of more than sevenfold within a period of 2 months to low but detectable levels of TNFR:Fc protein in the circulation (Fig. 6b). Quantitative analysis of TNFR:Fc mRNA from whole lungs of AAV[2/2] vector-treated animals that were later given either vehicle or a second dose of AAV[2/5] showed less than 1 and 75 TNFR:Fc mRNA molecules per cell, respectively (Fig. 7a). There was no significant difference in the amount of TNFR:Fc mRNA copies per cell in lungs of AAV[2/2]-treated animals that were given two more doses of AAV[2/5] compared with animals treated twice with AAV[2/5] alone (Fig. 7a).
Taken together, these data demonstrate that preexisting anti-AAV2 capsid-neutralizing antibodies have no significant effect on the kinetics of transduction or the levels of transgene expression in the circulation, following delivery of a second dose of AAV[2/5]CMV-TNFR:Fc, since they appear to be comparable in both animal groups (Fig. 1a, 6b, and 7a).
Pulmonary administration of AAV[2/5]CMV-TNFR:Fc vector to AAV[2/1]CMV-TNFR:Fc-treated rats.
Next, we tested the effect of preexisting anti-AAV1 capsid-neutralizing antibodies on the levels of serum TNFR:Fc following a second pulmonary delivery of an AAV[2/5]CMV-TNFR:Fc vector. Rats that were previously treated with AAV[2/1]CMV-TNFR:Fc vector (Fig. 1b) and that elicited anti-AAV1 capsid-neutralizing antibody response (Fig. 2c) were given another dose of AAV[2/5]CMV-TNFR:Fc vector endotracheally on day 104; serum TNFR:Fc protein levels were monitored by ELISA. The results (Fig. 6c) show that within a period of 5 to 6 months from delivery of 1012 DRP of AAV[2/5]CMV-TNFR:Fc to AAV[2/1]CMV-TNFR:Fc-treated animals (at a dose of either 1011 or 1012 DRP), there was a marked increase to over 500 ng of serum TNFR:Fc protein/ml. These protein levels declined over fivefold but remained detectable at approximately 100 ng/ml for over a year. Quantitative analysis of TNFR:Fc mRNA from whole lungs of these animals showed 100 TNFR:Fc mRNA molecules per cell, whether the animals were treated first with 1011 or 1012 DRP of AAV[2/1]CMV-TNFR:Fc vector (Fig. 7c).
DISCUSSION
In this study, we examined the potential utility of AAV vector-mediated gene delivery to the lung as a means for achieving systemically circulating levels of soluble proteins with known therapeutic value. We delivered pseudotyped AAV-TNFR:Fc vectors containing capsid of serotypes 1, 2, or 5 to the lung and compared the efficiency and duration of TNFR:Fc protein secretion to the systemic circulation. The results in rats show that AAV[2/5]-treated animals achieved significantly higher levels of TNFR:Fc protein in the blood than those treated with AAV[2/1], while AAV[2/2]-treated rats expressed no (when AAVCMV-TNFR:Fc was used) or very little (when AAVCC10-TNFR:Fc was used) TNFR:Fc protein in the circulation. However, AAV[2/2]- and AAV[2/5]-treated mice achieved similar transduction efficencies but with different kinetics. The earlier onset of TNFR:Fc transgene expression following pulmonary delivery of AAV[2/5] compared with AAV[2/2] may be due to differences in the interaction of type 2 or type 5 capsids with their distinct receptor and/or a coreceptor(s) on target cells and/or due to distinct capsid-mediated postentry pathways. For example, while AAV[2/2] and AAV[2/5] may bind to and infect similar type of cells in the murine lung, AAV[2/5] may bind these target cells with greater affinity than AAV[2/2] and mediate a more-efficient gene transfer. Consistent with this proposal, Zabner and colleagues demonstrated that differentiated airway epithelia bind AAV5-derived vectors approximately sevenfold more efficiently than AAV2 vectors in vitro (52). In addition, while AAV2 showed a time-dependent increase in gene transfer to human airway epithelia, maximal AAV5 transduction was rapid and resulted in similar levels of gene transfer after short or prolonged incubations (52). Alternatively, there may be more receptors or a coreceptor(s) for type 5 capsid on target cells than for type 2 capsid in the murine lung. This could result in the rapid generation of double-stranded AAV[2/5]-derived genomes, due to annealing of complementary (plus and minus) single-stranded vector genomes, resulting in early onset of transgene expression (36). Finally, AAV[2/5] viral particles may undergo more rapid uncoating and conversion to double-stranded DNA upon entry into the nucleus of the target cell, whereas the majority of AAV[2/2] genomes may persist as encapsidated single-stranded molecules within the nucleus for a longer period of time (47). Singly or in combination, these mechanisms may explain the differences between AAV[2/2] and AAV[2/5] lung transduction in rats, although in rats they may be more pronounced than in mice, thus affecting not only the kinetics but the overall transduction efficiency of AAV[2/2].
In the study of Auricchio and colleagues (4), which compared pseudotyped AAV vector-mediated transduction of human placental alkaline phosphatase (hPALP) or erythropoietin of the murine lung, there was little or no detectable transgene expression after AAV[2/2] administration. These results are inconsistent with our data with mice and perhaps reflect differences in the pulmonary administration between the two studies. In the Auricchio study, the vector was administered to mice by nasal instillation, while in our study, endotracheal administration of the vector was employed. It is possible that the majority of AAV[2/2] vector delivered intranasally was trapped and bound to the nasal epithelium, thus significantly limiting transduction of the lung. Alternatively, it may be using different mouse strains that accounts for the disparity in the data. In our study, immunodeficient Rag-1−/− mice were employed while in the Auricchio study, immunocompetent C57BL/6 mice were used. Taken together, the different results in rats and mice and between strains of mice highlight the limitations of animal studies and call for caution when drawing conclusions from studies carried in a single animal species.
What are the cells that are responsible for secretion of TNFR:Fc protein to the systemic circulation? Administration of AAV[2/2]CMV-EGFP or AAV[2/2]CMV-EGFP vector to the rat lung demonstrated differences in transduction efficiencies on a per-cell basis with AAV[2/5] being superior, but similar cell types were transduced by the two pseudotyped vectors. These included airway epithelial cells in trachea, bronchi, and bronchioles and cells in the alveoli, vascular endothelium, and vascular smooth muscle. The data in Fig. 1a show that a twofold decline in serum TNFR:Fc levels from peak expression at day 40 stretched over a period of 3 months, which is generally consistent with the half-life of airway epithelia. In addition, limiting TNFR:Fc expression primarily to the airway epithelia by substitution of the CC10 promoter for the CMV enhancer/promoter did not significantly change the level and duration of TNFR:Fc expression (Fig. 4). These results are also in agreement with positive immunohistochemical detection of hPALP in both the gas-exchanging alveolar and the conducting airway epithelial cells after pulmonary administration of an AAV[2/5] pseudotyped vector carrying the hPALP gene under the CC10 promoter (4). Following the decline of bulk secretion of TNFR:Fc protein, it was still detectable in the serum at low levels for an extended period. It is possible that the more long-lived cells in the lung, such as vascular smooth muscle cells, that are also transduced by AAV[2/5] are responsible for the low level of long-term production of TNFR:Fc protein in the lung.
Taken together, these results suggest that airway epithelia, either the conducting airways, the epithelial cells in the alveoli, or both, can serve as an efficient depot for production and secretion of TNFR:Fc protein to the bloodstream. Other cells with a longer half-life, such as vascular smooth muscle cells, may contribute to lower levels but longer-term production and secretion of TNFR:Fc protein to the circulation.
The studies reported here show that all pseudotyped vectors elicited serum anti-AAV capsid-neutralizing antibody response and that there was no cross-neutralization between AAV2 and AAV5 pseudotyped vectors. Nonetheless, AAV[2/5] could be successfully readministered to animals that were treated earlier with a vector of either the same serotype (AAV[2/5]) (Fig. 6a), another serotype (AAV[2/2] or AAV[2/1]) (Fig. 6b and c), or both (AAV[2/2], AAV[2/5], and AAV[2/5]) (Fig. 6b) and reinduce production and secretion of TNFR:Fc protein to levels similar to those in rats that were treated with AAV[2/5] for the first time. These results demonstrate that serum-neutralizing antibodies against AAV5, AAV2, and AAV1 capsids do not present a major barrier to repeat administration of AAV[2/5] vector to the lung. Our observation of successful readministration of AAV vectors of the same serotype to the lung in the presence of anti-capsid serum-neutralizing antibodies is consistent with those of previously published studies (4, 6). As suggested by Beck and colleagues, the anti-capsid neutralization assay measures a total serum antibody response, of which IgG is the predominant Ig species. In the absence of inflammation and capillary leakage of serum proteins, IgG is generally not available on the large airway surface to neutralize foreign antigens (6). The predominant surface immunoglobulin is secretory IgA (31). In this study, we did not directly measure secretory IgA, but the observation of successful readministration suggests the absence of such a response.
Following the initial pulmonary administration of AAV[2/5], peak serum TNFR:Fc protein expression was reached after 40 days (Fig. 1a). However, upon readministration, the kinetics of transgene expression appeared different, since maximum serum TNFR:Fc protein levels were realized only after 203 days (Fig. 6a). One explanation that may account for these differences is that the first administration of AAV[2/5] was carried out in young animals (5 to 7 weeks old), while readministration took place almost 9 months later. It is possible that the overall number or metabolic state of the lung target cells may differ with age, which may affect pre- or postentry pathways of vector metabolism, including receptor binding and internalization, intracellular trafficking, capsid uncoating, and conversion of the single-stranded DNA genome to a transcriptional competent double-stranded DNA template. This might ultimately affect the kinetics of serum TNFR:Fc protein expression.
Previously, it was demonstrated that both local (intra-articular) or systemic (intramuscular) administration of AAV[2/2]CMV-TNFR:Fc vector to rats with streptoccocal cell wall-induced arthritis resulted in marked suppression of disease parameters (9). Given the potential utility of AAV vector-mediated lung delivery of TNFR:Fc, we are currently employing a similar model of experimental arthritis in rats to evaluate the feasibility of AAV[2/5]TNFR:Fc vector delivery to the lung as a means to produce and secrete systemic therapeutic concentrations of TNFR:Fc protein to efficiently suppress chronic inflammatory arthritis.
Acknowledgments
We greatly appreciate the support of personnel at the Targeted Genetics Corporation Animal Facilities. We are also indebted to Simon Godwin and his dedicated team for their invaluable technical assistance in AAV vector production. We thank Barrie Carter and Ralph Paul for helpful discussions and for critically reading the manuscript.
REFERENCES
- 1.Acland, G. M., G. D. Aguirre, J. Ray, Q. Zhang, T. S. Aleman, A. V. Cideciyan, S. E. Pearce-Kelling, V. Anand, Y. Zeng, A. M. Maguire, S. G. Jacobson, W. W. Hauswirth, and J. Bennett. 2001. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 28:92-95. [DOI] [PubMed] [Google Scholar]
- 2.Afione, S. A., C. K. Conrad, W. G. Kearns, S. Chunduru, R. Adams, T. C. Reynolds, W. B. Guggino, G. R. Cutting, B. J. Carter, and T. R. Flotte. 1996. In vivo model of adeno-associated virus vector persistence and rescue. J. Virol. 70:3235-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aitken, M. L., R. B. Moss, D. A. Waltz, M. E. Dovey, M. R. Tonelli, S. C. McNamara, R. L. Gibson, B. W. Ramsey, B. J. Carter, and T. C. Reynolds. 2001. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther. 12:1907-1916. [DOI] [PubMed] [Google Scholar]
- 4.Auricchio, A., E. O'Connor, D. Weiner, G.-P. Gao, M. Hildinger, L. Wang, R. Calcedo, and J. M. Wilson. 2002. Noninvasive gene transfer of the lung for systemic delivery of therapeutic proteins. J. Clin. Investig. 110:449-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bantel-Schaal, U., B. Hub, and J. Kartenbeck. 2002. Endocytosis of adeno-associated virus type 5 leads to accumulation of virus particles in the Golgi compartment. J. Virol. 76:2340-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beck, S. E., L. A. Jones, K. Chesnut, S. M. Walsh, T. C. Reynolds, B. J. Carter, F. B. Askin, T. R. Flotte, and W. B. Guggino. 1999. Repeated delivery of adeno-associated virus vectors to the rabbit airway. J. Virol. 73:9446-9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck, S. E., B. L. Laube, C. I. Barberena, A. C. Fischer, R. J. Adams, K. Chesnut, T. R. Flotte, and W. B. Guggino. 2002. Deposition and expression of aerosolized rAAV vectors in the lungs of Rhesus macaques. Mol. Ther. 6:546-554. [DOI] [PubMed] [Google Scholar]
- 8.Bennett, J., D. Duan, J. Englehardt, and A. M. Maguire. 1997. Real-time non-invasive in vivo assessment of adeno-associated virus-mediated retinal transduction. Investig. Ophthalmol. Vis. Sci. 38:2857-2863. [PubMed] [Google Scholar]
- 9.Chan, J. M. K., G. Villareal, W. W. Jin, T. Stepan, H. Burstein, and S. M. Wahl. 2002. Intraarticular gene transfer of TNFR:Fc suppresses experimental arthritis with reduced systemic distribution of the gene product. Mol. Ther. 6:727-736. [DOI] [PubMed] [Google Scholar]
- 10.Chao, H., Y. Liu, J. Rabinowitz, C. Li, R. J. Samulski, and C. E. Walsh. 2000. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2:619-623. [DOI] [PubMed] [Google Scholar]
- 11.Chiorini, J. A., L. Yang, Y. Lui, B. Safer, and R. M. Kotin. 1997. Cloning of adeno-associated virus type 4 (AAV4) and generation of recombinant AAV4 particles. J. Virol. 71:6823-6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiorini, J. A., F. Kim, L. Yang, and R. M. Kotin. 1999. Cloning and characterization of adeno-associated virus type 5. J. Virol. 73:1309-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conrad, C. K., S. S. Allen, S. A. Afione, T. C. Reynolds, S. E. Beck, M. Fee-Maki, X. Barrazza-Ortiz, R. Adams, F. B. Askin, B. J. Carter, W. B. Guggino, and T. R. Flotte. 1996. Safety of single-dose administration of an adeno-associated virus (AAV)-CFTR vector in the primate lung. Gene Ther. 3:658-668. [PubMed] [Google Scholar]
- 14.Davidson, B. L., C. S. Stein, J. A. Heth, I. Martins, R. M. Kotin, T. A. Derksen, J. Zabner, A. Ghodsi, and J. A. Chiorini. 2000. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc. Natl. Acad. Sci. USA 97:3428-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Pasquale, G., B. L. Davidson, C. S. Stein, I. Martins, D. Scudiero, A. Monks, and J. A. Chiorini. 2003. Identification of PDGFR as a receptor for AAV-5 transduction. Nat. Med. 9:1306-1312. [DOI] [PubMed] [Google Scholar]
- 16.Duan, D., Y. Yue, P. B. McCray, and J. F. Englehardt. 1998. Polarity influences the efficiency of recombinant adeno-associated virus infection in differentiated airway epithelia. Hum. Gene Ther. 9:2761-2776. [DOI] [PubMed] [Google Scholar]
- 17.Fischer, A. C., S. E. Beck, C. I. Smith, B. L. Laube, F. B. Askin, S. E. Guggino, R. J. Adams, T. R. Flotte, and W. B. Guggino. 2003. Successful transgene expression with serial doses of aerosolized rAAV2 vectors in rhesus macaques. Mol. Ther. 8:918-926. [DOI] [PubMed] [Google Scholar]
- 18.Flotte, T. R., P. L. Zeitlin, T. C. Reynolds, A. E. Heald, P. Pedersen, S. Beck, C. K. Conrad, L. Brass-Ernst, M. Humphries, K. Sullivan, R. Wetzel, G. Taylor, and B. J. Carter. 2003. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients; a two part clinical study. Hum. Gene Ther. 14:1079-1088. [DOI] [PubMed] [Google Scholar]
- 19.Gao, G. P., G. Qu, L. Z. Faust, R. K. Engdahl, W. Xiao, J. V. Hughes, P. W. Zoltick, and J. M. Wilson. 1998. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum. Gene Ther. 9:2353-2362. [DOI] [PubMed] [Google Scholar]
- 20.Gao, G. P., M. R. Alvira, L. Wang, R. Calcedo, J. Johnston, and J. M. Wilson. 2002. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 99:11854-11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gey, G. O., W. D. Coffman, and M. T. Kubicek. 1952. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 12:264-265. [Google Scholar]
- 22.Graham, F. L. J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59-74. [DOI] [PubMed] [Google Scholar]
- 23.Halbert, C. L., T. A. Standaert, M. L. Aitken, I. E. Alexander, D. W. Russel, and A. D. Miller. 1997. Transduction by adeno-associated virus vectors in the rabbit airway: efficiency, persistence, and readministration. J. Virol. 71:5932-5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halbert, C. L., T. A. Standaert, M. L. C. B. Wilson, and A. D. Miller. 1998. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 72:9795-9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildinger, M., A. Auricchio, G. Gao, L. Wang, N. Chirmule, and J. M. Wilson. 2001. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J. Virol. 75:6199-6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jooss, K., Y. Yang, K. J. Fisher, and J. M. Wilson. 1998. Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J. Virol. 72:4212-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laughlin, C. A., J. D. Tratschin, H. Coon, and B. J. Carter. 1983. Cloning of infectious adeno-associated virus genomes in bacterial plasmids. Gene 23:65-73. [DOI] [PubMed] [Google Scholar]
- 28.Kaludov, N., K. E. Brown, R. W. Walters, J. Zabner, and J. A. Chiorini. 2001. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 75:6884-6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaludov, N., B. Handelman, and J. A. Chiorini. 2002. Scalable purification of adeno-associated virus type 2, 4, or 5 using ion-exchange chromatography. Hum. Gene Ther. 13:1235-1243. [DOI] [PubMed] [Google Scholar]
- 30.Kaplitt, M. G., P. Leone, R. J. Samulski, X. Xiao, D. W. Pfaff, K. L. O'Malley, and M. J. During. 1994. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 8:148-154. [DOI] [PubMed] [Google Scholar]
- 31.Merril, W. W., G. P. Naegel, J. J. Olchowski, and H. Y. Reynolds. 1985. Immunoglobulin G subclass proteins in serum and lavage fluid of normal subjects. Quantitation and comparison with immunoglobulins A and E. Am. Rev. Respir. Dis. 131:584-587. [DOI] [PubMed] [Google Scholar]
- 32.Mingozzi, F., J. Schuttrumpf, V. R. Arruda, Y. Liu, Y. L. Liu, K. A. High, W. Xiao, and R. W. Herzog. 2002. Improved hepatic gene transfer by using an adeno-associated virus serotype 5 vector. J. Virol. 76:10497-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss, R. B., D. Rodman, L. T. Spencer, M. L. Aitken, P. L. Zeitlin, D. Waltz, C. Milla, A. S. Brody, J. P. Clancy, B. Ramsey, N. Hamblett, and A. E. Heald. 2004. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest 125:509-521. [DOI] [PubMed] [Google Scholar]
- 34.Muramatsu, S., H. Mizukami, N. S. Young, and K. E. Brown. 1996. Nucleotide sequencing and generation of an infectious clone of adeno-associated virus 3. Virology 221:208-217. [DOI] [PubMed] [Google Scholar]
- 35.Nakai, H., R. W. Herzog, J. N. Hagstrom, J. Walter, S. H. Kung, E. Y. Yang, S. J. Tai, Y. Iwaki, G. J. Kurtzman, K. J. Fisher, P. Colosi, L. B. Couto, and K. A. High. 1998. Adeno-associated viral vector-mediated gene transfer of human blood coagulation factor IX into mouse liver. Blood 91:4600-4607. [PubMed] [Google Scholar]
- 36.Nakai, H., S. Fuess, T. A. Storm, L. A. Meuse, and M. A. Kay. 2003. Free DNA ends are essential for concatamerization of synthetic double-stranded adeno-associated virus vector genomes transfected into mouse hepatocytes in vivo. Mol. Ther. 7:112-121. [DOI] [PubMed] [Google Scholar]
- 37.Qing, K., C. Mah, J. Hansen, S. Zhou, V. Dwarki, and A. Srivastava. 1999. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus type 2. Nat. Med. 5:71-77. [DOI] [PubMed] [Google Scholar]
- 38.Rabinowitz, J. E., F. Rolling, C. Li, H. Conrath, W. Xiao, X. Xiao, and R. J. Samulski. 2002. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 76:791-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rutledge, E. A., C. L. Halbert, and D. W. Russel. 1998. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 72:309-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkar, R., R. Tetreault, G. Gao, L. Wang, P. Bell, R. Chandler, J. M. Wilson, and H. H. Kazazian. 2004. Total correction of hemophilia A mice with canine FVIII using an AAV8 serotype. Blood 103:1253-1260. [DOI] [PubMed] [Google Scholar]
- 41.Skorupa, A. F., K. J. Fisher, J. M. Wilson, M. K. Parente, and J. H. Wolfe. 1999. Sustained production of beta-glucuronidase from localized sites after AAV vector gene transfer results in widespread distribution of enzyme and reversal of lysosomal storage lesions in a large volume of brain in mucopolysaccharidosis VII mice. Exp. Neurol. 160:17-27. [DOI] [PubMed] [Google Scholar]
- 42.Snyder, R. O., C. H. Miao, G. A. Patijn, S. K. Spratt, O. Danos, D. Nagy, A. M. Gown, B. Winther, L. Meuse, L. K. Cohen, A. R. Thompson, and M. A. Kay. 1997. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat. Genet. 16:270-276. [DOI] [PubMed] [Google Scholar]
- 43.Srivastava, A., E. W. Lusby, and K. I. Berns. 1983. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 45:555-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stripp, B. R., P. L. Sawaya, D. S. Luse, K. A. Wikenheiser, S. E. Wert, J. A. Huffman, D. L. Lattier, G. Singh, S. L. Katyal, and J. A. Whitsett. 1992. cis-Acting elements that confer lung epithelial cell expression of CC10 gene. J. Biol. Chem. 267:14703-14712. [PubMed] [Google Scholar]
- 45.Summerford, C., and R. J. Samulski. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Summerford, C., J. S. Bartlett, and R. J. Samulski. 1999. αVβ5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med. 5:78-82. [DOI] [PubMed] [Google Scholar]
- 47.Thomas, C. E., T. A. Storm, Z. Huang, and M. A. Kay. 2004. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J. Virol. 78:3110-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walters, R. W., S. M. P. Yi, S. Keshavjee, K. E. Brown, M. J. Welsh, J. A. Chiorini, and J. Zabner. 2001. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 276:20610-20616. [DOI] [PubMed] [Google Scholar]
- 49.Xiao, X., J. Li, and R. J. Samulski. 1996. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol. 70:8098-8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao, W., N. Chirmule, S. C. Berta, B. McCullough, G. Gao, and J. M. Wilson. 1999. Gene therapy vectors based on adeno-associated virus type 1. J. Virol. 73:3994-4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao, W., N. Chirmule, M. A. Schnell, J. Tazelaar, J. V. Hughes, and J. M. Wilson. 2000. Route of administration determines induction of T-cell-independent humoral responses to adeno-associated virus vectors. Gene Ther. 1:323-329. [DOI] [PubMed] [Google Scholar]
- 52.Zabner, J., M. Seiler, R. Walters, R. M. Kotin, W. Fulgerus, B. L. Davidson, and J. A. Chiorini. 2000. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway ephithelia and facilitates gene transfer. J. Virol. 74:3852-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zaiss, A. K., Q. Liu, G. P. Bowen, N. C. W. Wong, J. S. Bartlett, and D. A. Muruve. 2002. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 76:4580-4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zolotukhin, S., M. Potter, I. Zolotukhin, Y. Sakai, S. Loiler, T. J. Fraites, V. A. Chiodo, T. Phillipsberg, N. Muzyczka, W. W. Hauswirth, T. R. Flotte, B. J. Byrne, and R. O. Snyder. 2002. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods 28:158-167. [DOI] [PubMed] [Google Scholar]