

Abstract
Non-sorbitol-fermenting, β-glucuronidase-negative Escherichia coli O157:H7 strains are regarded as a clone complex, and populations from different geographical locations are believed to share a recent common ancestor. Despite their relatedness, high-resolution genotyping methods can detect significant genome variation among different populations. Phylogenetic analysis of high-resolution genotyping data from these strains has shown that subpopulations from geographically unlinked continents can be divided into two primary phylogenetic lineages, termed lineage I and lineage II, and limited studies of the distribution of these lineages suggest there could be differences in their propensity to cause disease in humans or to be transmitted to humans. Because the genotyping methods necessary to discriminate the two lineages are tedious and subjective, these methods are not particularly suited for studying the large sets of strains that are required to systematically evaluate the ecology and transmission characteristics of these lineages. To overcome this limitation, we have developed a lineage-specific polymorphism assay (LSPA) that can readily distinguish between the lineage I and lineage II subpopulations. In the studies reported here, we describe the development of a six-marker test (LSPA-6) and its validation in a side-by-side comparison with octamer-based genome scanning. Analysis of over 1,400 O157:H7 strains with the LSPA-6 demonstrated that five genotypes comprise over 91% of the strains, suggesting that these subpopulations may be widespread.
The enterohemorrhagic Escherichia coli (EHEC) have emerged as a leading cause of bloody diarrhea (hemorrhagic colitis) in the United States and other countries (11). The predominant serotype of EHEC in the United States, Canada, Japan, and the United Kingdom is O157:H7; however, several other serotypes, including O26:H11 and O111:H2, O111:H8, and O111:H− are also common, particularly in continental Europe and Australia (4, 26). Despite differences in serotypes and genetic backgrounds, the three primary EHEC serotypes share a common set of virulence genes which include the stx1 and stx2 genes encoding the Shiga toxins (19, 21), several genes located within the locus of enterocyte effacement that encode a specialized attachment system (6, 7), and the plasmid-borne ehxA gene encoding a hemolysin (29, 30). Phylogenetic analyses indicate that O157:H7 strains comprise a single phylogenetic lineage while O26:H11 and O111:H8 strains comprise a second lineage (41) and that the two lineages evolved through parallel pathways of virulence gene acquisition (25).
Phylogenetic analysis of EHEC O157:H7 and O157:H− strains found worldwide has shown that they are highly related and comprise a clone complex (9, 41). A stepwise evolutionary model has been proposed on the basis of molecular genetic and phylogenetic studies according to which the contemporary β-glucuronidase negative, non-sorbitol-fermenting EHEC O157:H7 clone descended from an O55:H7-like enteropathogenic E. coli ancestor (9). The sequence of events includes lysogenization of the ancestor by Shiga toxin-converting phages, a serotype switch conferred by acquisition of genes within the gnd region, acquisition of the large pO157 plasmid, and loss of the β-glucuronidase and sorbitol fermentation characteristics (9, 38). Despite the relatedness of non-sorbitol-fermenting, β-glucuronidase-negative O157:H7 strains, significant genome diversity can be observed among individual isolates by methods such as pulse-field gel electrophoresis (PFGE) (8, 10, 32). Indeed, genome sequencing analysis of two different non-sorbitol-fermenting, β-glucuronidase-negative strains demonstrated that substantial strain-level variation can be detected in genome content, including differences in prophage content and genomic islands (23, 24). Recent genome-based studies also support the observation that variation in prophage content accounts for significant diversity among populations of O157:H7 (28, 31).
Studies to examine the phylogenetic relationships among non-sorbitol-fermenting, β-glucuronidase-negative O157:H7 strains have determined that the strains comprise two highly related but distinct populations that are globally spread (16, 17). Although the strain sets studied thus far have been relatively small, a biased distribution of the two lineages among human- and bovine-derived isolates was observed in one of the studies (16), suggesting that the two lineages could have unique transmissibility or virulence characteristics. Phenotypic studies of strains from human clinical samples and bovine production environments are also consistent with the hypothesis that O157:H7 strains may display differences in virulence characteristics (1, 20).
Because animal models to test the virulence of O157:H7 strains are limited, systematic testing of the differential virulence hypothesis requires the use of multiple approaches to examine virulence characteristics and transmission patterns of the different O157:H7 subpopulations. In order to study transmission patterns, a high-throughput genotyping method is necessary to allow large-scale analysis of strain sets from epidemiological studies. In this report, we describe incorporation of six of these markers into a multilocus genotyping assay, termed lineage-specific polymorphism assay-6 (LSPA-6). We demonstrate the validation of the assay and its capacity for high throughput by analysis of a large strain set comprising 1,429 O157:H7 strains from human clinical samples and bovine production environments.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Characteristics of the E. coli O157:H7 strains used in this study are reported online (http://foodsci.unl.edu/homepage/faculty/strain%20sets.xls). The strains comprising the USA 40 set, the Australian set (AU6 to AU1823), and the Francis set have been described previously (16, 17). Strains from the downer set were derived from a study of O157:H7 in downer cattle in the Midwestern United States (3). Additional human clinical isolates (CDC241 to CDC265) were derived from the Centers for Disease Control and Prevention (CDC; N. Strockbine). Strains in the Moxley 60, Moxley 387, Moxley W00, Moxley W01, and Moxley S01 collections were isolated from bovine feces and environmental sampling (rope) devices (35) in cross-sectional and longitudinal studies of O157:H7 in Midwestern feedlots during the periods of June to September 1999 (Moxley 00), February to March 2000 (Moxley 60), May to November 2000 (Moxley 387), January to June 2001 (Moxley W01), and May to July 2001 (Moxley S01) (15, 33). Human clinical isolates of O157:H7 strains were derived from the Nebraska Public Health Laboratory (NPHL). The NPHL strains were collected from sporadic cases and outbreaks in Nebraska, and each represents a unique XbaI PFGE genotype. Included among these genotypes are the XbaI PFGE genotypes most commonly reported in the United States by the CDC. All strains were maintained as frozen stock preparations and were minimally propagated on Luria-Bertani broth.
Primer design.
Primers for the LSPA-6 were designed to flank lineage-specific polymorphisms by using the PRIME program of the Genetics Computer Group package. Each primer combination consisted of a fluorescence-labeled forward primer and an unlabeled reverse primer (Table 1). The LSPA-6 forward primers were labeled with IRDye 700 (LI-COR Biosciences, Lincoln, Nebr.). The primer sequences are shown in Table 1.
TABLE 1.
PCR primers used in this study
| Marker name | Primer | Primer sequence | Tma (°C) |
|---|---|---|---|
| folD-sfmA | Forward | TACGTAGGTCGAAGGG | 51.0 |
| Reverse | CCAGATTTACAACGCC | 51.5 | |
| Z5935 | Forward | GTGTTCCCGGTATTTG | 50.9 |
| Reverse | CTCACTGGCGTAACCT | 50.3 | |
| yhcG | Forward | CTCTGCAAAAAACTTACGCC | 50.3 |
| Reverse | CAGGTGGTTGATCAGCG | 50.3 | |
| rbsB | Forward | AGTTTAATGTTCTTGCCAGCC | 51.2 |
| Reverse | ATTCACCGCTTTTTCGCC | 51.1 | |
| rtcB | Forward | GCGCCAGATCGATAAAGTAAG | 51.3 |
| Reverse | GCCGTTGTAAACGTGATAAAG | 50.3 | |
| arp-iclR | Forward | GCTCAATCTCATAATGCAGCC | 51.7 |
| Reverse | CACGTATTACCGATGACCG | 50.1 |
Tm, melting temperature.
Multiplex assay.
Multiplex PCR amplification of LSPA-6 primer combinations was accomplished in single reaction mixtures. Template DNA was prepared from overnight cultures that had been heated at 100°C for 10 min and centrifuged at 16,000 × g for 5 min. For each reaction, 1 μl of the boiled and centrifuged culture supernatant (template DNA) was combined with 1× PCR buffer (20 mM Tris-HCl [pH 8.4], 50 mM KCl; Invitrogen), a 200 μM concentration of each deoxynucleoside triphosphate (0.2 mM [each] dATP, dTTP, dCTP, and dGTP), 3 mM MgCl2, 1 U of Taq DNA Polymerase (Invitrogen), and a 0.3 nM concentration of each forward and reverse primer for all six markers. PCR thermocycler conditions were 1 cycle at 94°C for 4 min; 11 cycles of 94°C for 30 s, 50°C (decreasing 1°C/cycle) for 45 s, and 72°C for 1 min; 20 cycles of 94°C for 30 s, 52°C for 45 s, and 72°C for 1 min; and 1 cycle at 72°C for 5 min. After completion of the cycling, a one-half volume of loading dye (0.012% bromphenol blue-0.1 mM EDTA, [pH 8.0] in 100% formamide) was added, and the reactions were denatured at 94°C for 3 min prior to electrophoresis on an NEN Global Edition IR2 DNA Analyzer (LI-COR Biosciences). A portion (1 μl) of each reaction mixture was loaded onto a 6.5% denaturing polyacrylamide gel (length, 25 cm). Control reactions, derived from the K-12 strain MC1061 and the lineage I and lineage II O157:H7 control strains (93-001 and FRIK2000, respectively), were included on all gels.
Data analysis.
Printed copies of images from the electrophoresis runs were produced by an Alden Electronics 9315CTP photographic quality thermal printer (Westborough, Mass.). Alleles shared with the lineage I control strain were designated allele 1, and those common to the lineage II control strain were designated allele 2. Unique alleles (those migrating faster or slower than allele 1 or allele 2) were designated allele 3. If no band was apparent, a zero character state was given. Allele combinations were compiled in Microsoft Excel. Phylogenetic relationships were then assessed by using the unweighted pair group method with arithmetic mean (UPGMA) in PAUP version 4.0 (37) with a weight of 2 for the folD allele.
The nonrandom distribution of genotypes was tested by Z-test statistics and by the index of association (IA) test of Smith et al. (36). The Z test was calculated as
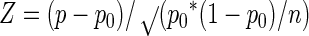 |
where p is the measured frequency of one genotype, p0 is the expected frequency of the genotype, assuming a random distribution of all alleles, and n is the number of strains tested. The IA value, which measures the observed variance in allele distributions versus that expected at randomness, was calculated in Microsoft Excel by using the method described by Smith et al. (36).
RESULTS
Marker identification.
In order to identify candidate polymorphisms that could discriminate the lineages, lineage-specific genome alterations were identified from a large-scale comparative genome analysis of 40 E. coli O157:H7 strains (20 lineage I strains and 20 lineage II strains) representing the genetic diversity of the two lineages (16, 17). The polymorphisms were identified by high-density octamer-based genome scanning (OBGS) analysis by using 174 different OBGS primer combinations on each of the strains in independent reactions. Polymorphic OBGS products that were specific to lineage I or lineage II strains were identified by electrophoresis of the labeled reaction products on automated DNA sequencers. A total of 95 lineage-specific OBGS products were purified, cloned, sequenced, and mapped onto the strain EDL933 (24) and strain Sakai (23) genome sequences as previously described (17). Each of the polymorphisms was confirmed by PCR analysis across the corresponding genome segment and DNA sequence analysis of the resulting cloned PCR product from each lineage. Details of this analysis are to be published elsewhere.
Candidate polymorphisms for development of the lineage-specific genotyping test met the following criteria: (i) the polymorphisms are conserved or nearly conserved in members of a lineage, (ii) the polymorphisms are derived from short insertion or deletion events (1 to 100 nucleotides), (iii) the polymorphisms occur in noncoding regions or within apparently nonessential genes or genes that would not be expected to confer selectable phenotypes in intestinal environments, and (iv) the polymorphisms are not within prophage, insertion sequences, or plasmid sequences. As shown in Table 2, six polymorphisms meeting these criteria were subsequently chosen for development of the assay. The first marker corresponds to a 9-base insertion in the intergenic region spanning the folD-sfmA genes and has been described previously (17). The insertion is found in all lineage II strains examined to date and serves as a primary marker for lineage determination. The other five markers are derived from alleles that are nearly conserved in lineage II strains and serve to further subdivide lineage II into several subpopulations. Relative to the EDL933 genome sequence (a lineage I strain), these markers include a 9-base insertion in the Z5935 coding region, a 78-base insertion in the yhcG gene, a 9-base deletion in the rbsB gene, a 9-base insertion in the rtcB gene, and an 18-base insertion in the intergenic region spanning the arp-iclR genes.
TABLE 2.
Polymorphisms used in the LSPAa
| Lineage | folD-sfmA | Z5935 | yhcG | rbsB | rtcB | arp-iclR |
|---|---|---|---|---|---|---|
| I | WT | WT | WT | WT | WT | WT |
| II | 9-base insertion (−129 of folD) | 9-base insertion (+1546) | 78-base insertion (+339) | 9-base deletion (+661-669) | 9-base insertion (+492) | 18-base insertion (−200 of arp) |
Wild-type (WT) allele corresponds to allele present in the EDL933 genome sequence (24). Residue numbers of insertions and deletions are given in parentheses.
Development of an LSPA.
As shown in Table 3, primer combinations were designed such that the amplicons from each of the alleles from each lineage would be of unique but defined length to allow multiplex amplification and subsequent resolution of amplicons from each of the corresponding loci regardless of the allele that is present. This assay is referred to as LSPA-6. When tested on DNA extracted from representative E. coli O157:H7 strains (Fig. 1), the six markers are efficiently amplified under the PCR conditions used and the amplicons are well resolved from one another, allowing unambiguous scoring of allele number and assignment of genotype. As expected, all lineage I strains produced products that were identical in length to the alleles in the K-12 control strain at the folD-sfmA, rbsB, rtcB, and arp-iclR1 loci. Only at the yhcG locus did strains in lineage II and in K-12 share the same allele. At the Z5935 locus, which is absent in K-12, no product was observed from the K-12 control strain. Based on these observations, we therefore arbitrarily assigned the allele at this locus from the lineage I strain 93-001 as allele 1 and those from the lineage 2 strain FRIK 2000 as allele 2 to serve as controls. Additional alleles are designated allele 3, and so on as new alleles are identified.
TABLE 3.
Allele sizes of lineage-specific polymorphisms in the LSPA-6
| Allele | Size (bp)
|
|||
|---|---|---|---|---|
| O157:H7 lineage Ia | O157:H7 lineage IIa | K-12b MG1655 | CFT073 UPECb | |
| folD-sfmA1 | 161 | 161 | Absent | |
| folD-sfmA2 | 170 | |||
| Z5935-1 | 133 | Absent | Absent | |
| Z5935-2 | 142 | |||
| yhcG1 | 394 | Absent | ||
| yhcG2 | 472 | 472 | ||
| rbsB1 | 218 | 218 | 218 | |
| rbsB2 | 209 | |||
| rbsB3 | 214c | |||
| rtcB1 | 270 | 270 | 270 | |
| rtcB2 | 279 | |||
| arp-iclR1 | 315 | 315 | Absent | |
| arp-iclR2 | 333 | |||
| arp-iclR3 | 324c | |||
FIG. 1.

Electrophoretic separation of LSPA-6 reaction products. E. coli O157:H7 strains from OBGS lineage I and lineage II and E. coli K-12 were subjected to LSPA-6. The reaction products were resolved on a LI-COR 4200 global analysis system by using a 25-cm gel. The positions of the products from the folD-sfmA, Z5395, yhcG, rtcB, rbsB, and iclR-arp loci are indicated on the left side of the image. Molecular size markers are loaded in the first, middle, and last lanes; sizes are indicated on the right-hand side of the image, and strain designations are indicated at the bottom of the image. H, strains derived from human clinical samples; C, strains derived from cattle.
Validation of the LSPA-6.
To validate the phylogeny inferred from LSPA-6 versus OBGS analysis, a total of 167 E. coli O157 isolates were tested by both LSPA-6 and OBGS. The strain sets included the USA 40 set, Australian set, Francis set, and the Moxley 60 set. The assignment of lineage from the OBGS data was performed by cluster analysis by the neighbor-joining method as previously described (16). Control strains, which had previously been assigned to a lineage by OBGS analysis, were included in the strain set to facilitate assignment from the OBGS data. The assignment of lineages from the LSPA-6 data was performed by using cluster analysis. For the analysis, the K-12 genotype of 110112 (folD-sfmA, rbsB, Z5935, rtcB, arp-iclR1, and yhcG) was used as an outgroup, and the data were clustered by using the UPGMA algorithm. Because the folD1 and folD2 alleles are conserved in every lineage I and lineage II strain, respectively, tested to date and because the other loci are only partially conserved in lineage II, the folD locus is weighted 2 relative to the other markers in the distance calculation to facilitate clustering. Lineage assignment predicted from OBGS analysis was then superimposed onto the phenogram. As shown in Fig. 2, the phenogram resulting from UPGMA analysis of the LSPA-6 data revealed two main clusters corresponding to the two OBGS lineages. All strains typing lineage I by OBGS comprised a single cluster of LSPA-6 genotype 111111, while all of the other LSPA-6 genotypes comprised a second cluster consistent with assignment to lineage II by OBGS, indicating that the two methods provide highly concordant data. Given the simplicity of the LSPA-6 and the excellent degree of correlation with OBGS results, we propose that the LSPA-6 can be implemented broadly as a simple tool for monitoring transmission patterns of O157:H7 subpopulations.
FIG. 2.

Comparison of LSPA-6- and OBGS-derived phylogenies. The phenogram was produced by UPGMA analysis of LSPA-6 data from 168 strains by using PAUP version 4.0 (tree length with six markers, 17 steps; consistency index, 0.5882; homoplasy index, 0.4118; retention index, 0.9711). The tree was rooted with E. coli MC1061 (a K-12 derivative [5]). Branches containing the predominant genotypes are shaded in different colors, and the allele combination is indicated to the right of the cluster in the corresponding cluster (allele order is folD, Z5935, yhcG, rtcB, rbsB, and arp-iclR). Strain MC1061 has the genotype 102111 relative to the six markers since it lacks the Z5935 gene. Strain sets include the USA 40 set, Australian set, Francis set, and the Moxley 60 set (strain information and data set are available from http://foodsci.unl.edu/homepage/faculty/strain%20sets.xls).
Linkage disequilibrium.
Given that the LSPA-6 markers were not arbitrarily chosen, we next evaluated linkage disequilibrium among the markers to formally test whether they may have undergone independent assortment during divergence of the different subpopulations. Assuming the simplest case of two possible alleles at each locus (binary character state at each locus), 64 different allele combinations are possible in the LSPA-6 analysis. Among the LSPA-6 genotypes derived from the 167-strain validation study, the genotypes could be divided into 20 of the 64 different possible genotypes. Four of the 20 genotypes that were observed were predominant (111111, 211111, 212111, and 222222), accounting for 93% of the samples, while 5 genotypes accounted for 92% of the strains from the larger collection of 1,429 human and bovine isolates described below. Z-test statistics indicate that the observed distribution of these genotypes deviates significantly from that expected if the alleles were distributed randomly (P < 0.05). As shown in Table 4, calculation of the IA value of Smith et al. (36), which compares the observed variance in allele combinations in a population with the variance that is predicted if randomized through recombination, shows that the IA ratio from the entire data set or the IA ratios from different subsets are all significant, indicating that the loci display strong linkage disequilibrium. Thus, similar to IA calculations on OBGS data (16), the LSPA-6 data also demonstrate significant linkage of the markers, suggesting that although genome diversity is significant, the multilocus linkages remain intact.
TABLE 4.
Measures of association between loci in E. coli O157
| Strain set | No. of strains | No. of loci | Mean genetic distance between strainsa | Expected value of the variance of distance between two strainsb | Observed value of the variance of distance between two strainsc | IAd |
|---|---|---|---|---|---|---|
| USA 40, Moxley 60, CDC, Francis, Australia, Downer | 215 | 6 | 2.22 | 1.36 | 4.23 | 2.10 |
| Moxley summer 00 | 360 | 6 | 0.81 | 0.58 | 1.39 | 1.41 |
| Moxley summer 01 + winter 01 | 714 | 6 | 1.31 | 0.88 | 2.20 | 1.49 |
| NPHL human | 140 | 6 | 0.88 | 0.61 | 1.26 | 1.05 |
| All strains | 1429 | 6 | 1.33 | 0.92 | 2.58 | 1.78 |
The mean number of loci at which the strains differ.
The variance of distance between the two strains assuming no independent assortment of the markers (e.g., no linkage disequilibrium).
Calculated variance of distance between two strains from the data.
Ratio of the observed variance to the variance expected if loci are independently assorting.
Identification of common genotypes in bovine- and human-derived O157:H7 strains.
Comparison of the LSPA-6 genotypes among the strains in the validation study showed that four genotypes accounted for most of the strains, with the most frequent genotype being 111111 (Fig. 2). Because this group of strains is quite diverse in temporal and spatial origin, this finding suggests that these genotypes could be the most common in bovine production environments and in human clinical samples. To test this hypothesis, a larger set of human- and bovine-derived isolates was tested. The strain sets were derived from several different studies and geographies. The human isolates comprised previously examined strains from the United States and Australia (16, 17) as well as strains from sporadic cases and outbreaks collected by the CDC, the NPHL, and the University of Wisconsin (10). The bovine-derived isolates originated from previous studies of dairy herds (8, 10, 17, 18, 32), as well as longitudinal studies of production feedlots (15). Collectively, the strains comprise a set of 1,429 isolates, each of which was tested by LSPA-6. As shown in Table 5, nearly 92% of the strains comprised only five different genotypes, including the four that were the most prevalent in the validation study.
TABLE 5.
E. coli O157 LSPA-6 genotypes observeda
| Genotype | No. of isolates |
|---|---|
| 111111 | 776 |
| 111121 | 1 |
| 111211 | 1 |
| 112111 | 44 |
| 113113 | 1 |
| 113131 | 1 |
| 113311 | 1 |
| 121111 | 1 |
| 122111 | 4 |
| 122211 | 8 |
| 122212 | 1 |
| 133311 | 1 |
| 133331 | 1 |
| 211111 | 341 |
| 211131 | 1 |
| 212111 | 108 |
| 212112 | 1 |
| 212212 | 1 |
| 212222 | 2 |
| 213111 | 3 |
| 221111 | 21 |
| 221212 | 4 |
| 221213 | 2 |
| 221222 | 4 |
| 222111 | 15 |
| 222211 | 1 |
| 222212 | 17 |
| 222213 | 6 |
| 222222 | 43 |
| 222223 | 3 |
| 222312 | 1 |
| 222322 | 2 |
| 223111 | 1 |
| 223213 | 3 |
| 231111 | 2 |
| 231233 | 1 |
| 232111 | 1 |
| 232221 | 1 |
| 232222 | 1 |
| 232233 | 1 |
| 232312 | 1 |
The character string in the genotype indicates the allele number at the folD, Z5935, yhcG, rtcB, rbsB, and arp-iclR loci. Total number of isolates, 1,429.
Because the sets of strains comprised human and bovine strains, we next compared the distribution of these five genotypes among human- and bovine-derived isolates. As shown in Fig. 3, when the genotypes were categorized into six groups, three of the five most common genotypes represented nearly identical proportions of the isolates. Only genotypes 111111 and 22222 showed any statistically significant difference in frequency. Chi-square analysis of the distribution of the six groups shows a significant difference between the bovine and human isolates, primarily due to biased distribution of the 111111 and 22222 genotypes. However, because this strain collection includes large numbers of bovine strains derived from longitudinal studies—and therefore collected from repeated sampling of the same pens and the same cattle over time—the importance of the chi-square statistic is uncertain. Nonetheless, the approach demonstrated here allows us to begin developing the sampling design and statistical approaches for testing the hypothesis of a nonhomogenous distribution of the genotypes between human- and bovine-derived strains.
FIG. 3.

Comparison of LSPA-6 genotype frequencies among bovine-derived and human clinical EHEC O157:H7 and O157:H− strains. The LSPA-6 genotypes from human clinical and bovine strains were grouped into six different categories according to the five most common LSPA-6 genotypes (111111, 211111, 212111, 222222, and 112111, with the allele order folD, Z5935, yhcG, rtcB, rbsB, and arp-iclR) and a sixth category that includes all other genotypes. The percentage of strains from each genotypic category was calculated from strains of bovine or human origin. The genotypes corresponding to the different color bars are indicated to the right of the graph. Strain information and data sets are available from http://foodsci.unl.edu/homepage/faculty/benson.htm.
DISCUSSION
At least three different studies have provided some genotypic or phenotypic evidence to support the hypothesis that different O157:H7 populations display distinct virulence or transmissibility characteristics (1, 16, 20). Testing this hypothesis is difficult due to the absence of a good animal model and the effort that is necessary to discriminate the O157:H7 populations. Although OBGS and amplified fragment length polymorphism analysis provide excellent discriminatory power (12, 14, 34, 42), they suffer from low throughput and difficulty with gel-to-gel comparisons. PFGE, the standard for epidemiological analyses, suffers from the fact that meaningful phylogenetic relationships are difficult to predict from the data. This problem arises because relatively small events can cause substantial changes in the pattern, and these events can occur rapidly on the evolutionary time scale. Another high-throughput method, multilocus sequence typing, recently was shown to be unable to readily distinguish O157:H7 strains from one another (22). Thus, a method is needed to provide accurate, reliable, standardizable, and high-throughput discrimination of the populations identified by OBGS.
Our studies presented here demonstrate that the LSPA-6 provides a simple and reliable multilocus assay with very high throughput. Our results show strong correlation between the phylogenetic assignments inferred from either LSPA-6 or OBGS data. Since the LSPA-6 is based on allele sizes relative to a set of lineage I and lineage II control strains, the assay can be standardized easily, even in laboratories that use different types of automated sequencers. Moreover, the data can be easily reported, compiled, and analyzed by several independent laboratories or teams. This will allow several independent laboratories to compare data and to participate in large-scale collaborative or independent studies to examine the distribution of the LSPA genotypes in different environments.
Stability of LSPA-6 markers.
PFGE analyses have consistently shown that substantial diversity can be detected in the genome of O157:H7 strains, even when they are derived from limited geographic regions (8, 32). Genome sequence analysis (13, 16, 23, 24), genotyping studies (16, 17, 27), and studies of phages isolated from O157:H7 strains (28, 39) all indicate that diversity in phage content, and perhaps in phage-mediated genome events, contributes substantially to the diversity that can be observed. Genome sequences also show that a substantial number of insertion elements and transposons exist in the genome as well. Based on these studies, it seems reasonable that the vast majority of the instances of diversity that are observed among O157:H7 strains can be accounted for by integration, excision, and recombination among different prophage or cryptic prophage within the genome and by movement of insertion elements and transposons. Although this characteristic is desirable in terms of trace-back studies, such diversity can obscure true phylogenetic relationships and impede the interpretation of broad geographic transmission patterns or ecological characteristics of populations.
In order to provide stable markers for the LSPA-6, the target polymorphisms were chosen on the basis of their potential for stability and their conservation within a lineage. Accordingly, the majority of the strains we have tested carry one of the two primary alleles (allele 1 or allele 2) at each of the loci. We have only observed rare instances (less than 0.1% of strains tested) where no signal is generated from a given strain at any one of these loci. Moreover, serial passage of strains (up to 10 times) in the laboratory did not lead to loss or detectable change in any of the LSPA-6 markers. Thus, the markers appear to be stably maintained in the populations and are, therefore, reliable population markers.
Origin of the LSPA-6 polymorphisms.
Although the nature of the LSPA-6 polymorphisms themselves is not informative about the evolutionary pathways of the different subpopulations, comparison of the alleles at the LSPA-6 loci to other E. coli strains does provide additional insight into the relative evolutionary relationship of lineage I and lineage II. Comparison of the LSPA-6 alleles from lineage I and II O157:H7 strains to the K-12 strain MG1655 (2) and the uropathogenic E. coli strain CFT073 (40) genome sequences (Table 3) showed that most alleles from lineage I are likely to be ancestral. Relative to the K-12 strain MG1655, which is believed to have last shared a common ancestor with the O157:H7 lineage 4 to 5 million years ago (25), the alleles found in four of the loci of lineage I O157:H7 strains are conserved in the K-12 genome. The exceptions are the Z5395 gene, which is unique to O157:H7, and the yhcG locus, where both K-12 and the O157:H7 OBGS lineage II strains carry the same 78-base insertion at nucleotide 339 of the yhcG coding region relative to the lineage I EDL933 O157:H7 genome sequence. The fact that OBGS lineage I O157:H7 strains share alleles at four of the five loci with K-12 implies that the alleles in lineage II strains are derived states. Relative to the CFT073 uropathogenic E. coli genome, only two of the six marker genes are present; however, the alleles in these two genes (rtcB and rbsB) are also conserved among lineage I O157:H7 strains and the K-12 strain. Collectively, these findings are consistent with our previous hypothesis that lineage I is ancestral and lineage II comprises derived populations (17).
As shown in Table 5, the most common LSPA-6 genotype in our strain set is 111111, which carries a lineage I allele at all loci. If lineage I is, indeed, the ancestral state, then one explanation for its predominance in the strain collections examined could be due to the founder effect, whereby a newly evolved population spreads rapidly in a new niche prior to substantial genetic differentiation. Moreover, its frequency in human-derived samples further implies that the ancestral state was virulent and that the underrepresented genotypes among human-derived strains would be indicative of a loss of virulence characteristics. Clearly, these speculations must be tempered because the strain sets in our studies were from temporally and geographically limited regional collections, and in some cases the isolates were from studies designed with repeated observations of animal and place. Nonetheless, broad application of the LSPA-6 genotyping method will now provide a convenient means for testing hypotheses about distribution of the genotypes among various types of samples and different types of environments.
Acknowledgments
This research was funded by U.S. Department of Agriculture National Research Initiative Competitive Grants Program grant 2001-35201-10115 to A.K.B., by funding provided by Nebraska's legislative bill LB1206, and by funding from the American Meat Institute.
Footnotes
Journal series paper 14771 of the Nebraska Agricultural Research Experimental Station.
REFERENCES
- 1.Baker, D. R., R. A. Moxley, and D. H. Francis. 1997. Variation in virulence in the gnotobiotic pig model of O157:H7 Escherichia coli strains of bovine and human origin. Adv. Exp. Med. Biol. 412:53-58. [DOI] [PubMed] [Google Scholar]
- 2.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 3.Byrne, C. M., I. Erol, J. E. Call, C. W. Kaspar, D. R. Buege, C. J. Hiemke, P. J. Fedorka-Cray, A. K. Benson, F. M. Wallace, and J. B. Luchansky. 2003. Characterization of Escherichia coli O157:H7 from downer and healthy dairy cattle in the upper Midwest region of the United States. Appl. Environ. Microbiol. 69:4683-4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caprioli, A., and A. E. Tozzi. 1998. STEC infections in continental Europe. ASM Press, Washington, D.C.
- 5.Casadaban, M. J., and S. N. Cohen. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179-207. [DOI] [PubMed] [Google Scholar]
- 6.Donnenberg, M. S., S. Tzipori, M. L. McKee, A. D. O'Brien, J. Alroy, and J. B. Kaper. 1993. The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J. Clin. Investig. 92:1418-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dytoc, M., R. Soni, F. Cockerill III, J. De Azavedo, M. Louie, J. Brunton, and P. Sherman. 1993. Multiple determinants of verotoxin-producing Escherichia coli O157:H7 attachment-effacement. Infect. Immun. 61:3382-3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faith, N. G., J. A. Shere, R. Brosch, K. W. Arnold, S. E. Ansay, M. S. Lee, J. B. Luchansky, and C. W. Kaspar. 1996. Prevalence and clonal nature of Escherichia coli O157:H7 on dairy farms in Wisconsin. Appl. Environ. Microbiol. 62:1519-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng, P., K. A. Lampel, H. Karch, and T. S. Whittam. 1998. Genotypic and phenotypic changes in the emergence of Escherichia coli O157:H7. J. Infect. Dis. 177:1750-1753. [DOI] [PubMed] [Google Scholar]
- 10.Gouveia, S., M. E. Proctor, M. S. Lee, J. B. Luchansky, and C. W. Kaspar. 1998. Genomic comparisons and Shiga toxin production among Escherichia coli O157:H7 isolates from a day care center outbreak and sporadic cases in southeastern Wisconsin. J. Clin. Microbiol. 36:727-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffin, P. M., and R. V. Tauxe. 1991. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol. Rev. 13:60-98. [DOI] [PubMed] [Google Scholar]
- 12.Hahm, B. K., Y. Maldonado, E. Schreiber, A. K. Bhunia, and C. H. Nakatsu. 2003. Subtyping of foodborne and environmental isolates of Escherichia coli by multiplex-PCR, rep-PCR, PFGE, ribotyping and AFLP. J. Microbiol. Methods 53:387-399. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi, T., K. Makino, M. Ohnishi, K. Kurokawa, K. Ishii, K. Yokoyama, C. G. Han, E. Ohtsubo, K. Nakayama, T. Murata, M. Tanaka, T. Tobe, T. Iida, H. Takami, T. Honda, C. Sasakawa, N. Ogasawara, T. Yasunaga, S. Kuhara, T. Shiba, M. Hattori, and H. Shinagawa. 2001. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8:11-22. [DOI] [PubMed] [Google Scholar]
- 14.Iyoda, S., A. Wada, J. Weller, S. J. Flood, E. Schreiber, B. Tucker, and H. Watanabe. 1999. Evaluation of AFLP, a high-resolution DNA fingerprinting method, as a tool for molecular subtyping of enterohemorrhagic Escherichia coli O157:H7 isolates. Microbiol. Immunol. 43:803-806. [DOI] [PubMed] [Google Scholar]
- 15.Khaitsa, M. L., D. R. Smith, J. A. Stoner, A. M. Parkhurst, S. Hinkley, T. J. Klopfenstein, and R. A. Moxley. 2003. Incidence, duration, and prevalence of Escherichia coli O157:H7 fecal shedding by feedlot cattle during the finishing period. J. Food Prot. 66:1972-1977. [DOI] [PubMed] [Google Scholar]
- 16.Kim, J., J. Nietfeldt, and A. K. Benson. 1999. Octamer-based genome scanning distinguishes a unique subpopulation of Escherichia coli O157:H7 strains in cattle. Proc. Natl. Acad. Sci. USA 96:13288-13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim, J., J. Nietfeldt, J. Ju, J. Wise, N. Fegan, P. Desmarchelier, and A. K. Benson. 2001. Ancestral divergence, genome diversification, and phylogeographic variation in subpopulations of sorbitol-negative, β-glucuronidase-negative enterohemorrhagic Escherichia coli O157. J. Bacteriol. 183:6885-6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, M. S., C. W. Kaspar, R. Brosch, J. Shere, and J. B. Luchansky. 1996. Genomic analysis using pulsed-field gel electrophoresis of Escherichia coli O157: H7 isolated from dairy calves during the United States National Dairy Heifer Evaluation Project (1992-1992). Vet. Microbiol. 48:223-230. [DOI] [PubMed] [Google Scholar]
- 19.McDaniel, T. K., K. G. Jarvis, M. S. Donnenberg, and J. B. Kaper. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 92:1664-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNally, A., A. J. Roe, S. Simpson, F. M. Thomson-Carter, D. E. Hoey, C. Currie, T. Chakraborty, D. G. Smith, and D. L. Gally. 2001. Differences in levels of secreted locus of enterocyte effacement proteins between human disease-associated and bovine Escherichia coli O157. Infect. Immun. 69:5107-5114. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Newland, J. W., N. A. Stockbrine, F. F. Miller, A. D. O'Brien, and R. K. Holmes. 1985. Cloning of shiga-like toxin genes from a toxin converting phage of Escherichia coli. Science 230:179-181. [DOI] [PubMed] [Google Scholar]
- 22.Noller, A. C., M. C. McEllistrem, O. C. Stine, J. G. Morris, Jr., D. J. Boxrud, B. Dixon, and L. H. Harrison. 2003. Multilocus sequence typing reveals a lack of diversity among Escherichia coli O157:H7 isolates that are distinct by pulsed-field gel electrophoresis. J. Clin. Microbiol. 41:675-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohnishi, M., T. Hayashi, and K. Kurokawa. 2001. [Determination of the whole genome sequence of enterohemorrhagic Escherichia coli O157:H7]. Tanpakushitsu Kakusan Koso. 46:1862-1867. [PubMed] [Google Scholar]
- 24.Perna, N. T., G. Plunkett III, V. Burland, B. Mau, J. D. Glasner, D. J. Rose, G. F. Mayhew, P. S. Evans, J. Gregor, H. A. Kirkpatrick, G. Posfai, J. Hackett, S. Klink, A. Boutin, Y. Shao, L. Miller, E. J. Grotbeck, N. W. Davis, A. Lim, E. T. Dimalanta, K. D. Potamousis, J. Apodaca, T. S. Anantharaman, J. Lin, G. Yen, D. C. Schwartz, R. A. Welch, and F. R. Blattner. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529-533. [DOI] [PubMed] [Google Scholar]
- 25.Reid, S. D., C. J. Herbelin, A. C. Bumbaugh, R. K. Selander, and T. S. Whittam. 2000. Parallel evolution of virulence in pathogenic Escherichia coli. Nature 406:64-67. [DOI] [PubMed] [Google Scholar]
- 26.Robins-Browne, R., E. Elliott, and P. Desmarchelier. 1998. Shiga toxin-producing Escherichia coli in Australia. ASM Press, Washington, D.C.
- 27.Samadpour, M., L. M. Grimm, B. Desai, D. Alfi, J. E. Ongerth, and P. I. Tarr. 1993. Molecular epidemiology of Escherichia coli O157:H7 strains by bacteriophage lambda restriction fragment length polymorphism analysis: application to a multistate foodborne outbreak and a day-care center cluster. J. Clin. Microbiol. 31:3179-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato, T., T. Shimizu, M. Watarai, M. Kobayashi, S. Kano, T. Hamabata, Y. Takeda, and S. Yamasaki. 2003. Distinctiveness of the genomic sequence of Shiga toxin 2-converting phage isolated from Escherichia coli O157:H7 Okayama strain as compared to other Shiga toxin 2-converting phages. Gene 309:35-48. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt, H., B. Henkel, and H. Karch. 1997. A gene cluster closely related to type II secretion pathway operons of gram-negative bacteria is located on the large plasmid of enterohemorrhagic Escherichia coli O157 strains. FEMS Microbiol. Lett. 148:265-272. [DOI] [PubMed] [Google Scholar]
- 30.Schmitt, C. K., K. C. Meysick, and A. D. O'Brien. 1999. Bacterial toxins: friends or foes? Emerg. Infect. Dis. 5:224-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaikh, N., and P. I. Tarr. 2003. Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 185:3596-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shere, J. A., K. J. Bartlett, and C. W. Kaspar. 1998. Longitudinal study of Escherichia coli O157:H7 dissemination on four dairy farms in Wisconsin. Appl. Environ. Microbiol. 64:1390-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith, D., M. Blackford, S. Younts, R. Moxley, J. Gray, L. Hungerford, T. Milton, and T. Klopfenstein. 2001. Ecological relationships between the prevalence of cattle shedding Escherichia coli O157:H7 and characteristics of the cattle or conditions of the feedlot pen. J. Food Prot. 64:1899-1903. [DOI] [PubMed] [Google Scholar]
- 34.Smith, D., G. Willshaw, J. Stanley, and C. Arnold. 2000. Genotyping of verocytotoxin-producing Escherichia coli O157: comparison of isolates of a prevalent phage type by fluorescent amplified-fragment length polymorphism and pulsed-field gel electrophoresis analyses. J. Clin. Microbiol. 38:4616-4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith, D. R., J. T. Gray, R. A. Moxley, S. M. Younts-Dahl, M. P. Blackford, S. Hinkley, L. L. Hungerford, C. T. Milton, and T. J. Klopfenstein. 2004. A diagnostic strategy to determine the Shiga toxin-producing Escherichia coli O157 status of pens of feedlot cattle. Epidemiol. Infect. 132:297-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith, J. M., N. H. Smith, M. O'Rourke, and B. G. Spratt. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. USA 90:4384-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swofford, D. 2002. PAUP v. 4.0 beta 10: phylogenetic analysis using parsimony. Sinauer and Associates, Sunderland, Mass.
- 38.Tarr, P. I., L. M. Schoening, Y. L. Yea, T. R. Ward, S. Jelacic, and T. S. Whittam. 2000. Acquisition of the rfb-gnd cluster in evolution of Escherichia coli O55 and O157. J. Bacteriol. 182:6183-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unkmeir, A., and H. Schmidt. 2000. Structural analysis of phage-borne stx genes and their flanking sequences in Shiga toxin-producing Escherichia coli and Shigella dysenteriae type 1 strains. Infect. Immun. 68:4856-4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welch, R. A., V. Burland, G. Plunkett III, P. Redford, P. Roesch, D. Rasko, E. L. Buckles, S. R. Liou, A. Boutin, J. Hackett, D. Stroud, G. F. Mayhew, D. J. Rose, S. Zhou, D. C. Schwartz, N. T. Perna, H. L. Mobley, M. S. Donnenberg, and F. R. Blattner. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 99:17020-17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whittam, T. S., M. L. Wolfe, I. K. Wachsmuth, F. Orskov, I. Orskov, and R. A. Wilson. 1993. Clonal relationships among Escherichia coli strains that cause hemorrhagic colitis and infantile diarrhea. Infect. Immun. 61:1619-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao, S., S. E. Mitchell, J. Meng, S. Kresovich, M. P. Doyle, R. E. Dean, A. M. Casa, and J. W. Weller. 2000. Genomic typing of Escherichia coli O157:H7 by semi-automated fluorescent AFLP analysis. Microbes Infect. 2:107-113. [DOI] [PubMed] [Google Scholar]