

Abstract
Quantifying the multiple processes which control and modulate the extent of oral bioavailability for drug candidates is critical to accurate projection of human pharmacokinetics (PK). Understanding how gut wall metabolism and hepatic elimination factor into first-pass clearance of drugs has improved enormously. Typically, the cytochrome P450s, uridine 5′-diphosphate-glucuronosyltransferases and sulfotransferases, are the main enzyme classes responsible for drug metabolism. Knowledge of the isoforms functionally expressed within organs of first-pass clearance, their anatomical topology (e.g. zonal distribution), protein homology and relative abundances and how these differ across species is important for building models of human metabolic extraction. The focus of this manuscript is to explore the parameters influencing bioavailability and to consider how well these are predicted in human from animal models or from in vitro to in vivo extrapolation. A unique retrospective analysis of three AstraZeneca molecules progressed to first in human PK studies is used to highlight the impact that species differences in gut wall metabolism can have on predicted human PK. Compared to the liver, pharmaceutical research has further to go in terms of adopting a common approach for characterisation and quantitative prediction of intestinal metabolism. A broad strategy is needed to integrate assessment of intestinal metabolism in the context of typical DMPK activities ongoing within drug discovery programmes up until candidate drug nomination.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-016-9889-y) contains supplementary material, which is available to authorized users.
Keywords: animal models, drug-metabolising enzymes, first-pass oral clearance, gut wall metabolism, oral bioavailability
INTRODUCTION
Drug discovery and development is a costly and often time-consuming activity. It is widely accepted that prescription of orally formulated drugs is the preferred method of administration, both in terms of maximising patient compliance and convenience of dosing (1). Consequently, most small-molecule drug programs pursued by pharmaceutical companies aspire to develop candidate drugs (CDs) for oral administration in humans. Key to their success is the design and optimisation of novel compounds with acceptable oral pharmacokinetic (PK) properties. This is to facilitate target engagement within the relevant tissue, for the requisite duration, that elicits the desired pharmacodynamic (PD) effect and in vivo efficacy. Poor oral bioavailability (F oral) has been established as a major reason for the failure of drug candidates in pre-clinical and clinical development (2). A lead compound should therefore have adequate F oral to achieve the necessary drug plasma concentration time profile efficiently from the standpoint of a commercially viable dose size and regimen. It also needs to be predictable, given that low F oral is associated with greater interpatient variability which predisposes the patient to a higher risk of exposure to undesirable toxic or sub-therapeutic drug plasma concentrations (3).
The absolute F oral of a drug is defined as the rate and extent to which it becomes available to the systemic circulation and is a function of absorption and first-pass elimination. This is expressed mathematically in Eq. 1 (4).
| 1 |
The fraction of dose entering the cellular space of the enterocytes from the intestinal lumen is given as F a. The fraction of the drug entering the enterocytes that escapes first-pass metabolism is given as F G. The fraction of the drug that escapes first-pass hepatic metabolism and biliary secretion is given as F H.
Note that the lung, heart and blood are also tissues where first-pass metabolism can occur but these are generally viewed as less important in oral drug exposure. Assuming that clearance (CL) remains the same, their contributions cancel out if the oral plasma exposure is compared to the plasma exposure following intravenous administration. This is a reasonable assumption if systemic drug exposure from intravenous (IV) and oral administration remain close to each other (Eq. 2, (4)).
| 2 |
Several approaches for quantitative prediction of human oral PK profiles and F oral have been developed with mixed success. Some utilise physiologically based pharmacokinetic (PBPK) models linked with in vitro to in vivo extrapolation (IVIVE) of kinetic parameters. These have typically been determined from in vitro experiments and animal PK data (5–7) although allometry has also been used (8–10). Recently, a PhRMA initiative evaluated how accurately a range of models, including allometry, predicted the plasma concentration time profiles in humans for a diverse set of blinded clinical lead compounds (n = 108). These had been collected across several member companies (11). It is not within the scope of this review to detail observations and conclusions drawn within this series of manuscripts or indeed its prediction success in relation to other reported industry approaches (7,12,13). Nevertheless, it is worth highlighting that a high percentage of simulated IV profiles could be categorised as achieving a medium (44%), or medium to high (25%), degree of accuracy when compared to observed plasma PK profiles for a common set of compounds. However, simulated oral PK profiles were less accurate with only 20% achieving a moderate categorisation. The authors noted that the phenomenon appeared to be more commonly associated with compounds receiving a biopharmaceutical classification system (BCS) II categorization (high permeability, low solubility according to criteria outlined in (14)) and may have been due to an underestimation of the total fraction absorbed. This may have resulted from transporter mechanisms, intestinal metabolism, particle size effects from the oral formulation or inaccurate estimation of intrinsic solubility/dissolution rate. It is assumed that absence of relevant input data prevented modelling of the non-solubility-related parameters.
In an earlier publication, prediction of human F oral had been reasonably successful, in spite of an assumption that only F H limited F oral (8). However, the criterion used in this evaluation was less precise. Successful prediction was defined only in terms of being able to correctly categorise F oral for the purposes of drug development decision making (e.g. ability to differentiate compounds according to criteria of <10%, 10 to <30% or >30% F oral) rather than making quantitative predictions or accurately simulating oral PK profiles. Whether F oral can be adequately predicted at all from pre-clinical in vivo models has been questioned (15–17). Taken at face value, the published correlation is weak between absolute F oral of various drugs in rodents, dogs and primates versus that reported in humans. A reanalysis of the data used in many of these studies was recently undertaken (18). Musther et al. employed more stringent inclusion and exclusion criteria to improve the integrity of the dataset. In so doing, they highlighted important limitations impacting the quality of previous data analyses. In keeping with previous findings, there was a lack of agreement between human and animal F oral for all species. This was quantified as the concordance correlation coefficient and was 0.444, 0.470, 0.605 and 0.698 for mouse, rat, dog and monkey, respectively. The correlation (slope of the regression line) between animal and human F oral was also low, e.g. 0.25, 0.29, 0.37 and 0.69 for mouse, rat, dog and monkey, respectively (18).
However, as exemplified in Eq. 1, F oral is a multi-parametric endpoint. Perhaps a more telling assessment would be to examine how well each independent parameter can be measured across species to predict the corresponding value in humans. Does this spotlight parameters that are more or less well understood and predictable in humans? Clearly, any species differences in absorption, distribution, metabolism and excretion (ADME) can greatly affect the correlation of F oral. In subsequent sections, an examination will be made of how successfully F a, F H and F G can be predicted from pre-clinical models and in vitro data.
Until relatively recently, the liver has been perceived as the major site of first-pass clearance. This is principally because of its size and capacity for drug metabolism and elimination (19). It is frequently cited that CYP3A4, a major contributor to the drug-metabolising capacity of the small intestine (ca. 80% of the total cytochrome CYP450 (CYP450) content according to Paine et al., (20)), is only expressed at relatively low levels compared to the liver (ca. 1% (21)). However, the intestine is positioned anterior to the liver, in a serial relationship. As such, it is the first organ exposed to drug following oral dosing. Therefore, high concentration of drug in the enterocytes during the absorption phase may lead to substantial metabolic extraction before the drug enters the liver. Indeed, a growing body of evidence demonstrates that the gastrointestinal (GI) tract not only contributes to low F oral, through restricting the fraction absorbed, but also by metabolism that can occur as a drug transits through the gut wall (22–24). It was noted from an analysis of 309 drugs with IV and oral clinical PK data that around 30% showed greater than 20% intestinal extraction (25). Predictive tools have been developed ranging in complexity from minimal models like the static Q gut model to more complex, integrative PBPK models such as the segmental segregated flow model. These have enabled simulation of the extent of first-pass gut wall metabolism furthering our understanding of the importance of the small intestine as an eliminating organ (22,23,26–29).
Projections of human PK properties and efficacious human dose, the maximal absorbable dose (MAD), the potential to cause adverse drug-drug interactions (DDI) and the drug therapeutic margin are scientific cornerstones supporting project investment decisions to either stop or continue development of CDs for first in human (FIH) clinical trials. It is no surprise then, given the prohibitive cost of bringing a drug to market, that the accuracy and certainty of these predictions face considerable scrutiny. The purpose of this article is to discuss the importance of understanding and accounting for species differences in intestinal metabolism when making projections of human F oral and dose for CDs based on in vitro and pre-clinical PK data typically available during the drug discovery/early drug development phases. A retrospective analysis of three AstraZeneca case studies are used to highlight the impact of species differences in gut wall extraction on the accurate projection of human PK, as determined from FIH clinical PK studies. Particular emphasis is given to detailing current understanding of the CYP450, sulfatransferases (SULTs) and UDP-glucuronosyltransferases (UGTs) expressed within the gut wall and liver in humans and pre-clinical models in two companion papers. Consideration of the potential for DDIs falls outside the scope of this manuscript. However, the reader is directed to a number of excellent review articles detailing the models and considerations for risk assessment of potential clinical DDIs arising from the interplay between drug-metabolising enzymes (DMEs) and transporters during pre-systemic metabolism (30,31).
CAN HUMAN ORAL ABSORPTION BE ACCURATELY PREDICTED FROM PRE-CLINICAL MODELS AND/OR IN VITRO DATA?
According to scientific and regulatory definitions, F a is the fraction of the dose absorbed across the apical cell membrane into the cellular space of the enterocyte. There are a number of factors influencing this complex in vivo process. These can be categorised as being (i) specific to the drug molecule itself and thereby governed by its physico-chemical properties (e.g. pKa and degree of ionisation, solubility and dissolution rate from the solid form, intestinal permeability, substrate affinity for transporter proteins, chemical degradation or metabolism within the intestinal lumen and luminal complex binding), (ii) related to its pharmaceutical properties (e.g. choice of formulation excipients) and (iii) physiological, genetic or biochemical in nature (e.g. gastrointestinal pH, transporter protein abundance, membrane porosity, gastric emptying rate and intestinal motility which govern GI transit).
The fundamental principles associated with F a have been comprehensively reviewed elsewhere (4,28,32). Despite its considerable complexity, a number of qualitative as well as quantitative approaches have been successfully employed for estimation of human F a, either from animal models (33,34) or from IVIVE of data from in vitro systems such as Caco-2 monolayers or Ussing chamber preparations (14,35–37). Perhaps suited to late stage discovery compounds, due to the level of compound-specific information required, commercial software such as Simcyp® and GastroPlusTM are available to facilitate predictions of F a through integration of permeability and solubility data into mathematical models alongside appropriate physiological parameters (38–40). Quantitative structure-activity relationship (QSAR) models have been devised to guide compound design during the discovery phase, effectively targeting structure-property space (e.g. values for certain molecular descriptors and physico-chemical properties such as lipophilicity) associated with a higher likelihood of achieving good oral absorption (41).
Several mechanisms of oral drug absorption have been shown in small intestinal regions and include passive transcellular diffusion, paracellular transport and carrier-mediated active transport. Of these, passive diffusion is recognised as the main mechanism for absorption of most lipophilic compounds (16). Good correlations between permeability and F a in the same species have been demonstrated for drugs with no significant solubility or dissolution limitations (35). Building on this, a strong overall correlation (R 2=0.97) was reported between rat and human F a for 64 drugs with varying physico-chemical properties and absolute F oral (42). Further work showed that rats may serve as a good in vivo model for predicting dose-dependent (when dose was normalised to body weight) as well as dose-independent oral absorption properties in humans (16,33). Some may consider this surprising given that the rat small intestine has ca. fourfold lower surface area than humans, once normalised to body surface area (43). Whilst monkeys also appear to be a good predictor of human F a (R 2 = 0.974, n = 43 drugs), cost and ethical concerns limit their applicability within drug discovery (34). The dog on the other hand has frequently been regarded as an inferior in vivo model (R 2 = 0.51, n = 43) for prediction of human Fa (44). In these studies, the higher absorption reported for many drugs in dogs compared to humans could be explained in several ways. For example, weakly basic compounds with pH-dependent solubility would show more efficient absorption in dogs than humans due to the higher intestinal pH (ca. 1 unit) measured in fasted dogs (45). However, human data published more recently suggests that the intestinal pH values may be similar in both species (46). It is also possible given that many water-soluble, low molecular weight, neutral compounds show greater absorption in dogs, that the size and frequency of tight junction for paracellular transport may be greater in dogs than humans (47). The absorption of poorly water-soluble drugs may be enhanced in dogs due to a higher bile salt secretion rate which may have a solubilising effect on the drug residing within the intestine (44).
However, experience within AstraZeneca suggests for CDs absorbed via the transcellular route that prediction of human F a from pre-clinical in vivo data is more achievable using the dog (48). It is the authors’ view that sufficient understanding of a CDs permeability and solubility can often be gleaned from in vitro experimentation, when coupled with PK understanding from in vivo models, affording a good level of confidence in predictions of human F a (36,40,48). The safety of an orally intended drug must be evaluated in animals prior to dosing in humans. Animal models also provide a fuller representation of the complexities of the in vivo situation and, as detailed above, can be predictive of human F a. As such, pharmaceutical companies will continue to focus part of their prediction strategy on the ability of animal models to predict human F a (5).
IS HUMAN HEPATIC CLEARANCE AND FIRST-PASS EXTRACTION SUFFICIENTLY PREDICTABLE?
For most drugs, total systemic clearance in humans can often be described by a hepatic (metabolism and biliary elimination) and renal (active and passive) component (49). With most CDs, it is likely that hepatic metabolism will be the major route of elimination, as has been shown for oral marketed drugs (50). Accurate prediction of in vivo hepatic CL is still a key priority within drug discovery. It is a major determinant of a drugs’ oral exposure as well as half-life, which in turn help define the size of dose and dosing interval. Given that there is no reliable means to predict elimination pathways in humans from in silico or in vitro methods, a combination of establishing clearance routes in pre-clinical species, and use of human in vitro systems, is required to predict human CL (5,48). In practice, confidence in the ability to make projections of human CL from in vitro data is explored during lead optimisation. Individual compounds or compound series can be prioritised on the basis of demonstrating acceptable IVIVE of CL in pre-clinical models (51,52). Those compounds for which in vivo CL cannot be adequately described by simple, hepatic metabolic elimination would be poorly predicted and require further investigation. If the accuracy of the CL prediction did not improve after factoring in alternative routes identified through follow-up studies in rat or dog, the compound would carry greater uncertainty in terms of its human CL prediction and likely be de-prioritized (5). Thus, for compounds demonstrating acceptable IVIVE of CL in pre-clinical species, and that are allowed to progress, likelihood of success can be high in terms of the human hepatic clearance prediction (48).
Key to the success of this approach is the existence of robust, well-understood in vitro systems to investigate a compound’s metabolic pathways and kinetics in the liver, through application of well-characterized in vitro-in vivo physiological scaling factors and mathematical models (53–55). When isolated and handled correctly, hepatocytes provide an intact cellular system containing a full complement of DMEs, transporters and co-factors, making them well suited for studying rates of drug metabolism (56). There have been mixed successes with quantitative prediction of hepatic clearance from microsomal- and hepatocyte-based assays. Typically, extrapolation of hepatocyte-derived intrinsic metabolic clearances (CLint) commonly results in an underestimation of the in vivo value, despite incorporation of established physiological scaling factors and the unbound fractions in both blood and in vitro matrix (57). There are a number of plausible explanations for this observation such as the in vitro incubation conditions, which can greatly influence the rate of drug metabolism (54). However, refinement of these models and incorporation of empirical correction factors to account for the systematic under prediction can reliably enhance predictions of human CL (51,52,58). Typically, when human CL was scaled from hepatocyte data using the regression correction approach, ∼76% of drugs were predicted within twofold, with an ‘average absolute fold error’ of 1.6 (51). Hepatic uptake transporters may modulate the rate of metabolism for certain drugs by elevating the free intracellular concentration relative to that in the plasma (59). In such cases, standard approaches for IVIVE of CL may not work. However, IVIVE may still be established from a range of specialized hepatocyte-based assays such as the “media loss” or “oil-spin” methods, accepting the extrapolation process is far less well established than from standard assays (59).
IS THE EXTENT OF INTESTINAL METABOLISM PREDICTABLE AND CAN IT HELP TO RATIONALISE SPECIES DIFFERENCES IN Foral?
In Vivo Evidence Supporting Importance of Gut Wall Metabolism
The importance of the intestine as a site for first-pass metabolism has received growing attention since its infancy, well over 20 years ago. Our knowledge of the DMEs present and functioning in the gut wall has improved greatly. In vivo, enterocytes constitute approximately 90% of the cells within the epithelium (60) and contain a complement of phase I DMEs including CYP450s, esterases and amidases, epoxide hydrolase and alcohol dehydrogenase (20,61,62). Conjugating enzymes have also been identified including the UGTs, SULTs, N-acetyl transferases and glutathione S-transferases (63,64). Seminal work on drugs such as cyclosporine A and midazolam in anhepatic patients has clearly established the role of the intestine in limiting oral exposure of certain human CYP3A substrates (65,66). Similar findings have been reported with other CYP3A substrates including tacrolimus (67), verapamil (68) and felodipine (69). However, information on human intestinal drug metabolism from in vivo studies is scarce, principally because these studies are technically and ethically challenging. Multiple dose and sampling routes have been explored in pre-clinical models such as the rat. However, the labour-intensive and low throughput nature of these studies mean they are not routinely employed (70). There are a range of in vivo and in situ approaches for estimation of F G, and their advantages and limitations have been detailed elsewhere (71). Care must be taken when comparing in vivo estimates of F G from different methodologies. This is due to a number of underlying assumptions that can lead to contributions from the intestine being overemphasised (19). The indirect measurement of F G from total plasma clearance and F oral data is often the favoured approach within pharmaceutical companies. However, this can be prone to error if left uncorrected in the event of notable extrahepatic systemic clearance (72) or if the blood:plasma ratio deviates significantly from an assumed value of one (73). Calculation of F G can also be sensitive to the hepatic blood flow (HBF) rate employed (23,73) as well as dose if this leads to intestinal drug concentrations that exceed K m of the relevant DMEs. Given that decoupling F a and F G is experimentally difficult, intestinal availability (F a × F G) is often presented from in vivo PK data, assuming that there are no complications in the estimation of F H.
A comparison of intestinal availability has been made across species for a range of drugs predominantly metabolised by human CYP3A, CYP2C, CYP2D or UGT enzymes (Fig. 1, (15,25,74–81) and references included therein). With the CYP450 substrates, excepting tacrolimus (F a ∼15%), most of the drugs assessed are believed to exhibit good oral absorption in man (≥80% (25) data supplemental). Drugs such as dexamethasone, alprazolam, flupiritine and quinidine appear largely unaffected by gut wall metabolism in humans. Drugs including cyclosporine A, midazolam, diltiazem, verapamil, sildenafil and nifedipine showed moderate extractions whereas extensive intestinal metabolism was evident with tacrolimus, saquinavir, nicardipine, domperidone and also nisoldipine (data not shown).
Fig. 1.
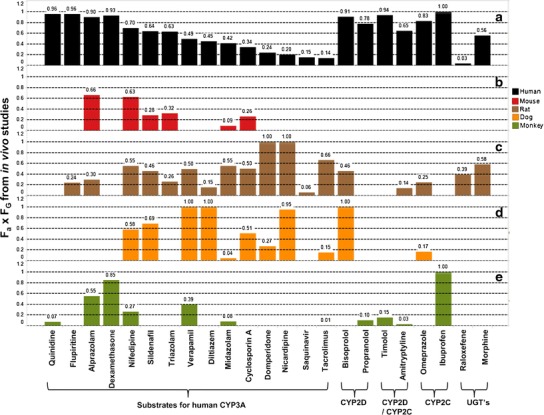
In vivo intestinal availability determined across species for selected human CYP3A, CYP2C, CYP2D and UGT substrates. Human data is presented in a (15,25,75,79). Mouse data is presented in b (79,81). Rat data is presented in c (75,79). Dog data is presented in d (74,76–81) (AstraZeneca unpublished data). Note that for diltiazem, midazolam and verapamil clearance approached or exceeded liver blood flow (LBF) in the dog; therefore, significant uncertainty and error is expected in the calculation of intestinal availability. Monkey data is presented in e (15,79,83)
In contrast, CYP2C and CYP2D substrates such as bisoprolol, propranolol, timolol, amitriptyline, omeprazole and ibuprofen generally showed good intestinal availability. One might anticipate a similar extraction across species if orthologous enzymes of human CYP3A4 expressed in rat, dog, monkey and mouse were highly conserved and followed similar expression patterns along the GI tract. Sildenafil showed comparable F a × F G in mice, rats, dogs and humans as did nifedipine, albeit with a slightly higher intestinal extraction in monkeys. Intriguingly, marked species differences were noted for tacrolimus and midazolam. The former with highest F a × F G values reported in rat, of the order rat>>human>dog>monkey. The latter showed a similar F a × F G in rat and human which was much higher than in other species, e.g. rat∼human>>monkey∼mouse>dog. With regard to dogs, intestinal CYP450 enzymes are generally less active than in humans (82). Although monkeys are genetically similar to humans, several of the exemplified drugs have shown remarkably lower intestinal availability in the monkey. It has been postulated that this may be a reflection of higher DME and efflux transporter activities in monkey intestine than those in human (15,83). Others have postulated, through experimentation with midazolam in Ussing chamber type studies, that asymmetric localisation of metabolic activity in the cynomolgus monkey small intestine, toward the apical side, may lead to extensive metabolism during uptake from the apical cell surface (84). This may be partly driven by close proximity of CYP3A to the extracellular efflux transporter P-glycoprotein (P-gp), both of which possess overlapping substrate specificities. The coordinated effect of P-gp and CYP3A distribution along the human small intestine has been investigated. It has been suggested for certain drugs (high rates of metabolism, high efflux and low F a) that the presence of P-gp may help to de-saturate CYP3A resulting in a reduced F G (85).
In vivo studies comparing species differences in gut wall extraction mediated through UGT enzymes are limited. However, it is clear from comparison across rat and human F a × F G that profound differences are possible depending upon the substrate. With raloxifene, very high extraction was observed in human intestines whereas moderate extraction was reported in rat (86). Conversely, with morphine, moderate extractions were seen in both rats and humans (79). Recently, Furukawa and co-workers assessed the in vivo intestinal availability of several human UGT substrates across rat, dog, monkey and humans (87). No obvious correlation was observed between F a × F G measured indirectly from PK studies in humans and rats (R 2 = 0.1). Rat was also poorly correlated with dogs and monkeys whereas a reasonable correlation (R 2 = 0.8) was observed between humans and dogs, albeit with higher values generally seen for dog. Additionally, a good correlation (R 2 = 0.99) was observed between humans and monkeys (87).
The contrasting extractions noted across species for the drugs evaluated in Fig. 1 could point to a lack of selectivity of these human substrates in other species. Alternatively, it may reflect significant differences in DMEs expressed across species in the gut wall. Certainly, metabolism studies in pre-clinical species have reported marked differences when compared to human, depending upon the CYP450 subfamily of interest (79). This highlights an ongoing challenge associated with interpretation of complex in vivo data, in particular, quantifying the exact contribution of intestinal metabolism indirectly from more conventional IV and oral dosing strategies (30,71). Regardless, taken at face value, there is little evidence in vivo that any one animal is sufficiently predictive of human F G, or indeed F a × F G, to be used as a standalone model to predict human oral exposures for novel chemical entities (NCEs). If feasible, a more mechanistic ‘bottom up’ approach to understanding organ-specific roles in metabolism, based on in vitro data, is desirable.
In Vitro Approaches to Assess Gut Wall Metabolism
Application of in vitro systems for the study of intestinal metabolism has grown in popularity during recent times (88). These include precision cut tissue slices, everted gut sacs, Ussing chamber preparations, enterocyte preparations and intestinal microsomes (71). Several offer the speed and capacity amenable to high throughput screening, allowing investigators to address two key areas. Firstly, to mechanistically probe the role that intestinal metabolism plays in mediating poor F oral in animal PK models that are integral to drug discovery programmes. For instance, facilitating troubleshooting of ‘compound series’ focussed issues such as the underlying causes and consequences of poor oral exposure in the rat (5,48). Secondly, to understand the human relevance of species differences in intestinal DME expression and rates of metabolism. Here, the goal is to extrapolate intestinal availability in humans from the most relevant animal model, or if necessary directly from human intestinal metabolism data that has been generated in vitro (22,23,27,29). The latter consideration is particularly important given that patterns of phase I and II DME expression in the intestine can differ markedly between species (63,79,87). Although research into IVIVE of intestinal metabolism data is evolving (88), it is still some way behind the established models used for the liver (22,23,28,29). This is due in part to the heterogeneous expression of enzymes along the GI tract and the fact that in vitro techniques for isolating the enzymes affects their quantification, in turn making comparison of data between laboratories difficult (24). Additionally, unlike the liver (53,89), little is known about the physiological scalars necessary for extrapolation of data generated from the various in vitro systems (88,90). In relative terms, more information is known about sub-cellular fractions and published values are available for rat, dog and human (90). However, the limited number of studies and frequent failure to correct for losses during sub-cellular fraction preparation (90) preclude confidence in IVIVE using microsomal scaling factors typified for the liver (53,55). As a result, other strategies have been utilised to scale intestinal CLint, for example based on CYP3A abundance (22). It is noteworthy that these values come from samples prepared by mucosal scraping, which can bias the estimate due to the highly mechanical nature of the procedure which is known to dilute or deteriorate the CYP450s (20,91).
RETROSPECTIVE ANALYSIS USING AstraZeneca CASE STUDIES: IMPACT OF GUT WALL METABOLISM ON HUMAN ORAL PK PREDICTIONS
In the following section, a retrospective analysis of three AstraZeneca case studies provides pharmaceutical based insight into species differences in gut wall extraction and the impact this can have on accurate projection of human PK, as determined from FIH clinical PK studies.
Case Study 1: Differential Intestinal Metabolism Across Species and Impact on AZ12470164 Clinical Oral PK
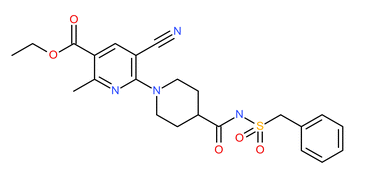
AZ12470164 (Figure S1 in Supplementary Materials) was a discovery compound from AstraZeneca’s Oncology portfolio that was taken into phase 1 clinical development. A summary of the pertinent physico-chemical and in vitro ADME properties are reported in Table I along with the pre-clinical PK parameters. This discovery data supported the human PK prediction. The biological effective concentration was translated from the PK/PD efficacy relationship developed in tumour-bearing mice models. Combined together, they informed the human dose prediction. Taken with other key considerations, such as the safety profile and pharmaceutical properties, a positive clinical investment decision was made to enter into phase I clinical trials.
Table I.
DMPK Properties for AZ12470164 Prior to its Nomination into Clinical Development and Following FIH Phase I Trials
| Parameter | AZ12470164 |
|---|---|
| Molecular weight (Da) | 399.4 |
| logD7.4 | >3 |
| Binding to plasma (% free) | <3 across mouse, rat, dog and human |
| Solubility at pH7.4 (μmol/L) | 46 |
| Caco-2 P app in apical to basolateral direction, pH 6.5 to 7.4 (10−6 cm/s) | 18 to 27, no evidence of efflux |
| Hepatocyte CLint (μL/min/106 cells); mouse/rat/dog | 7/43/<1 |
| Human liver microsomal CLint (μL/min/mg protein) | 18 |
| Total plasma clearance (mL/min/kg); CD-1 mouse/Han Wistar rat/Beagle dog | 125/22/8.6 |
| F oral (%); mouse/rat/dog | 56/14/44 to ∼100a |
| Calculated in vivo F a × F G (%); mouse/rat/dog | >100/20/50 to ∼100c |
| Predicted human F a (%) | 60 |
| Predicted MAD (mg) | 800 |
| Predicted human clearance (mL/min/kg) | 5.6 |
| Predicted human F oral (%) | 46 |
| Predicted biologically effective dose from once daily schedule (mg) | 154 |
| CL/F oral (L/h) ± Stdev | 2790 ± 2960b |
| Vz/F oral (L) ± Stdev | 12,400 ± 862b |
| Revised biologically effective dose for once daily schedule (mg) | >3000 |
Metabolism studies in hepatocytes from mouse, rat, dog and human revealed that AZ12470164 underwent many oxidative reactions as well as direct glucuronidation. No information was available on the phase II enzyme isoforms responsible for metabolism of AZ12470164, but CYP2C19, and to a lesser degree CYP3A4, mediated the phase I oxidative processes
ND not determined
aThe F oral was approximately 50% from low oral doses, but was complete at 100 mg using the formulation identified for the first in human studies. Phase I clinical PK data for a patient cohort receiving 80 mg orally
bThe clearance and terminal volume of distribution (V z) are reported as CL/F oral and V z/F oral as they are derived from oral dosing
cThe in vivo F a × F G was calculated from IV and oral PK data using the indirect method given by F oral/F H = F a × F G
In brief, AZ12470164 received internally a tentative BCS II classification based on its good Caco-2 intrinsic permeability (concentration and active transport-independent passive epithelial permeability), absence of efflux, but solubility limited absorption. At face value, the calculated MAD of 800 mg appeared adequate in the context of the predicted biologically effective dose (154 mg once daily or 43 mg twice daily). At that time, no consideration had been given to the potential impact of gut wall metabolism. The predicted human F oral was built largely from consideration of the likely fraction absorbed and the hepatic first-pass clearance. The former was predicted using solubility and Caco-2 permeability data (40) plus consideration of the F a achieved in pre-clinical models. The latter was guided principally by allometry rather than from in vitro to in vivo scaling of human in vitro CLint data (51). With hindsight, it could be argued that the prediction of human CL and F oral was overly optimistic. The metabolic fate of AZ12470164 was assessed in hepatocytes. Species differences in metabolism were evident with the major biotransformation in humans reported as a product of direct glucuronidation. By contrast, in the rat and dog, the major biotransformations were products of phase I oxidative metabolism. In the discovery phase of the project, the rate of metabolism had been assessed in human liver microsomes. Only later were cryopreserved human hepatocyte incubations carried out revealing a much higher CLint. There were also species differences in the intestinal availability (F a × F G) which could be interpreted as a signal for differences in intestinal loss. Complete F a × F G was reported in the mouse and dog, but this was much lower in the rat (20%).
AZ12470164 was progressed into phase I clinical trials. The oral pharmacokinetics was assessed in patients following single and multiple ascending doses (20 to 80 mg once daily). The predicted PK parameters have been compared against the clinical data from a patient cohort receiving 80 mg (Table I). The mean oral PK profile (n = 3) at this dose is shown in Fig. 2. It was noted that the clinical exposures were non-linear between 20, 40 and 80 mg, highly variable and much lower than anticipated. The calculated CL/F oral was 2790 ± 2960 L/h equating to approximately 664 mL/min/kg (e.g. 33-fold above liver blood flow (LBF) using a value of 20 mL/min/kg).
Fig. 2.
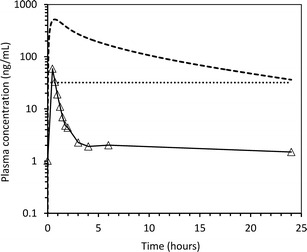
Phase 1 clinical PK data for AZ12470164. The open triangles represent geometric mean plasma concentrations determined from patients (n = 3) who received a single oral 80-mg dose. The dotted line is the biological effective target concentration derived from the quantitative PKPD-efficacy relationship in tumour-bearing mice. The dashed line is simulated steady-state oral PK profile for a 154-mg dose
At the time, it was felt that continuous cover above the effective concentration was necessary for biological activity. Unsurprisingly, factoring in the clinical exposure data using a rather crude linear extrapolation led to a revised dose (>3000 mg) that was much higher than the original prediction (154 mg once daily) and exceeded the calculated MAD (∼800 mg). It was questionable whether either the biologically effective dose for proof of mechanism, or the maximum well-tolerated dose, could be achieved. This made the clinical development of AZ12470164 as an oral agent, in the cancer disease setting, a high risk. In the context of other project concerns and business drivers, the decision was subsequently taken to halt further work on this development programme.
In order to understand the significant underprediction associated with the clinical PK, additional in vitro data was generated to complement the original discovery DMPK package. When the human hepatocyte CLint was measured, it was much higher than the liver microsomal CLint (Table II). The hepatocyte CLint scaled to give a predicted clearance approximating 75% LBF (51). This gave a much higher hepatic extraction ratio than had previously been estimated by allometry. However, this higher predicted clearance still could not account for the very high clinical CL/F oral values. Therefore, the rate of metabolism in intestinal microsomes was investigated. Reaction phenotyping, in recombinant expressed human CYP450’s, revealed that a number of CYP450s were involved in the metabolism of AZ12470164, including CYP2C19, CYP3A4 and, to a lesser extent, CYP2D6 and CYP3A5. Only later on were a range of commercially available UGTs assessed where it was shown that at least UGT1A9 was involved in the metabolism of AZ12470164. This isoform is expressed in the liver, and there is equivocal evidence that it is functionally expressed in the intestine (63). It is known to catalyze glucuronidation of primary and secondary amines (92) in addition to bulky phenols (93). Alerted to the potential for extra-hepatic metabolism, AZ12470164 was incubated in line with published methodology (94) in rat, dog and human intestinal microsomes to assess oxidative metabolism and glucuronidation. The intestinal microsomes employed were prepared within AstraZeneca, and in vitro physiological scalars were determined (manuscript in preparation: Hatley O, Jones C, Galetin A, Rostami-Hodjegan A. Critical assessment and optimisation of intestinal microsomal preparation using rat as a model species). The CLint values were scaled to an estimated F G using the Q gut model (29). Despite challenges of scaling in vitro CLint data for UGT metabolism (24,63) in intestinal preparations (79,91), intestinal availability in rat and dog estimated from the in vitro data (Table II) compared well with those estimated from PK data. Taking the same approach with the human in vitro data yielded a much lower F G value (15%) suggestive of high extraction in the human gut wall. In combination with the revised F H predicted from hepatocytes, a much lower F oral (2.3%) was estimated compared with the original estimate (46%). Accounting for this in the estimation of systemic plasma CL, using the clinical oral AUC data, gave a more realistic assessment of the human systemic CL (∼15 mL/min/kg), as opposed to 664 mL/min/kg (>33-fold LBF) deduced with a dose based on F oral set at 46% (e.g. CL = (Dose × F oral)/AUCoral).
Table II.
Data Generated on AZ12470164 During the Early Clinical Development Phase
| Parameters considered in retrospective analysis | AZ12470164 |
|---|---|
| Human hepatocyte CLint (μL/min/106 cells) | 100 |
| Predicted clearance from human hepatocytes (mL/min/kg) | 14.3a |
| Intestinal microsomal CLint (μL/min/mg); rat/dog/human | 17/54/334 |
| F G (%); mouse/rat/dog/human | ND/74/51/15b |
| Calculated F H (%); mouse/rat/dog/human | 69/84/25c |
| Calculated in vivo F a × F G (%); mouse/rat/dog/human | >100/20/50 to 120/NDd |
| Predicted F a × F G from in vitro data (%); mouse/rat/dog/human | ND/26/51/10 |
aThe predicted clearance from hepatocytes was scaled using the well-stirred model and a lab-specific empirical correction factor according to (51)
b F G was scaled from activated intestinal microsomes using the Q gut model (29)
cThe pre-clinical F H was calculated from IV PK studies whereas the human value was predicted from scaled cryopreserved human hepatocytes
dThe in vivo F a × F G was calculated from IV and oral PK data using the indirect method given by F oral/F H = F a × F G
This case study highlights the importance of considering species differences in gut wall metabolism for the prediction of human F oral and dose. With the benefit of hindsight, a closer inspection of the rat and dog PK data was needed. Despite AZ12470164 appearing to have excellent in vitro permeability, marked species differences in the apparent in vivo F a (more appropriately considered as F a × F G) were evident signalling variable intestinal loss. Assessment of the underlying causes for this intestinal loss and direct assessment in a relevant human matrix would have been of significant value to the human PK risk assessment. Firstly, because intestinal extraction was much higher in humans, of the order: human>>dog>rat. Secondly, metabolite identification studies showed phase II glucuronidation as the major clearance route in humans. Given that AZ12470164 has solubility limited absorption, it would potentially be very difficult to increase exposures sufficiently to saturate glucuronidation in the gut wall.
Key lessons that can be taken from this case study include:
Investigate underlying causes of low in vivo F a × F G reported in one or more pre-clinical PK models to rule out involvement of gut wall metabolism, particularly if the in vitro ADME properties of the compound predict that it should have good absorption potential.
Metabolism data generated from intestinal microsomes can offer a valuable, high throughput approach, to predict and design against liabilities arising from gut wall metabolism. However, in vitro intestinal metabolism data can only be applied in a truly meaningful way, for quantitative prediction, if the in vitro physiological scalars are known and used with an appropriate model describing extraction from the intestine.
Be mindful of structural motifs that make a molecule susceptible to direct phase II glucuronidation. This is important given the marked differences in expression levels of the individual enzyme isoforms across species and organs (63,79,87). Compounds falling outside the BCS I classification may be at greater risk of intestinal glucuronidation. Their solubility and/or permeability limitations may preclude reaching sufficiently high local gut concentrations to saturate these high capacity enzymes.
Case Study 2: Metabolism and Transporter Data from Human Intestine in the Ussing Chamber Model Could Have Prevented the Progression of AZD1283 into Clinical Studies
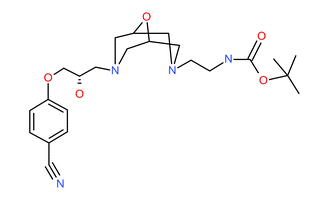
AZD1283 (Figure S2 in Supplementary Materials) was a development compound from AstraZeneca’s Cardiovascular portfolio (95). A summary of the pertinent compound properties are presented (Table III).
Table III.
Pertinent Physico-Chemical and ADME Properties Known at the Time of AZD1283 Nomination into Clinical Development
| Parameter | AZD1283 |
|---|---|
| Molecular weight (Da) | 470.6 |
| logDpH7.4 | 1.4 |
| pKa of acidic ionisation centre | 4.6 |
| Binding to plasma (% free); mouse/dog/cynomolgus monkey/human | 1.11/0.54/0.94/0.59a |
| Solubility (μmol/L) | 0.2 to 346b |
| Caco-2 permeability in apical to basolateral direction, pH 6.5 to 7.4 (10−6 cm/s) | 18 |
| Hepatocyte CLint (μL/min/106 cells); mouse/rat/dog/monkey/human | ND/35/<4/ND/15 |
| Liver microsomal CLint (μL/min/mg); mouse/rat/dog/monkey/human | ND/285c/<12/<12/<5 |
| Total plasma clearance (mL/min/kg); female mouse/female Sprague-Dawley rat/Beagle dog/cynomolgus monkey | 85/119/0.67/5 |
| Hepatocyte CLint (μL/min/106 cells); mouse/rat/dog/monkey/human | ND/35/<4/ND/15 |
| Liver microsomal CLint (μL/min/mg); mouse/rat/dog/monkey/human | ND/285c/<12/<12/<5 |
| Total plasma clearance (mL/min/kg); female mouse/female Sprague-Dawley rat/Beagle dog/cynomolgus monkey | 85/119/0.67/5 |
| In vitro human blood:plasma ratio | 0.63 |
| F oral (%); mouse/rat/dog/monkey | 42/24/100/66 |
| Calculated in vivo F a × F G (%); mouse/rat/dog/monkey | ND/ND/100/80d |
aAZD1283 is stable in dog, monkey and human plasma up to 3 h at 37°C. Ester hydrolysis accounted for 43% losses observed in mouse plasma. This could be inhibited by co-incubation with 4-(2(aminoethyl)benzene sulfonyl fluoride hydrochloride. AZD1283 is chemically stable across a full pH range
bThe aqueous solubility of AZD1283 is pH dependent and increases at pH values above its pKa. In aqueous solutions, from pH 1.1 to 8.0, the solubility ranges from 0.2 to 346 μmol/L
cRat microsomal CLint is high in the presence and absence of NADPH
dThe in vivo F a × F G was calculated from IV and oral PK data using the indirect method according to equation F oral/F H = F a × F G. The LBF values used at the time for calculation of F h in mouse, rat, dog, monkey and human were 152, 80, 33, 44 and 21 mL/min/kg, respectively
This discovery DMPK data supported the human PK prediction. The biological effective concentration (target trough concentrations ∼1 μmol/L, Fig. 3) came from translation of the PK/PD efficacy relationship built in the anaesthetised dog anti-thrombotic model.
Fig. 3.
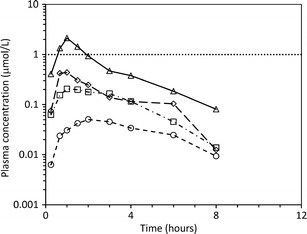
Geometric mean PK profiles from clinical single ascending dose studies with AZD1283. The open circles, squares, diamonds and triangles represent geometric mean plasma concentrations of AZD1283 determined in cohorts (n = 2 to 6 male healthy volunteers) receiving 50, 250, 750 or 2000 mg. The dotted line is the estimated biological effective target concentration derived from the quantitative PK/PD efficacy relationship in the anaesthetized dog anti-thrombotic model
Taken together, they informed the human dose prediction used as part of the clinical investment decision (Table IV). In brief, AZD1283 contains an ester functional group as well as an acidic acylated suphonamide. Subsequently, it is susceptible to ester hydrolysis in certain species. Stability was confirmed in human, dog and cynomolgus monkey plasma. However, AZD1283 showed instability in mouse and rat plasma precluding these species for purposes of predicting human PK. AZD1283 was stable at low acidic pH and within human intestinal fluid. Low to moderate rates of metabolism were reported from CLint incubations in dog, monkey and human liver microsomes and hepatocytes. AZD1283 has a low fraction unbound in plasma across species (≤1% free). The scaled in vitro data predicted that AZD1283 would have a low hepatic extraction. A high rate of metabolism was observed in rat microsomes in the presence and absence of NADPH. This pointed to the involvement, at least in rodents, of non-CYP450 mediated hepatic (and potentially extra-hepatic) metabolic processes.
Table IV.
Predicted human PK properties supporting nomination of AZD1283 into clinical development versus select clinical oral PK parameters from 250 mg dose cohort
| Parameter | AZD1283 |
|---|---|
| Predicted human F a | 76 to 100 |
| Predicted human clearance (mL/min/kg) | 0.4 to 3.3a |
| Predicted human V ss (L/kg) | 0.3 to 1.1 |
| Predicted human F oral (%) | 65 to 100 |
| Predicted biologically effective dose twice daily (mg/dose) | 250 |
| CL/F oral (L/h) | 601b |
| Projected CL/F oral (mL/min/kg) | 3.5c |
| V z/F oral (L) | 1436b |
| Projected V z/F oral (L/kg) | 0.5c |
| Oral half-life (hours) | 1.65 |
| Estimated F oral (%) | <5d |
aAllometry performed using dog and monkey PK, mouse and rat excluded due to plasma stability issues with AZD1283. Separate allometric predictions were made from dog and monkey, respectively, factoring in correction for species differences in plasma protein binding
bThe clearance and volume of distribution were reported as CL/F oral and V z/F oral as they were derived from oral dosing
cProjected CL/F oral and Vz/F oral with bioavailability estimate set at 2.5%
dEstimated bioavailability at all clinical doses
Although products of amide hydrolysis were detected in mouse, dog and human hepatocytes, ester hydrolysis was the major route of metabolism. Predicting human PK for molecules containing ester structural motifs can be challenging. This is due to large species differences associated with ester hydrolysis (96–98). Poor allometric correlation between dog and cynomolgus monkey meant that two species scaling was not appropriate (the slope of the unbound CL relationship was ∼0.3 with a low correlation coefficient ∼0.15). Instead, the human CL was predicted using allometry from single species scaling, correcting for species differences in plasma protein binding. It was anticipated, from modelling in GastroPlusTM, that solubility should not limit oral absorption at relatively low doses (<250 mg). Caco-2 permeability was high (18×10−6 cm/s in the apical to basolateral direction (pH 6.5/7.4) despite significant efflux (Caco-2 efflux ratio = 43)). A pH dependency was noted with a lower P app reported when the assay was run at pH 7.4. Good F oral and a high calculated fraction absorbed were observed in dog and monkey; therefore, at likely pharmacologically active doses, complete absorption was expected. The estimated human PK properties are captured in Table IV. A consequence of uncertainty in the predicted CL and half-life meant that the project had to accept a wide ranging dose prediction going forwards (40 to 500 mg). However, at the time, the project believed that there was a realistic potential of achieving the requisite target cover profile in humans from a midpoint dose prediction of 250 mg twice daily.
Disappointingly, clinical PK data from the single ascending dose studies (Fig. 3) showed that oral exposures of AZ1283 were much lower than projected from the predicted human PK parameters. Importantly, in light of the target concentration and dose range already explored, it was highly unlikely that the necessary clinical exposure profile could be achieved. It was difficult to identify the primary parameters that had been poorly predicted, highlighting a key limitation to working with just oral PK data. After reviewing the clinical data, it was felt that the oral half-life had been adequately predicted and the volume of distribution was likely to fall within the predicted range (Table IV). The systemic CL may have fallen above the predicted range but was still thought to have been relatively low (∼15% LBF) pointing to a low hepatic extraction compound (Table IV). Thus, the low F oral (estimated at <5% for all clinically tested doses) was unlikely to have been unduly limited by hepatic first-pass clearance. So other possibilities needed consideration.
Knowing the affinity of AZD1283 for efflux transporters and the potential for ester hydrolysis, it became increasingly apparent that low human intestinal availability was the likely culprit. Surprisingly, given that ester hydrolysis was identified as the major biotransformation in hepatocytes, in vitro work in intestinal S9 fractions did not yield evidence of intestinal metabolism. Assuming that functional activity of the cytosolic carboxylesterases had been retained in the S9 fraction, one might have expected to have detected evidence of this metabolic pathway. It is possible that significant carboxylesterase activity was lost from the intestinal S9 fractions given the susceptibility of DMEs such as these to degradation by proteolytic enzymes released during tissue preparation (24).
Regardless, experiments with intact human jejunal and colon tissue in the Ussing Chamber model demonstrated intestinal metabolism working in concert with transporter mediated efflux to efficiently limit availability of AZD1283 (Fig. 4). This elegant approach, utilising radio-labelled compound, has been published in detail elsewhere (36). Briefly, incubating with radio-labelled compound in Ussing chamber tissue studies allows measurement of parent as well as metabolites. Interpreted together, such data permits consideration of the separate contributions of F a (driven by intrinsic permeability and efflux as defined by measurement of parent plus metabolites) and F G (driven by metabolism as defined by the extraction ratio calculated from the differences in parent versus parent and metabolites Papp) to the intestinal availability. A comparison was made between the total P app for AZD1283 (red bars in Fig. 4a) and the P app for parent compound alone (black bars in Fig. 4a). The P app was ca. two- to threefold higher at lower incubation concentrations (10 and 30 μM). This indicated a high apparent extraction ratio for AZD1283, 78 and 49%, respectively (panel C). Whereas at higher concentrations (70 and 100 μM), the P app values for parent and total levels (parent plus metabolites) increased markedly suggesting saturation of efflux. Interestingly, at these higher concentrations, the relative difference between parent and total P app values diminishes, suggesting a lower extraction ratio. This could be attributed to saturation of the DMEs. Monitoring P app in both directions was also revealing. The lower P app observed at the higher concentrations in the serosal to mucosal direction (Fig. 4b) inferred possible involvement of a basal uptake transporter that may be saturated at these higher concentrations. However, this remains speculative as no further work was done to elucidate the putative transporter. One can conclude that at low concentrations, the apparent P app was low due to significant efflux and metabolism. AZD1283 shows a clear concentration-dependent absorption profile in the 10 to 100 μM range due to saturation of the efflux mechanisms. This effect may also be contributing synergistically to the lower metabolic extraction evident at these higher concentrations (e.g. reduced residence time within the enterocyte for metabolism to occur).
Fig. 4.
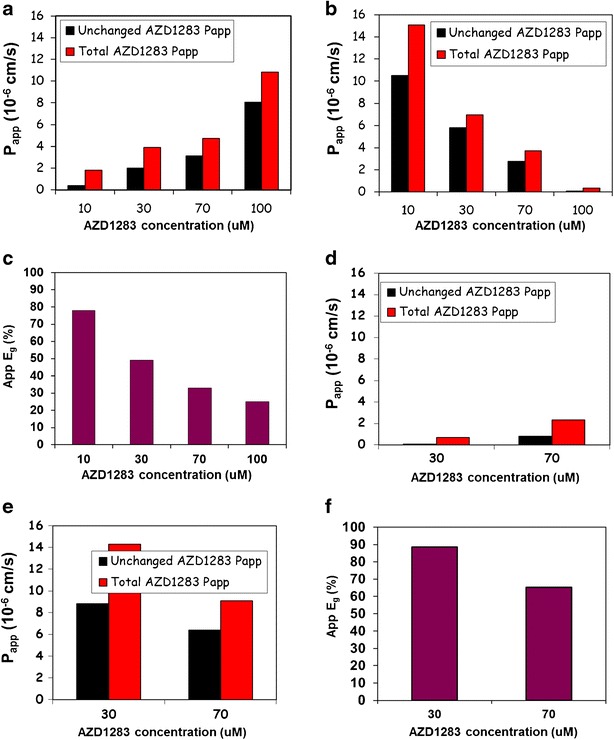
Effect of intestinal metabolism on apparent permeability of AZD1283 in human jejunal tissue (n = 2) in Ussing chamber. Differences in permeability between AZD1283 and 14C radio-labelled AZD1283 is shown. a P app in the mucosa to serosa direction at 10, 30, 70 and 100 uM for unchanged AZD1283 (black bars) and for total 14C radio-labelled AZD1283, e.g. contributions from unchanged parent and its metabolites (red bars). b P app in the serosa to mucosa direction at identical concentrations of unchanged AZD1283 (black bars) and total 14C radio-labelled AZD1283 (red bars). c Apparent extraction ratio (App E g) calculated from the equation App E g = (P total − P unchanged) / (P total). The methodology and approach have been described elsewhere (36). d–f Analogous permeability plots to a–c tested at 30 and 70 uM and from the colon (n = 1) rather than the jejunal tissue
Given the intestinal concentrations, anticipated at the projected therapeutic doses (250 mg through to the top dose tested), it is reasonable to assume that efflux and metabolism within the enterocytes may be working efficiently, in concert, to limit the systemic exposures. These findings meant that the options going forward were very limited for the project. Although saturating metabolism by carboxylesterases is potentially achievable, the limited solubility profile of AZD1283 likely precluded attaining the necessary concentrations to test this orally. An extended release formulation was considered but this was stopped in light of additional Ussing Chamber data indicating that absorption in the colon would in all likelihood be even lower (Fig. 4d–f).
Overall, this case study highlights that examination of the F G component cannot be ignored when extrapolating across species to estimate human F a and F oral. This is especially the case for compounds with tentative BCS II, III or IV classifications with sub-optimal physico-chemical properties for complete absorption. As such, they are inherently more sensitive to intestinal metabolism either because sufficiently high concentrations of free compound cannot be achieved to saturate the metabolic processes, or the residence time is extended sufficiently to favour extensive metabolic extraction. Although dog and monkey are often considered better pre-clinical models for estimation of human F a, in the case of AZD1283, these species did not accurately reflect the human intestinal availability. In this case, it is likely that intestinal availability was severely restricted due to intestinal losses arising from transporter-metabolism interplay in the gut wall, a limitation not evident in dog or monkey for this compound. With benefit of hindsight, the Ussing Chamber data would have been the most appropriate data for risk assessment and would likely have stopped AZD1283 being progressed into phase I clinical trials.
Again, key lessons can be taken from this case study and include:
Both dog and cynomolgus monkey turned out to be poor models of human intestinal availability. This may be a reflection of differences between species in expression of the enzymes mediating ester hydrolysis in the gut wall.
Only data from the human Ussing chamber model using radio-labelled compound was sufficiently detailed to provide mechanistic insight into the processes-limiting clinical exposures. Indeed, an in-house retrospective analysis of AstraZeneca compounds indicated that this in vitro approach was the only one consistently able to identify compounds at risk of achieving poor human exposure due to intestinal loss.
The in vitro data demonstrated that the processes governing intestinal loss could be saturated but the solubility profile and impact of metabolism-efflux transporter interplay on the molecule (characteristic of BDDCS class II) was not good enough to take advantage of this.
A balanced approach for assessment of intestinal metabolism is needed in order to support the number of projects typically run within pharmaceutical companies from lead identification and optimisation phases through to clinical development. Costly, low throughput assays requiring fresh intact human tissue cannot realistically support the level of demand. It is important, therefore, to develop integrated approaches utilising higher throughput, sub-cellular fractions wherever possible.
Case Study 3: Application of a PBPK Model to Mechanistically Interpret Discrepancies in Oral PK Profiles of AZD7009
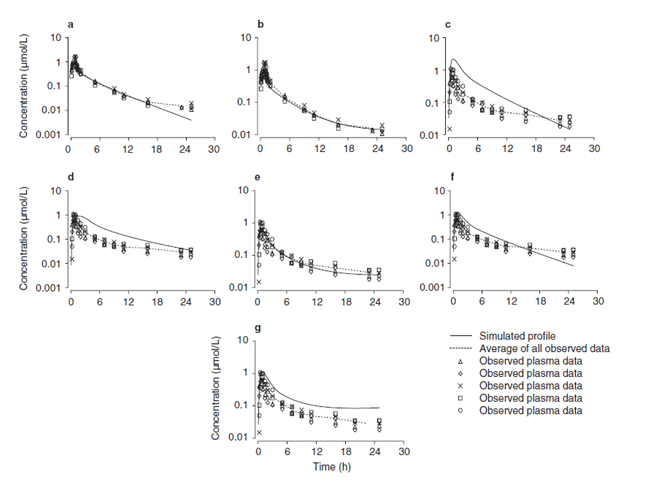
AZD7009 (Figure S3 in Supplementary Materials) was another development compound from AstraZeneca’s cardiovascular portfolio that was progressed into phase I clinical trials. Perhaps a rarity, but in addition to the oral PK data from single ascending oral studies, clinical IV PK data was also generated. This enabled human PK parameters such as CL, apparent steady-state volume of distribution (V ss), half-life and F oral to be described with greater accuracy. Increased confidence in these measured parameters was of great value to the PK modelling activities. Simulation can be used to help develop mechanistic understanding of the processes governing the observed PK profiles through line shape analysis (99). It is not our intention to cover this case study in detail as it has been disclosed previously. The reader is directed to the original work for explanation of the simulation approach and in-depth analysis performed therein (100). The example is still worthy of inclusion here as it highlights the broader value of introducing simulation work to probe potential anomalies/disconnects in PK. Simulation work such as this can really help address the ‘what if’ scenarios that often guide the direction of subsequent experimentation. For the readers’ benefit, the fundamental physico-chemical and pre-clinical ADME properties of the compound have been reproduced in Table S1 (in Supplementary Materials). The predicted human PK properties are also included for comparison to the phase I clinical PK data.
From consideration of the physico-chemical properties and IV PK parameters of AZD7009, the oral PK data can be simulated. If processes determining the IV profile (clearance and tissue distribution) are well-described, solubility and permeability are the only parameters governing the oral line shape, and the simulated oral PK profiles should show reasonable agreement with the observed. Any mismatch between the simulated and observed PK could reflect additional factors that may not have been considered such as gut wall metabolism, P-gp efflux, chemical degradation or enterohepatic re-circulation. In the case of AZD7009, the observed rat PK data inferred a high fraction absorbed and showed a good fit to the simulated data (Figure S4 in Supplementary Materials).
However, unlike the case in rat, the F oral (16%) in healthy male patients was not consistent with what one might expect solely from hepatic first-pass clearance (45%). In contrast to the rat, the human oral PK data revealed a poor fit between observed and simulated profiles, if intestinal loss was ignored (Figure S5, panel c in Supplementary Materials). Thus, simulation was employed to help explore several ‘what if’ scenarios to see if the apparent intestinal loss could be rationalised. At the time, the source of intestinal loss could not be established through in vitro experimentation. Nevertheless, the author noted that it was highly likely that gut wall metabolism was involved. Simulation exploring impact of P-gp indicated that it was unlikely to be playing a significant role (Figure S5, panels c and d in Supplementary Materials).
However, allowing for enterohepatic re-circulation and introduction of intestinal loss rate constants into the simulations achieved a much improved fit (Figure S5, panel e in Supplementary Materials). Supporting this view, there was an equivocal evidence from comparison of differences in metabolite:parent drug ratios (Table S2 in Supplementary Materials) calculated from both IV and oral dosing (100).
In summary, key lessons to be taken from this case study include:
The rat did not exhibit intestinal loss and so was a poor in vivo model for prediction of the human oral PK.
Using simulation, the potential processes underlying intestinal losses could be identified. It also helped prioritize follow-up experimentation by eliminating mechanisms not probable for the compound of interest.
Even those well-established in vitro assays, generally considered suitable as models for extrapolation of in vivo intestinal availability, are not always accurate predictors of in vivo outcome.
CONCLUSION
Our knowledge of intestinal metabolism has increased substantially over recent years. Both in vitro and in vivo data have clearly demonstrated that the gastrointestinal tract can play a significant role in mediating the extent of first-pass elimination of xenobiotics under certain situations. Evidence overwhelmingly points to lower protein and catalytic activity for the majority of phase 1 and phase II DMEs within the gut wall, compared to the liver. However, anatomical positioning and physiology of this organ means that metabolism in the small intestine can substantially impact F oral. This effect in humans has been highlighted using three AstraZeneca case studies in which lower than anticipated oral exposures were reported from the FIH clinical trials. Improved understanding of hepatic and intestinal expression profiles of DMEs across species should help to rationalise differences in F oral between animal models and humans. In turn, this should lead to more informed judgements about projected human oral PK based on in vitro and animal PK data. Whilst substantial progress has been made in the field of intestinal metabolism, there is still much to be done in terms of improving quantitative prediction of pre-clinical oral PK and understanding relevance to human PK predictions. A broad strategy is needed to integrate assessment of intestinal metabolism in context of typical DMPK activities ongoing within drug discovery programmes.
Key learnings to be taken from this review include:
Resource efforts should be focused on optimizing compound properties that lead to improved exposure in humans. Therefore, underlying causes of intestinal loss in animal models should be investigated with a view to better understanding their relevance to prediction of human oral PK.
Structural motifs in molecules that introduce metabolic liabilities within the gut wall, such as direct phase II glucuronidation, require careful consideration. This is especially important for NCEs that are substrates for DMEs selectively expressed in the human small intestine, such as UGT1A8, UGT1A10 and SULT1A3.
In vitro models should be established that are amenable to quantitative IVIVE of F G and have sufficient capacity to profile lead compound series. Potential CDs should be further profiled, using more physiologically relevant models, to establish interplay between transporters and DMEs which may limit F G. Currently, within AstraZeneca, intestinal microsomes that have been activated for phase II metabolism are preferred for assessment of compound series during lead optimisation. Experimentation with human intestinal tissue in the Ussing chamber model being preferred for evaluation of potential CDs.
Gaps remain in our understanding of physiological scalers for various in vitro systems used to evaluate intestinal metabolism across species. Emerging mass spectrometry-based technologies are beginning to address this through provision of robust, quantitative data on protein abundances in various tissues including the intestine. Although progress has been made with some in vitro systems, consensus is lacking on best practise to ensure consistent, high recoveries of functional DMEs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Structure features of AZ12470164. (GIF 3 kb)
{kind=link}
Structure of AZD1283. (GIF 4 kb)
{kind=link}
Structure of AZD7009. (GIF 3 kb)
{kind=link}
{kind=link}
Physiologically based PK simulations of PK profiles of AZD7009 in humans. (a) Simulation of the intravenous profile without enterohepatic recirculation. (b) Simulation of the intravenous profile with inclusion of enterohepatic recirculation. (c) Simulated oral profile against the observed when intestinal loss was not considered; the permeability used was 5.8 cm/s. (d) Simulated oral profile against the observed when intestinal loss was not considered; the permeability used was 1.9 cm/s. (e) Simulation of the oral profile with intestinal loss rate constants introduced and refinement with enterohepatic recirculation. (f) Simulation of the oral profile excluding enterohepatic recirculation and including intestinal loss. (g) Simulation of the oral profile including enterohepatic recirculation and excluding intestinal loss. Reprinted with permission (100). (GIF 35 kb)
Acknowledgments
This work was contributed to the OrBiTo project (http://www.imi.europa.eu/content/orbito) as side ground.
O.J.D.Hatley was funded by a PhD grant awarded through the CASE award scheme, receiving support from both the MRC and AstraZeneca.
All in vivo work conducted within AstraZeneca was subject to internal ethical review and conducted in accordance with Home Office requirements under the Animals Scientific Procedures Act (1986).
References
- 1.Liu G, Franssen E, Fitch MI, Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol. 1997;15(1):110–5. doi: 10.1200/JCO.1997.15.1.110. [DOI] [PubMed] [Google Scholar]
- 2.Lesko LJ, Rowland M, Peck CC, Blaschke TF. Optimizing the science of drug development: opportunities for better candidate selection and accelerated evaluation in humans. Pharm Res. 2000;17(11):1335–44. doi: 10.1023/A:1007574217260. [DOI] [PubMed] [Google Scholar]
- 3.Hellriegel ET, Bjornsson TD, Hauck WW. Interpatient variability in bioavailability is related to the extent of absorption: implications for bioavailability and bioequivalence studies. Clin Pharmacol Ther. 1996;60(6):601–7. doi: 10.1016/S0009-9236(96)90208-8. [DOI] [PubMed] [Google Scholar]
- 4.Hurst S, Loi C-M, Brodfuehrer J, El-Kattan A. Impact of physiological, physicochemical and biopharmaceutical factors in absorption and metabolism mechanisms on the drug oral bioavailability of rats and humans. Expert Opin Drug Metab Toxicol. 2007;3(4):469–89. doi: 10.1517/17425255.3.4.469. [DOI] [PubMed] [Google Scholar]
- 5.Ballard P, Brassil P, Bui KH, Dolgos H, Petersson C, Tunek A, et al. The right compound in the right assay at the right time: an integrated discovery DMPK strategy. Drug Metab Rev. 2012;44(3):224–52. doi: 10.3109/03602532.2012.691099. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Jin JY, Mukadam S, Malhi V, Kenny JR. Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: strategy and approach during the drug discovery phase with four case studies. Biopharm Drug Dispos. 2012;33(2):85–98. doi: 10.1002/bdd.1769. [DOI] [PubMed] [Google Scholar]
- 7.Jones HM, Parrott N, Jorga K, Lavé T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45(5):511–42. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 8.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, Macintyre F, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283(1):46–58. [PubMed] [Google Scholar]
- 9.Van den Bergh A, Sinha V, Gilissen R, Straetemans R, Wuyts K, Morrison D, et al. Prediction of human oral plasma concentration-time profiles using preclinical data. Clin Pharmacokinet. 2011;50(8):505–17. doi: 10.2165/11587230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Zou P, Yu Y, Zheng N, Yang Y, Paholak HJ, Lawrence XY, et al. Applications of human pharmacokinetic prediction in first-in-human dose estimation. AAPS J. 2012;14(2):262–81. doi: 10.1208/s12248-012-9332-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poulin P, Jones R, Jones HM, Gibson CR, Rowland M, Chien JY, et al. PHRMA CPCDC initiative on predictive models of human pharmacokinetics, part 5: prediction of plasma concentration-time profiles in human by using the physiologically‐based pharmacokinetic modeling approach. J Pharm Sci. 2011;100(10):4127–57. doi: 10.1002/jps.22550. [DOI] [PubMed] [Google Scholar]
- 12.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–80. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 13.Zhang T, Heimbach T, Lin W, Zhang J, He H. Prospective predictions of human pharmacokinetics for eighteen compounds. J Pharm Sci. 2015;104(9):2795–806. doi: 10.1002/jps.24373. [DOI] [PubMed] [Google Scholar]
- 14.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 15.Akabane T, Tabata K, Kadono K, Sakuda S, Terashita S, Teramura T. A comparison of pharmacokinetics between humans and monkeys. Drug Metab Dispos. 2010;38(2):308–16. doi: 10.1124/dmd.109.028829. [DOI] [PubMed] [Google Scholar]
- 16.Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin H-C, et al. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm Res. 2006;23(8):1675–86. doi: 10.1007/s11095-006-9041-2. [DOI] [PubMed] [Google Scholar]
- 17.Grass GM, Sinko PJ. Physiologically-based pharmacokinetic simulation modelling. Adv Drug Deliv Rev. 2002;54(3):433–51. doi: 10.1016/S0169-409X(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 18.Musther H, Olivares-Morales A, Hatley OJ, Liu B, Hodjegan AR. Animal versus human oral drug bioavailability: do they correlate? Eur J Pharm Sci. 2014;57:280–91. doi: 10.1016/j.ejps.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin JH, Chiba M, Baillie TA. Is the role of the small intestine in first-pass metabolism overemphasized? Pharmacol Rev. 1999;51(2):135–58. [PubMed] [Google Scholar]
- 20.Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 “pie”. Drug Metab Dispos. 2006;34(5):880–6. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Tucker GT, Rostami‐Hodjegan A. Cytochrome P450 3A expression and activity in the human small intestine. Clin Pharmacol Ther. 2004;76(4):391. doi: 10.1016/j.clpt.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Gertz M, Houston JB, Galetin A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab Dispos. 2011;39(9):1633–42. doi: 10.1124/dmd.111.039248. [DOI] [PubMed] [Google Scholar]
- 23.Kadono K, Akabane T, Tabata K, Gato K, Terashita S, Teramura T. Quantitative prediction of intestinal metabolism in humans from a simplified intestinal availability model and empirical scaling factor. Drug Metab Dispos. 2010;38(7):1230–7. doi: 10.1124/dmd.109.029322. [DOI] [PubMed] [Google Scholar]
- 24.Kaminsky LS, Fasco MJ. Small intestinal cytochromes P450. Crit Rev Toxicol. 1992;21(6):407–22. doi: 10.3109/10408449209089881. [DOI] [PubMed] [Google Scholar]
- 25.Varma MV, Obach RS, Rotter C, Miller HR, Chang G, Steyn SJ, et al. Physicochemical space for optimum oral bioavailability: contribution of human intestinal absorption and first-pass elimination. J Med Chem. 2010;53(3):1098–108. doi: 10.1021/jm901371v. [DOI] [PubMed] [Google Scholar]
- 26.Fan J, Chen S, Chow EC, Pang SK. PBPK modeling of intestinal and liver enzymes and transporters in drug absorption and sequential metabolism. Curr Drug Metab. 2010;11(9):743–61. doi: 10.2174/138920010794328931. [DOI] [PubMed] [Google Scholar]
- 27.Furukawa T, Yamano K, Naritomi Y, Tanaka K, Terashita S, Teramura T. Method for predicting human intestinal first-pass metabolism of UGT substrate compounds. Xenobiotica. 2012;42(10):980–8. doi: 10.3109/00498254.2012.680620. [DOI] [PubMed] [Google Scholar]
- 28.Pang SK. Modeling of intestinal drug absorption: roles of transporters and metabolic enzymes (for the Gillette Review Series) Drug Metab Dispos. 2003;31(12):1507–19. doi: 10.1124/dmd.31.12.1507. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8(7):676–84. doi: 10.2174/138920007782109733. [DOI] [PubMed] [Google Scholar]
- 30.Galetin A, Gertz M, Houston JB. Potential role of intestinal first-pass metabolism in the prediction of drug-drug interactions. Expert Opin Drug Metab Toxicol. 2008;4(7):909–22. doi: 10.1517/17425255.4.7.909. [DOI] [PubMed] [Google Scholar]
- 31.Tang C, Prueksaritanont T. Use of in vivo animal models to assess pharmacokinetic drug-drug interactions. Pharm Res. 2010;27(9):1772–87. doi: 10.1007/s11095-010-0157-z. [DOI] [PubMed] [Google Scholar]
- 32.Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42(6):620–43. doi: 10.1177/00970002042006005. [DOI] [PubMed] [Google Scholar]
- 33.Chiou W, Ma C, Chung S, Wu T, Jeong H. Similarity in the linear and non-linear oral absorption of drugs between human and rat. Int J Clin Pharmacol Ther. 2000;38(11):532–9. doi: 10.5414/CPP38532. [DOI] [PubMed] [Google Scholar]
- 34.Chiou WL, Buehler PW. Comparison of oral absorption and bioavailability of drugs between monkey and human. Pharm Res. 2002;19(6):868–74. doi: 10.1023/A:1016169202830. [DOI] [PubMed] [Google Scholar]
- 35.Lennernäs H. Human intestinal permeability. J Pharm Sci. 1998;87(4):403–10. doi: 10.1021/js970332a. [DOI] [PubMed] [Google Scholar]
- 36.Sjöberg Å, Lutz M, Tannergren C, Wingolf C, Borde A, Ungell A-L. Comprehensive study on regional human intestinal permeability and prediction of fraction absorbed of drugs using the Ussing chamber technique. Eur J Pharm Sci. 2013;48(1):166–80. doi: 10.1016/j.ejps.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Ungell AL, Artursson P. An overview of Caco‐2 and alternatives for prediction of intestinal drug transport and absorption. In: Van de Waterbeemd H, Testa B, editors. Drug Bioavailability: Estimation of Solubility, Permeability, Absorption and Bioavailability. 40. 2nd ed. KGaA, Weinheim, Germany: Wiley-VCH Verlag GmbH & Co; 2009. p. 133–59.
- 38.Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:300–21. doi: 10.1016/j.ejps.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Parrott N, Lave T. Applications of physiologically based absorption models in drug discovery and development. Mol Pharm. 2008;5(5):760–75. doi: 10.1021/mp8000155. [DOI] [PubMed] [Google Scholar]
- 40.Sjögren E, Westergren J, Grant I, Hanisch G, Lindfors L, Lennernäs H, et al. In silico predictions of gastrointestinal drug absorption in pharmaceutical product development: application of the mechanistic absorption model GI-Sim. Eur J Pharm Sci. 2013;49(4):679–98. doi: 10.1016/j.ejps.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 41.Van de Waterbeemd H. In silico models to predict oral absorption. In: Taylor JB, Triggle DJ, editors. Comprehensive medicinal chemistry II. 5. Oxford: Elsevier Science Ltd; 2007. pp. 669–97. [Google Scholar]
- 42.Chiou WL, Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm Res. 1998;15(11):1792–5. doi: 10.1023/A:1011981317451. [DOI] [PubMed] [Google Scholar]
- 43.DeSesso JM, Williams AL. Contrasting the gastrointestinal tracts of mammals: factors that influence absorption. Annu Rep Med Chem. 2008;43:353–71. doi: 10.1016/S0065-7743(08)00021-3. [DOI] [Google Scholar]
- 44.Chiou WL, Jeong HY, Chung SM, Wu TC. Evaluation of using dog as an animal model to study the fraction of oral dose absorbed of 43 drugs in humans. Pharm Res. 2000;17(2):135–40. doi: 10.1023/A:1007552927404. [DOI] [PubMed] [Google Scholar]
- 45.Dressman JB. Comparison of canine and human gastrointestinal physiology. Pharm Res. 1986;3(3):123–31. doi: 10.1023/A:1016353705970. [DOI] [PubMed] [Google Scholar]
- 46.Becker D, Zhang J, Heimbach T, Penland RC, Wanke C, Shimizu J, et al. Novel orally swallowable IntelliCap® device to quantify regional drug absorption in human GI tract using diltiazem as model drug. AAPS PharmSciTech. 2014;15(6):1490–7. doi: 10.1208/s12249-014-0172-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He YL, Murby S, Warhurst G, Gifford L, Walker D, Ayrton J, et al. Species differences in size discrimination in the paracellular pathway reflected by oral bioavailability of poly (ethylene glycol) and D‐peptides. J Pharm Sci. 1998;87(5):626–33. doi: 10.1021/js970120d. [DOI] [PubMed] [Google Scholar]
- 48.Grime KH, Barton P, McGinnity DF. Application of in silico, in vitro and preclinical pharmacokinetic data for the effective and efficient prediction of human pharmacokinetics. Mol Pharm. 2013;10(4):1191–206. doi: 10.1021/mp300476z. [DOI] [PubMed] [Google Scholar]
- 49.McGinnity DF, Collington J, Austin RP, Riley RJ. Evaluation of human pharmacokinetics, therapeutic dose and exposure predictions using marketed oral drugs. Curr Drug Metab. 2007;8(5):463–79. doi: 10.2174/138920007780866799. [DOI] [PubMed] [Google Scholar]
- 50.Thomas VH, Bhattachar S, Hitchingham L, Zocharski P, Naath M, Surendran N, et al. The road map to oral bioavailability: an industrial perspective. Expert Opin Drug Metab Toxicol. 2006;2(4):591–608. doi: 10.1517/17425255.2.4.591. [DOI] [PubMed] [Google Scholar]
- 51.Sohlenius-Sternbeck A-K, Afzelius L, Prusis P, Neelissen J, Hoogstraate J, Johansson J, et al. Evaluation of the human prediction of clearance from hepatocyte and microsome intrinsic clearance for 52 drug compounds. Xenobiotica. 2010;40(9):637–49. doi: 10.3109/00498254.2010.500407. [DOI] [PubMed] [Google Scholar]
- 52.Sohlenius-Sternbeck A-K, Jones C, Ferguson D, Middleton BJ, Projean D, Floby E, et al. Practical use of the regression offset approach for the prediction of in vivo intrinsic clearance from hepatocytes. Xenobiotica. 2012;42(9):841–53. doi: 10.3109/00498254.2012.669080. [DOI] [PubMed] [Google Scholar]
- 53.Barter ZE, Bayliss MK, Beaune PH, Boobis AR, Carlile DJ, Edwards RJ, et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human micro-somal protein and hepatocellularity per gram of liver. Curr Drug Metab. 2007;8(1):33–45. doi: 10.2174/138920007779315053. [DOI] [PubMed] [Google Scholar]
- 54.Grime K, Riley R. The impact of in vitro binding on in vitro-in vivo extrapolations, projections of metabolic clearance and clinical drug-drug interactions. Curr Drug Metab. 2006;7(3):251–64. doi: 10.2174/138920006776359266. [DOI] [PubMed] [Google Scholar]
- 55.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6(2):140–8. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 56.Hewitt NJ, Gómez Lechón MJ, Houston JB, Hallifax D, Brown HS, Maurel P, et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39(1):159–234. doi: 10.1080/03602530601093489. [DOI] [PubMed] [Google Scholar]
- 57.Riley RJ, McGinnity DF, Austin RP. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab Dispos. 2005;33(9):1304–11. doi: 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]
- 58.Ito K, Houston JB. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 2005;22(1):103–12. doi: 10.1007/s11095-004-9015-1. [DOI] [PubMed] [Google Scholar]
- 59.Soars MG, Webborn PJ, Riley RJ. Impact of hepatic uptake transporters on pharmacokinetics and drug-drug interactions: use of assays and models for decision making in the pharmaceutical industry. Mol Pharm. 2009;6(6):1662–77. doi: 10.1021/mp800246x. [DOI] [PubMed] [Google Scholar]
- 60.FDA Approved Drug Products (2012) 2012. Retrieved 27th September 2012 from]. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm.
- 61.De Waziers I, Cugnenc P, Yang C, Leroux J, Beaune P. Cytochrome P450 isoenzymes, epoxide hydrolase and glutathione transferases in rat and human hepatic and extrahepatic tissues. J Pharmacol Exp Ther. 1990;253(1):387–94. [PubMed] [Google Scholar]
- 62.Nishimuta H, Nakagawa T, Nomura N, Yabuki M. Significance of reductive metabolism in human intestine and quantitative prediction of intestinal first-pass metabolism by cytosolic reductive enzymes. Drug Metab Dispos. 2013;41(5):1104–11. doi: 10.1124/dmd.113.051177. [DOI] [PubMed] [Google Scholar]
- 63.Ritter JK. Intestinal UGTs as potential modifiers of pharmacokinetics and biological responses to drugs and xenobiotics. Expert Opin Drug Metab Toxicol. 2007;3(1):93–107. doi: 10.1517/17425255.3.1.93. [DOI] [PubMed] [Google Scholar]
- 64.Thummel KE, Kunze KL, Shen DD. Enzyme-catalyzed processes of first-pass hepatic and intestinal drug extraction. Adv Drug Deliv Rev. 1997;27(2):99–127. doi: 10.1016/S0169-409X(97)00039-2. [DOI] [PubMed] [Google Scholar]
- 65.Kolars JC, Watkins P, Merion RM, Awni W. First-pass metabolism of cyclosporin by the gut. Lancet. 1991;338(8781):1488–90. doi: 10.1016/0140-6736(91)92302-I. [DOI] [PubMed] [Google Scholar]
- 66.Paine MF, Shen DD, Kunze KL, Perkins JD, Marsh CL, McVicar JP, et al. First‐pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther. 1996;60(1):14–24. doi: 10.1016/S0009-9236(96)90162-9. [DOI] [PubMed] [Google Scholar]
- 67.Floren LC, Bekersky I, Benet LZ, Mekki Q, Dressler D, Lee JW, et al. Tacrolimus oral bioavailability doubles with coadministration of ketoconazole. Clin Pharmacol Ther. 1997;62(1):41–9. doi: 10.1016/S0009-9236(97)90150-8. [DOI] [PubMed] [Google Scholar]
- 68.Glaeser H, Drescher S, Hofmann U, Heinkele G, Somogyi AA, Eichelbaum M, et al. Impact of concentration and rate of intraluminal drug delivery on absorption and gut wall metabolism of verapamil in humans. Clin Pharmacol Ther. 2004;76(3):230–8. doi: 10.1016/j.clpt.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 69.Lown KS, Bailey DG, Fontana RJ, Janardan SK, Adair CH, Fortlage LA, et al. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J Clin Investig. 1997;99(10):2545. doi: 10.1172/JCI119439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mistry M, Houston JB. Quantitation of extrahepatic metabolism. Pulmonary and intestinal conjugation of naphthol. Drug Metab Dispos. 1985;13(6):740–5. [PubMed] [Google Scholar]
- 71.van de Kerkhof EG, de Graaf IA, Groothuis GM. In vitro methods to study intestinal drug metabolism. Curr Drug Metab. 2007;8(7):658–75. doi: 10.2174/138920007782109742. [DOI] [PubMed] [Google Scholar]
- 72.Cubitt HE, Houston JB, Galetin A. Relative importance of intestinal and hepatic glucuronidation—impact on the prediction of drug clearance. Pharm Res. 2009;26(5):1073–83. doi: 10.1007/s11095-008-9823-9. [DOI] [PubMed] [Google Scholar]
- 73.Galetin A, Gertz M, Houston JB. Contribution of intestinal cytochrome P450-mediated metabolism to drug-drug inhibition and induction interactions. Drug Metab Pharmacokinet. 2010;25(1):28–47. doi: 10.2133/dmpk.25.28. [DOI] [PubMed] [Google Scholar]
- 74.Beddies G, Fox PR, Papich MD, Kanikanti V-R, Krebber R, Keene BW. Comparison of the pharmacokinetic properties of bisoprolol and carvedilol in healthy dogs. Am J Vet Res. 2008;69(12):1659–63. doi: 10.2460/ajvr.69.12.1659. [DOI] [PubMed] [Google Scholar]
- 75.Bueters T, Juric S, Sohlenius-Sternbeck A-K, Hu Y, Bylund J. Rat poorly predicts the combined non-absorbed and presystemically metabolized fractions in the human. Xenobiotica. 2012;43(7):607–16. doi: 10.3109/00498254.2012.752117. [DOI] [PubMed] [Google Scholar]
- 76.Furukawa H, Imventarza O, Venkataramanan R, Suzuki M, Zhu Y, Warty VS, et al. The effect of bile duct ligation and bile diversion on FK506 pharmacokinetics in dogs. Transplantation. 1992;53(4):722. doi: 10.1097/00007890-199204000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heykants J, Knaeps A, Meuldermans W, Michiels M. On the pharmacokinetics of domperidone in animals and man I. Plasma levels of domperidone in rats and dogs. Age related absorption and passage through the blood brain barrier in rats. Eur J Drug Metab Pharmacokinet. 1981;6(1):27–36. doi: 10.1007/BF03189513. [DOI] [PubMed] [Google Scholar]
- 78.Higuchi S, Shiobara Y. Comparative pharmacokinetics of nicardipine hydrochloride, a new vasodilator, in various species. Xenobiotica. 1980;10(6):447–54. doi: 10.3109/00498258009033779. [DOI] [PubMed] [Google Scholar]
- 79.Komura H, Iwaki M. In vitro and in vivo small intestinal metabolism of CYP3A and UGT substrates in preclinical animals species and humans: species differences. Drug Metab Rev. 2011;43(4):476–98. doi: 10.3109/03602532.2011.597401. [DOI] [PubMed] [Google Scholar]
- 80.Le Traon G, Burgaud S, Horspool L. Pharmacokinetics of cimetidine in dogs after oral administration of cimetidine tablets. J Vet Pharmacol Ther. 2009;32(3):213–8. doi: 10.1111/j.1365-2885.2008.01026.x. [DOI] [PubMed] [Google Scholar]
- 81.Walker D. Pharmacokinetics and metabolism of sildenafil in mouse, rat, rabbit, dog and man. Xenobiotica. 1999;29(3):297–310. doi: 10.1080/004982599238687. [DOI] [PubMed] [Google Scholar]
- 82.Prueksaritanont T, Gorham LM, Hochman JH, Tran LO, Vyas KP. Comparative studies of drug-metabolizing enzymes in dog, monkey, and human small intestines, and in Caco-2 cells. Drug Metab Dispos. 1996;24(6):634–42. [PubMed] [Google Scholar]
- 83.Takahashi M, Washio T, Suzuki N, Igeta K, Yamashita S. The species differences of intestinal drug absorption and first‐pass metabolism between cynomolgus monkeys and humans. J Pharm Sci. 2009;98(11):4343–53. doi: 10.1002/jps.21708. [DOI] [PubMed] [Google Scholar]
- 84.Nishimura T, Amano N, Kubo Y, Ono M, Kato Y, Fujita H, et al. Asymmetric intestinal first-pass metabolism causes minimal oral bioavailability of midazolam in cynomolgus monkey. Drug Metab Dispos. 2007;35(8):1275–84. doi: 10.1124/dmd.106.013037. [DOI] [PubMed] [Google Scholar]
- 85.Darwich A, Neuhoff S, Jamei M, Rostami-Hodjegan A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the “Advanced Dissolution, Absorption, Metabolism (ADAM)” model. Curr Drug Metab. 2010;11(9):716–29. doi: 10.2174/138920010794328913. [DOI] [PubMed] [Google Scholar]
- 86.Jeong EJ, Liu Y, Lin H, Hu M. Species-and disposition model-dependent metabolism of raloxifene in gut and liver: role of UGT1A10. Drug Metab Dispos. 2005;33(6):785–94. doi: 10.1124/dmd.104.001883. [DOI] [PubMed] [Google Scholar]
- 87.Furukawa T, Naritomi Y, Tetsuka K, Nakamori F, Moriguchi H, Yamano K, et al. Species differences in intestinal glucuronidation activities between humans, rats, dogs and monkeys. Xenobiotica. 2013;44(3):205–16. doi: 10.3109/00498254.2013.828362. [DOI] [PubMed] [Google Scholar]
- 88.Fisher MB, Labissiere G. The role of the intestine in drug metabolism and pharmacokinetics: an industry perspective. Curr Drug Metab. 2007;8(7):694–9. doi: 10.2174/138920007782109788. [DOI] [PubMed] [Google Scholar]
- 89.Wilson Z, Rostami‐Hodjegan A, Burn J, Tooley A, Boyle J, Ellis S, et al. Inter‐individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol. 2003;56(4):433–40. doi: 10.1046/j.1365-2125.2003.01881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hatley OJD. Mechanistic prediction of intestinal first-pass metabolism using in vitro data in preclinical species and in man [PhD thesis]. Manchester: The University of Manchester; 2014.
- 91.Cubitt HE, Houston JB, Galetin A. Prediction of human drug clearance by multiple metabolic pathways: integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab Dispos. 2011;39(5):864–73. doi: 10.1124/dmd.110.036566. [DOI] [PubMed] [Google Scholar]
- 92.Orzechowski A, Schrenk D, Bock-Hennig BS, Bock KW. Glucuronidation of carcinogenic arylamines and their N-hydroxy derivatives by rat and human phenol UDP-glucuronosyltransferases of the UGT1 gene complex. Carcinogenesis. 1994;15(8):1549–53. doi: 10.1093/carcin/15.8.1549. [DOI] [PubMed] [Google Scholar]
- 93.Ebner T, Burchell B. Substrate specificities of two stably expressed human liver UDP-glucuronosyltransferases of the UGT1 gene family. Drug Metab Dispos. 1993;21(1):50–5. [PubMed] [Google Scholar]
- 94.Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP-glucuronosyltransferase cofactors in alamethicin-activated human liver microsomes. Drug Metab Dispos. 2009;37(1):82–9. doi: 10.1124/dmd.108.023853. [DOI] [PubMed] [Google Scholar]
- 95.Bach P, Antonsson T, Bylund R, Björkman J-A. Österlund, Giordanetto F, et al. Lead optimization of ethyl 6-aminonicotinate acyl sulfonamides as antagonists of the P2Y12 receptor. Separation of the antithrombotic effect and bleeding for candidate drug AZD1283. J Med Chem. 2013;56(17):7015–24. doi: 10.1021/jm400820m. [DOI] [PubMed] [Google Scholar]
- 96.Nishimuta H, Houston JB, Galetin A. Hepatic, intestinal, renal, and plasma hydrolysis of prodrugs in human, cynomolgus monkey, dog, and rat: implications for in vitro-in vivo extrapolation of clearance of prodrugs. Drug Metab Dispos. 2014;42(9):1522–31. doi: 10.1124/dmd.114.057372. [DOI] [PubMed] [Google Scholar]
- 97.Crow JA, Borazjani A, Potter PM, Ross MK. Hydrolysis of pyrethroids by human and rat tissues: examination of intestinal, liver and serum carboxylesterases. Toxicol Appl Pharmacol. 2007;221(1):1–12. doi: 10.1016/j.taap.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rudakova E, Boltneva N, Makhaeva G. Comparative analysis of esterase activities of human, mouse, and rat blood. Bull Exp Biol Med. 2011;152(1):73–5. doi: 10.1007/s10517-011-1457-y. [DOI] [PubMed] [Google Scholar]
- 99.Peters SA. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet. 2008;47(4):261–75. doi: 10.2165/00003088-200847040-00004. [DOI] [PubMed] [Google Scholar]
- 100.Peters SA. Identification of intestinal loss of a drug through physiologically based pharmacokinetic simulation of plasma concentration-time profiles. Clin Pharmacokinet. 2008;47(4):245–59. doi: 10.2165/00003088-200847040-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structure features of AZ12470164. (GIF 3 kb)
Structure of AZD1283. (GIF 4 kb)
Structure of AZD7009. (GIF 3 kb)
Physiologically based PK simulations of PK profiles of AZD7009 in humans. (a) Simulation of the intravenous profile without enterohepatic recirculation. (b) Simulation of the intravenous profile with inclusion of enterohepatic recirculation. (c) Simulated oral profile against the observed when intestinal loss was not considered; the permeability used was 5.8 cm/s. (d) Simulated oral profile against the observed when intestinal loss was not considered; the permeability used was 1.9 cm/s. (e) Simulation of the oral profile with intestinal loss rate constants introduced and refinement with enterohepatic recirculation. (f) Simulation of the oral profile excluding enterohepatic recirculation and including intestinal loss. (g) Simulation of the oral profile including enterohepatic recirculation and excluding intestinal loss. Reprinted with permission (100). (GIF 35 kb)