

Abstract
Antibody-drug conjugates (ADCs) are a promising class of cancer therapeutics that combine the specificity of antibodies with the cytotoxic effects of payload drugs. A quantitative understanding of how ADCs are processed intracellularly can illustrate which processing steps most influence payload delivery, thus aiding the design of more effective ADCs. In this work, we develop a kinetic model for ADC cellular processing as well as generalizable methods based on flow cytometry and fluorescence imaging to parameterize this model. A number of key processing steps are included in the model: ADC binding to its target antigen, internalization via receptor-mediated endocytosis, proteolytic degradation of the ADC, efflux of the payload out of the cell, and payload binding to its intracellular target. The model was developed with a trastuzumab-maytansinoid ADC (TM-ADC) similar to trastuzumab-emtansine (T-DM1), which is used in the clinical treatment of HER2+ breast cancer. In three high-HER2-expressing cell lines (BT-474, NCI-N87, and SK-BR-3), we report for TM-ADC half-lives for internalization of 6–14 h, degradation of 18–25 h, and efflux rate of 44–73 h. Sensitivity analysis indicates that the internalization rate and efflux rate are key parameters for determining how much payload is delivered to a cell with TM-ADC. In addition, this model describing the cellular processing of ADCs can be incorporated into larger pharmacokinetics/pharmacodynamics models, as demonstrated in the associated companion paper.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-016-9892-3) contains supplementary material, which is available to authorized users.
KEY WORDS: antibody-drug conjugate, cellular trafficking, pharmacokinetics/pharmacodynamics, T-DM1, trastuzumab emtansine
INTRODUCTION
Antibody-drug conjugates (ADCs) are an emerging modality for cancer treatment, designed to selectively deliver chemotherapeutic payload drugs to tumor cells and reduce systemic toxicity. ADCs are comprised of an antibody specific to a cancer-associated antigen, a chemotherapeutic drug, and a linker to connect the antibody and drug payload. There are currently two FDA-approved ADCs available in the USA, brentuximab vedotin (Adcetris) and trastuzumab emtansine (T-DM1, Kadcyla) (1), with more than 30 ADCs in clinical trials (2). Key ADC design parameters include target antigen, antigen expression level (in normal tissue and tumor), linker type, conjugation site, conjugation chemistry, drug-to-antibody ratio (DAR), and payload drug potency (3,4).
Previous studies have shown that an ADC will traffic through the body very similarly to its parent antibody, unless the ADC has a high DAR (5). When an ADC reaches a tumor, the ADC binds its target antigen on the cancer cell surface. Next, the ADC is internalized via receptor-mediated endocytosis. Inside the endosomal/lysosomal compartments, the ADC is degraded and the payload is released from the antibody. The payload can then bind its intracellular target, resulting in cell death. These processing steps are widely accepted in the field (3, 6, 7), but they have not been combined in a complete quantitative model. Some pharmacokinetic/pharmacodynamic models for ADCs have been previously established (8–12); however, the focus of the current work is to develop a cellular level model that incorporates physiological processing of ADCs.
In order to build our model, we used a trastuzumab-maytansinoid antibody-drug conjugate (TM-ADC), similar to T-DM1, as the model ADC. The antibody component of T-DM1 is the antibody trastuzumab (Herceptin), which binds HER2, a member of the human epidermal growth factor receptor family that is often overexpressed on breast cancer cells (13). T-DM1 takes advantage of the therapeutic nature of the antibody itself; upon trastuzumab binding to HER2, downstream growth signaling is blocked. Additional cytotoxic effects are achieved with the payload component of T-DM1, emtansine (DM1), which is a potent microtubule-binding maytansine drug. DM1 is conjugated to lysine residues in trastuzumab via a non-cleavable linker.
A number of models have been developed previously to describe T-DM1 pharmacokinetics/pharmacodynamics (PK/PD) (14–19). However, these models have focused on PK/PD at an organism or tissue-specific level and do not incorporate the cellular-level mechanisms of ADC processing. For our model, we have focused on the cellular processing of ADCs, an area which is fundamental to the design and efficacy of ADCs. Understanding which intracellular processing steps influence ADC payload delivery, as well as how ADC design parameters affect the rate of these processing steps, may enable more rational design of safe and effective ADCs. The established model and parameters for TM-ADC intracellular processing described here have also been incorporated into a larger-scale PK/PD model as described in a companion paper.
MATERIALS AND METHODS
Cell Lines and Materials
BT-474, NCI-N87 (N87), and SK-BR-3 cell lines were obtained from ATCC. BT-474 and N87 cells were grown in RPMI 1640 medium (Corning) supplemented with 10% FBS and 1% penicillin-streptomycin. SK-BR-3 cells were grown in McCoy’s 5A Medium Modified, with l-Glutamine (Lonza) supplemented with 10% FBS and 1% penicillin-streptomycin. Trastuzumab labeled with Alexa Fluor 647 (Tras-647) was prepared as described previously (20). The trastuzumab-maytansinoid ADC (TM-ADC), which is structurally similar to T-DM1, was also prepared as described previously (21, 22). MATLAB software (Mathworks) was used for model predictions and parameter fits. GraphPad Prism software was also used for parameter fits. Flow cytometry was performed using a BD Accuri C6 Flow Cytometer.
Alexa Fluor 647 Labeling of TM-ADC (TM-ADC-647)
TM-ADC was labeled using an Alexa Fluor 647 Protein Labeling Kit (Life Technologies) following the product manual recommendations, with purification on an AKTA size exclusion chromatography system (GE Healthcare). The fluorophore to antibody ratio was 2–7.5 based on absorbance at 280 and 647 nm.
Model Development
We used standard biomolecular kinetic methods (23) to develop material balances for each species as given in Eqs. (1)–(6). The variables used in the model are as follows:
- [Ab]
Concentration of ADC in cell growth media (M)
- R
Number of free surface receptors (HER2) per cell (#/cell)
- C
Number of ADC-receptor complexes per cell (#/cell)
- I
Number of internalized, intact ADCs per cell (#/cell)
- D
Number of degraded ADCs per cell (#/cell)
- N
Concentration of cells in well (# cells/L)
The model parameters are as follows:
- kon
Association rate constant (h−1 M−1)
- koff
Dissociation rate constant (h−1)
- KD
Equilibrium dissociation constant (M)
- ke
Net internalization rate constant (h−1)
- kdeg
Degradation rate constant (h−1)
- kout
Efflux rate constant (h−1)
- μ
Cell growth rate (h−1)
- Vs
Receptor synthesis rate (#/(cell h))
- HER2
Total number of HER2 receptors per cell (#/cell)
- Nav
Avogadro’s number (6.02 × 1023 #/mol)
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
The terms k on[Ab]R and k off C represent the association of ADC with the surface receptor (HER2) and dissociation of ADC from receptor, respectively. The equilibrium dissociation constant, K D, is equal to k off/k on. The internalization of receptor or antibody-receptor complex is given by k e R or k e C, respectively. Note that there may be recycling of the receptor or antibody-receptor complex back to the cell surface; however, the internalization rate used here is the net internalization, i.e., the internalization in excess of that rapidly recycled back to the cell surface. As cells grow, their cellular contents are diluted with each cell division. The terms μR, μC, μI, and μD represent this dilution by growth. The degradation of the intact ADC and release of the payload is given by k deg I.
Once the payload is released from the antibody, the payload must escape the endosomal/lysosomal compartment before it can bind its intracellular target. Once in the cytosol, the payload may bind its intracellular target or may leave the cell. Within the parameters of the current experimental system, we could not directly measure payload escape from endosomal/lysosomal compartments. Thus, the model developed here is simplified and does not distinguish between payload in the cytosol and payload in endosomal/lysosomal compartments.
The term k out D represents the efflux of payload from the cell. The receptor synthesis rate, V s, is determined assuming a constant HER2 expression level and the steady state material balance (from Eq. (1)) for receptor with no ADC present; thus, V s = (μ + k e)HER2. Note that most of the species are described in units of “number per cell” to correspond with per cell measurements made by flow cytometry. Equations (1)–(4) can be converted to concentrations based on the concentration of cells in a manner similar to Eq. (5). Antibody in the media is described as a concentration (M) rather than a per cell basis.
Determination of KD and koff
To determine the apparent K D of trastuzumab, we treated fixed SK-BR-3 cells with a range (0.6–320 pM) of Tras-647 overnight at 37°C. Cells were fixed to prevent internalization. Cells were washed twice with 1 mL cold stain buffer (PBS, pH 7.4, 0.2% BSA, 0.09% sodium azide, filtered), and fluorescence signal was read via flow cytometry. We minimized depletion effects using a minimal number of cells and large suspension volumes.
To determine k off, we treated fixed cells (BT-474, N87, and SK-BR-3) with 10 nM TM-ADC-647 at least overnight at 37°C. At each time point (between 0 and 78 h), cells were washed with cold stain buffer and resuspended in stain buffer with 100 nM trastuzumab in order to compete with any TM-ADC-647 that had dissociated from cells. After the time course, all cells were washed with cold stain buffer and read on the flow cytometer.
Determination of HER2 Expression Levels
The HER2 expression levels for each cell line were determined using Quantum Simply Cellular anti-Human IgG Quantitation beads (Bangs Lab). Beads were prepared following the product manual and stained with 10 μL of Tras-647 to give a final concentration of 0.8 μM. Fixed cells were stained with 10 nM Tras-647 overnight at 37°C. Fixation was performed using Cytofix Buffer (BD Biosciences) at 4°C for 25 min as described in the product manual. The fluorescence signals for beads and cells (triplicate per cell line) were read via flow cytometry. Using the calibration spreadsheet provided by Bangs Lab, the average fluorescence intensity for each cell line was converted to number of HER2 receptors on the surface of each cell.
Determination of Cell Growth Rate
Cell growth rates for untreated cells were determined by plating 2 × 105 cells per well in six-well plates. At each time point, cells were washed with PBS, detached from the plate using 0.25% Trypsin/EDTA (Corning), pelleted, and resuspended in 250 μL of PBS supplemented with 5% FBS. To each sample, 50 μL of CountBright Absolute Counting Beads (Life Technologies) was added. The cell counts were determined via flow cytometry using gating on forward scatter (FSC) and side scatter (SSC). The average of the triplicates for each time point was used to fit an exponential growth rate.
Determination of Net Internalization Rate
The methods used to measure the net internalization rates were adapted from those published previously (24–26). To determine what fraction of the total signal from Tras-647 or TM-ADC-647 was from surface-bound antibody rather than internalized antibody, we used an antihuman antibody rather than acid stripping or quenching antibodies. In 24-well plates, 105 cells per well were plated and left to adhere overnight. Cells were treated with 10–20 nM of Tras-647 or TM-ADC-647 for time points between 0–9 h. Based on the dissociation and association rates, this concentration range ensures a rapid equilibration rate, with the resulting equilibrium favoring saturated surface receptors. After treatment, cells were washed once with PBS and then detached from the plate using 0.25% Trypsin/EDTA. Cells were pelleted at 1000×g for 5 min and then resuspended in stain buffer with 10 μL of Alexa Fluor 488 Goat anti-Human IgG (H+L) (Life Technologies). Cells were incubated at 4°C on a rotator for 30 min and then washed twice with 500 μL of stain buffer. The mean fluorescence intensity (MFI) was measured via flow cytometry. This MFI was normalized as described in the next paragraph.
In order to determine the Alexa Fluor 647 signal which corresponds to fully saturated surface receptors, an additional 105 cells per cell line were fixed to prevent internalization. The fixed cells were then stained with 10–20 nM Tras-647 or TM-ADC-647 for at least 1 h at 37°C. The difference in MFI of the stained fixed cells versus unstained fixed cells was used to normalize the Alexa Fluor 647 signal for cells treated for internalization. New cells were fixed and stained at the same time as each experimental replicate to account for any variations in HER2 expression level. To normalize the Alexa Fluor 488 signal, the average of the Alexa Fluor 488 signal (besides the initial time point) was considered a fully saturated surface. The internalized fraction was determined by subtracting the normalized Alexa Fluor 488 signal (surface-bound antibody) from the normalized Alexa Fluor 647 signal (total antibody). A global fit of the data from triplicate independent experiments was used to determine the net internalization rate. Equation 7 demonstrates the linear function used for the fit.
| 7 |
To test whether non-specific uptake is significant, cells were treated for at least 20 min with 800 nM (40-fold excess) or 500 nM (25-fold excess) of unlabeled trastuzumab or unlabeled TM-ADC, respectively. After pre-treatment, Tras-647 or TM-ADC-647 was added to a final concentration of 20 nM. At various time points, the cells were washed and the Alexa Fluor 647 MFI was measured using flow cytometry.
Determination of Degradation Rate
Degradation rate was measured using a time course of cell lysate samples prepared from cells treated with TM-ADC-647. In six-well tissue culture plates, 105 cells were plated and allowed to adhere overnight. Then cells were treated for 30 min with 10 nM TM-ADC-647 at 37°C. Cells were washed twice with PBS, and media were replaced with fresh media. At each time point, cells were washed once with PBS, and 100 μL of ice-cold cell lysis buffer (150 nM NaCl, 50 mM Tris-HCl, 1% Triton X-100 plus freshly added proteases inhibitors, “cOmplete, mini, EDTA-free Protease Inhibitor Cocktail Tablets” (Roche), with one tablet per 10 mL buffer) was added to each well. Cells were scraped from the well, and the suspension of cells in lysis buffer was transferred to a micro-centrifuge tube. Samples were placed on a rotator at 4°C for 30 min, centrifuged at 12,000 rpm for 20 min, and the resulting supernatant was stored at 4°C.
After all time points were collected, 12 μL of each sample was mixed with 3 μL of non-reducing, no dye SDS loading buffer (0.125 M Tris-HCl, 0.35 M sodium dodecyl sulfate, 50% by volume glycerol). From this mixture, 10 μL was added to each lane in a 4–12% Bis-Tris Protein Gel (Life Technologies). Gels were run in MOPS buffer at 250 V for 15 min. They were then imaged for Alexa Fluor 647 signal using a Typhoon Imager (GE). Intact antibody bands were quantified using ImageJ software (NIH). Data were normalized to the initial time point, which was taken immediately after the treatment period. Using the model described in the model development section, the degradation rate was fit by minimizing the difference between data and model predictions for the sum of C, intact antibody in complex with HER2 on the surface of the cell, and I, the intact (non-degraded) antibody inside the cell. Since the cell lysate samples measure from the population of cells rather than individual cells, the total intact antibody from all cells (C × N, #/L) was used to compare the model predictions and data.
Determination of Efflux Rate
The efflux rate was determined using the total fluorescence signal in cells over time as measured by flow cytometry. Cells were plated in six-well tissue culture plates (105 cells per well) and allowed to adhere overnight. Then cells were treated for 30 min with 10 nM TM-ADC-647 at 37°C. Cells were washed twice with PBS, and media were replaced with fresh media. At each time point, cells were washed once with PBS, detached from the plate using 0.25% Trypsin/EDTA, pelleted, and resuspended in PBS supplemented with 5% FBS. Total Alexa Fluor 647 fluorescence signal was read via flow cytometry and normalized to the fluorescence signal at the initial time point, immediately after treatment. Using the complete model described in the “Model Development” section, the efflux rate was fit by minimizing the measured normalized total fluorescence signal and the normalized total amount of TM-ADC in cells from the model. The total amount of TM-ADC is the sum of TM-ADC in complex with HER2 on the surface of the cell (C), internalized intact TM-ADC (I), and degraded products (D).
Loss of fluorescence signal in cells is mainly due to efflux of degraded products and dilution by growth. To ensure an accurate fit of the efflux rate constant, independent of dilution by growth, we measured the cell growth rate (μ) during each experiment using counting beads and fit using an exponential growth model.
Sensitivity Analysis
To determine the model sensitivity to each of the model parameters, we calculated the local sensitivity based on 10% perturbations from the established parameters as described by Eq. (8). The area under the curve (AUC) for the degraded products (payload) at different parameter values, k i, was calculated and the difference normalized to the AUC at the established parameter values. The treatment regimen used for determining AUC was 10 days at surface saturating concentrations of ADC (10 nM ADC).
| 8 |
The parameters k e and HER2 were analyzed as one parameter since these parameters do not act independently under saturating antibody conditions.
To define the length of time required to reach steady state, we used the time at which the concentration of degraded antibody inside the cell was equal to 95% of the concentration of degraded antibody after 100 days of treatment, with antibody concentration in the media maintained at 10 nM (saturating for the cell surface) and no cell growth.
Incorporation of Payload Binding to Target
Payload binding to target can be incorporated in the model as shown in Eq. (9), where k PL ‐ Targeton is the association rate constant for payload (DM1) binding to its intracellular target (tubulin) in (#/cell)−1 h−1, k PL ‐ Targetoff is the dissociation rate constant in h−1, T is the amount of target (tubulin) in cells in #/cell, and Q is the number of drug-target complexes per cell.
| 9 |
For these analyses, we used the following previously reported values (8, 27): K PL ‐ TargetD(=k PL ‐ Targeton/k PL ‐ Targetoff) of 930 nM, k PL ‐ Targeton of 0.44 M−1 h−1, and T of 65 nM. To convert the amount of payload drug (D) from #/cell to an intracellular concentration, we assumed the cell volume was 1000 μm3.
RESULTS
Model Development
Figure 1 illustrates the model schema for this work. With the model equations established, we proceeded to parameterize the model. Parameters were measured in a sequential manner in order to guide the design of experiments for rate constant measurements for later processing steps. The apparent equilibrium binding constant, K D, measured via a cell-based assay was 38 ± 16 pM, as illustrated in Supplemental Fig. 1A. The measured dissociation rate constant, k off, was 0.014 ± 0.016 h−1, as illustrated in Supplemental Fig. 1B. Flow cytometry quantitation beads were used with Tras-647 to determine the HER2 expression levels. The measured HER2 expression levels for each cell line were 2.71 × 106, 3.25 × 106, and 3.55 × 106 HER2/cell for BT-474, N87, and SK-BR-3 cells, respectively. We observed some variability in the precise expression level with time in culture. These HER2 expression levels are similar to those reported previously for these cell lines (28–30). In addition, the untreated cell growth rate was 0.013 ± 0.003, 0.019 ± 0.007, and 0.011 ± 0.002 h−1 for BT-474, N87, and SK-BR-3 cells, respectively, as shown in Supplemental Fig. 2A.
Fig. 1.
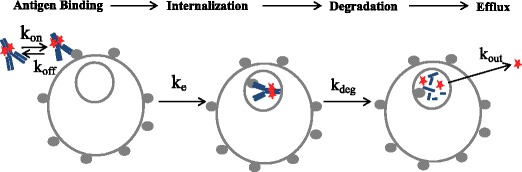
Schematic of kinetic model for ADC cellular processing, including ADC association, dissociation, internalization, degradation, and efflux. Model parameter descriptions are provided in the “MATERIALS AND METHODS” section, under “Model Development”
Determination of Internalization Rate Constant
The net internalization rate constant, k e, was determined for both trastuzumab and TM-ADC, using Tras-647 and TM-ADC-647, respectively. The Alexa Fluor 647 signal from labeled trastuzumab or TM-ADC was used as a measure of total antibody in the cell, i.e., both on the surface and internalized within cells. The amount of surface-bound antibody was detected using an Alexa Fluor 488 antihuman antibody. In order to correlate the Alexa Fluor 647 and Alexa Fluor 488 signal, both signals were normalized to that of cells with saturated surface receptors. The difference in the normalized signal between the total antibody and surface-bound antibody is the signal arising from internalized antibody.
Figure 2a depicts a representative example of the total, surface-bound, and internalized signal versus time for cells treated with TM-ADC-647. The unbound HER2 and TM-ADC quickly equilibrate between the initial time point and the 1.5-h time point. The surface-bound signal remains constant after 1.5 h, indicating there is little downregulation of HER2 during this time period, as observed previously (31), and that there is no depletion of ADC in the media. Within the 9-h time course, we assume the rate of degradation is negligible compared to the rate of internalization. Tests of non-specific uptake showed that less than 2% of the total Alexa Fluor 647 signal measured for unblocked cells was observed with cells that were pre-blocked with unlabeled trastuzumab or unlabeled TM-ADC.
Fig. 2.
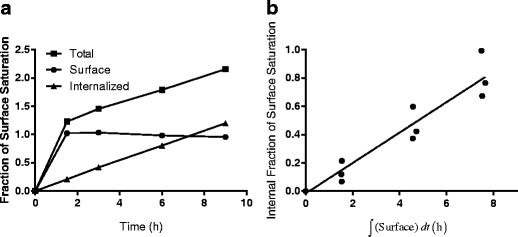
Determination of internalization rate constant, k e. a Representative plot of the normalized Alexa Fluor 647 signal (total antibody), normalized Alexa Fluor 488 signal (surface-bound antibody), and internalized (total–surface) antibody versus time for BT-474 cells treated with 10 nM TM-ADC-647 and stained with an Alexa Fluor 488 antihuman antibody. The y-axis is fraction of the normalized surface saturation level, which is either Alexa Fluor 647 or Alexa Fluor 488 MFI normalized as described in the “MATERIALS AND METHODS” section. b Fit of internalization rate using the internalized fraction of TM-ADC-647 versus surface integral as given by Eq. 7. A representative plot for TM-ADC-647 internalization in BT-474 cells is shown here. The equivalent plots for other cell lines and Tras-647 are shown in Supplemental Fig. 3. Fit values for the internalization rate constants for Tras-647 and TM-ADC-647 are presented in Table I
Figure 2b illustrates the global fit of triplicate experiments for BT-474 cells treated with TM-ADC-647 based on the surface integral and internalized fraction from plots such as Fig. 2a. The equivalent graphs for other cell lines are shown in Supplemental Fig. 3. A summary of the net internalization rates, k e (±95% confidence intervals), measured for three different cell lines are shown in Table I. The half times, t 1/2, for internalization, which were calculated using t 1/2 = ln(2)/k e, are also shown. The range spans the 95% confidence intervals of the net internalization rate.
Table I.
Net Internalization Rates (k e) and Half-Lives (t 1/2) for Tras-647 and TM-ADC-647
| Tras-647 | TM-ADC-647 | Significantly different? p value | |||
|---|---|---|---|---|---|
| Cell line | k e(h− 1) | t 1/2(h) | k e(h− 1) | t 1/2(h) | |
| BT-474 | 0.054 ± 0.007 | 12.8 | 0.11 ± 0.02 | 6.3 | <0.0001 |
| NCI-N87 | 0.035 ± 0.008 | 19.8 | 0.051 ± 0.006 | 13.6 | <0.01 |
| SK-BR-3 | 0.043 ± 0.005 | 16.1 | 0.09 ± 0.01 | 7.7 | <0.000001 |
Determination of Degradation Rate Constant
In TM-ADC, DM1 is conjugated to trastuzumab via a non-cleavable linker, succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC). Thus, the drug metabolite of TM-ADC is lysine-N ε-SMCC-DM1, which is the payload, linker, and residual amino acid (lysine) to which the linker payload was conjugated (32,33). This metabolite results from complete proteolytic degradation of the antibody component of TM-ADC in lysosomal compartments after internalization. Thus, the degradation rate we measure describes the rate of proteolytic degradation of the antibody, which results in release of the payload.
In order to measure the degradation rate constant, k deg, we developed a gel-based imaging assay. Cell lysate samples were collected at different time points (0–130 h) after cells were treated for 30 min with 10 nM TM-ADC-647. These samples were then run on a non-reducing SDS-PAGE gel, which was imaged for fluorescence. The fluorescence signal from the intact antibody was quantified. Figure 3a depicts a typical gel image with BT-474 cell lysate samples collected from different time points (0–130 h) after treatment. The higher band corresponds to full antibody, as confirmed by running samples in a gel with a protein ladder, as illustrated in Supplemental Fig. 4. The main band at approximately 150 kDa seen in Supplemental Fig. 4 corresponds to intact full antibody, based on comparison to the protein ladder and the positive control of TM-ADC-647 in lysis buffer (lane 4). The signal at the very bottom runs at the small molecule front and includes Alexa Fluor 647 lysine that has been released via degradation of the ADC. In addition, some minor bands are seen which correspond to aggregates (>200 kDa) and the dissociated heavy (50 kDa) and light (25 kDa) chains of the antibody.
Fig. 3.
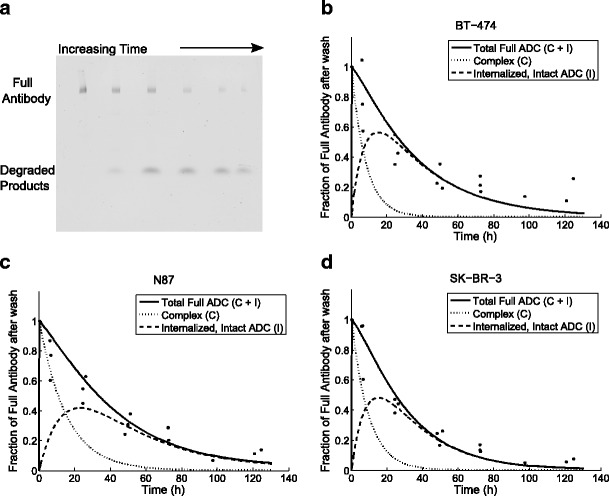
Determination of degradation rate constant, k deg. Image of native SDS-PAGE gel with cell lysate samples over 0–130 h after BT-474 cells were treated for 30 min with 10 nM TM-ADC-647 (a). The full antibody at each time point was quantified from images such as this. The decay over time of the full antibody signal was used to fit the degradation rate constant for BT-474 (b), N87 (c), and SK-BR-3 cells (d). The full antibody signal is the sum of the full antibody in complex with receptors on the cell surface and the intact antibody that has been internalized into the cell but not yet degraded. The model predictions for these two species are shown in dashed lines as indicated by the legend. Data points are from triplicate independent experiments
Only the total full antibody was quantified from gels such as Fig. 3a. The total full antibody is the sum of both antibody on the cell surface in complex with HER2 and intact antibody that has been internalized. The predicted contributions of both of these components to the total antibody signal are shown in dashed lines in Fig. 3b, c, d. The amount of internalized, intact ADC in the cells increases initially due to internalization of ADC in complex with HER2 and then decreases due to degradation of the ADC. The antibody in complex on the cell surface decreases due to antibody internalization and dissociation. The experimental setup was chosen to isolate the process of degradation as much as possible. By briefly dosing cells with TM-ADC-647, we quickly saturate the HER2 receptors on the cell surface. At later time points, there is no longer ADC on the surface to be internalized and the decay in signal comes from degradation. In Fig. 3b, c, d, the fit curves for BT-474, N87, and SK-BR-3, respectively, are shown. The degradation rate was fit using the total intact antibody signal, normalized to the initial signal from cells collected immediately after wash at the end of the 30-min treatment period. The degradation rate constants and half-lives are shown in Table II. The degradation rate of TM-ADC-647 is similar across the three cell lines tested, with half-lives on the order of 1 day.
Table II.
Degradation Rates (k deg) and Half-Lives (t 1/2) for TM-ADC-647
| Cell line | k deg(h− 1) | t 1/2(h) |
|---|---|---|
| BT-474 | 0.03 ± 0.01 | 23.3 |
| NCI-N87 | 0.027 ± 0.008 | 25.4 |
| SK-BR-3 | 0.038 ± 0.009 | 18.0 |
Determination of Efflux Rate Constant
With the internalization and degradation rate constants established, we next turned to measurement of the efflux rate constant, k out, which describes the rate at which the payload metabolite exits the cell after the ADC is internalized and degraded. This model parameter encompasses a number of possible mechanisms for payload release from the cell, including passive efflux, such as diffusion of payload across the cell membrane, and active efflux, such as pumping of the payload out of the cell via multidrug resistance pumps. Since endosomal/lysosomal escape was not included as a separate parameter in this model, the efflux rate includes this escape rate in series with either passive or active efflux. Efflux of payload from the cell may also be due to lysosomal fusion with the cell membrane (34) or exosomes (35–37). A recent study of residualization rates showed a surprising similarity of efflux rate for a number of different fluorophores (38), suggesting that fluorophore efflux mechanisms may be independent of fluorophore structure and characteristics.
To determine the efflux rate constant, we tracked the total cell fluorescence over time using flow cytometry following a 30-min treatment period with TM-ADC-647 to saturate the surface receptors. The loss of total fluorescence signal over time is due to dissociation of surface-bound ADC, efflux of fluorophore metabolites from degraded ADCs, and dilution by growth. Internalization and degradation change the form of the ADC, but do not decrease the total fluorescence signal due to ADC in the cell. Using the complete model, which takes into account the contributions from dissociation and dilution by growth, we fit the efflux rate based on decay of the total cell fluorescence over time. Here, we tracked efflux of the fluorophore metabolite as a proxy for the maytansinoid metabolite. Figure 4a, b, c shows the curves used to fit the efflux rate constant for degraded products from cells treated with TM-ADC-647. The cell growth rate was measured during each experimental replicate as illustrated in Supplemental Fig. 2B-D. The fit efflux rate constants and corresponding half-lives are listed in Table III.
Fig. 4.
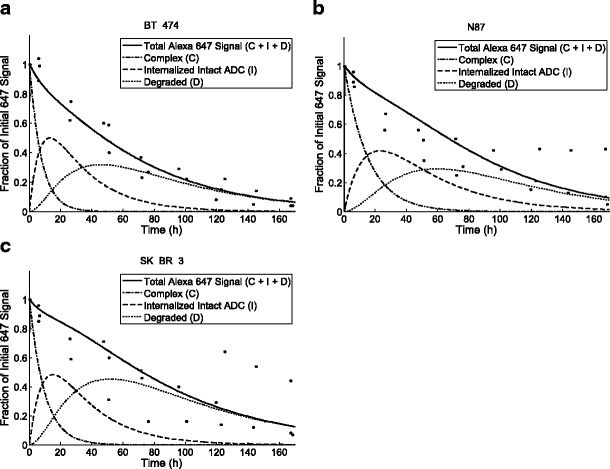
Determination of efflux rate constant, k out. The decay over time of the total fluorescence signal as measured by flow cytometry from cells treated with 10 nM TM-ADC-647. The fit curves are shown for BT-474 (a), N87 (b), and SK-BR-3 cells (c). The total fluorescence signal is the sum of the signal from antibody in complex with receptors on the cell surface (c), intact ADC (I), and degraded products (d). The model predictions for these species are shown as indicated in the legend for each graph. Data points are from triplicate independent experiments
Table III.
Efflux Rates (k out) and Half-Lives (t 1/2) of Metabolites for TM-ADC-647
| Cell line | k out(h− 1) | t 1/2(h) |
|---|---|---|
| BT-474 | 0.009 ± 0.004 | 75.3 |
| NCI-N87 | 0.022 ± 0.009 | 31.7 |
| SK-BR-3 | 0.015 ± 0.006 | 45.3 |
Sensitivity Analysis
Once we established all of the model parameters, we performed a local sensitivity analysis in order to determine which parameters have the largest impact on the amount of payload delivered into cells. Figure 5 illustrates the model sensitivity for each of the model parameters for cells treated with TM-ADC for 10 days at surface saturating conditions, which is physiologically relevant for cancer patients treated with tumor-targeting antibodies (8, 9). Figure 5a includes dilution by cell growth assuming a growth rate equal to that of untreated cells. Alternatively, if a sufficiently large quantity of payload is delivered, then cell growth would cease; Fig. 5b presents the same sensitivity analysis, but with no cell growth (μ = 0). In both cases, the internalization rate (k eHER2) and efflux rate (k out) are key parameters for determining how much payload is delivered to cells.
Fig. 5.
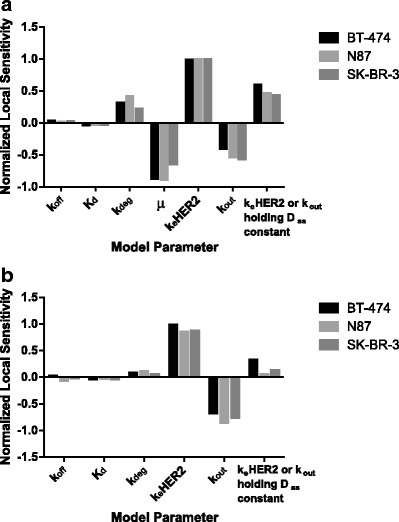
Local sensitivity analysis for model parameters (a) with cell growth rate (μ) equal to untreated cell growth rate or (b) with no cell growth. Sensitivity was calculated based on variations in the area under the curve for released payload after 10 days of treatment with 10 nM TM-ADC-647 with 10% perturbations in the indicated model parameter
Another way to evaluate how effectively an ADC delivers payload to a cell is to consider the payload concentration within cells at steady state with constant exposure to ADC. Assuming sufficiently high ADC concentration to saturate HER2 receptors on the cell surface, the expression for steady state payload concentration is given in Eq. (10).
| 10 |
Assuming no cell growth in addition to sufficiently high ADC concentration to saturate HER2 receptors on the cell surface, the steady state expression of payload drug is simplified to Eq. (11).
| 11 |
Equation 11 illustrates the crucial balance between the amount of drug that enters the cell via internalization and that which leaves the cell. This expression also demonstrates that expression level and internalization rate do not act independently of one another, rather the product of the two dictates the amount of ADC internalized. Although the amount of payload at steady state (D ss) captures the key parameters, it is important to note that it would take 8–15 days for cells to reach steady state with continuous exposure to surface saturating levels of ADC, based on the parameters measured for TM-ADC-647 in the three cell lines tested as described in the “MATERIALS AND METHODS” section. Supplemental Fig. 5 illustrates the amount of each species in the cell over time to reach steady state. The number of slow processing steps results in this long approach to steady state. Figure 5a, b also includes the model sensitivity to modifications of k eHER2 and k out when holding D ss constant. For the case with no cell growth (Fig. 5b), although the model is sensitive to the internalization rate (k eHER2) and efflux rate (k out) independently, it is relatively insensitive to changes to these parameters if D ss is held constant.
Incorporation of Payload Binding to Target
Another processing step we have incorporated into the model is payload binding to its intracellular target. DM1 binding to its target, tubulin, provides an additional sink that could reduce the amount of payload that effluxes from cells. The balance between target binding and efflux has been demonstrated previously with d and l isomers of the maytansinoid DM4 (32). The K D for DM1 binding to microtubules has been measured experimentally (27), and the on rate and concentration of tubulin in a tumor have been estimated via a large scale PK/PD model (8).
Based on the developed model and parameter estimates, the concentration of payload metabolites in the cell reaches 1–3 μM after 1 day of treatment at surface saturating concentrations of TM-ADC. This concentration of payload metabolite is in the range of previously reported IC50 values for DM1 inhibition of microtubule growth (27) and experimentally determined catabolite concentrations for other antibody-SMCC-DM1 conjugates (39). At these concentrations, the quantity of DM1 present in a cell is 50–2500 times greater than the number of tubulin-binding sites, which is on the order of 1000–10,000 sites per cell (8, 40). Thus, accounting for payload binding to target does not dramatically affect the free payload concentration in the cell. However, it is important to note these calculations assume all of the drug payload catabolite escapes the lysosome and is in the cytosol. As others have suggested (39, 41), it is possible that some payload metabolite may be trapped in endosomal/lysosomal compartments. In addition, the payload may non-specifically bind to other intracellular proteins. Thus, free payload concentration in the cytosol may be lower than the concentration of degraded ADC species in this model; however, free payload concentration in the cytosol is the relevant value to dictating how much payload ultimately reaches its target.
DISCUSSION
In this work, we have developed a model for the cellular processing of ADCs, and we have reported generalizable methods to measure the model parameters. A trastuzumab-maytansinoid ADC (TM-ADC), which is similar to a clinically relevant ADC, T-DM1 (Kadcyla), was used to establish this model. For TM-ADC, we found the internalization rate to be moderately relative to other antibodies (42) (half-life of 6–14 h), the degradation rate to be slower than internalization (half-life of 18–25 h), and the efflux rate to be the slowest rate (half-life of 32–75 h).
The association rate constant (k on) and equilibrium dissociation constant (K D) are parameters that can be tuned based on the antibody component of the ADC. Typical values for k on for a protein-protein interaction are 105 M−1s−1, and K D ranges from 10−12 to 10−6 (43). On the other hand, the net internalization rate constant (k e) depends on both the antigen target as well as the antibody itself. For example, trastuzumab internalizes based on natural HER2 internalization and recycling, whereas other antibodies induce rapid HER2 downregulation due to internalization upon binding (31). The net internalization rate can range from 10−3 to 1 h−1 (43). The degradation rate constant (k deg), which describes how quickly the payload is released from the antibody, is highly dependent on the linker design. For instance, an ADC with a pH-sensitive or protease-cleavable linker will likely degrade more quickly than a non-cleavable linker.
The receptor expression level (HER2) and receptor synthesis rate (Vs) both vary with antigen target. Receptor expression level can range from 103 to 106 (3). Often, high receptor expression is considered necessary for an ADC to be effective. From the cellular processing perspective, the product of receptor expression level and net internalization (k eHER2) drives how much drug is being delivered into a cell. Thus, a lower receptor expression level could be compensated for by more rapid internalization. However, it is also important to note the impact that antigen expression and internalization have on tumor penetration (42).
Recent work has shown that internalization is not required to effectively deliver payload via an ADC (44, 45). Rather than payload entering a cell via receptor-mediated endocytosis of the antibody component of the ADC, the payload may be released from the ADC outside the cell and then enter the cell via passive diffusion or active uptake via transporters. In this model, we did not account for free payload diffusion into the cell and instead focused on classical receptor-mediated delivery. Since ADC treatment periods were brief-pulse treatments, excess ADCs in the culture media that could generate large amounts of free payload were not present. Depending on the stability of the ADC in the extracellular space as well as the concentration of ADC in tumor, diffusion of the payload into the cell could contribute significantly to the amount of payload delivered to a cell. The permeability of the payload catabolite, as well as the catabolite’s interactions with transporters, will dictate how readily the payload enters the cell from the extracellular space.
The chemical structure of the payload catabolite may differ depending on whether the ADC is degraded in endosomal/lysosomal compartments within the cell or in the extracellular space. The structure is also highly dependent on the linker design. As previous studies have demonstrated (32), different linker designs can result in different catabolites for the same payload; these payload catabolites may, in turn, have widely different abilities to penetrate surrounding cells via the bystander effect. Payload catabolite permeability may also affect the payload’s ability to escape from endosomal/lysosomal compartments. Although a minimally permeable payload may diffuse more slowly out of a cell, thus improving the chances of cell killing, it may also become trapped in the endosomal/lysosomal compartments, thus reducing the bystander effect.
The model developed here provides a framework to compare the rates of cellular processing of ADCs in order to determine what the rate-limiting steps are for payload delivery via an ADC. When considering how to optimize ADC efficacy, it is crucial to understand how these various cellular processing steps relate to one another, as the relationships may be non-intuitive. This work highlights the importance of evaluating cellular processing steps in the context of the entire system rather than individually. The framework developed here could help guide decisions during the drug development process in order to optimize the performance of a candidate ADC; importantly, the methods developed here are generalizable for any ADC candidate.
In order to track the processing of TM-ADC, we used Alexa Fluor 647-labeled TM-ADC. The use of a fluorescent label offers a number of advantages: the label enables tracking of the ADC in a quantitative manner; fluorescent labels can be easily applied to different ADCs of interest; fluorescence signal can be measured using multiple approaches; and fluorescent labeling is safer than radiolabeling, a common alternative. On the other hand, fluorescence labeling also has disadvantages, including susceptibility to photobleaching and environmental sensitivity; however, Alexa fluorophores are relatively stable and environmentally insensitive. An additional caveat to note is that the addition of any type of label may perturb the structure and behavior of an ADC.
At a single-cell level, efflux of payload from cells is not ideal, considering that the desired outcome after ADC treatment is the payload binding to its target to cause cell death. However, on the scale of a whole tumor, efflux of payload could be beneficial due to the so-called bystander effect (32, 46). Cell killing via the bystander effect involves a tumor cell taking up an ADC, then releasing free drug payload into the surroundings, where it can diffuse freely into nearby cells. The bystander effect can affect both tumor cells and stroma.
We hypothesize that the escape of an ADC drug payload from endosomes and lysosomes is a key factor that affects how much payload actually reaches its intracellular target. Our analysis of intracellular payload concentrations indicates that if endosomal escape is not limited, then the concentration of DM1 in the cell is similar to the IC50 for DM1 binding to tubulin when cells are treated for 1 day with T-DM1 at cell surface saturating conditions. However, if only 10% of the payload metabolite escapes endosomes, then it would take ∼four times longer for cells to reach intracellular payload concentrations equal to the IC50. A more detailed understanding of how different payloads escape the endosomal/lysosomal compartments could improve ADC design for more efficient payload delivery. Recent studies demonstrate that transporters can be involved in payload escape from endosomal/lysosomal compartments (47) and present methods to enrich for lysosomes in cellular fractions in order to study payload concentrations in lysosomes (48).
One limitation of our analysis is that we were unable to track the payload, DM1, itself once it was separated from the antibody component of TM-ADC. Instead, we tracked efflux of the fluorophore metabolite as a proxy for the DM1 metabolite. This assumption is reasonable given that the molecular weight and hydrophobicity of the fluorophore metabolite and DM1 metabolite are similar; in TM-ADC-647, both DM1 and the Alexa Fluor 647 dye were attached to trastuzumab via lysine residues. The use of fluorescent drug payloads or fluorescent drug analogs could be better suited for studying payload trafficking. However, fluorescent drug analogs could be processed differently by cells than the parent drugs depending on the modifications, and they are generally challenging to access synthetically. In ongoing work, we are studying ADCs bearing fluorescent drug payloads to enable tracking of the actual payload metabolite.
In conclusion, a quantitative understanding of ADC cellular processing allows one to compare the rates at which different processing steps occur and appreciate how these rates are related to one another. This level of understanding may be useful for improving ADC design. The cellular mechanisms of ADC processing can be integrated into larger PK/PD models, as described in the associated companion paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Curve fits for (A) the apparent KD of Tras-647 on SK-BR-3 cells and (B) koff for Tras-647 on BT-474, N87, and SK-BR-3 cells. (GIF 26 kb)
{kind=link}
Cell growth rates for (A) untreated cells and (B-D) treated cells during efflux rate experiments. For the treated cells, a cell growth rate was only fit for BT-474 cells because the other cell lines did not demonstrate growth. (GIF 47 kb)
{kind=link}
Determination of internalization rate constants for Tras-647 and TM-ADC-647 in three cell lines (BT-474, N87, and SK-BR-3). The linear fit equations are reported for each fit at the top of the graph. Data points represent triplicate independent experiments. (GIF 125 kb)
{kind=link}
Image of native SDS-PAGE gel with cell lysate samples from cells treated for 30 min with 10 nM TM-ADC-647. Lanes are as follows: L – ladder, 1, 2, 3 – cell lysate from BT-474, N87, SK-BR-3 cells (respectively) 19 h after treatment with TM-ADC-647, 4 – positive control of TM-ADC-647 in cell lysis buffer. (GIF 14 kb)
{kind=link}
Plot of species quantity in cells over time as steady state is approached. The species (antibody in complex on the cell surface, internalized antibody, and degraded antibody) over time are shown for three cell lines: (A) BT-474, (B) N87, and (C) SK-BR-3. The vertical dashed line corresponds to the time at which steady state is reached as defined in the methods section. Note that one degraded antibody corresponds to the release of the DAR of drug molecules, i.e. one degraded antibody equals release of two drug molecules if the ADC has a DAR of 2. (GIF 50 kb)
Acknowledgments
We thank Lindsay King, Nahor Haddish-Berhane, and members of the Wittrup Lab for their technical suggestions. For the gift of the trastuzumab-maytansinoid ADC (TM-ADC), we are grateful to the Pfizer Oncology Bioconjugation group, including William Hu, Ellie Muszynska, Nadira Prashad, Kiran Khandke, and Frank Loganzo. K.F.M. was supported by a Hertz Foundation Fellowship and a National Science Foundation Graduate Research Fellowship. C.K. was supported by the Pfizer Worldwide Research & Development Post-Doctoral Program. This work was also supported by a research grant from Pfizer and in part by the Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute. We thank the Koch Institute Swanson Biotechnology Center for the technical support, specifically the Flow Cytometry Core.
References
- 1.Zolot RS, Basu S, Million RP. Antibody–drug conjugates. Nat Rev Drug Discov [Internet] 2013;12(4):259–260. doi: 10.1038/nrd3980. [DOI] [PubMed] [Google Scholar]
- 2.Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. mAbs. 2014;6(1):34–45. doi: 10.4161/mabs.27022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14(3):154–169. doi: 10.1097/PPO.0b013e318172d704. [DOI] [PubMed] [Google Scholar]
- 4.McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J [Internet]. 2015;(6). Available from: http://link.springer.com/10.1208/s12248-014-9710-8 [DOI] [PMC free article] [PubMed]
- 5.Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res [Internet]. 2004 Oct 15 [cited 2012 Mar 31];10(20):7063–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15501986. [DOI] [PubMed]
- 6.Kovtun Y V, Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett [Internet]. 2007 Oct 8 [cited 2012 Apr 26];255(2):232–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17553616. [DOI] [PubMed]
- 7.Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res [Internet]. 2014 Jan [cited 2014 Oct 9];16(2):209. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4058749&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 8.Shah DK, Haddish-Berhane N, Betts A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab-vedotin. J Pharmacokinet Pharmacodyn [Internet]. 2012 Dec [cited 2012 Dec 10];39(6):643–59. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23151991. [DOI] [PubMed]
- 9.Shah DK, King LE, Han X, Wentland J-A, Zhang Y, Lucas J, et al. A priori prediction of tumor payload concentrations: preclinical case study with an auristatin-based anti-5t4 antibody-drug conjugate. AAPS J [Internet]. 2014 Mar 1 [cited 2014 Apr 1]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24578215. [DOI] [PMC free article] [PubMed]
- 10.Sukumaran S, Gadkar K, Zhang C, Bhakta S, Liu L, Xu K, et al. Mechanism-based pharmacokinetic/pharmacodynamic model for THIOMABTM drug conjugates. Pharm Res [Internet]. 2014;1884–93. Available from: http://link.springer.com/10.1007/s11095-014-1582-1 [DOI] [PubMed]
- 11.Singh AP, Shin YG, Shah DK. Application of pharmacokinetic-pharmacodynamic modeling and simulation for antibody-drug conjugate development. Pharm Res [Internet]. 2015;32(11):3508–25. Available from: http://link.springer.com/10.1007/s11095-015-1626-1 [DOI] [PubMed]
- 12.Sadekar S, Figueroa I, Tabrizi M. Antibody drug conjugates: application of quantitative pharmacology in modality design and target selection. AAPS J [Internet]. 2015;17(4):828–36. Available from: http://link.springer.com/10.1208/s12248-015-9766-0 [DOI] [PMC free article] [PubMed]
- 13.Burris H a, Tibbitts J, Holden SN, Sliwkowski MX, Lewis Phillips GD. Trastuzumab emtansine (T-DM1): a novel agent for targeting HER2+ breast cancer. Clin Breast Cancer [Internet]. 2011 Oct [cited 2012 Jul 15];11(5):275–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21729661. [DOI] [PubMed]
- 14.Wada R, Erickson HK, Lewis Phillips GD, Provenzano C a, Leipold DD, Mai E, et al. Mechanistic pharmacokinetic/pharmacodynamic modeling of in vivo tumor uptake, catabolism, and tumor response of trastuzumab maytansinoid conjugates. Cancer Chemother Pharmacol [Internet]. 2014 Nov 4 [cited 2014 Oct 1];74(5):969–80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25186956. [DOI] [PubMed]
- 15.Erickson HK, Lewis Phillips GD, Leipold DD, Provenzano C a, Mai E, Johnson H a, et al. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther [Internet]. 2012 May [cited 2012 Aug 6];11(5):1133–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22408268. [DOI] [PubMed]
- 16.Jumbe NL, Xin Y, Leipold DD, Crocker L, Dugger D, Mai E, et al. Modeling the efficacy of trastuzumab-DM1, an antibody drug conjugate, in mice. J Pharmacokinet Pharmacodyn [Internet]. 2010 Jun [cited 2014 Jan 12];37(3):221–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20424896. [DOI] [PubMed]
- 17.Chudasama VL, Schaedeli Stark F, Harrold JM, Tibbitts J, Girish SR, Gupta M, et al. Semi-mechanistic population pharmacokinetic model of multivalent trastuzumab emtansine in patients with metastatic breast cancer. Clin Pharmacol Ther [Internet]. 2012;92(4):520–7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3745717&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 18.Lu D, Joshi A, Wang B, Olsen S, Yi JH, Krop IE, et al. An integrated multiple-analyte pharmacokinetic model to characterize trastuzumab emtansine (T-DM1) clearance pathways and to evaluate reduced pharmacokinetic sampling in patients with HER2-positive metastatic breast cancer. Clin Pharmacokinet. 2013;52(8):657–72 [DOI] [PubMed]
- 19.Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69(5):1229–1240. doi: 10.1007/s00280-011-1817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maass KF, Kulkarni C, Quadir MA, Hammond PT, Betts AM, Wittrup KD. A flow cytometric clonogenic assay reveals the single-cell potency of doxorubicin. J Pharm Sci [Internet]. 2015 Sep;n/a – n/a. Available from: http://doi.wiley.com/10.1002/jps.24631. [DOI] [PMC free article] [PubMed]
- 21.Chari RVJ, Martell BA, Gross JL, Gross L, McKenzie SJ, Goldmacher VS, et al. Immunoconjugates containing novel maytansinoids: promising anticancer drugs. Cancer Res. 1992;52(1):127–131. [PubMed] [Google Scholar]
- 22.Loganzo F, Tan X, Sung M, Jin G, Myers JS, Melamud E, et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol Cancer Ther [Internet]. 2015;14(April):952–64. Available from: http://mct.aacrjournals.org/cgi/doi/10.1158/1535-7163.MCT-14-0862 [DOI] [PubMed]
- 23.Linderman J, Lauffenburger D. Receptors: models for binding, trafficking, and signaling. Oxford: Oxford University Press; 1993. [Google Scholar]
- 24.Lund KA, Opresko LK, Starbuck C, Walsh BJ, Wiley HS. Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J Biol Chem. 1990. [PubMed]
- 25.Schmidt MM, Thurber GM, Wittrup KD. Kinetics of anti-carcinoembryonic antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer Immunol Immunother [Internet]. 2008 Dec [cited 2011 Jun 29];57(12):1879–90. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2840397&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed]
- 26.Harper J, Mao S, Strout P, Kamal A. Selecting and optimal antibody for antibody-drug conjugate therapy: internalization and intracellular localization. In: Ducry L, editor. Methods in Molecular Biology [Internet]. Totowa, NJ: Humana Press; 2013 [cited 2014 Apr 1]. p. 41–9. Available from: http://link.springer.com/10.1007/978-1-62703-541-5. [DOI] [PubMed]
- 27.Lopus M, Oroudjev E, Wilson L, Wilhelm S, Widdison W, Chari R, et al. Maytansine and cellular metabolites of antibody-maytansinoid conjugates strongly suppress microtubule dynamics by binding to microtubules. Mol Cancer Ther [Internet]. 2010 Oct [cited 2014 Apr 1];9(10):2689–99. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2954514&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 28.Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin a-M, et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif [Internet]. 2007 Aug;40(4):580–94. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17635524. [DOI] [PMC free article] [PubMed]
- 29.Hendriks BS, Klinz SG, Reynolds JG, Espelin CW, Gaddy DF, Wickham TJ. Impact of tumor HER2/ERBB2 expression level on HER2-targeted liposomal doxorubicin-mediated drug delivery: multiple low-affinity interactions lead to a threshold effect. Mol Cancer Ther [Internet]. 2013 Sep [cited 2014 Jan 12];12(9):1816–28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23723124. [DOI] [PubMed]
- 30.Pillow TH, Tien J, Parsons-reponte KL, Bhakta S, Li H, Staben LR, et al. Site-specific trastuzumab maytansinoid antibody–drug conjugates with improved therapeutic activity through linker and antibody engineering. 2014. [DOI] [PubMed]
- 31.Austin CD, Maziere AM, Pisacane PI, van Dijk SM, Eigenbrot C, Sliwkowski MX, et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell. 2004;15:5268–5282. doi: 10.1091/mbc.E04-07-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erickson HK, Park PU, Widdison WC, Kovtun Y V, Garrett LM, Hoffman K, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res [Internet]. 2006 Apr 15 [cited 2012 Mar 30];66(8):4426–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16618769. [DOI] [PubMed]
- 33.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res [Internet]. 2008 Nov 15 [cited 2014 Jan 10];68(22):9280–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19010901. [DOI] [PubMed]
- 34.Rodríguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2 + -regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137(1):93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safaei R, Larson BJ, Cheng TC, Gibson M a, Otani S, Naerdemann W, et al. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol Cancer Ther. 2005;4(10):1595–604 [DOI] [PubMed]
- 36.Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS One. 2014;9(2). [DOI] [PMC free article] [PubMed]
- 37.Brinton LT, Sloane HS, Kester M, Kelly KA. Formation and role of exosomes in cancer. Cell Mol Life Sci [Internet] 2014;72(4):659–671. doi: 10.1007/s00018-014-1764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cilliers C, Liao J, Atangcho L, Thurber GM. Residualization rates of near-infrared dyes for the rational design of molecular imaging agents. Mol Imaging Biol [Internet]. 2015; Available from: http://link.springer.com/10.1007/s11307-015-0851-7 [DOI] [PMC free article] [PubMed]
- 39.Singh R, Salomon PL. A sensitive ELISA method for the measurement of catabolites of antibody-drug conjugates (ADCs) in target cancer cells. Mol Pharm [Internet]. 2015;150304113735005. Available from: http://pubs.acs.org/doi/abs/10.1021/acs.molpharmaceut.5b00028 [DOI] [PubMed]
- 40.Spiegelman BM, Lopata M a, Kirschner MW. Multiple sites for the initiation of microtubule assembly in mammalian cells. Cell. 1979;16(2):239–52 [DOI] [PubMed]
- 41.Loganzo F, Tan X, Sung M, Jin G, Myers JS, Melamud E. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol Cancer Ther. 2015. [DOI] [PubMed]
- 42.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev [Internet]. 2008 Sep [cited 2014 Dec 11];60(12):1421–34. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2820307&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 43.Thurber GM, Dane Wittrup K. A mechanistic compartmental model for total antibody uptake in tumors. J Theor Biol [Internet]. Elsevier; 2012 Dec 7 [cited 2012 Dec 3];314:57–68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22974563. [DOI] [PMC free article] [PubMed]
- 44.Perrino E, Steiner M, Krall N, Bernardes GJL, Pretto F, Casi G, et al. Curative properties of non-internalizing antibody-drug conjugates based on maytansinoids. Cancer Res [Internet]. 2014 Feb 11 [cited 2014 Apr 1]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24520075 [DOI] [PubMed]
- 45.Casi G, Neri D. Noninternalizing targeted cytotoxics for cancer therapy. Mol Pharm [Internet] 2015;12(6):1880–1884. doi: 10.1021/mp500798y. [DOI] [PubMed] [Google Scholar]
- 46.Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res [Internet]. 2010 Feb 1 [cited 2014 Mar 28];16(3):888–97. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20086002. [DOI] [PubMed]
- 47.Hamblett KJ, Jacob AP, Gurgel JL, Tometsko ME, Rock BM, Patel SK, et al. SLC46A3 is required to transport catabolites of noncleavable antibody maytansine conjugates from the lysosome to the cytoplasm. Cancer Res [Internet] 2015;75(24):5329–5340. doi: 10.1158/0008-5472.CAN-15-1610. [DOI] [PubMed] [Google Scholar]
- 48.Rock BM, Tometsko ME, Patel SK, Hamblett KJ, Fanslow WC, Rock DA. Intracellular catabolism of an antibody drug conjugate with a noncleavable linker. Drug Metab Dispos [Internet] 2015;43(9):1341–1344. doi: 10.1124/dmd.115.064253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Curve fits for (A) the apparent KD of Tras-647 on SK-BR-3 cells and (B) koff for Tras-647 on BT-474, N87, and SK-BR-3 cells. (GIF 26 kb)
Cell growth rates for (A) untreated cells and (B-D) treated cells during efflux rate experiments. For the treated cells, a cell growth rate was only fit for BT-474 cells because the other cell lines did not demonstrate growth. (GIF 47 kb)
Determination of internalization rate constants for Tras-647 and TM-ADC-647 in three cell lines (BT-474, N87, and SK-BR-3). The linear fit equations are reported for each fit at the top of the graph. Data points represent triplicate independent experiments. (GIF 125 kb)
Image of native SDS-PAGE gel with cell lysate samples from cells treated for 30 min with 10 nM TM-ADC-647. Lanes are as follows: L – ladder, 1, 2, 3 – cell lysate from BT-474, N87, SK-BR-3 cells (respectively) 19 h after treatment with TM-ADC-647, 4 – positive control of TM-ADC-647 in cell lysis buffer. (GIF 14 kb)
Plot of species quantity in cells over time as steady state is approached. The species (antibody in complex on the cell surface, internalized antibody, and degraded antibody) over time are shown for three cell lines: (A) BT-474, (B) N87, and (C) SK-BR-3. The vertical dashed line corresponds to the time at which steady state is reached as defined in the methods section. Note that one degraded antibody corresponds to the release of the DAR of drug molecules, i.e. one degraded antibody equals release of two drug molecules if the ADC has a DAR of 2. (GIF 50 kb)