

Abstract
The human diseases melioidosis and glanders are caused by the bacteria Burkholderia pseudomallei and B. mallei respectively, and both species are regarded as potential biowarfare agents. We used B. pseudomallei DNA microarrays to compare the genomes of several clinical and environmental isolates of B. pseudomallei, B. mallei, and B. thailandensis, a closely related but avirulent species. Open reading frames (ORFs) deleted between the three species were associated with diverse cellular functions, including nitrogen and iron metabolism, quorum sensing, and polysaccharide production. Deleted ORFs in B. mallei exhibited significant genomic clustering, whereas deletions in B. thailandensis were more uniformly dispersed, suggesting that B. mallei and B. thailandensis may have diverged from B. pseudomallei and each other via distinct mechanisms. The genomes of independent B. pseudomallei isolates were highly conserved with a large-scale variance of less than 3% between isolates, and at least three distinct molecular subtypes could be defined. An analysis of subtype-specific genomic regions suggests that DNA loss has played an important role in the evolutionary radiation of B. pseudomallei in the natural environment. Our results raise several hypotheses concerning the possible mechanisms underlying the diverse biological properties exhibited by members of the Burkholderia family.
The Gram-negative bacterium Burkholderia pseudomallei is an environmental saprophyte endemic to many parts of South East Asia and Northern Australia. Infections by this organism can cause the human disease melioidosis—a clinically serious condition characterized by severe pulmonary distress with frequent progression to septicemia and death (Dance 1991, 2000; Dharakul and Songsivilai 1999). In areas where this bacterium is wide-spread, B. pseudomallei infections have been estimated to be responsible for up to 40% of all mortalities due to septicemia (Suputtamongkol et al. 1994). Related to B. pseudomallei is B. mallei, the causative agent of glanders—a primarily soliped (horses, mules, and donkeys) disease which can also occur in human subjects with occupational exposure to glanderous animals (McGilvray 1944). In addition to being public health hazards, B. pseudomallei and B. mallei are also potential biowarfare agents (USA CDC, Category B) (Rotz et al. 2002), and B. mallei may already have been used in this regard (Wheelis 1998). Although B. pseudomallei possesses a comparatively large bacterial genome size (7.2 Mb), there is currently a relative lack of knowledge regarding the molecular mechanisms that underlie B. pseudomallei behavior and virulence. There is thus an urgent need to better understand the Burkholderia family, in particular B. pseudomallei and B. mallei, and the biological mechanisms that underlie their interaction with the environment and with human hosts.
In recent years, DNA microarrays coupled with comparative genomics has emerged as a powerful experimental approach to address questions of strain evolution, pathogen detection, and transcriptional changes resulting from genetic and environmental perturbations (Wilson et al. 1999; Laub et al. 2000; Wei et al. 2001; Dziejman et al. 2002). In the present study, we used B. pseudomallei DNA microarrays to perform comparative genomic hybridization experiments on various natural isolates of B. pseudomallei, B. mallei, and the related but avirulent species B. thailandensis (Brett et al. 1998) to elucidate their genomic relationships. Our analysis yielded several novel discoveries. First, we identified a large number of open reading frames (ORFs) that were deleted in B. mallei and B. thailandensis compared to B. pseudomallei. These ORFs were associated with a wide variety of metabolic and cellular functions, which might contribute to the various species-specific behaviors exhibited by these bacteria. Second, we discovered that deletions in B. mallei exhibited a significantly higher tendency to cluster in the genome compared to B. thailandensis, suggesting that B. mallei may have diverged from B. pseudomallei via the loss of large chromosomal segments. Third, we found that the B. pseudomallei strains in our study could be divided into at least three distinct molecular subtypes, and identified several gene clusters (12) that could discriminate between the subtypes. As 10 of these 12 clusters are also present in both B. mallei and B. thailandensis, this suggests that DNA loss has played an important role in the evolutionary radiation of B. pseudomallei in the natural environment. In addition to being the first reported use of B. pseudomallei microarrays in the literature, our results provide insights into the genetic relationships among the various Burkholderia species, and constitute a useful framework upon which subsequent molecular work on B. pseudomallei and other members of the Burkholderia species can be interpreted.
Results
Creation of a B. pseudomallei DNA microarray
Beginning with unannotated shotgun sequence information, we designed and fabricated a B. pseudomallei microarray consisting of amplified DNA fragments representing ∼6800 putative ORFs in the B. pseudomallei genome (see Methods). This number most likely represents a slight overestimate of the number of transcribed genes in B. pseudomallei (Songsivilai and Dharakul 2000). To minimize potential cross-hybridization artifacts, each array probe was designed to be highly specific to a particular ORF with minimal homology to other predicted ORFs. After primer design and synthesis, genomic DNA from B. pseudomallei reference strain K96243 was used as a template to PCR-amplify all 6895 probes. Using a uniform set of PCR amplification conditions, we achieved a success rate of ∼92%, where the amplification reaction yielded a single discrete product of the expected molecular size (Supplemental material). The success rate of PCR amplification is comparable to that obtained for other bacterial and eukaryotic species (DeRisi et al. 1997; Dziejman et al. 2002).
The genomic coverage of the B. pseudomallei microarray was determined by `tiling' the array probes against the assembled B. pseudomallei genome sequence (completed in late 2002). On average, there were 9–12 array probes for every 10 kb of chromosomal DNA, corresponding to ∼1 array probe every 1000 bp. There were only eight 10-kb regions containing only a single array probe, and the largest `uncovered' region of the genome was a single 18-kb region on Chromosome 2 (position 2207216 to 2225527) (Supplemental material). This finding indicates that with these few exceptions, the DNA microarray provides relatively dense and uniform saturation of the entire B. pseudomallei genome.
Identification of ORFs which are differentially present among B. pseudomallei, B. mallei, and B. thailandensis
The first major objective of this project was to define general patterns of genomic variance among the different Burkholderia species, and specifically to identify genes exhibiting variation between B. mallei or B. thailandensis compared to B. pseudomallei. We isolated genomic DNAs from 23 Burkholderia strains (Table 1), including 18 strains of B. pseudomallei (excluding K96243), two strains of B. mallei, and three strains of the related but clinically avirulent species B. thailandensis, and performed microarray comparative genomic hybridizations using genomic DNA from a `test' strain and the reference strain K96243 (see Methods). The individual microarray hybridizations were grouped according to their respective species, and a multistep procedure was implemented to identify ORFs deleted between the different species at high confidence with minimal false positives (i.e., ORFs that are erroneously called `deleted'; Fig. 1). Throughout this manuscript, the phrases `deleted' and `deletions' have been used in the generic sense to reflect the status of various ORFs in B. mallei and B. thailandensis with respect to the B. pseudomallei genome, and not in the specific evolutionary sense to imply that the differential presence of these ORFs among the three species arose through gene loss.
Table 1.
Burkholderia strains and species used
| Species | Strain name | Source of isolation | Sex/age/race, others |
|---|---|---|---|
| B. pseudomallei | K96243 (Reference) | Thai human clinical isolate | — |
| B. pseudomallei | #3 | Singapore human clinical isolate (Blood) | Male/55/Chinese, Alive |
| B. pseudomallei | #9 | Singapore human clinical isolate (Pus) | Male/19/Malay, NS man, Alive |
| B. pseudomallei | #11 | Singapore human clinical isolate (Blood) | Male/10/Chinese, Died |
| B. pseudomallei | #14 | Singapore human clinical isolate (Pus) | Male/64/Malay, Alive |
| B. pseudomallei | #22 | Singapore human clinical isolate (Sputum) | Male/19/Chinese/NS man, Died |
| B. pseudomallei | #23 | Singapore human clinical isolate (Blood) | Male/19/Chinese/NS man, Died |
| B. pseudomallei | #33 | Singapore human clinical isolate (Sputum) | Male/20/Malay, Alive |
| B. pseudomallei | #59 | Singapore human clinical isolate (Urine) | Male/77/Chinese, Died |
| B. pseudomallei | ATCC 23343 | Type strain (Human) | Unknown |
| B. pseudomallei | 15—10 | Singapore soil isolate | — |
| B. pseudomallei | 78/96 | Singapore soil isolate | — |
| B. pseudomallei | ATCC 15682 | Type strain (Monkey) | — |
| B. pseudomallei | 5/96 | Pig | — |
| B. pseudomallei | 15/96 | Pig | — |
| B. pseudomallei | 20/96 | Pig | — |
| B. pseudomallei | 21/96 | Pig | — |
| B. pseudomallei | 497/96 | Pig | — |
| B. pseudomallei | 567/96 | Pig | — |
| B. thailandensis | ATCC 700388 | Type strain Thai soil isolate (rice field) | |
| B. thailandensis | S95019 | Thai human clinical isolate | |
| B. thailandensis | TRF 681 (Thai A) | Thai soil isolate | |
| B. mallei | ATCC 23344 | Type strain | |
| B. mallei | ATCC 10399 | Type strain |
Figure 1.

Identification on differentially present ORFs across different Burkholderia species and strains. (A–C) Log-log scatter plots of fluorescence ratios of (A) the average B. pm (Burkholderia pseudomallei) profile (x-axis) vs. an individual B. pm sample (y-axis), (B) the average B. pm profile vs. the average B. mallei profile, and (C) the average B. pm profile vs. the average B. thailandensis profile. Red spots denote deletion with 99.99% confidence, green (amplification with 99.99% confidence), and blue (deletion or amplification with 99% confidence, deleted spots lying to the left of the diagonal axis, and amplified spots to the right). (D,A) Fluorescence ratios of various Burkholderia species plotted against chromosomal position. Red denotes the average B. pm, green (B. mallei), and blue (B. thailandensis). The left figure indicates a genomic region that is present in all three species. The right figure illustrates a separate genomic region that is completely deleted in both B. mallei and B. thailandensis.
Briefly, we first utilized a t-statistic at various confidence levels (95%–99.99%) to identify ORFs whose fluorescence ratios were significantly different between the groups being compared (e.g., B. pseudomallei vs. B. thailandensis). Second, the experimental variance associated with repeated microarray measurements was computed using the hybridization data of the 18 individual B. pseudomallei isolates, and the maximum experimental variance was used to define appropriate fluorescence ratio threshold values for deleted, present, and amplified ORFs. Third, an ORF was classified as `deleted' or `amplified' if its fluorescence ratio in the two groups being compared was both significantly different by t-statistic, and within the appropriate threshold zones (i.e., a `deleted' or `amplified' ORF could only be compared to a `present' one; see Methods). Advantages of this combined approach include the assignment of a specific confidence value to each ORF, and also the use of fluorescence threshold boundaries that are based on true experimental variance. This approach, however, also carries two important caveats. First, by deliberately focusing on identifying commonly deleted ORFs in isolates of each species compared to B. pseudomallei, a subset of ORFs that vary between different isolates of the same species may have been missed (the entire microarray data set is downloadable for readers interested in such intraspecies genetic variation). Second, the `appropriate' confidence threshold to be used can often depend upon the specific analytical method used. In addition to using t-tests, we also analyzed the microarray data set using intensity dependent z-scores, an alternative methodology (Sprinthall 1996), and found that although both approaches (t-test and z-scores) identified many of the same ORFs as being differentially present, higher confidence levels were invariably assigned to these ORFs using the z-scores method (Supplemental material). In this report, we confined any in-depth analysis to ORFs that are differentially deleted at a >99.99% confidence threshold, so as to minimize the number of potential false-positive results (i.e., ORFs that are falsely called `deleted').
To experimentally validate this strategy, several randomly selected ORFs identified as being significantly different at 99.99% confidence in different comparisons were selected and tested for their absence or presence in the various Burkholderia strains using conventional PCR. As shown in Figure 2, there was good agreement between the array predictions and the PCR assay (see Supplemental material for additional validations). In addition, although this report also does not focus on the issue of `amplified' ORFs, preliminary quantitative PCR experiments suggest that these may represent regions of genome duplication (Supplemental material).
Figure 2.

Experimental and phenotypic validation of array results. (A) Deletions in B. mallei. Lanes 1–8: B. pseudomallei K96243, #3, KHW22, ATCC15682, and ATCC23343 (P), B. thailandensis ATCC700388 (T), B. mallei ATCC10399 and ATCC23344 (M). Left gel shows the deletion of ORF 3548902 in both B. mallei isolates ATCC10399 and ATCC23344 only. Right gel shows deletion of ORF 3551702 in both B. mallei isolates. (B) Deletions in B. thailandensis.. Lanes 1–8: B. pseudomallei K96243, #3, KHW22, ATCC15682, and ATCC23343 (P), B. thailandensis ATCC700388 (T), B. mallei ATCC10399 and ATCC23344 (M). Left gel shows the deletion of the ORF 3534302 in B. thailandensis ATCC700388 only. Right gel shows deletion of ORF 3534002 in B. thailandensis isolate ATCC700388 only. (C) Deletions found in both B. mallei and B. thailandensis. Lanes 1–8: B. pseudomallei K96243, #3, KHW22, ATCC15682, and ATCC23343 (P), B. thailandensis ATCC700388 (T), B. mallei ATCC10399 and ATCC23344 (M). Left gel shows the deletion of ORF 3490802 in B. thailandensis ATCC700388 and both B. mallei isolates ATCC10399 and ATCC23344. Right gel shows deletion of ORF 3528002 in B. thailandensis ATCC700388 and B. mallei ATCC10399 only. (D) B. pseudomallei intraspecies variations. Lanes 1–5: B. pseudomallei K96243, #3, KHW22, ATCC15682, and ATCC23343. Left gel shows the deletion of ORF 3433202 in all four B. pseudomallei isolates except B. pseudomallei K96243. Right gel shows deletion of ORF 3542302 in B. pseudomallei isolates #3, KHW22, and ATCC15682 but not in K96243 and ATCC23343. (E) Iron metabolism in Burkholderia species. Siderophore production was assessed via CAS assays (27) for bacteria grown in iron-deprived (left, –Fe) and iron-supplemented (right, + Fe) media. From left to right: Control with CAS solution and growth media (–), B. pseudomallei strains (ATCC 23343 and ATCC 15682) (P), B. mallei strains (ATCC 23344 and ATCC 10399) (M), and B. thailandensis strain ATCC 700388 (T). Color changes from blue to orange/pink indicate the presence of siderophores. The strongest color changes are observed in B. pseudomallei followed by B. thailandensis, with B. mallei exhibiting comparatively weaker activity.
Deleted ORFs in B. mallei exhibit preferential genomic clustering compared to B. thailandensis
A comparable number of ORFs were deleted in both B. mallei and B. thailandensis compared to B. pseudomallei (Table 2). At a `high-confidence' threshold of 99.99%, 344 and 304 ORFs were deleted in B. mallei and B. thailandensis, respectively, while 812 (B. mallei) versus 969 (B. thailandensis) ORFs were deleted at a lower confidence level of 95%. The difference in the numbers of deleted ORFs between the two species (B. mallei and B. thailandensis) was not significant (P=0.603, by Student's t-test). Consistent with other reports, several genes which were previously known to be absent in B. mallei compared to B. pseudomallei, such as the Type III secretion system cluster TTSS1 (Rainbow et al. 2002), and a serine metalloprotease (Lee and Liu 2000) were also reflected as `deleted' in the microarray data (P. Tan, unpubl.). In addition, genes that had been previously reported to be deleted in B. thailandensis compared to B. pseudomallei, such as TTSS1 (Rainbow et al. 2002), and a Type I O-PS polysaccharide capsular gene cluster (Reckseidler et al. 2001) were also reported as `deleted' by the microarray as well (P. Tan, unpubl.), indicating that there is good agreement between the microarray data and the published literature.
Table 2.
Analysis of flanking probe deletions at >95% and >99.99% confidence levels for B. mallei and B. thailandensis, compared to B. pseudomallei, using t-tests.
|
Probability (%) of flanking deletions
|
|||||
|---|---|---|---|---|---|
| Species | Coverage (% confidence) | No. of probes | Two sides | One side | Isolated |
| B. mallei | Whole genome (>95%) | 812 | 59.24 | 32.51 | 8.25 |
| Whole genome (>99.99%) | 344 | 59.01 | 33.72 | 7.27 | |
| Chr 1 (>95%) | 246 | 48.78 | 37.40 | 13.82 | |
| Chr 1 (>99.99%) | 106 | 45.28 | 42.45 | 12.26 | |
| Chr 2 (>95%) | 566 | 63.78 | 30.39 | 5.83 | |
| Chr 2 (>99.99%) | 238 | 65.13 | 29.83 | 5.04 | |
| B. thailandensis | Whole genome (>95%) | 969 | 41.49 | 35.09 | 23.43 |
| Whole genome (>99.99%) | 304 | 49.01 | 33.22 | 17.76 | |
| Chr 1 (>95%) | 380 | 41.84 | 30.53 | 27.63 | |
| Chr 1 (>99.99%) | 109 | 46.79 | 32.11 | 21.10 | |
| Chr 2 (>95%) | 589 | 41.26 | 38.03 | 20.71 | |
| Chr 2 (>99.99%) | 195 | 50.26 | 33.85 | 15.90 | |
For intensity-dependent z-score results, please see Supplemental material for a more detailed analysis
The B. pseudomallei genome consists of two chromosomes (1 and 2). For both B. mallei and B. thailandensis, there was a strong preference for deleted ORFs to be associated with Chromosome 2 rather than Chromosome 1 (70% of all deleted ORFs for B. mallei and 61% for B. thailandensis, at 95% deletion confidence). In the case of B. mallei, an initial visual inspection of the microarray data revealed that the majority of these deletions appeared to cluster together in the genome, rather than being uniformly dispersed (P. Tan, unpubl.). To quantitate this observation, we calculated the frequency at which ORFs flanking a `high-confidence' deleted ORF were also themselves deleted for both B. mallei and B. thailandensis. This was done in a reiterative fashion, such that if an ORF flanking a deletion was itself `deleted,' then its secondary flanking ORFs were also assessed in a similar manner. This analysis was repeated at increasingly relaxed levels of stringency until the lowest confidence threshold was finally reached (lowest confidence limit = 95%). We found that deleted ORFs in B. mallei exhibited a significantly higher likelihood of being flanked by deleted ORFs on both sides than on one side (59.2% for two-sided deletions vs. 32.5% for one-sided deletions, P = 1.15 × 10–8). In contrast, this bias was much less apparent for B. thailandensis (41.5% for two-sided deletions vs. 35.1% for one-sided deletions, P = 0.007, compared to P = 1.15 × 10–8 for B. mallei) (Table 2). These results suggest that deleted ORFs in B. mallei have a higher tendency to exhibit genomic coclustering than deleted ORFs in B. thailandensis. A χ2 analysis confirmed that the distribution of ORF deletions in B. mallei was indeed significantly different from that of B. thailandensis (P = 2.15 × 10–20), with an ORF deleted in B. mallei having a twofold higher chance of being flanked on both sides by other deleted ORFs than B. thailandensis (Supplemental material). These results suggest that the loss of large continuous segments of genomic DNA might be one possible mechanism contributing to the evolutionary divergence of B. mallei from B. pseudomallei. This phenomenon is particularly striking on Chromosome 2, where deletions of >12 kb are detected in several places (Fig. 3). In contrast, as the tendency for deleted ORFs to cocluster was not as striking in B. thailandensis, this raises the possibility that B. thailandensis may have utilized a different mechanism of divergence from B. pseudomallei and B. mallei.
Figure 3.

Regions of deletion in B. mallei and B. thailandensis along the B. pseudomallei genome, using t-tests. Chromosome 1 (A) and Chromosome 2 (B) in terms of 106 base pairs. White gaps represent regions deleted at >99% confidence. Orange areas represent deleted regions at the lower level of confidence, 95%. (C,D) Higher-resolution figures for regions II-a and II-b. For z-score versions of A–D, please see Supplemental material.
ORFs deleted in B. mallei and B. thailandensis are associated with diverse cellular functions
ORFs deleted in B. mallei and B. thailandensis compared to B. pseudomallei were associated with a wide variety of diverse cellular processes. Because it would be beyond the scope of this report to provide a comprehensive analysis of every cellular pathway used by the different Burkholderia species, we have chosen to highlight various `interesting' ORFs whose identity logically suggests specific testable predictions regarding phenotypic differences among the three species. We focused on five major groups (metabolism, transcription, membrane-associated, cellular signaling, secreted proteins), which are discussed below. We note that the ORFs listed here were those found to be deleted at an extremely high confidence level (>99.99%), and it is thus likely that other deleted ORFs remain to be discovered at lower confidence levels. In addition, independent PCR validation of many of these ORFs is provided in the Supplemental material.
Metabolism
B. mallei and B. pseudomallei possess distinct ecological niches and host selectivities (Howe 1950; Dance 2000), but little is known about the molecular basis underlying these differences. In comparison to B. pseudomallei, B. mallei lacks several ORFs related to nitrogen metabolism, such as 2-aminobenzoate CoA ligase, narD (a putative nitrate/nitrite transporter), and several subunits of nitrate reductase, which may be important for B. pseudomallei being able to exist in the rhizosphere (Dharakul and Songsivilai 1999). Also deleted in B. mallei were a cluster of cytochrome-related ORFs (CYC4, Cytochrome C oxidase chain II, and cytochrome C oxidase subunit 1), which may be important for energy production and redox control, and ORFs involved in riboflavin metabolism and barbamide (a chlorinated lipopeptide) synthesis (Chang et al. 2002). We also found that B. mallei lacks a specific Class D β-lactamase implicated in the development of resistance of B. pseudomallei to ceftazidime, an antibiotic frequently used to treat melioidosis (Niumsup and Wuthiekanun 2002). We speculate that the absence of this enzyme in B. mallei might explain why ceftazidime-resistant strains of B. pseudomallei have been observed (Dance 1991; Kenny et al. 1999), but similarly resistant strains of B. mallei have not yet been reported. Finally, previous studies have shown that B. pseudomallei possesses a siderophore which is capable of scavenging iron in vitro (Yang et al. 1991, 1993). We found that a homolog of the P. aeruginosa fptA gene, which encodes the precursor to the Fe3+-pyochelin receptor and functions to transport iron into the bacterium, was present in B. pseudomallei but deleted in B. mallei. Notably, the successful establishment of a bacterial infection is often dependent upon the ability of the pathogen to scavenge iron from host sources (Cox 1982; Takase et al. 2000).
Unlike B. mallei, B. thailandensis is commonly found in the same ecological niches as B. pseudomallei (Smith et al. 1997; Dance 2000) but is considered to be avirulent. One ORF deleted in B. thailandensis which might contribute to this important clinical distinction is wbiH, an undecaprenyl phosphate N-acetylglucosaminyltransferase which resides in a cluster of ORFs regulating the biosynthesis of type II O-antigenic polysaccharides (DeShazer et al. 1998). Previous experiments have shown that B. pseudomallei mutants with disruptions in this cluster (e.g., apaH, wbiA, wbiG, wbiI) exhibit decreased virulence in various animal models of melioidosis (DeShazer et al. 1998). However, as B.thailandensis has been reported to express type II O-PS (Brett et al. 1998), and is also apparently resistant to serum-mediated killing (DeShazer et al. 1998), it is likely that the lack of wbiH in B. thailandensis causes a more subtle effect than the complete abolition of Type II OPS production. Two potential gene clusters related to polyketide synthesis were also deleted in B. thailandensis—the first cluster contained ORFs with strong similarities to enoyl-CoA hydratase and polyketide synthase from Bacillus subtilis, and TaC (an acyl enzyme) from Myxococcus xanthus (Paitan et al. 1999). The second cluster contained two ORFs with similarity to nonribosomal peptide synthetases (NRPSs) and the polyketide synthetase epoD, which functions in Streptomyces coelicolor to synthesize the bactericidal polyketide epothilone (Tang et al. 2000).
Transcription
One ORF which was deleted in B. mallei exhibited strong homology to the P. aeruginosa transcriptional regulator PchR, which regulates expression of the pyochelin biosynthesis gene cluster (Heinrichs and Poole 1993). The chromosomal location of this PchR homolog in B. pseudomallei was in close proximity to the fptA homolog described in the previous section, suggesting that ORFs in this region may represent an ORF cluster related to iron metabolism. Supporting this idea, another nearby deleted ORF exhibited homology to ATP-binding cassette (ABC) transport proteins, a genomic organization highly similar to the P. aeruginosa pyochelin biosynthesis cluster (Reinmann et al. 2001). The finding that this ORF cluster is present in B. pseudomallei but not in B. mallei raises the possibility that iron metabolism may be more severely compromised in the latter. To experimentally test this possibility, we compared siderophore production in various Burkholderia strains grown in either iron-supplemented or irondeprived media. Supporting the microarray data, the B. mallei strains displayed decreased siderophore production compared to B. pseudomallei and B. thailandensis in both media types (Fig. 2E). This finding demonstrates how the microarray data can be used to discover phenotypic differences between species.
B. pseudomallei and B. thailandensis are biochemically distinct in their ability to assimilate arabinose (B. pseudomallei is Ara(–) whereas B. thailandensis is Ara(+); Brett et al. 1998), but the molecular mechanisms that underlie this phenotypic difference are unclear. One transcriptional regulator that was deleted in B. thailandensis contained weak homology to the AraC family of transcription factors. In Escherichia coli, AraC regulates arabinose metabolism by controlling both positive and negative expression of the araABD operon (Gallegos et al. 1997), and AraC-family members in numerous bacteria have also been implicated in carbon metabolism and virulence through the transcriptional control of Type III secretion systems (Francis et al. 2002). This observation raises the intriguing possibility that the well known correlation between arabinose-metabolizing ability and pathogen virulence in B. pseudomallei and B. thailandensis may be more functionally interrelated than previously suspected.
Membrane-associated and cell-surface components
Proteins in this class may mediate important cellular processes such as pathogen attachment to host cells and immune recognition by host defense systems. Several putative transmembrane and membrane-associated proteins were differentially present between B. pseudomallei and B. mallei, in particular ORFs related to the production and maintenance of fimbriae and pili, which have been implicated in multiple aspects of bacterial pathogenesis (Shi and Sun 2002). For example, one ORF deleted in both B. mallei and B. thailandensis possessed weak homology to the Streptococcus parasanguis Fap1 gene, a fimbriae-associated glycoprotein which has been shown to be functionally important for biofilm production and pathogen adherence to host surfaces (Froeliger and Fives-Taylor 2001; Stephenson et al. 2002). We also detected in B. thailandensis the deletion of a cluster of ORFs with similarity to fimbrial biosynthesis proteins of Y. pestis, and another surface-associated protein related to the Bordatella pertussis fhaC gene, which functions as an outer-membrane pore-forming protein through which the filamentous hemagglutinin (FHA) virulence factor is secreted (Jacob-Dubuisson et al. 1999). Potentially related to this finding, another ORF in this category that was deleted in B. thailandensis exhibited strong similarity to hemagglutinins as well.
Cellular signaling
Proteins in this class play essential roles in the integration and translation of environmental signals into discrete biological responses. Of particular interest were two ORFs deleted in B. mallei that were related to quorum sensing, a system used by numerous bacterial species to relate the local density of the bacterial population to specific biological activities. These ORFs included a regulator with homology to a Ralstonia solanacearum transcriptional autoinducer, and an acyl-homoserine lactone (AHL) synthase similar to that found in B. cepacia, another Burkholderia-related species that is an opportunistic pathogen. Intact quorum sensing in the related species B. cepacia is necessary for biofilm production (Huber et al. 2001), which may play an important role in its ability to establish a persistent infection in compromised individuals.
Secreted proteins
Secreted proteins are often deemed to be important for bacterial virulence, and B. pseudomallei may be capable of secreting a potent toxin (Haase et al. 1997; Haubler et al. 1998; Shang et al. 2001). For B. thailandensis, we identified two deleted ORFs involved in the production and secretion of toxins, one encoding a toxin secretion ATP-binding protein, and the other encoding a secretion protein belonging to the HlyD family, which is believed to play a role in toxin folding and trafficking. These two ORFs occurred in close proximity to each other, suggesting that they may represent an ORF cluster involved in toxin secretion. Supporting the hypothesis that these ORFs may represent a toxin secretion cluster in B. pseudomallei was the observation that their counterparts in V. cholera (V1080 and V1084) also lie in very close proximity to one another, being separated by only three hypothetical proteins (Heidelberg et al. 2000).
The large-scale genomic variance of B. pseudomallei isolates defines at least three distinct molecular subtypes
The second major objective of this project was to establish a molecular taxonomy for the comparison and classification of different natural isolates of B. pseudomallei. In this study, the B. pseudomallei isolates had been isolated from a variety of clinical (pediatric, aged, young) and environmental sources (soil, pigs, monkeys), mostly from within the South-East Asian region. In contrast to other subtyping techniques utilizing either a single or limited number of genetic loci, we attempted to classify the B. pseudomallei isolates in an unbiased manner using whole-genome similarities. An unsupervised average-linkage hierarchical clustering algorithm was first used to group the B. pseudomallei isolates to one another on the basis of their overall genomic similarity using data from all 6895 microarray probes. In addition to B. pseudomallei, isolates belonging to the other two species (B. mallei and B. thailandensis) were also included as `outlier subgroups' in this analysis. We found that the 18 B. pseudomallei strains could be segregated into three discernible groups or subtypes, designated G1, G2, and G3 respectively (Fig. 4A). The correlation distances separating the G1, G2, and G3 B. pseudomallei subtypes were comparable to or greater than the distances separating B. mallei and B. thailandensis, two bona fide distinct species, suggesting that the B. pseudomallei subtypes are quite distinct.
Figure 4.

Molecular subtypes of B. pseudomallei.(A) Hierarchical clustering of all Burkholderia strains. B. pseudomallei strains can be grouped into three distinct subtypes (G1, G2, G3), further confirmed by the plot of correlation between each strain and the `average' or `representative' profiles of B. mallei, B. thailandensis, and subtypes G1, G2, and G3 on the right. Numbers at the left of each node represent the number of ORFs that differentiate between the two branches of the node at a confidence level of 99.99% and above. (B) Principal components analysis (PCA) of B. pseudomallei strains based on the top 6000 most highly varying genetic elements. Axes P1, P2, and P3 refer to first, second, and third principal components, respectively. Groupings similar to those in (A) are observed. (C) PCA of B. pseudomallei strains based on P3 and P4. Circled `X's represent centroids of each subtype G1, G2, and G3. A circle is drawn from each centroid to the most deviant member of the corresponding group, showing that none of these circles overlap each other. Distributions similar to those in (A) are observed.
To confirm the robustness of the G1, G2, and G3 subtypes, we first generated a `representative' profile for each of the five groups (G1, G2, G3, B. mallei, and B. thailandensis) by averaging the profiles of individual members within each group, and computed the correlations among the five representative group profiles and profiles of the individual 23 isolates. As shown in Figure 4A, each isolate was most highly correlated to the representative profile of the group to which it was originally assigned, indicating that (1) the representative profile of each group is highly similar to the profiles of individual members within that group, and (2) the representative profiles are also sufficiently distinct from one another to robustly subdivide the individual isolates.
Second, we also grouped the 23 Burkholderia isolates using principal components analysis (PCA), an independent analytical technique. Once again using data from all 6895 ORFs, a PCA graph of principal components 1 and 2 (which represent the largest components of variance in the data set) segregated the Burkholderia isolates into their respective three species (Fig. 4B). A PCA graph of principal components 3 and 4 further subdivided the B. pseudomallei isolates into the same three subtypes (G1, G2, and G3) revealed by the hierarchical clustering analysis, with isolates of a particular subtype all lying closer in multidimensional `distance' to the average computed profile of that subtype than isolates of other subtypes. Third, we also employed K-means clustering, another independent technique, to regroup the profiles. Under a specification of k=5 clusters, the k-means analysis created two clusters consisting of the B. mallei and B. thailandensis isolates, and three clusters grouping the B. pseudomallei strains into the same G1, G2, and G3 subtypes identified by the hierarchical clustering analysis. Similar results were also obtained using a self-organizing map (SOM) algorithm as well (P. Tan, unpubl.). Taken collectively, these results support the hypothesis that natural isolates of B. pseudomallei can be divided into at least three distinct molecular subtypes.
Strains belonging to subtype G1 (strains 5/96, 21/96, 497/96, 567/96) had all been isolated from pigs that had been housed in a Malaysian abattoir, whereas all of the G2 strains (strains 14, 22, 23, 33) were clinical isolates from patients with melioidosis. Isolates in subtype G3, however, were associated with highly diverse origins. For example, two G3 strains (strains 15/96 and 20/96) had also been isolated from pigs at a similar time and location as strains belonging to subtype G2. These data indicate that there were at least two distinct molecular subtypes of B. pseudomallei in the pig population (G2 and G3 strains) at the time the sampling was performed. In addition to pigs, subtype G3 also contained strains that had been isolated from soil, monkeys, and patients with melioidosis. The strain diversity observed in subtype G3 raises the possibility that, with the accumulation of more strains, this subtype (G3) might be further resolved into finer subclassifications. Addressing this issue and determining whether clinical infections caused by G2 strains are clinically distinct from those caused by G3 strains are important tasks for future research.
Besides addressing questions of strain evolution and molecular phylogeny (Behr et al. 1999; Dziejman et al. 2002), the identification of genetic elements to discriminate between bacterial subtypes is also highly relevant for the development of strain-typing applications for use in environmental monitoring or medical and forensic diagnostics. Using the G1, G2, and G3 subtypes as a reference, we adopted a supervised approach to identify genetic elements that could be used to discriminate between the subtypes, for use as strain-typing markers. By selecting genomic elements that were consistently present in one group, but which were lost in either one or both of the other two groups, we identified 160 ORFs that were differentially deleted across the G1–G3 subtypes at a 95% confidence level (Fig. 5; Supplemental material). This number, constituting about 2% of the total number of predicted ORFs, suggests that the genomes of individual B. pseudomallei isolates are highly conserved in terms of large-scale genetic variance. Of these 160 ORFs, 125 could be grouped into 12 distinct chromosomal regions, six localizing to Chromosome 1 and six to Chromosome 2. In keeping with previous studies, we refer to these regions as regions of difference (RDs) (Fitzgerald et al. 2001). Although the majority of the RD ORFs were of unknown cellular function, three RDs (2, 5, and 10) contained ORFs related to phage proteins whereas RDs 1, 3, 8, and 11 also contained ORFs with homology to transposase or integrase proteins, which is consistent with these genomic regions being areas of active DNA recombination. A summary representation of the 12 RDs and their presence or absence in the various B. pseudomallei isolates is presented in Figure 5. The differential status of one large RD (RD2) was also independently confirmed by PCR (Supplemental material). Although most of the RDs exhibited a strong subtype-specific character (e.g., RDs 3, 4, 8, and 9 are present primarily in G1 strains, whereas RDs 6, 7, and 12 are present in G3 strains), two RDs (2 and 10) exhibited a more complex pattern in which they were absent from a common subset of G2 and G3 strains. In this respect, it is interesting that RD2 and RD10 both contain ORFs with similarity to highly related bacteriophages (CTX and P2) (Christie et al. 1986; Nakayama et al. 1999). Thus, one possible explanation for the identical pattern of absence and presence for RD2 and RD10 across the B. pseudomallei isolates is that the molecular events that resulted in loss of either RD2 or RD10 may have also caused the simultaneous loss of the other.
Figure 5.

Hierarchical clustering of G1, G2, and G3 B. pseudomallei strains by ORFs that are maximally different between subtypes but minimally different within a subtype at ≥95% confidence level. ORFs are depicted according to their linear order along the B. pseudomallei genome. Color bars represent ORFs that exhibit genomic clustering (RD, regions of difference, see text for details). Fold change ratios are depicted by the “color scheme” bar, with a value of ≤0.6 representing a “deleted” ORF.
Discussion
There are many challenges associated with the successful diagnosis, treatment, and prevention of B. pseudomallei infections. First, the bacterium is remarkably versatile and adaptive— although moist soils and stagnant waters are its primary reservoirs (Ellison et al. 1969), B. pseudomallei can also be isolated from multiple tissues in an infected individual (Asche 1991), and can persist in the human body for decades in a latent form (Mays and Ricketts 1975). Second, the bacterium is resistant to many conventionally used antibiotics (Chaowagul 2000), and sophisticated laboratory techniques are generally required to achieve an accurate clinical diagnosis. Third, at the molecular level, very little is currently known about B. pseudomallei behavior and virulence. Consistent with its complex biology, B. pseudomallei possesses a genome of ∼7 Mb, one of the largest prokaryotic genomes that has been publicly sequenced to date. It is estimated that B. pseudomallei contains between 5000 and 6000 genes (Songsivilai and Dharakul 2000), a number comparable to the eukaryotic model organism Sacchromyces cerevisiae. Despite this large gene number, the dearth of molecular knowledge concerning B. pseudomallei is reflected in the fact that to date, less than 100 genes that have been molecularly cloned and functionally characterized from B. pseudomallei (NCBI GenBank database, Dec. 1, 2002).
In the present study, whole-genome B. pseudomallei DNA microarrays were used to compare the genomic content of three related Burkholderia species: B. pseudomallei (the causative agent of melioidosis), B. mallei (the causative agent of glanders), and B. thailandensis (a clinically avirulent species highly related to B. pseudomallei). Using a multi-tier analytical approach combining statistical metrics and fluorescence fold-change thresholds, we identified several genetic elements that were differentially present or absent in the various species being compared. A major advantage of microarray technology is that it allows dozens of related strains and species to be rapidly compared, and thus functions as a strong complement to the alternative approach of comparing microbial genomes based on direct sequence data, as in the latter case such data are typically only available for one or two genomes for a particular species. In addition, as the B. thailandensis genome has yet to be sequenced, microarray technology currently represents the most convenient approach to interrogate the genomic content of this species as well.
It is important to note, however, that the analysis described in this report carries a number of assumptions and caveats. Most importantly, the comparisons reported here are unidirectional— as the DNA microarray used in this study was based exclusively on B. pseudomallei strain K96243 genomic sequence, we are unable to comment on whether B. mallei or B. thailandensis might contain any species-specific genes that are absent in B. pseudomallei, as such genes would not be represented on the microarray. Another basic assumption used in this analysis was that the genome sequences of the species being compared should be sufficiently similar such that nucleotide sequence variations among the species should not significantly affect the hybridization strengths of genomic sequences to the array probes, and that the chromosomal order of genes in the three species should be relatively similar. We believe that these assumptions are supported by several independent observations: (1) The three species have historically been treated as extremely closely related species. (2) Previous studies have successfully utilized subtractive molecular techniques to identify differentially present genetic elements between B. thailandensis, B. pseudomallei, and B. mallei, suggesting that there is a high degree of sequence conservation among the species (DeShazer et al. 2001; Reckseidler et al. 2001). (3) The genome sequences of specific B. pseudomallei and B. mallei strains are available (Sanger Institute, www.sanger.ac.uk or TIGR, The Institute for Genomic Research, www.tigr.org), and preliminary BLAST analyses by ourselves and others (Woods et al. 2002; P. Tan, unpubl.) have indicated that many genes in the two species are highly conserved both in terms of nucleotide identity and gene order. (4) Although the genome of B. thailandensis has not been sequenced, preliminary comparisons reveal that large regions of genomic synteny (>400 kb) can be found between the genomes of B. pseudomallei and B. cepacia, another Burkholderia species (P. Tan, unpubl.; B. cepacia genome sequence is available from the Sanger Centre). As B. cepacia, by ribosomal typing, is believed to be even more distantly related to B. pseudomallei than B. thailandensis or B. mallei, this result supports the assumption that the linear order of most genes in B. pseudomallei is also conserved in B. thailandensis. (5) For the array probe sizes used in this report (300–1000 bp), previous work has also demonstrated that sequences of up to 88% sequence homology can still hybridize at comparable strengths (DeRisi et al. 1997). Thus, it is reasonable to assume that the vast majority of the `deleted' ORFs identified in the present study will correspond to DNA sequences that are physically present or absent in the genomes of the species being compared, rather than elements of hypervariable sequence.
One interesting finding that emerged from the analysis was that although B. mallei and B. thailandensis possess a comparable number of genes that were deleted with respect to B. pseudomallei (344 vs. 304 respectively, at 99.99% confidence), the physical locations of the deletions were qualitatively different. Using the B. pseudomallei genome as a reference, we found that deletions in B. mallei tend to occur in the form of large contiguous chromosomal stretches (particularly on Chromosome 2), whereas deletions in B. thailandensis exhibit a more uniform distribution pattern. One possible concern might be that some of the scattered `deletions' observed in B. thailandensis may not be bona fide gene deletions, but artifacts resulting from high levels of sequence variation in intergenic areas (causing low hybridization signals). Because such latter `pseudogenes,' which represent false positives of the gene prediction algorithm, are usually associated with short ORF lengths and/or unusual codon usage frequencies, we also confirmed that there are no significant differences in ORF length and codon usage frequencies between ORFs designated as `deleted' or `present' in B. thailandensis (Supplemental material). Thus, although we cannot entirely eliminate the possibility that some of the ORFs on the microarray may not represent true genes, this result indicates that the presence of such `pseudogenes' is unlikely to significantly affect the overall pattern of scattered deletions observed in B. thailandensis. Our results raise the possibility that B. mallei and B. thailandensis may have utilized distinct mechanisms of variation to diverge from B. pseudomallei, or may indicate that B. mallei is more closely related to B. pseudomallei in evolutionary age than B. thailandensis. An alternative hypothesis might be that B. pseudomallei is the more recently arisen species, being derived from either B. mallei or B. thailandensis via the acquisition of novel sequences, perhaps by lateral gene transfer. This latter hypothesis can be tested because nucleotide sequences acquired via lateral gene transfer often contain a G+C content that differs from the rest of the recipient genome (Ochman et al. 2000). We compared the G+C nucleotide content of the deleted genomic regions to the rest of the overall B. pseudomallei genome, and found no substantial differences in G+C content between the `absent' and `present' regions (P. Tan, unpubl.). Thus, at this point, we favor the former hypothesis that B. mallei and B. thailandensis are derivatives of an ancestral B. pseudomallei (or B. pseudomallei-like) progenitor.
By comparing multiple independent isolates of B. pseudomallei to one another, we also found that the genomes of the B. pseudomallei isolates used in this study were highly conserved, with an average large-scale genomic variance of 2%–3% across strains (P. Tan, unpubl.). This is in contrast to other bacterial species such as Staphylococcus aureus, Helicobacter pylori, and E. coli), in which the genomes of individual strains have been reported to be highly variant (Ochman and Jones 2000; Salama et al. 2000; Fitzgerald et al. 2001). The high conservation of the B. pseudomallei genome among strains may indicate that it is either a relatively recently derived species, or that the diverse environmental pressures faced by the bacterium necessitate the preservation of a large and versatile gene complement. Based on their large-scale genomic variance, we found that the B. pseudomallei isolates could be grouped into at least three distinct molecular subtypes, referred to as G1, G2, and G3. We note that our classification of B. pseudomallei into the G1, G2, and G3 subtypes is based on whole-genome similarity matching, and thus it is quite possible that alternative classifications of B. pseudomallei might exist if one were to utilize a more restricted subset of genes. For example, a recent study using multilocus sequence typing (MLST), which compares nucleotide sequence variations in seven highly conserved housekeeping genes, was able to define at least 71 distinct `sequence types' of B. pseudomallei (Goday et al. 2003). One issue with MLST is that the assayed gene(s) must be present in all the strains studied, and it does not consider genes that are differentially present or absent; this may explain why the MLST study defined B. mallei as a `clone' of B. pseudomallei rather than a distinct species, a conclusion which is not supported by conventional 16s rRNA sequence comparisons (Coenye and Vandamme 2003). Intriguingly however, both MLST and pulsed-field gel electrophoresis (PDGE) assays commonly found that a set of 17 B. pseudomallei isolates derived from a geographical region close to ours (Hong Kong) could also be divided into three distinct subtypes (Goday et al. 2003). It will be of great interest to see whether these three subtypes match the G1, G2, and G3 subtypes defined in our study.
We identified 12 RDs (regions of difference) that could be used to discriminate between the G1, G2, and G3 subtypes. Using the microarray data, we found that 10 of these RDs are also present in B. mallei and B. thailandensis (P. Tan, unpubl.), indicating that the differential presence of these 10 RDs in the B. pseudomallei strains is most likely due to loss rather than acquisition of DNA (although it is formally possible that some of these ORFs may represent cross-hybridizing phage genes in B. mallei and B. thailandensis). The two remaining RDs (4 and 11) contain ORFs with homology to transposases, and RD4 also contains ORFs with homology to several plant bacteria, raising the possibility that RD4 may have been introduced by lateral transfer. However, we are not able to rule out the alternative possibility that RD4 and RD11 were lost from each of the three different Burkholderia species in an independent fashion. Further work, perhaps by using more strains of each of the three species, must be performed to explore this further.
In conclusion, we performed a genome-wide comparative analysis of three highly related Burkholderia species. Two of these (B. pseudomallei and B. mallei) are recognized human pathogens and potential biowarfare agents, and the third (B. thailandensis)is clinically avirulent. An analysis of their overall genomic relationships led to the identification of intriguing differences in their patterns of divergence, and an analysis of ORFs that were differentially present among the various groups led to the formulation of many testable hypotheses regarding mechanisms that might underlie their environmental and clinical differences. These findings show that DNA microarrays can facilitate a better understanding of the genetic differences between closely related organisms, providing useful information for the identification of virulence factors, exploration of molecular phylogeny, improvement of diagnostics, and the development of vaccines. Despite this success, it should not be forgotten that a significant number of the ORFs identified in this study could not be ascribed to a known cellular or biochemical function. Clearly, there is a huge amount of work to be yet performed before the complex biology of the Burkholderias will be fully understood.
Methods
Strains and media
B. pseudomallei, B. mallei, and B. thailandensis strain isolates used in this study are described in Table 1, and were obtained from the American Type Culture Collection (ATCC), DMRI laboratory stocks, or as gifts (see Acknowledgments). All strains were grown on TSA plates and stored as frozen stocks in LB broth with 20% glycerol.
Prediction of B. pseudomallei genes (ORFs) and microarray fabrication
Details of the B. pseudomallei microarray are provided in the Supplemental material. Briefly, the ORF prediction program GLIMMER (v2.0) was used to identify potential genes (ORFs) in partially assembled sequence data of B. pseudomallei strain K96243. Thus, 6895 ORF `clusters' were defined, and Primer 3 software was used to design oligonucleotide primer pairs to amplify 300–1000-bp fragments for each cluster. Comparisons of ORF prediction results using partially assembled (PA) and fully assembled (FA) B. pseudomallei genome sequences also revealed that 81% of the PA ORFs (5553 of 6895) was mappable to the FA set within a tolerance of ±100 bp, and an additional 756 (11%) could be matched to an FA ORF of at least 50% sequence similarity. Only 110 FA ORFs failed to be convincingly assigned to a partner entity in the PA ORF data set (Supplemental material). The ORFs identified in this study have been linked to their official corresponding entities in the ongoing B. pseudomallei genome annotation project performed at the Sanger Centre. Early access to these data was provided to our group for this purpose. Online mapping between both annotated sets, at www.omniarray.com/pseudomallei.html, will be made possible upon publication of the official genome annotation.
Genomic DNA from B. pseudomallei strain K96243 was used as a template to PCR-amplify array probes, and each array probe was printed in duplicate on commercially available microarray slides (Full Moon Biosystems) using an SDDC-2 Microarrayer (Virtek). The final array contains ∼13,500 features, which represent >90% of the predicted ORFs in the B. pseudomallei genome.
Sample hybridization and data acquisition
Genomic DNA from test bacterial strains was labeled with Cy3-dCTP using nick translation, and cohybridized to microarrays with Cy5-dCTP-labeled reference DNA (B. pseudomallei strain K96243). Reciprocal dye-swap hybridizations (i.e., where the test strains were labeled with Cy5 and the reference DNA with Cy3) were also performed for all 23 isolates. After hybridization and washing, fluorescent microarray images were acquired using a Genepix Scanner (Axon), and raw fluorescence data corresponding to individual array probes were uploaded into a centralized database for storage and subsequent analysis.
Data processing and analysis
Each array was internally normalized between the Cy3 and Cy5 channels by the factor N as follows, where D1 and D2 are the intensity values (background subtracted) for the Cy5 and Cy3 channels, respectively:
 |
and the log ratio for a spot being:
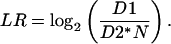 |
The entire data set was also normalized by mean-centering each array. Although not strictly essential, this operation can reduce potential skewing of the mean array fluorescence ratio that might be caused by outlier values in a single channel. Mean fluorescence ratios associated with each ORF were determined by averaging the fluorescence ratio values associated with replicate probes within an array and across duplicate experiments. As each ORF is represented on the microarray by two replicate array probes, the average ORF fluorescence ratio ultimately corresponds to the average of four (including replicate hybridizations) independent fluorescence ratio readings. The final data set of 23 isolates by 6895 ORFs can be obtained at http://www.omniarray.com/bioinformatics/BPM_SupData/.
A multistep procedure was conducted to identify differentially deleted ORFs at high confidence. First, Student's t-test was used to identify ORFs whose fluorescence ratios were significantly different between the two groups being compared (e.g., B. mallei vs. B. pseudomallei) at various levels of confidence (95%, 99%, 99.9%, 99.99%). Second, to define appropriate deletion and amplification cut-off thresholds, the general experimental variability associated with different microarray hybridizations was computed from the global standard deviations (SDs) associated with each of the 18 B. pseudomallei isolate hybridizations. The SDs, each based upon 6895 individual measurements, can be taken as a measure of experimental variability as preliminary experiments indicated that the genomes of distinct B. pseudomallei isolates were highly conserved (P. Tan, unpubl.). The resultant range of SDs was 0.44–0.55, and the upper end of this range (0.6) was used to define the deletion and amplification thresholds D (–0.6) and A (+0.6). An ORF with a fluorescence ratio falling within these thresholds (–0.6 to +0.6) was considered as `present.' In a separate analysis, we also confirmed that the experimental variances associated with the B. mallei and B. thailandensis hybridizations are comparable to B. pseudomallei, as the fluorescence ratio SDs of each of the individual 6895 ORFs, across hybridizations of isolates belonging to the same species, exhibited highly similar average, median, and mod ranges (Supplemental material). Third, an ORF was classified as `deleted' or `amplified' only if (1) the fluorescence values were significantly different in the two groups being compared (step 1), and (2) one ORF fluorescence value exceeded the D or A threshold, and the other was within the `present' zone (i.e., -0.6 to +0.6). This approach ensures that a `deleted' or `amplified' ORF is always compared to a `present' one, and reduces the possibility of false-positive calls (i.e., ORFs that are falsely classified as `deleted' and `amplified'). In cases where the number of groups being compared is >2 (i.e., Q groups), this process was repeated QC2 times to obtain all possible pairwise comparisons. In addition to this approach, we also analyzed the data set using the intensity-dependent z-scores methodology (Sprinthall 1996), and confirmed that both approaches result in a large majority of the same ORFs being identified as differentially present (Supplemental material). Hierarchical clustering was performed using average linkage clustering, with uncentered correlation as the similarity metric. Similarities between two strains X and Y were calculated using a “modified” Pearson correlation coefficient:
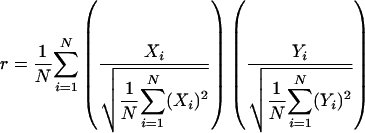 |
where Xi and Yi are the hybridization values for gene i in strains X and Y, respectively. The distance between two strains, two nodes, or a strain and a node was then taken as 1–r. Figure 4A reflects these distances. Software programs used were CLUSTER and TREEVIEW, both from Stanford University.
Analysis of array probes flanking a deleted ORF
To determine the probability that a deleted ORF would be flanked by another deleted ORF, a series of four data sets were first created representing ORFs deleted at various confidence levels (>99.99%, >99.9%, >99%, >95). Every member in each of the four groups was then analyzed to determine whether its flanking ORFs were also deleted at the lowest confidence limit (95%) on either one or both sides. The percentages of deleted ORFs with (1) deleted neighbors at both sides, (2) only one deleted neighbor, or (3) no deleted neighbor were then calculated by dividing the number of ORFs belonging to the various categories with the total number of deleted ORFs in that particular confidence level (“flanking probabilities”). There was no significant variation in flanking probabilities due to the use of different confidence levels (Supplemental material).
Measurement of siderophore production
Starter cultures were first grown at 37°C without shaking for ∼20 h. After centrifugation (3500g for 5 min), the bacterial pellet was washed and resuspended in 5 mL of 0.85% NaCl saline solution. One mL of resuspended bacteria was transferred to culture tubes containing either 4 mL of non-iron-supplemented chemically defined minimal (CDM) media or CDM supplemented with 0.02 mM FeCl3.6H2O. All cultures were incubated at 37°C for approximately another 20 h without shaking. Overnight cultures were centrifuged at 3500g for 5 min, and 4.5 mL of supernatant was added to 800 μL of CAS solution. Color changes were observed at 60 min, 120 min, 24 h, and 48 h. A color change from blue to orange or pink indicates that siderophores are present. Cell viability counts were performed using the Miles and Misra method on resuspended cells (after transfer of the supernatant to the CAS solution) confirmed that equivalent numbers of viable cells were used for each strain. Experiments were performed in duplicate.
Acknowledgments
We thank Lionel Lee, Eric Yap, and Hui Kam Man for their advice and encouragement, Michael Laub, Tan Boon Huan, and Julian Parkhill for a critical reading of this manuscript, Prof. Yap Eu Hian (National Univ. of Singapore) for providing the B. pseudomallei clinical isolates, and Drs. Sirirurg Songsivilai, Tararaj Dharakul, and Visaru Thamlikitkul (Mahidol Univ., Bangkok) for the B. thailandensis isolates. This work was supported by a DSTA Technology Development Plan grant (P.T., M.A.L.) and an NCC Core Grant (P.T.). Unannotated shotgun sequence data for B. pseudomallei were obtained in March 2001 from the Sanger Centre (www.sanger.ac.uk/Projects/B_pseudomallei/) and permission was obtained to use these data for this project.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1608904.
Footnotes
[Supplemental material is available online at www.genome.org and Microarray data sets are available at http://www.omniarray.com/bioinformatics/BPM_SupData/.]
References
- Asche, V. 1991. Melioidosis—A disease for all organs. Today's Life Sci. June: 34-40.
- Behr, M.A., Wilson, M.A., Gill, W.P., Salamon, H., Schoolnik, G.K., Rane, S., and Small, P.M. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520-1523. [DOI] [PubMed] [Google Scholar]
- Brett, P.J., DeShazer, D., and Woods, D.E. 1998. Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int. J. Syst. Bacteriol. 48: 317-320. [DOI] [PubMed] [Google Scholar]
- Chang, Z., Flatt, P., Gerwick, W., Nguyen, V., Willis, C., and Sherman, D. 2002. The barbamide biosynthetic gene cluster: A novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual tricholroleucyl starter unit. Gene 296: 235. [DOI] [PubMed] [Google Scholar]
- Chaowagul, W. 2000. Recent advances in the treatment of severe melioidosis. Acta. Trop. 74: 133-137. [DOI] [PubMed] [Google Scholar]
- Christie, G.E., Haggard-Ljungquist, E., Feiwell, R., and Caldendar, R. 1986. Regulation of bacteriophage P2 late-gene expression: The ogr gene. Proc. Natl. Acad. Sci. 83: 3238-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenye, T. and Vandamme, P. 2003. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiology 5: 719-729. [DOI] [PubMed] [Google Scholar]
- Cox, C.D. 1982. Effect of pyochelin on the virulence of Pseudomonas aeruginosa. Infect. Immun. 36: 17-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance, D.A. 1991. Melioidosis: The tip of the iceberg? Acta. Trop. 74: 115-119. [Google Scholar]
- Dance, D.A. 2000. Ecology of Burkholderia pseudomallei and the interactions between environmental Burkholderia spp. and human-animal hosts. Acta. Trop. 74: 159-168. [DOI] [PubMed] [Google Scholar]
- DeRisi, J.L., Iyer, V.R., and Brown, P.O. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680-686. [DOI] [PubMed] [Google Scholar]
- DeShazer, D., Brett, P.J., and Woods, D.E. 1998. The type II O-antigenic polysaccharide moiety of Burkholderia pseudomallei lipopolysaccharide is required for serum resistance and virulence. Mol. Micro. 30: 1081-1100. [DOI] [PubMed] [Google Scholar]
- DeShazer, D., Waag, D.M., Fritz, D.K., and Woods, D.E. 2001. Identification of a Burkholderia mallei polysaccharide gene cluster by subtractive hybridization and demonstration that the encoded capsule is an essential virulence determinant. Micro. Pathog. 30: 253-269. [DOI] [PubMed] [Google Scholar]
- Dharakul, T. and Songsivilai, S. 1999. The many facets of melioidosis. Trends Microbiol. 7: 138-140. [DOI] [PubMed] [Google Scholar]
- Dziejman, M., Balon, E., Boyd, D., Fraser, C., Heidelberg, J.F., and Mekalanos, J.J. 2002. Comparative genomic analysis of Vibrio cholerae: Genes that correlate with cholera epidemic and pandemic disease. Proc. Natl. Acad. Sci. 99: 1556-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison, D.W., Baker, H.J., and Mariappan, M. 1969. Melioidosis in Malaysia. I. A method for isolation of Pseudomonas pseudomallei from soil and surface water. Am. J. Trop. Med. Hyg. 18: 694-697. [PubMed] [Google Scholar]
- Fitzgerald, J.R., Sturdevant, D.E., Mackie, S.M., Gill, S.R., and Musser, J.M. 2001. Evolutionary genomics of Staphylococcus aureus: Insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc. Natl. Acad. Sci. 98: 8821-8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, M.S., Wolf-Watz, H., and Forsberg, A. 2002. Regulation of type III secretion systems. Curr. Opin. Microbiol. 5: 166-172. [DOI] [PubMed] [Google Scholar]
- Froeliger, E.H. and Fives-Taylor, P. 2001. Streptococcus paraganguis fimbria-associated adhesin fap1 is required for biofilm formation. Infect. Immun. 69: 2512-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos, M.T., Schleif, R., Bairoch, A., Hofmann, K., and Ramos, J.L. 1997. AraC/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61: 393-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy, D., Randle, G., Simpson, A.J., Aanensen, D.M., Pitt, T.L., Kinoshita, R., and Spratt, B.G. 2003. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J. Clin. Microbiol. 41: 2068-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase, A., Janzen, J., Barrett, S., and Currie, B. 1997. Toxin production by Burkholderia pseudomallei strains and correlation with severity of melioidosis. J. Med. Microbiol. 46: 557-563. [DOI] [PubMed] [Google Scholar]
- Haubler, S., Nimtz, M., Domke, T., Wray, V., and Steinmetz, I. 1998. Purification and characterization of a cytotoxic exolipid of Burkholderia pseudomallei. Infect. Immun. 66: 1588-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg, J.F., Eisen, J.A., Nelson, W.C., Clayton, R.A., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Umayam, L. et al. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406: 477-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs, D.E., and Poole, K. 1993. Cloning and sequence analysis of a gene (pchR) encoding an AraC family activator of pyochelin and ferripyochelin receptor synthesis in Pseudomonas aeruginosa. J. Bacteriol. 175: 5882-5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, C. 1950. Glanders. In: The Oxford medicine (ed. H.A. Christian), pp. 185-202. Oxford University Press, New York.
- Huber, B., Riedel, K., Hentzer, M., Heydorn, A., Gotschlich, A., Givskov, M., Molin, S., and Eberl, L. 2001. The cep quorum-sensing system of Burkholderia cepacia H111 controls biofilm formation and swarming motility. Microbiology 147: 2517-2528. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson, F., El-Hamel, C., Saint, N., Guedin, S., Willery, E., Molle, G., and Locht, C. 1999. Channel formation by FhaC, the outer membrane protein involved in the secretion of the Bordetella pertussis filamentous hemagglutinin. J. Biol. Chem. 274: 37731-37735. [DOI] [PubMed] [Google Scholar]
- Kenny, D.J., Russell, P., Rogers, D., Eley, S.M., and Titball, R.W. 1999. In vitro susceptibilities of Burkholderia mallei in comparison to those of other pathogenic Burkholderia spp. Antimicrob. Agents Chemother. 43: 2773-2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laub, M.T., McAdams, H.H., Feldblyum, T., Fraser, C.M., and Shapiro, L. 2000. Global analysis of the genetic network controlling a bacterial cell cycle. Science 290: 2144-2148. [DOI] [PubMed] [Google Scholar]
- Lee, M.A. and Liu, Y. 2000. Sequencing and characterization of a novel serine metalloprotease from Burkholderia pseudomallei. FEMS Microbiol. Lett. 192: 67-72. [DOI] [PubMed] [Google Scholar]
- Mays, E.E. and Ricketts, E.A. 1975. Melioidosis: Recrudescence associated with bronchogenic carcinoma twenty-six years following initial geographic exposure. Chest 68: 261-263. [DOI] [PubMed] [Google Scholar]
- McGilvray, C.D. 1944. The transmission of glanders from horse to man. Can. J. Public Health 35: 268-275. [Google Scholar]
- Nakayama, K., Kanaya, S., Ohnishi, M., Terawaki, T., and Hayashi, T. 1999. The complete nucleotide sequence of phiCTX, a cytotoxin-converting phage of Pseudomonas aeruginosa: Implications for phage evolution and horizontal gene transfer via bacteriophages. Mol. Microbiol. 31: 399-419. [DOI] [PubMed] [Google Scholar]
- Niumsup, P. and Wuthiekanun, V. 2002. Cloning of the class D β-lactamase gene from Burkholderia pseudomallei and studies on its expression in ceftazidime-susceptible and -resistance strains. J. Antimicrob. Chemother. 50: 445-455. [DOI] [PubMed] [Google Scholar]
- Ochman, H. and Jones, I.B. 2000. Evolutionary dynamics of full genome content in Escherichia coli. EMBO J. 19: 6637-6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman, H., Lawrence, J.G., and Groisman, E.A. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405: 299-304. [DOI] [PubMed] [Google Scholar]
- Paitan, Y., Orr, E., Ron, E.Z., and Rosenberg, E. 1999. Genetic and functional analysis of genes required for the post-modification of the polyketide antibiotic TA of Myxococcus xanthus. Microbiology 145: 3059-3067. [DOI] [PubMed] [Google Scholar]
- Rainbow, L., Hart, C.A., and Winstanley, C. 2002. Distribution of type III secretion gene clusters in Burkholderia pseudomallei, B. thailandensis and B. mallei. J. Med. Microbiol. 51: 374-384. [DOI] [PubMed] [Google Scholar]
- Reckseidler, S.L., DeShazer, D., Sokol, P.A., and Woods, D.E. 2001. Detection of bacterial virulence genes by subtractive hybridization: Identification of capsular polysaccharide of Burkholderia pseudomallei as a major virulence determinant. Infect. Immun. 61: 34-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinmann, C., Patel, H.M., Serino, L., Barone, M., Walsh, C.T., and Hass, D. 2001. Essential PchG-dependent reduction in pyochelin biosynthesis of Pseudomonas aeruginosa. J. Bacteriol. 183: 813-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotz, L.D., Khan, A.S., Lillibridge, S.R., Ostroff, S.M., and Hughes, J.M. 2002. Public health assessment of potential biological terrorism agents. Emerg. Infect. Dis. 8: 225-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama, N., Guillemin, K., McDaniel, T.K., Sherlock, G., Tompkins, L., and Falkow, S. 2000. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc. Natl. Acad. Sci. 97: 14668-14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, H., Thong, J.T., Gopalakrishnakone, P., Lee, M.A., Yap, E.P., Moochhala, S., and Yap, E.H. 2001. Production of oxalate in the culture supernate of Burkholderia pseudomallei. Med. Microbiol. 50: 655-656. [DOI] [PubMed] [Google Scholar]
- Shi, W.Y. and Sun, H. 2002. Type IV pilus-dependent motility and its possible role in bacterial pathogenesis. Infect. Immun. 70: 1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M.D., Angus, B.J., Wuthiekanun, V., and White, N.J. 1997. Arabinose assimilation defines a nonvirulent biotype of Burkholderia pseudomallei. Infect. Immun. 65: 4319-4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songsivilai, S. and Dharakul, T. 2000. Multiple replicons constitute the 6.5-megabase genome of Burkholderia pseudomallei. Acta Tropica 74: 169-179. [DOI] [PubMed] [Google Scholar]
- Sprinthall, R.C. 1996. Basic statistical analysis, 5th ed. Allyn & Bacon, Boston.
- Stephenson, A.E., Wu, H., Novak, J., Tomana, M., Mintz, K., and Fives-Taylor, P. 2002. The Fap1 fimbridal ahesin is a glycoprotein: Antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol. Micro. 43: 147-157. [DOI] [PubMed] [Google Scholar]
- Suputtamongkol, Y., Hall, A.J., Dance, D.A.B., Chaowagul, W., and Wajchanuvong, M.D. 1994. The epidemiology of melioidosis in Ubon Ratchatani, Northeast Thailand. Int. J. Epidemiol. 23: 1082-1090. [DOI] [PubMed] [Google Scholar]
- Takase, H., Nitanai, H., Hoshino, K., and Otani, T. 2000. Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect. Immun. 68: 1834-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, L., Shah, S., Chung, L., Carney, J., Katz, L., Khosla, C., and Julien, B. 2000. Cloning and heterologous expression of the epothilone gene cluster. Science 287: 640-642. [DOI] [PubMed] [Google Scholar]
- Wei, Y., Lee, J., Richmond, C., Blattner, F.R., Rafalski, J.A., and LaRossa, R.A. 2001. High-density microarray mediated gene expression profiling of Escherichia coli. J. Bacteriol. 183: 545-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelis, M. 1998. First shots fired in biological warfare. Nature 395: 213. [DOI] [PubMed] [Google Scholar]
- Wilson, M., DeRisi, J., Kristensen, H.H., Imboden, P., Rane, S., Brown, P.O., and Schoolnik, G.K. 1999. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc. Natl. Acad. Sci. 96: 12833-12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, D.E., Jeddeloh, J.A., Fritz, D.L., and DeShazer, D. 2002. Burkholderia thailandensis E125 harbors a temperate bacteriophage specific for Burkholderia mallei. J. Bacteriol. 184: 4003-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H.M., Chaowagul, W., and Sokol, P.A. 1991. Siderophore production by Pseudomonas pseudomallei. Infect. Immun. 56: 776-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Kooi, C.D., and Sokol, P.A. 1993. Ability of Pseudomonas pseudomallei malleobactin to acquiretransferrin-bound, lactoferrin-bound, and cell-derived iron. Infect. Immun. 61: 656-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- www.sanger.ac.uk/Projects/B_pseudomallei/; Web page containing the official genome annotation of B. pseudomallei strain K96243.
- www.tigr.org; The Institute for Genomic Research.
- www.omniarray.com/bioinformatics/BPM_SupData/; Web page containing the Supplemental material for this report, including downloadable microarray data sets.
- www.omniarray.com/pseudomallei.html; Web page matching microarray probes to official gene annotations.