

New roles of an epigenetic regulator in RNA processing are discovered, which could be targeted for cancer treatment.
Keywords: Lysine demethylase 5, Jhd2, KDM5, JARID1, alternative polyadenylation, CCND1, DICER1, KDM5A, KDM5B, KDM5C
Abstract
The complexity by which cells regulate gene and protein expression is multifaceted and intricate. Regulation of 3′ untranslated region (UTR) processing of mRNA has been shown to play a critical role in development and disease. However, the process by which cells select alternative mRNA forms is not well understood. We discovered that the Saccharomyces cerevisiae lysine demethylase, Jhd2 (also known as KDM5), recruits 3′UTR processing machinery and promotes alteration of 3′UTR length for some genes in a demethylase-dependent manner. Interaction of Jhd2 with both chromatin and RNA suggests that Jhd2 affects selection of polyadenylation sites through a transcription-coupled mechanism. Furthermore, its mammalian homolog KDM5B (also known as JARID1B or PLU1), but not KDM5A (also known as JARID1A or RBP2), promotes shortening of CCND1 transcript in breast cancer cells. Consistent with these results, KDM5B expression correlates with shortened CCND1 in human breast tumor tissues. In contrast, both KDM5A and KDM5B are involved in the lengthening of DICER1. Our findings suggest both a novel role for this family of demethylases and a novel targetable mechanism for 3′UTR processing.
INTRODUCTION
Epigenetics is an ever-evolving field that focuses on heritable phenotypes that do not involve changes of the underlying DNA sequence. Histone modification, DNA methylation, and microRNA (miRNA) regulation are three of the most commonly studied epigenetic phenomena. As the body of knowledge surrounding the field of epigenetics increases, it becomes more apparent that epigenetic regulators play important roles in development and disease. Histone-modifying enzymes are crucial to maintaining the balance of posttranslational modifications on histones. Histone methyltransferases and demethylases have been shown to be involved in cancer, development, and aging [reviewed by Greer and Shi (1)]. This study reveals the role of a histone demethylase, Jhd2, in 3′ untranslated region (UTR) processing. Jhd2 (also referred to as KDM5) is the only known H3K4me3 demethylase in Saccharomyces cerevisiae (2–5). It has not been found to be associated with any known complexes. Here, we show that it recruits polyadenylation machinery to chromatin and promotes alteration of the 3′UTR.
Most eukaryotic mRNA transcripts are cotranscriptionally modified with a 5′ guanosine cap and a 3′ polyadenine (polyA) tail. These modifications aid in preventing degradation and targeting mRNA export. The polyA tail can be added at various places after the stop codon without affecting the coding sequence (CDS) of the resulting transcript. Most transcripts have a canonical polyA site, but many have one or more alternative polyadenylation (APA) sites (6–8). Usage of these alternative sites is common in certain tissues in mammals and during certain stresses in yeast (8–10). APA is also associated with varying disease states, such as cancer, and with important cell processes, such as differentiation in stem cells (7, 10). Upstream or “promoter-proximal” sites are usually the alternative sites and preferred in many tumors, whereas distal sites are globally preferred during development and differentiation (7, 11).
The exact mechanism by which alternative polyA sites are chosen over canonical polyA sites is not well understood. Current literature suggests that the same core machinery is used for both canonical and alternative polyA sites (12). In eukaryotes, cleavage of mRNA is tightly coupled with polyadenylation (11). Although the association between polyadenylation and disease is well known, core polyadenylation machinery is essential to the cell and therefore has not been a feasible target for drug development. Here, we outline the impact of Jhd2 and its human homologs, KDM5B and KDM5A, on 3′UTR length and polyA site choice, suggesting that KDM5 enzymes are novel drug targets to modulate 3′UTR length.
RESULTS
Jhd2 physically interacts with polyadenylation machinery
Extensive classification of a complex containing budding yeast demethylase, Jhd2, has not previously been reported. We set out to identify proteins associated with Jhd2 by performing an affinity purification using a tandem affinity purification (TAP)–tagged Jhd2 strain, in which the TAP tag was inserted into the endogenous Jhd2 locus. The protein A portion of the TAP tag binds tightly to immunoglobulin G (IgG). We used IgG-coated beads to pull down TAP-tagged Jhd2. Mass spectrometry results indicated that many RNA processing components copurify with Jhd2 (Fig. 1A and fig. S1A), and we verified these results by tandem mass spectrometry (fig. S1, B and C). Many of these associated proteins are part of the 3′UTR processing machinery (Fig. 1B). We then verified our mass spectrometry results using affinity purification, followed by Western blotting. Strains containing TAP-tagged polyadenylation machinery components at the endogenous loci were engineered to overexpress FLAG-Jhd2. The TAP-tagged proteins were affinity-purified from these strains using IgG beads, and the immunoprecipitates were probed with an antibody against Jhd2 (Fig. 1, C and D, and fig. S2).
Fig. 1. Jhd2 interacts physically with polyadenylation machinery.
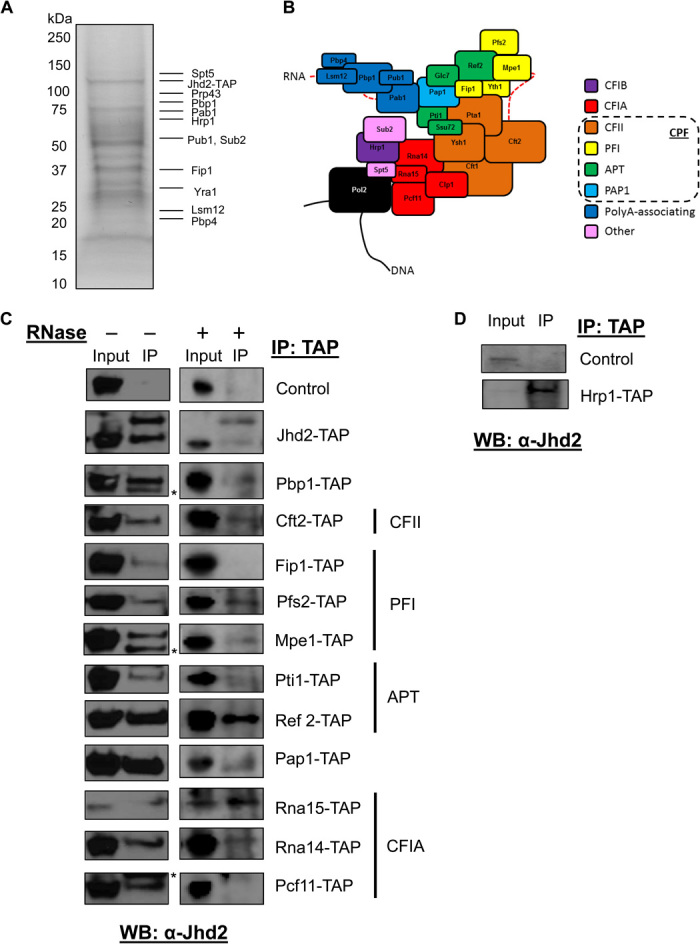
(A) Coomassie staining of TAP-Jhd2–associated protein complex. Selected proteins identified by tandem mass spectrometry are indicated. (B) Known components of the yeast polyadenylation complex and their associated subcomplexes. CPF, cleavage and polyadenylation factor. (C) Western blot analysis of TAP immunoprecipitates from the indicated TAP-tagged strains overexpressing FLAG-Jhd2 with a Jhd2 antibody. The Jhd2 antibody recognizes both Jhd2-TAP (top band, 105 kDa) and FLAG-Jhd2 (bottom band, 86 kDa) (row 2). Nonspecific bands are indicated by asterisks. Right: Copurifications performed as in the left panel but with the addition of RNase. Control indicates a yeast strain with no TAP-tagged protein. (D) Western blot analysis of TAP immunoprecipitates from Hrp1-TAP or wild-type strains expressing endogenous levels of Jhd2 with a Jhd2 antibody.
We found that Jhd2 interacts with many of the polyadenylation complex components from various subcomplexes (Fig. 1, C and D). The CPF complex is involved in recognition of polyA consensus sequences and cleavage of mRNA before polyadenylation by the polyA polymerase 1 (13). All available components of the CPF complex were shown to interact with Jhd2. The canonical cleavage factor 1A (CF1A) complex consists of Rna14, Rna15, Pcf11, and Clp1 (Fig. 1B, red) and is involved in recognition of a positioning element bound directly by Rna15. Pcf11 has also been shown to interact with phosphorylated serine 2 on RNA polymerase II (Pol II), a modification that is present during elongation and termination in yeast (14). Binding of the positioning element by CF1A allows for placement of CPF along the polyA signal. We found that Rna14, Rna15, and Pcf11 interact with Jhd2. Clp1 has been shown to be exchanged for Yra1 when weaker polyadenylation sites are chosen (15, 16). This is interesting because our mass spectrometry data showed that Yra1, but not Clp1, copurifies with Jhd2 (Fig. 1A and fig. S1C). The sole component of subcomplex CF1B, Hrp1, binds to the efficiency element and interacts with Rna14 and Rna15 to direct selection of the polyA site. Consistent with our mass spectrometry results, Hrp1 was pulled down with endogenous Jhd2 (Fig. 1, A and D, and fig. S2B). We also noted that Jhd2 interacts with itself because we could detect both TAP-tagged and FLAG-tagged Jhd2 in our Jhd2-TAP pulldown (Fig. 1C and fig. S2A).
The RNA processing complex has an intricate interaction with RNA. This led us to hypothesize that Jhd2 binding of polyA machinery could be mediated by interaction with RNA. To examine this possibility, we set out to determine whether these interactions were maintained in the absence of RNA. To accomplish this, we repeated the coaffinity purifications in the presence of ribonuclease (RNase). The absence of RNA did not have any significant effect on the interaction of Jhd2 with most 3′UTR processing machinery components or with itself but abolished its interaction with Fip1 and Pcf11 (Fig. 1C, right).
Jhd2 recruits the CF1A complex to 3′UTRs
Jhd2 is a histone demethylase known to bind chromatin at promoters (17). We established that Jhd2 associates with polyadenylation machinery, which led us to hypothesize that it also binds chromatin in the genomic region corresponding to the 3′UTR.
Chromatin immunoprecipitation (ChIP) experiments were performed in cells expressing TAP-tagged Jhd2 or Pcf11. Pcf11 was used as a representative core component of the polyadenylation complex. Consistent with the role of Jhd2 in transcriptional regulation, we found that Jhd2 binds not only to the PMA1 promoter but also to the genomic regions near the polyA sites, similar to Pcf11 (Fig. 2A). We then performed ChIP sequencing (ChIP-seq) experiments to examine the genome-wide binding patterns of Jhd2 and Pcf11 using the TAP-tagged strains mentioned above. Consistent with a previous report, which showed that polyadenylation factors are recruited near the promoters and terminator regions (18), we found that Jhd2 and Pcf11 binding peaks are very broad and are enriched both near the promoters and at certain 3′UTRs (tables S1 and S2). No ChIP-seq data for Jhd2 have been published, but our results parallel those found previously for Pcf11 (19, 20). We found that genes bound by Jhd2 and genes bound by Pcf11 overlapped significantly (Fig. 2B; P = 7.9 × 10−141, hypergeometric test).
Fig. 2. Jhd2 recruits CF1A to 3′UTRs.
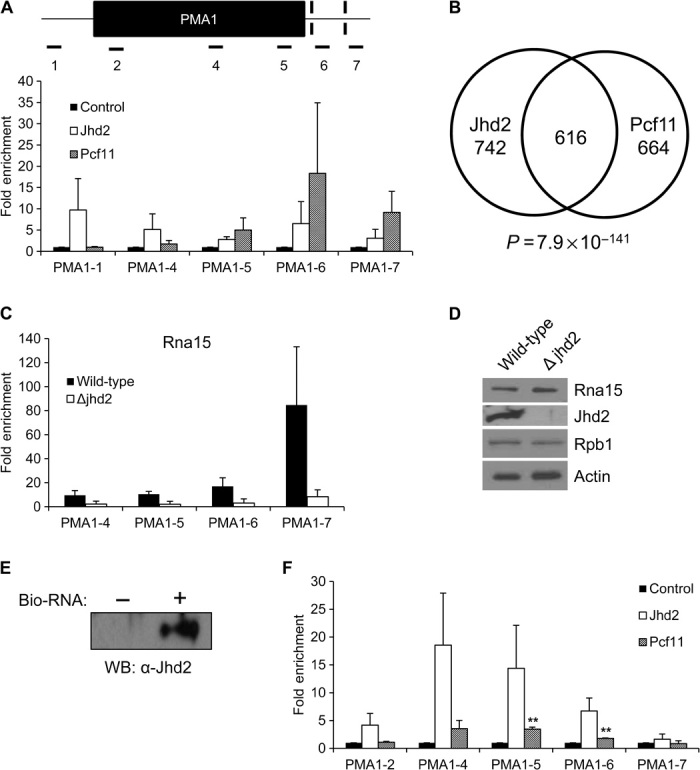
(A) Jhd2 and Pcf11 ChIP experiments of yeast expressing endogenous levels of Jhd2-TAP or Pcf11-TAP. DNA corresponding to noted locations on PMA1 was quantified using quantitative polymerase chain reaction (qPCR) with the indicated primers (table S6). Data are represented as fold enrichment over the control ChIP using beads only. Error bars represent SEM for biological triplicate experiments. Note the trend, although the differences are not statistically different. (B) Venn diagram showing genes bound by both Jhd2 and Pcf11. (C) Rna15 ChIP experiments using wild-type and Jhd2 deletion strains. Data are represented as fold enrichment over the control. Error bars represent SEM for biological duplicate experiments. Note the trend, although the differences are not statistically different. (D) Western blot analysis of wild-type and Jhd2 deletion strains with the indicated antibodies. (E) RNA binding experiments were performed using a biotinylated RNA 20-mer conjugated to magnetic beads. Recombinant Jhd2 bound to RNA was detected using Western blotting. Beads not conjugated to RNA served as a negative control. (F) RIP experiments of yeast expressing endogenous levels of TAP-tagged Jhd2 or TAP-tagged Pcf11. RNA associated with these proteins was converted to complementary DNA (cDNA) and quantified using qPCR. Data are shown as fold enrichment over the control. Error bars represent SEM for biological duplicate experiments. **P < 0.01. Note the trend, although the other differences are not statistically different.
The above results led us to hypothesize that Jhd2 recruits the CF1A complex (Pcf11, Clp1, Rna14, and Rna15) to chromatin and/or RNA. To determine whether this phenomenon was true, we performed ChIP of Rna15, a core component of CF1A that binds tightly to Pcf11, in wild-type and Jhd2 deletion yeast strains. As expected, in wild-type strains, Rna15 bound chromatin at the 3′UTR, as shown previously for other components of the CF1A complex (Fig. 2C) (19, 20). Consistent with our hypothesis, Jhd2 deletion led to a significant decrease in Rna15 recruitment to chromatin at 3′UTRs (Fig. 2C), and this change is not due to the decreased expression level of Rna15 (Fig. 2D).
Jhd2 binds RNA in vitro and in vivo
The large number of RNA processing proteins that copurified with Jhd2 led us to hypothesize that Jhd2 also binds RNA and affects RNA processing. No previous studies have addressed the RNA binding capabilities of Jhd2 in vitro or in vivo. To determine whether Jhd2 binds RNA in vitro, we performed an RNA binding assay using recombinant Jhd2 and a randomly generated, biotinylated RNA 20-mer that contains no predicted secondary structure (fig. S3, A to C). Although Jhd2 has no known RNA binding domains, it does have a zinc finger/PHD (plant homeodomain) domain, which could possibly bind nucleic acids (fig. S3D). Our results show that recombinant Jhd2 has nonsequence-specific RNA binding activity (Fig. 2E).
We then performed RNA immunoprecipitation (RIP) experiments to determine whether Jhd2 binds nascent mRNA transcript in vivo. The same TAP-tagged Jhd2 and Pcf11 strains used for ChIP were used for RIP. Again, Pcf11 was used as a representative polyadenylation machinery component because it is part of the CF1A complex known to bind sites upstream of polyadenylation signals (Fig. 1B). Similar to Pcf11, Jhd2 was enriched from the middle of the transcript and continued to be enriched to just after the first polyA site (Fig. 2F).
Jhd2 promotes 3′UTR shortening in a demethylase-dependent manner
Our affinity purification studies showed that Jhd2 interacts with the polyadenylation machinery. We have also shown that Jhd2 recruits the CF1A complex and binds RNA, which led us to hypothesize that Jhd2 is also involved in the selection of polyadenylation sites. The point at which polyA is added to the mRNA precursor is determined by the cleavage step of 3′ end processing. To determine whether Jhd2 directly affects cleavage, we performed an in vitro reaction using a synthetic RNA substrate containing the GAL7 polyA site (fig. S4A). Extracts lacking Jhd2 were as efficient for cleavage as those prepared from wild-type cells, suggesting that Jhd2 is not a constitutive polyadenylation factor and may only promote processing during active transcription. To test this idea, we used an in vitro transcription assay that measures how well transcription proceeds beyond the polyA site on exogenously added DNA. In this assay, recognition and cleavage at a polyA site result in less accumulation of RNA containing sequence downstream of the polyA site. However, there was no increase in transcription downstream of a polyA site in extract from the jhd2 deletion strain (fig. S4B).
These results suggested that Jhd2 may only modulate 3′ end processing when transcription occurs in vivo from a chromatin template. To examine this possibility, we performed initial in vivo experiments using transcripts that had well-defined and well-studied polyadenylation sites. The S. cerevisiae gene PMA1 is known to code for two distinct polyadenylation sites (15, 21). It is also expressed endogenously at high levels and is therefore a good candidate for studying transcript length.
All core polyadenylation components in yeast are essential; therefore, none of them can be deleted. To establish the effect of core polyadenylation machinery on 3′UTR length, we used pcf11-2, a temperature-sensitive strain that codes for a nonfunctional Pcf11 protein when grown at 37°C (22, 23). RNA was extracted from wild-type and pcf11-2 strains and subjected to reverse transcription qPCR (RT-qPCR). Polyadenylation typically occurs 10 to 30 base pairs (bp) downstream from the polyadenylation signal. The length of the 3′UTR of PMA1 was studied using primer sets amplifying the CDS (both the longer and shorter transcripts) and the transcript region downstream of the first polyA signal (the longer transcript only) (Fig. 3A). We found that inactivation of Pcf11 caused an increase in the longer transcript (Fig. 3B). Note that depletion of Pcf11 did not affect transcript levels of Jhd2 (fig. S5A).
Fig. 3. Jhd2 promotes 3′UTR shortening of PMA1 in a demethylase-dependent manner.
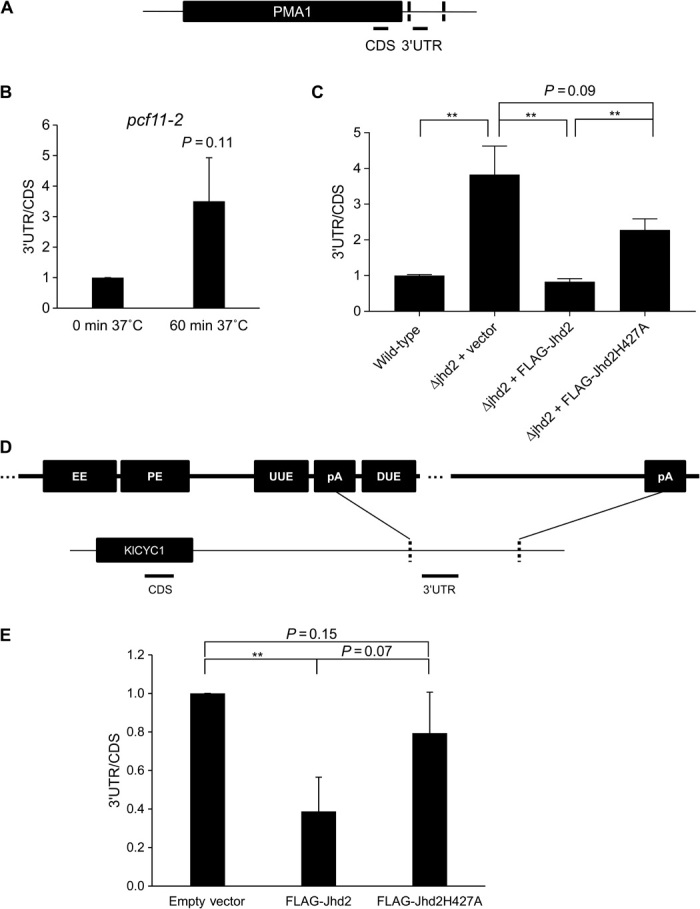
(A) Schematic representing amplicons used to study the 3′UTR region of PMA1. (B) Ratio of PMA1 3′UTR to CDS mRNA in a Pcf11 temperature-sensitive strain. Yeast cells grown at 30°C were transferred to 37°C for 60 min, resulting in a nonfunctional Pcf11 protein. Error bars represent SEM for biological duplicate experiments. (C) Ratio of PMA1 3′UTR to CDS mRNA in wild-type and Jhd2 deletion cells expressing empty vector (Δjhd2 + vector), FLAG-Jhd2 (Δjhd2 + FLAG-Jhd2), or demethylase-inactive FLAG-Jhd2 (Δjhd2 + FLAG-Jhd2H427A). Error bars represent SEM (n = 8). **P < 0.01. (D) Representation of the KlCYC1 3′UTR with conserved polyA motifs at its proximal polyadenylation site but with no conserved motifs at its distal polyadenylation site. EE, efficiency element; PE, positioning element; UUE, upstream U-rich enhancer element; DUE, downstream U-rich enhancer element. (E) RT-qPCR analysis of the ratio of KlCYC1 3′UTR to CDS mRNA in strains expressing KlCYC1 and either empty vector, FLAG-Jhd2, or demethylase-inactive FLAG-Jhd2H427A. All mRNA levels are normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Error bars represent SEM for biological triplicate experiments. **P < 0.01.
To determine whether loss of Jhd2 phenocopies Pcf11 inactivation, we obtained a strain with a Jhd2-deleted and inserted empty vector, FLAG-Jhd2 or FLAG-Jhd2H427A, a catalytically inactive Jhd2 mutant (fig. S5, B and C). Similar to pcf11-2 strains, loss of Jhd2 caused 3′UTR lengthening (Fig. 3C). Conversely, reintroduction of catalytically active Jhd2 partially reversed the 3′UTR-lengthening phenotype (Fig. 3C). In contrast, overexpression of the demethylase-deficient Jhd2 (Jhd2H427A) only had a partial effect (Fig. 3C).
In metazoans, distal polyadenylation sites tend to be canonical, whereas proximal sites are used less often. After noting that Jhd2 deletion causes lengthening in transcripts that can be rescued by reintroduction of a functional enzyme, we wanted to determine whether Jhd2 was aiding in selection for shorter 3′UTRs or whether it was specifically allowing selection of weaker and/or less conserved polyA sites. Because yeast uses both polyadenylation sites on PMA1 equally, we could not determine whether promotion of 3′UTR shortening by Jhd2 resulted in selection of polyA sites based on signal strength or distance from the CDS. For this reason, we chose to further our studies using a system that has the strong and weak polyadenylation sites in the opposite positioning, that is, a system with a well-defined “weak” downstream polyadenylation site (24, 25). This method involves using the Kluyveromyces lactis CYC1 (KlCYC1) gene, which was reported to have two polyadenylation sites that are roughly 400 bp apart in S. cerevisiae (Fig. 3D). The distal site of KlCYC1 has none of the canonical polyA motifs, whereas the proximal site has all the motifs (Fig. 3D). Unlike the Jhd2 deletion strains used above, all these strains contain the chromosomal copy of the JHD2 gene (fig. S6). We observed that overexpression of Jhd2 caused 3′UTR shortening of KlCYC1 (Fig. 3E). In contrast, overexpression of the demethylase-deficient Jhd2 did not have any effect (Fig. 3E). Together, these results suggest that Jhd2 can aid in the selection of proximal polyA sites of some genes in a demethylase-dependent manner.
Jhd2 affects 3′UTR length on a global level
The results above led us to hypothesize that Jhd2 deletion affects 3′UTR length on a global level. To this end, we used 3′ region extraction and deep sequencing (3′READS) to map all the polyadenylation sites in wild-type and Jhd2 deletion strains (26). Briefly, 3′READS involves fragmentation of RNA, enrichment for polyadenylated fragments using magnetic beads, digestion by RNase H, and ligation of sequencing adapters (Fig. 4A). Consistent with previous studies using direct RNA sequencing (RNA-seq), we found that most genes (3860 of 4979 or 78%) have at least two polyadenylation sites (27, 28).
Fig. 4. Jhd2 affects global polyadenylation site choice.
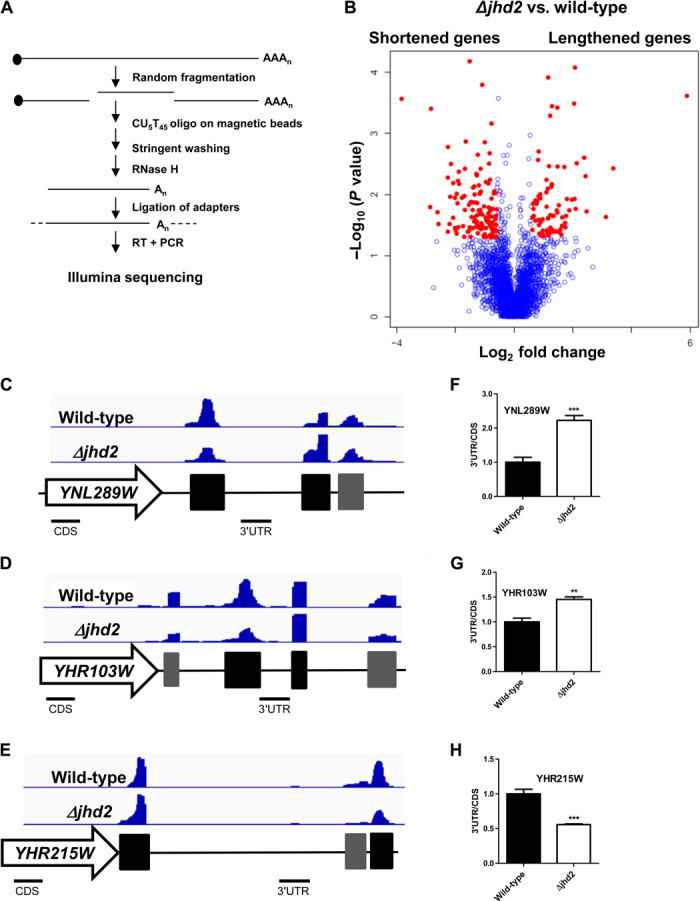
(A) Schematic of the 3′READS method used to enrich for 3′UTRs before RNA-seq. (B) Volcano plot showing P value and fold change of the two most abundant pA clusters in Jhd2 deletion versus wild-type strains. Red dots indicate genes with P < 0.05 and fold change >1.5. The Jhd2 deletion strain selects for distal polyA sites for 76 lengthened transcripts (right), whereas the wild-type strain selects for distal polyA sites in 125 shortened transcripts (left). (C to E) Integrative Genomics Viewer (IGV) snapshot showing polyA clusters (the two most abundant ones in black bars and the others in gray bars) of (C) YNL289W, (D) YHR103W, and (E) YHR215W from representative wild-type and Jhd2 deletion samples. (F to H) RT-qPCR analysis validating the ratio of 3′UTR to CDS mRNA for (F) YNL289W, (G) YHR103W, and (H) YHR215W in wild-type and Jhd2 deletion samples. Error bars represent SEM for biological triplicate experiments. **P < 0.01; ***P < 0.001.
Because many genes have multiple transcripts, we chose to compare the ratio of the two most abundant transcripts for each gene between Jhd2 deletion and wild-type strains. We found that among the 3510 genes with abundant reads, Jhd2 deletion led to increased distal transcripts for 76 genes (referred to as lengthened genes) and decreased distal transcripts for 125 genes (referred to as shortened genes) (Fig. 4B and table S3). For example, Jhd2 deletion results in increased usage of the distal polyA sites for YNL289W and YHR103W and of the proximal polyA site for YHR215W (Fig. 4, C to E), and these results have been validated with RT-qPCR (Fig. 4, F to H). These results support our hypothesis that Jhd2 is involved in maintenance of 3′UTR length and mRNA stability.
Low KDM5B level is associated with 3′UTR lengthening of oncogene CCND1
Humans have four KDM5 family proteins: KDM5A, KDM5B, KDM5C, and KDM5D (29). The human KDM5 histone demethylases have been associated with cancer progression and stem cell development (30–35) and function as transcriptional regulators of genes key to cell proliferation and development (36–38). KDM5B (also known as JARID1B or PLU1) has been shown to play critical roles in breast cancer and melanoma (39–43).
Here, the phenomena involving Jhd2 that we have noted suggest that its mammalian paralogs may play similar roles in human cancers. It has been discovered that, in cancer cells, oncogenes have shortened 3′UTRs (44). These shortened 3′UTRs lack miRNA binding sites that would normally lead to translational repression or mRNA degradation. As a result, the corresponding oncoproteins are overexpressed and contribute to cancer progression. A general shortening of oncogene 3′UTRs in breast cancer cell lines and tumors has since been corroborated (45, 46).
We hypothesized that KDM5B works through a mechanism similar to Jhd2 in promoting 3′UTR shortening of oncogenes. We chose to examine cyclin D1 (CCND1) as a candidate oncogene because it expresses long and short transcripts in multiple cancer cell lines (44). CCND1 has several polyadenylation sites with many miRNA binding sites in between (Fig. 5A). The distal polyA site is the canonical site used in MCF10A immortalized mammary epithelial lines (44). Use of the proximal sites causes loss of all or many miRNA binding sites, and an increase in usage of proximal polyA sites results in an increase in its expression (Fig. 5A) (44). To study the potential involvement of KDM5B in this phenomenon, we knocked down KDM5B expression using a short hairpin RNA (shRNA). We chose to use two breast cancer cell lines, T47D and MCF7 (Fig. 5, B and C), which are known to express high levels of KDM5B. Knockdown of KDM5B in both cell lines resulted in an increase in the long transcript (Fig. 5, B and C). In contrast, knockdown of KDM5A (also known as JARID1A or RBP2) in T47D cells showed no effect (Fig. 5D). Together, these results suggest that down-regulation of KDM5B, but not KDM5A, causes lengthening of the CCND1 3′UTR.
Fig. 5. KDM5B down-regulation leads to 3′UTR lengthening of CCND1.
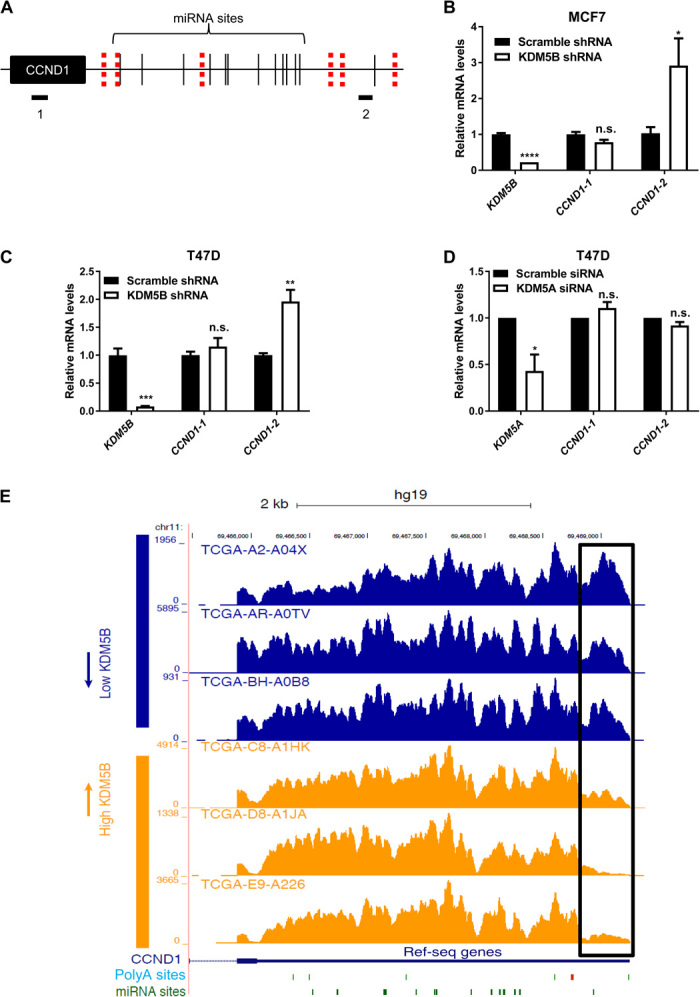
(A) Schematic representation of the amplicons used to study 3′UTR length of CCND1. Both polyA sites (red dashes) and miRNA sites (black lines) are noted. (B and C) RT-qPCR analysis of KDM5B and CCND1 with the indicated amplicons in (B) MCF7 and (C) T47D breast cancer cells infected with the indicated lentiviruses. Error bars represent SEM for biological triplicate experiments. *P < 0.05; ***P < 0.001; ****P < 0.0001; n.s., not statistically significant. (D) RT-qPCR analysis of KDM5A and CCND1 with the indicated amplicons in T47D breast cancer cells transfected with the indicated small interfering RNAs (siRNAs). Error bars represent SEM for biological duplicate experiments. *P < 0.05. (E) RNA-seq coverage of the 3′UTR region of CCND1 in patient tumors expressing low levels of KDM5B compared with patient tumors expressing high levels of KDM5B. Both polyA (green and red bars) and miRNA (green bars) sites are noted. The box highlights the difference of the two long isoforms of CCND1 between these two groups. TCGA tumor sample identifiers are listed.
The high levels of KDM5B noted in some breast tumor samples led us to wonder whether the above regulation can be validated in breast tumors. To this end, we investigated the correlation between KDM5B expression and CCND1 3′UTR length in patient samples using dynamic analysis of APA from RNA-seq (DaPars), which allows for detection of APA events in standard RNA-seq data (47). To study the effect of KDM5B levels on APA on oncogenes, we selected 10 breast tumors expressing the highest levels of KDM5B and 10 breast tumors expressing the lowest levels of KDM5B from The Cancer Genome Atlas (TCGA) breast cancer data set and compared APA levels of CCND1 in these two groups. We observed strong association of high KDM5B expression with selection for short CCND1 3′UTR (P = 0.004, Wilcoxon rank sum test; six representative samples in Fig. 5E). These results parallel those that we have found for KDM5B in breast cancer cell lines (Fig. 5, B and C).
KDM5s regulate 3′UTR length in both demethylase-dependent and demethylase-independent manners
As we have shown that Jhd2 promotes 3′UTR shortening of some genes in a demethylase-dependent manner in yeast, we further examined the contribution of the KDM5 demethylase activity to 3′UTR regulation in mammalian cells using a potent cell-permeable pan-KDM5 inhibitor, KDM5-C70 (48, 49). Although KDM5-C70 treatment increased global H3K4me3 levels in MCF7 cells as expected (Fig. 6A), it did not affect the length of 3′UTR for CCND1 (Fig. 6B), suggesting that demethylase activity of KDM5B is not required for APA regulation of CCND1. We also examined another well-characterized APA target, DICER1, which was shown to be shortened after CFIm25 down-regulation (50). Similarly, KDM5-C70 treatment increased global H3K4me3 levels in HeLa cells (Fig. 6C). In this case, KDM5-C70 also led to decreased 3′UTR length of DICER1 (Fig. 6D), supporting the notion that the demethylase activity of KDM5 family members is involved in APA regulation of DICER1. To determine which KDM5(s) contribute to this regulation, we deleted KDM5A, KDM5B, and KDM5C individually using an inducible CRISPR (clustered regularly interspaced short palindromic repeats) system (51, 52) and found that deletion of KDM5A or KDM5B, but not KDM5C, decreased 3′UTR length of DICER1 (Fig. 6E), suggesting that both KDM5A and KDM5B are involved in APA regulation.
Fig. 6. KDM5s are involved in selection of polyA sites.
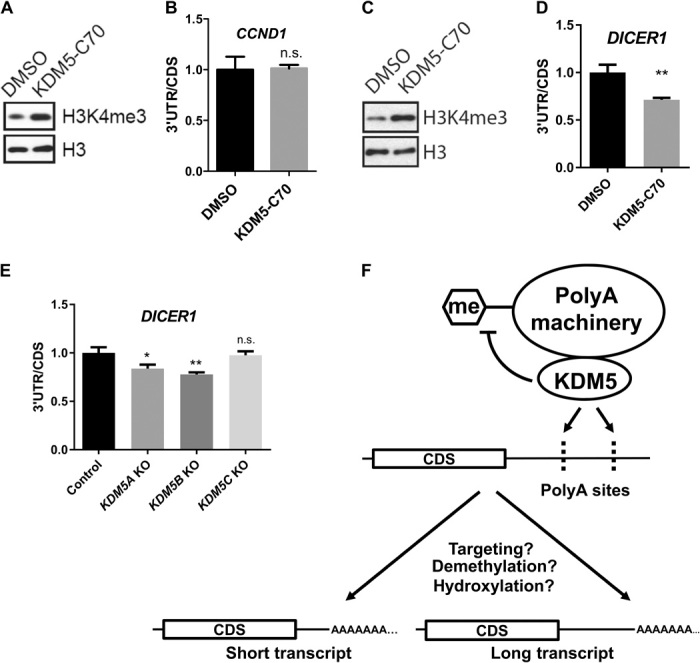
(A) Western blot and (B) RT-qPCR analyses of MCF7 cells treated with dimethyl sulfoxide (DMSO) or 10 μM KDM5-C70 for 3 days. The ratio of 3′UTR to CDS for CCND1 mRNA was plotted. Error bars represent SEM for biological triplicate experiments. (C) Western blot and (D) RT-qPCR analyses of HeLa cells treated with DMSO or 10 μM KDM5-C70 for 3 days. The ratio of 3′UTR to CDS for DICER1 mRNA was plotted. Error bars represent SEM for biological triplicate experiments. **P < 0.01. (E) RT-qPCR analysis of HeLa/iCas9-c1 cells transduced with lentiviruses carrying single-guide RNAs against KDM5A, KDM5B, KDM5C, or nontargeting control. The ratio of 3′UTR to CDS for DICER1 mRNA was plotted. KO, knockout. Error bars represent SEM for biological triplicate experiments. *P < 0.05; **P < 0.01. (F) Working model for KDM5 involvement in APA. KDM5 recruits the polyA machinery to nascent RNA to modulate polyA site choices. Demethylation or hydroxylation of certain subunits of the polyA machinery by KDM5 also contributes to selection of the polyA sites. The polyA sites (dashes) are noted on the nascent transcript.
DISCUSSION
Here, we found that in addition to its role in transcription regulation, the yeast KDM5 family protein, Jhd2, recruits polyadenylation machinery and promotes alteration of 3′UTRs. We showed that regulation of 3′UTR length is likely linked to the ability of Jhd2 to interact with both chromatin and mRNA transcript. We further showed consistent results in regulation of 3′UTR by its mammalian homologs KDM5B and KDM5A, indicating that this function is evolutionarily conserved. Thus, our work revealed a novel functional interaction of a chromatin regulator, KDM5, with mRNA processing machinery, suggesting that transcription regulation and mRNA processing are coupled through the function of the KDM5 family of enzymes.
The mechanism by which cells choose different polyA sites is one of the great mysteries of RNA processing. Here, we showed that KDM5 enzymes can alter usage of polyA sites, and our findings suggest several possible mechanisms (Fig. 6F). First, KDM5 enzymes could do this by targeting the polyA machinery to different polyA sites. By interacting with RNA and chromatin, Jhd2 could increase the amount of polyadenylation factors at proximal or distal polyA sites and promote 3′ end processing at these sites (Fig. 6F).
Second, Jhd2 may also affect polyA site usage by affecting RNA Pol II pausing and elongation, independent of its effect on polyadenylation factor recruitment. In this regard, it has been shown that the speed of elongation can affect polyadenylation site choice (53–55). Consistent with this idea, our affinity purification data also showed that Jhd2 can interact with several components involved in transcription elongation, such as Spt5 and Dst1 (Fig. 1A).
Third, our work on PMA1 and KlCYC1 also suggests that the demethylase activity of Jhd2 promotes usage of the proximal polyA sites for some genes. Previous studies showed that Jhd2 loss increased H3K4me3 levels near transcription termination sites during sporulation (56). We performed ChIP-seq using an anti-H3K4me3 antibody in wild-type and Jhd2 deletion strains (fig. S7) and observed a significant dip of H3K4me3 signal near proximal polyA sites (fig. S7A). Surprisingly, we found no major difference in H3K4me3 levels near the beginning of open reading frames (ORFs) [translational start site (TSS)], at either proximal or distal polyA sites genome-wide, and even of genes in which polyA site choice is affected by Jhd2. These results suggest that H3K4 methylation does not contribute to polyA site selection under normal growth conditions. Note that some components of polyadenylation machinery, including Pcf11, are methylated at lysines and arginines (57–59). It is possible that Jhd2 partly regulates 3′UTR choice by demethylating or hydroxylating factors involved in polyadenylation or transcription elongation (Fig. 6F).
Our results have important implications in the role of the KDM5 proteins in carcinogenesis. We showed that KDM5B knockdown increased the percentage of longer transcripts for a well-known oncogene, CCND1 (Fig. 5, B and C), whereas KDM5A and KDM5B deletion led to shortening of DICER1, a tumor suppressor gene critical for miRNA processing (60). These results suggest that targeting KDM5s in cancer could have beneficial effects through APA regulation. Using KDM5-C70, a potent pan-KDM5 inhibitor, we showed that the demethylase activity of KDM5s is required for the regulation of APA for DICER1, but not for CCND1. These data suggest that both the demethylase activity and the demethylase-independent functions of KDM5s are required for modulating APA. Previous studies also showed that KDM5s have roles beyond their demethylase activity in cancer and development (36, 48, 61, 62). Current KDM5 inhibitors are small molecules that inhibit KDM5 demethylase activity (48, 49, 63–67), and our results suggest that targeting other roles of KDM5A and KDM5B is also required to suppress the oncogenic roles of KDM5A and KDM5B.
MATERIALS AND METHODS
Plasmids, yeast strains, and primers
All plasmids, yeast strains, and primers are described in tables S4, S5, and S6, respectively.
Purification and mass spectrometric identification of proteins interacting with Jhd2
To determine protein-protein interactions, a strain of S. cerevisiae expressing a TAP-tagged version of Jhd2 was grown to mid-log phase and harvested by centrifugation. Cells were then frozen in liquid nitrogen, cryogenically lysed using a Retsch MM301 ball mill, and thawed in immunoprecipitation (IP) buffer [20 mM Hepes-NaOH (pH 7.4), 2 mM MgCl2, 300 mM NaCl, 0.1% Tween 20, and 1/100 protease inhibitor cocktail]. Deoxyribonuclease I (DNase I) was added to each lysate at 2% (w/v). Cells were incubated at 4°C for an hour and centrifuged at 1800g at 4°C for 10 min to create cell lysate. Cell lysate from 12 g of cells was incubated with IgG-coated M270 Epoxy Dynabeads (Invitrogen) at 4°C for 3 hours. A magnet was used to isolate the Dynabeads, and samples were washed five times in IP buffer [20 mM Hepes-NaOH (pH 7.5), 2 mM MgCl2, 300 mM NaCl, 0.1% Tween 20, and 1/100 protease inhibitor cocktail]. After the final wash, the beads were incubated with 500 mM NH4OH and 0.5 mM EDTA (pH 8.0) for 10 min at room temperature. Supernatant was flash-frozen and lyophilized in a SpeedVac. Samples were then resuspended in protein sample buffer (78 mM tris-HCl, 25 mM tris base, 12.5% glycerol, 6.25 mM tris(2-carboxyethyl)phosphine–HCl, 2.5% SDS, and 0.003% bromophenol blue), heated at 95°C for 5 min, and resolved by 4 to 20% tris-glycine SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels (Bio-Rad). Samples were stained with GelCode Blue Coomassie (Thermo Fisher). Gels were excised into 30 bands. Gel bands were destained and digested using trypsin (Sigma-Aldrich). Peptides were then collected using ZipTip (Waters). Spectra were collected on a Thermo LTQ-XL Linear Ion Trap Mass Spectrometer and analyzed using Scaffold software (Proteome Software). Protein identity was cut off at 99%, and peptide certainty was set at 95% with at least two peptides identified. Tandem spectra for proteins of interest were manually verified using Xcalibur software (Thermo).
Coaffinity purification and Western blotting of polyadenylation machinery
Coaffinity purification experiments were performed using TAP-tagged strains of polyadenylation machinery components. Strains were grown in appropriate media to mid-log phase and lysed in IP buffer with or without RNase A (75 ng/μl). Complexes were purified using IgG-Sepharose beads (Sigma-Aldrich) and washed four times in IP buffer. After the final wash, beads were incubated with 500 mM NH4OH and 0.5 mM EDTA (pH 8.0) for 10 min at room temperature. Complexes were then lyophilized, resuspended in SDS sample buffer, and resolved on an SDS-PAGE gel. Gels were transferred to nitrocellulose membranes and blotted with a Jhd2 antibody (Active Motif, 39263) or horseradish peroxidase (HRP)–IgG (Rockland Immunochemicals, 612-4302).
In vitro cleavage and transcription reactions
In cleavage assays, radioactive RNA containing the GAL7 polyA site was incubated with extracts prepared from yeasts in the presence of 3′-deoxyadenosine triphosphate, which blocks polyA addition. Preparation of extract and precursor and detailed reaction conditions are as described by Zhao et al. (68). RNAs were purified from samples, resolved on a denaturing 5% polyacrylamide gel, and visualized by phosphorimager analysis.
Extracts were prepared as described by Pearson and Moore (69) and used to transcribe 32P-labeled RNA from the DNA templates, which contains a promoter for RNA Pol II and five G-less cassettes of different lengths downstream of the transcription start site, with or without the CYC1 polyA site inserted between the second and third cassettes. RNA produced by transcription was digested with RNase T1, and the RNA fragments were resolved on a 6% polyacrylamide/7 M urea gel and visualized by phosphorimager analysis. The signal derived from upstream cassettes [84 and 100 nucleotides (nt)] was compared to that from the downstream cassettes (120, 131, and 145 nt) to determine the effect of a polyA site.
RNA extraction and RT-qPCR
Yeast RNA was extracted via a standard hot acid phenol procedure (70). Briefly, yeast cells were grown to mid-log phase, resuspended in TES buffer [10 mM tris-HCl (pH 7.5), 10 mM EDTA (pH 8.0), and 0.5% SDS] and acid phenol, and incubated at 65°C. Temperature-sensitive pcf11-2 mutants were grown at 30°C until mid-log and then transferred to 37°C for an hour before RNA was collected. RNA was then extracted twice in acid phenol and once in chloroform before it was resuspended in dimethyldicarbonate-treated water. Human cell line RNA was extracted using RNeasy Mini Kit (Qiagen). Quantification was performed on a NanoDrop spectrophotometer (Thermo Fisher). For mRNA expression analysis, RNA was converted into cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosciences) and was amplified using SYBR Green mix (Applied Biosciences) on a ViiA7 machine from Applied Biosystems. For quantification, mRNA levels were normalized to GAPDH and compared to wild-type levels. All studies involving relative mRNA levels were carried out in biological triplicate and represented as averages ± SEM. All the data were assessed with unpaired Student's t test.
APA analysis
For 3′READS analysis, wild-type yeast or yeast with Jhd2 deletion was grown to mid-log phase, and RNA was extracted from three biological replicates of Jhd2 deletion samples and two biological replicates of wild-type samples as mentioned above. cDNA libraries enriched for 3′UTRs were prepared as previously published and noted in Fig. 4A (26). Samples were then subjected to RNA-seq on the Illumina HiSeq 2000 using 50- and 100-bp single-end reads. 3′READS data were deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database under accession number GSE67212. Data were analyzed as previously described with the following modifications: Adapter sequences were trimmed off the single-end reads (26). The remaining reads longer than 15 bp were mapped to yeast genome SacCer3 using bowtie2 with the setting “-5 4 --local,” trimming off 4 bp from 5′ and allowing soft clipping at both ends (71). The alignments were then filtered by (i) mapping quality score ≥10, (ii) mismatches ≤5%, and (iii) unaligned 5′ Ts ≥2 to get the polyA site–supporting (PASS) reads. The last aligned positions of the PASS reads were grouped to pA clusters with a clustering size of 24 bp. Each pA cluster was assigned to one of the genes defined by the Saccharomyces Genome Database with verified ORF. pA assignment was conducted using a 5′-truncated gene model, starting from 100 bp downstream of the start of CDS, to 1000 bp downstream of the end of CDS or 100 bp downstream of the next CDS in the same direction, whichever is shorter. The pA clusters that were then filtered by contains (i) ≥10 PASS reads and (ii) ≥5% of all PASS reads mapped to the gene.
For each gene with more than one pA cluster, the two most abundant pA clusters with at least nine supporting PASS reads in all the samples combined were tested for APA. A linear mixed-regression model was used to examine whether the log2(distal ratio) was significantly different between three Jhd2 deletion and two wild-type samples. Distal ratio is defined as the ratio of PASS reads of the distal pA cluster over that of the proximal pA cluster. In the linear mixed model, the log2 count of PASS reads of each pA site of each sample was modeled with the lmerTest package in the following formula: logCount ~ pA + genotype + pA/genotype + batch + (1|sample), where pA is one of the two pA sites (distal and proximal), genotype is either wild-type or Jhd2-deleted, and the batch is one of two batches. The sample was modeled as a normalizing random parameter. The interaction term pA/geneotype was used to imply APA. The top list was selected using the following criteria: (i) P ≤ 0.05 and (ii) fold change of distal ratio ≥1.5.
ChIP and sequencing
For ChIP experiments, yeast cells were grown to mid-log phase and cross-linked using 1% formaldehyde for 15 min at room temperature. Samples were washed in phosphate-buffered saline and lysed by lysis buffer [50 mM Hepes (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate (DOC), 0.1% SDS, and protease inhibitors] with 500 μl of glass beads. Shearing of DNA was carried out using a Bioruptor sonicator (Diagenode) for 25 min with 30-s on and 30-s off cycles. TAP-tagged proteins were isolated from chromatin using rabbit IgG-Sepharose beads. CL-4B Sepharose beads (Sigma-Aldrich) were used for control IP. Data for H3K4me3 ChIP were attained using ChIP-grade antibodies against H3K4me3 (Abcam, ab8580) and Protein A-Sepharose beads (Sigma-Aldrich). IgG (Sigma-Aldrich, I5381) was used as a nonspecific control for H3K4me3 and Rna15 ChIP. All samples were washed in lysis buffer, washing buffer [10 mM tris-HCl (pH 8.0), 250 mM LiCl, 0.5% NP-40, 0.5% NaDOC, and 1 mM EDTA], and tris-EDTA (TE) buffer [10 mM tris-HCl (pH 8.0) and 1 mM EDTA (pH 8.0)]. Cross-links were reversed in TES [10 mM tris-Cl (pH 7.5), 1 mM EDTA, and 1.0% SDS] at 65°C, and samples were treated with proteinase K. DNA was precipitated using standard ethanol precipitation protocols and was resuspended in TE containing RNase. The same SYBR Green mix and ViiA7 machine, as mentioned above, were used for quantification of ChIP efficiency. ChIP data were analyzed using fold enrichment over the control ChIP. All ChIP experiments were carried out in biological triplicate. ChIP-seq experiments were performed using HiSeq 2000 (Illumina).
ChIP-seq analysis
The sequencing reads (50 bp, single-end) were trimmed for 3′ adapter sequences using Trimmomatic, and the reads shorter than 36 bp after trimming were excluded (72). The remaining sequencing reads were aligned to the S. cerevisiae reference genome (SacCer3) using bowtie (71). Up to one mismatch was allowed in the 28-bp seeding region for each aligned read. Reads mapping to multiple sites were excluded. ChIP-seq data were deposited in the NCBI Gene Expression Omnibus database under accession number GSE67212. Binding regions were identified using SICER with the following parameters: a effective genome size of 0.74, a window size of 200 bp, and a gap size of 200 bp (73). HOMER was used for peak annotation and coverage calculation (74). The IGV was used for visualization (75). Python and R scripts were used to obtain the metagene plots on the basis of the peak annotation and normalized coverage. The base pair coverage for each gene in the gene set was aggregated by sum at each position of the metagene region (±1000 bp relative to TSSs or polyA sites). The coverage was then normalized to the total coverage in the region for each sample (IP, input separately), followed by a log2 ratio calculation between IP and input.
Expression of recombinant Jhd2
Recombinant Jhd2-6xHis used for RNA binding experiments was cloned into the pRSF-duet vector using Sal I and Hind III restriction enzyme sites. Protein was expressed in BL21DE3 Escherichia coli cells, lysed in lysis buffer (pH 8.0) (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, and proteinase inhibitors), and purified using an ÄKTA pure 25 fast performance liquid chromatography instrument equipped with a HisTrap nickel column (GE Healthcare Life Sciences). The column was equilibrated and washed with lysis buffer, and His-tagged Jhd2 was eluted using elution buffer (pH 8.0) (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and proteinase inhibitors). Protein was verified by Western blot using both Jhd2 (Active Motif, 39263) and His tag antibodies (Cell Signaling Technology, 2365) (fig. S3A).
In vitro RNA binding assay
A biotinylated RNA 20-mer was designed using a random sequence generator and obtained from Integrated DNA Technologies. Three RNA structure prediction software tools (RNAfold, Sfold, and RNA structure) were used to determine the absence of secondary structure. Reactions were prepared containing 0.01 μg of RNA and 5.0 μg of recombinant His-Jhd2 in binding buffer [50 mM tris-HCl (pH 7.9), 10% glycerol, 100 mM KCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, and 0.1% NP- 40]. A control that contained no RNA was prepared. The reactions were incubated with rotating for 1 hour at room temperature. Streptavidin-coupled magnetic M-280 Dynabeads (Invitrogen) were added to the reaction, and samples were incubated while rotating for an additional 30 min at room temperature. The beads were washed four times with binding buffer. Samples were combined with SDS loading buffer and incubated at 95°C for 5 min. The proteins were resolved using SDS-PAGE and detected via Western blot using a Jhd2 antibody (Active Motif, 39263).
RNA immunoprecipitation
RIP experiments were performed as previously published by Selth et al. (76). Briefly, yeast cells were grown to mid-log phase and cross-linked using 1% formaldehyde for 15 min at room temperature. Samples were lysed in FA lysis buffer with glass beads. DNase was added before and after cell lysis. Shearing of DNA was carried out using a Bioruptor sonicator (Diagenode) for 7 min, with 15-s on and 60-s off cycles. TAP-tagged proteins were isolated from chromatin using IgG-Sepharose beads (Sigma-Aldrich). CL-4B Sepharose beads were used as a negative control (Sigma-Aldrich). Samples were then washed with FA lysis buffer, high-salt lysis buffer [50 mM Hepes (pH 7.5), 500 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton X-100, 0.1% sodium deoxycholate, and protease inhibitors], LiCl buffer [10 mM tris-Cl (pH 8.0), 250 mM LiCl, 1 mM EDTA (pH 8.0), 5% NP-40, 1% sodium deoxycholate, and protease inhibitors], and TE buffer, all containing RNase inhibitors. Samples were eluted in TE + 1% SDS. Cross-links were reversed at 65°C, and samples were treated with proteinase K. RNA was precipitated using the RNA extraction protocol outlined above. Samples were resuspended in RNase-free water containing DNase. RNA was converted into cDNA as mentioned above and analyzed using real-time PCR. Data analysis was performed using fold enrichment over beads alone. All RIP experiments were carried out in biological duplicate.
KDM5 knockdown and knockout cell lines
Breast cancer cell lines MCF7 and T47D and cervical cancer cell line HeLa were obtained from the American Type Culture Collection tissue bank. shRNAs targeting either scrambled sequence or KDM5B as described previously were introduced to these cell lines using lentiviral infection (63). Cell lines stably expressing these shRNAs were used for 3′UTR length studies as mentioned above. For KDM5A knockdown, siRNAs targeting scrambled sequence or KDM5A as described previously were transiently transfected into T47D cell lines (36). Knockout of KDM5 members in HeLa/iCas9-c1 cells was achieved using the inducible CRISPR/Cas9 system as previously described (51).
Analysis of CCND1 3′UTR in patient samples
Of the 1240 breast invasive carcinoma RNA-seq samples in TCGA, we selected 10 samples with highest KDM5B expression and another 10 samples with lowest KDM5B expression. These two groups were analyzed by DaPars to identify dynamic 3′UTR usage events. The 3′UTR usage of each sample was quantified by percentage of distal polyA site usage index (PDUI) (47). Finally, the change in PDUI between two groups (ΔPDUI ≥0.2) and t test of PDUI between two groups P ≤ 0.05 were used to identify significant APA events. On the basis of these parameters, the gene CCND1 was identified to have significantly different 3′UTR usage between the high–KDM5B expression group and the low–KDM5B expression group. The read densities of six selected samples are shown in Fig. 5E. Institutional Review Board guidelines were followed for this analysis.
Supplementary Material
Acknowledgments
We acknowledge P. Cramer for TAP-tagged Hrp1 strain, F. Lacroute for the pcf11-2 temperature-sensitive mutant, M. A. Freire-Picos for the KlCYC1 construct, Y. Zhang for FLAG-Jhd2 and FLAG-Jhd2H427A vectors, G. Shadel and D. F. Stern for use of equipment, and the University of Arkansas for Medical Sciences Proteomics Facility (supported by NIH grants UL1TR000039, P30GM103450, P20GM103625, and P20GM103429) for mass spectrometric support. We thank M. Zhong of the Yale Stem Cell Center Genomics Core facility for next-generation sequencing service. We thank J. Steitz and members of the Moore, Shadel, Steitz, Stern, and Yan laboratories for their help and valuable discussions. Funding: This work was supported by the American Cancer Society Research Scholar grant RSG-13-384-01-DMC (to Q.Y.); Department of Defense (DOD) Breast Cancer Research Program awards W81XWH-14-1-0308 and W81XWH-15-1-0117 (to Q.Y.); DOD Peer Reviewed Cancer Research Program award W81XWH-13-1-0235 (to Q.Y.); Breast Cancer Alliance Young Investigator Grant (to Q.Y.); NIH grants R21CA191548, R21CA187862 (to Q.Y.), R01GM106024 (to A.J.T.), R33CA173264 (to A.J.T.), R01GM084089 (to B.T.), and R01HG007538 (to W.L.); Cancer Prevention Research Institute of Texas grant RP150292 (to W.L.), NSF grant MCB-1244043 (to C.L.M.); Leslie H. Warner Postdoctoral Fellowship (to L.W.); Connecticut Regenerative Medicine Research Fund; and Li Ka Shing Foundation (Yale Stem Cell Center Genomics Core). Author contributions: L.P.B. and Q.Y. conceived the project and designed the experiments. L.P.B., R.L.D.L., L.W., D.Z., Z.X., E.L.P., F.I.N., J.C., S.M.L., and R.J.R. performed the experiments. S.G.M. and A.J.T. performed mass spectrometry. A.J.T. and C.L.M. contributed to design of yeast experiments. L.P.B., Z.L., R.L.D.L., Z.X., C.L.M., W.L., B.T., and Q.Y. analyzed the data. L.P.B. and Q.Y. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: ChIP-seq and 3′READS data were deposited in the NCBI Gene Expression Omnibus database under accession number GSE67212. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1501662/DC1
fig. S1. Mass spectrometry results.
fig. S2. Western blot analysis of TAP-tagged proteins.
fig. S3. Materials for in vitro RNA binding experiments.
fig. S4. Loss of Jhd2 does not affect processing or polyA-dependent termination in vitro.
fig. S5. RT-qPCR and Western blot analysis for Jhd2 mRNA and protein used in PMA1 studies.
fig. S6. RT-qPCR and Western blot analysis for Jhd2 mRNA and protein used in KlCYC1 studies.
fig. S7. Metagene plots of H3K4me3 levels.
table S1. Jhd2 bound peaks identified using SICER.
table S2. Pcf11 bound peaks identified using SICER.
table S3. List of genes with APA after Jhd2 deletion.
table S4. Plasmids used in this study.
table S5. Yeast strains used in this study.
table S6. Primers used in this study.
REFERENCES AND NOTES
- 1.Greer E. L., Shi Y., Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang G., Klose R. J., Gardner K. E., Zhang Y., Yeast Jhd2p is a histone H3 Lys4 trimethyl demethylase. Nat. Struct. Mol. Biol. 14, 243–245 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Seward D. J., Cubberley G., Kim S., Schonewald M., Zhang L., Tripet B., Bentley D. L., Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat. Struct. Mol. Biol. 14, 240–242 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Tu S., Bulloch E. M. M., Yang L., Ren C., Huang W.-C., Hsu P.-H., Chen C.-H., Liao C.-L., Yu H.-M., Lo W.-S., Freitas M. A., Tsai M.-D., Identification of histone demethylases in Saccharomyces cerevisiae. J. Biol. Chem. 282, 14262–14271 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mersman D. P., Du H.-N., Fingerman I. M., South P. F., Briggs S. D., Polyubiquitination of the demethylase Jhd2 controls histone methylation and gene expression. Genes Dev. 23, 951–962 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mata J., Genome-wide mapping of polyadenylation sites in fission yeast reveals widespread alternative polyadenylation. RNA Biol. 10, 1407–1414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Y., Li Z., Ozsolak F., Woo Kim S., Arango-Argoty G., Liu T. T., Tenenbaum S. A., Bailey T., Paula Monaghan A., Milos P. M., John B., An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 40, 8460–8471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y., Alternative polyadenylation: New insights from global analyses. RNA 18, 2105–2117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L., Dowell R. D., Yi R., Genome-wide maps of polyadenylation reveal dynamic mRNA 3′-end formation in mammalian cell lineages. RNA 19, 413–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boutet S. C., Cheung T. H., Quach N. L., Liu L., Prescott S. L., Edalati A., Iori K., Rando T. A., Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 10, 327–336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elkon R., Ugalde A. P., Agami R., Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 14, 496–506 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Tian B., Manley J. L., Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 38, 312–320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millevoi S., Vagner S., Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 38, 2757–2774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phatnani H. P., Greenleaf A. L., Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Johnson S. A., Cubberley G., Bentley D. L., Cotranscriptional recruitment of the mRNA export factor Yra1 by direct interaction with the 3′ end processing factor Pcf11. Mol. Cell 33, 215–226 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson S. A., Kim H., Erickson B., Bentley D. L., The export factor Yra1 modulates mRNA 3′ end processing. Nat. Struct. Mol. Biol. 18, 1164–1471 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang F., Chandrasekharan M. B., Chen Y.-C., Bhaskara S., Hiebert S. W., Sun Z.-W., The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J. Biol. Chem. 285, 24548–24561 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al Husini N., Kudla P., Ansari A., A role for CF1A 3′ end processing complex in promoter-associated transcription. PLOS Genet. 9, e1003722 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer A., Schreieck A., Lidschreiber M., Leike K., Martin D. E., Cramer P., The spt5 C-terminal region recruits yeast 3′ RNA cleavage factor I. Mol. Cell. Biol. 32, 1321–1331 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haddad R., Maurice F., Viphakone N., Voisinet-Hakil F., Fribourg S., Minvielle-Sébastia L., An essential role for Clp1 in assembly of polyadenylation complex CF IA and Pol II transcription termination. Nucleic Acids Res. 40, 1226–1239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim M., Ahn S.-H., Krogan N. J., Greenblatt J. F., Buratowski S., Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J. 23, 354–364 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amrani N., Minet M., Wyers F., Dufour M. E., Aggerbeck L. P., Lacroute F., PCF11 encodes a third protein component of yeast cleavage and polyadenylation factor I. Mol. Cell. Biol. 17, 1102–1109 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadowski M., Dichtl B., Hübner W., Keller W., Independent functions of yeast Pcf11p in pre-mRNA 3′ end processing and in transcription termination. EMBO J. 22, 2167–2177 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freire-Picos M. A., Lombardia-Ferreira L. J., Ramil E., González-Domínguez M., Cerdan M. E., The KlCYC1 gene, a downstream region for two differentially regulated transcripts. Yeast 18, 1347–1355 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Seoane S., Lamas-Maceiras M., Rodríguez-Torres A. M., Freire-Picos M. A., Involvement of Pta1, Pcf11 and a KlCYC1 AU-rich element in alternative RNA 3′-end processing selection in yeast. FEBS Lett. 583, 2843–2848 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Hoque M., Ji Z., Zheng D., Luo W., Li W., You B., Yeon Park J., Yehia G., Tian B., Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 10, 133–139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozsolak F., Kapranov P., Foissac S., Kim S. W., Fishilevich E., Monaghan A. P., John B., Milos P. M., Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143, 1018–1029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graber J. H., Nazeer F. I., Yeh P.-c., Kuehner J. N., Borikar S., Hoskinson D., Moore C. L., DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Res. 23, 1690–1703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blair L. P., Cao J., Zou M. R., Sayegh J., Yan Q., Epigenetic regulation by lysine demethylase 5 (KDM5) enzymes in cancer. Cancers (Basel) 3, 1383 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma S. V., Lee D. Y., Li B., Quinlan M. P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M. A., Wong K.-K., Brandstetter K., Wittner B., Ramaswamy S., Classon M., Settleman J., A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin W., Cao J., Liu J., Beshiri M. L., Fujiwara Y., Francis J., Cherniack A. D., Geisen C., Blair L. P., Zou M. R., Shen X., Kawamori D., Liu Z., Grisanzio C., Watanabe H., Minamishima Y. A., Zhang Q., Kulkarni R. N., Signoretti S., Rodig S. J., Bronson R. T., Orkin S. H., Tuck D. P., Benevolenskaya E. V., Meyerson M., Kaeli W. G., Yan Q., Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc. Natl. Acad. Sci. U.S.A. 108, 13379–13386 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dey B. K., Stalker L., Schnerch A., Bhatia M., Taylor-Papidimitriou J., Wynder C., The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol. Cell. Biol. 28, 5312–5327 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie L., Pelz C., Wang W., Bashar A., Varlamova O., Shadle S., Impey S., KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 30, 1473–1484 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz S. U., Albert M., Malatesta M., Morey L., Johansen J. V, Bak M., Tommerup N., Abarrategui I., Helin K., Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 30, 4586–4600 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang Y., Zhu Z., Han G., Ye X., Xu B., Peng Z., Ma Y., Yu Y., Lin H., Chen A. P., Chen C. D., JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 19226–19231 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao J., Liu Z., Cheung W. K. C., Zhao M., Chen S. Y., Chan S. W., Booth C. J., Nguyen D. X., Yan Q., Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 6, 868–877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou M. R., Cao J., Liu Z., Huh S. J., Polyak K., Yan Q., Histone demethylase jumonji AT-rich interactive domain 1B (JARID1B) controls mammary gland development by regulating key developmental and lineage specification genes. J. Biol. Chem. 289, 17620–17633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teng Y.-C., Lee C.-F., Li Y.-S., Chen Y.-R., Hsiao P.-W., Chan M.-Y., Lin F.-M., Huang H.-D., Chen Y.-T., Jeng Y.-M., Hsu C.-H., Yan Q., Tsai M.-D., Juan L.-J., Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 73, 4711–4721 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Lu P. J., Sundquist K., Baeckstrom D., Poulsom R., Hanby A., Meier-Ewert S., Jones T., Mitchell M., Pitha-Rowe P., Freemont P., Taylor-Papadimitriou J., A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J. Biol. Chem. 274, 15633–15645 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Catchpole S., Spencer-Dene B., Hall D., Santangelo S., Rosewell I., Guenatri M., Beatson R., Scibetta A. G., Burchell J. M., Taylor-Papadimitriou J., PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int. J. Oncol. 38, 1267–1277 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto S., Wu Z., Russnes H. G., Takagi S., Peluffo G., Vaske C., Zhao X., Vollan H. K. M., Maruyama R., Ekram M. B., Sun H., Kim J. H., Carver K., Zucca M., Feng J., Almendro V., Bessarabova M., Rueda O. M., Nikolsky Y., Caldas C., Liu X. S., Polyak K., JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell 25, 762–777 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roesch A., Fukunaga-Kalabis M., Schmidt E. C., Zabierowski S. E., Brafford P. A., Vultur A., Basu D., Gimotty P., Vogt T., Herlyn M., A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roesch A., Vultur A., Bogeski I., Wang H., Zimmermann K. M., Speicher D., Körbel C., Laschke M. W., Gimotty P. A., Philipp S. E., Krause E., Pätzold S., Villanueva J., Krepler C., Fukunaga-Kalabis M., Hoth M., Bastian B. C., Vogt T., Herlyn M., Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 23, 811–825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayr C., Bartel D. P., Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akman B. H., Can T., Erson-Bensan A. E., Estrogen-induced upregulation and 3′-UTR shortening of CDC6. Nucleic Acids Res. 40, 10679–16088 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lembo A., Di Cunto F., Provero P., Shortening of 3′UTRs correlates with poor prognosis in breast and lung cancer. PLOS ONE 7, e31129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Z., Donehower L. A., Cooper T. A., Neilson J. R., Wheeler D. A., Wagner E. J., Li W., Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 5, 5274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horton J. R., Liu X., Gale M., Wu L., Shanks J. R., Zhang X., Webber P. J., Bell J. S.K., Kales S. C., Mott B. T., Rai G., Jansen D. J., Henderson M. J., Urban D. J., Hall M. D., Simeonov A., Maloney D. J., Johns M. A., Fu H., Jadhav A., Vertino P. M., Yan Q., Cheng X., Structural basis for KDM5A histone lysine demethylase inhibition by diverse compounds. Cell Chem. Biol. 23, 769–781 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansson C., Velupillai S., Tumber A., Szykowska A., Hookway E. S., Nowak R. P., Strain-Damerell C., Gileadi C., Philpott M., Burgess-Brown N., Wu N., Kopec J., Nuzzi A., Steuber H., Egner U., Badock V., Munro S., LaThangue N. B., Westaway S., Brown J., Athanasou N., Prinjha R., Brennan P. E., Oppermann U., Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol. 12, 539–545 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Masamha C. P., Xia Z., Yang J., Albrecht T. R., Li M., Shyu A.-B., Li W., Wagner E. J., CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao J., Wu L., Zhang S.-M., Lu M., Cheung W. K. C., Cai W., Gale M., Xu Q., Yan Q., An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. gkw660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gale M., Sayegh J., Cao J., Norcia M., Gareiss P., Hoyer D., Merkel J. S., Yan Q., Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 7, 39931–39944 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwalds-Gilbert G., Prescott J., Falck-Pedersen E., 3′ RNA processing efficiency plays a primary role in generating termination-competent RNA polymerase II elongation complexes. Mol. Cell. Biol. 13, 3472–3480 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plant K. E., Dye M. J., Lafaille C., Proudfoot N. J., Strong polyadenylation and weak pausing combine to cause efficient termination of transcription in the human Gγ-globin gene. Mol. Cell. Biol. 25, 3276–3285 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yonaha M., Proudfoot N. J., Transcriptional termination and coupled polyadenylation in vitro. EMBO J. 19, 3770–3777 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu M., Soloveychik M., Ranger M., Schertzberg M., Shah Z., Raisner R., Venkatasubrahmanyan S., Tsui K., Gebbia M., Hughes T., van Bakel H., Nislow C., Madhani H. D., Meneghini M. D., Timing of transcriptional quiescence during gametogenesis is controlled by global histone H3K4 demethylation. Dev. Cell 23, 1059–1071 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu M. C., Bachand F., McBride A. E., Komili S., Casolari J. M., Silver P. A., Arginine methyltransferase affects interactions and recruitment of mRNA processing and export factors. Genes Dev. 18, 2024–2035 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moore K. E., Carlson S. M., Camp N. D., Cheung P., James R. G., Chua K. F., Wolf-Yadlin A., Gozani O., A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol. Cell 50, 444–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bremang M., Cuomo A., Agresta A. M., Stugiewicz M., Spadotto V., Bonaldi T., Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome. Mol. Biosyst. 9, 2231–2247 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Raaijmakers M. H. G. P., Mukherjee S., Guo S., Zhang S., Kobayashi T., Schoonmaker J. A., Ebert B. L., Al-Shahrour F., Hasserjian R. P., Scadden E. O., Aung Z., Matza M., Merkenschlager M., Lin C., Rommens J. M., Scadden D. T., Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464, 852–857 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klein B. J., Piao L., Xi Y., Rincon-Arano H., Rothbart S. B., Peng D., Wen H., Larson C., Zhang X., Zheng X., Cortazar M. A., Peña P. V., Mangan A., Bentley D. L., Strahl B. D., Groudine M., Li W., Shi X., Kutateladze T. G., The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 6, 325–335 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu X., Greer C., Secombe J., KDM5 interacts with Foxo to modulate cellular levels of oxidative stress. PLOS Genet. 10, e1004676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sayegh J., Cao J., Zou M. R., Morales A., Blair L. P., Norcia M., Hoyer D., Tackett A. J., Merkel J. S., Yan Q., Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem. 288, 9408–9417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L., Chang J., Varghese D., Dellinger M., Kumar S., Best A. M., Ruiz J., Bruick R., Peña-Llopis S., Xu J., Babinski D. J., Frantz D. E., Brekken R. A., Quinn A. M., Simeonov A., Easmon J., Martinez E. D., A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 4, 2035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kristensen L. H., Nielsen A. L., Helgstrand C., Lees M., Cloos P., Kastrup J. S., Helin K., Olsen L., Gajhede M., Studies of H3K4me3 demethylation by KDM5B/Jarid1B/PLU1 reveals strong substrate recognition in vitro and identifies 2,4-pyridine-dicarboxylic acid as an in vitro and in cell inhibitor. FEBS J. 279, 1905–1914 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Vinogradova M., Gehling V. S., Gustafson A., Arora S., Tindell C. A, Wilson C., Williamson K. E, Guler G. D., Gangurde P., Manieri W., Busby J., Flynn E. M., Lan F., Kim H.-j., Odate S., Cochran A. G., Liu Y., Wongchenko M., Yang Y., Cheung T. K., Maile T. M., Lau T., Costa M., Hegde G. V., Jackson E., Pitti R., Arnott D., Bailey C., Bellon S., Cummings R. T., Albrecht B. K., Harmange J.-C., Kiefer J. R., Trojer P., Classon M., An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 12, 531–538 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Bavetsias V., Lanigan R. M., Ruda G. F., Atrash B., McLaughlin M. G., Tumber A., Mok N. Y., Le Bihan Y.-V., Dempster S., Boxall K. J., Jeganathan F., Hatch S. B., Savitsky P., Velupillai S., Krojer T., England K. S., Sejberg J., Thai C., Donovan A., Pal A., Scozzafava G., Bennett J. M., Kawamura A., Johansson C., Szykowska A., Gileadi C., Burgess-Brown N. A., von Delft F., Oppermann U., Walters Z., Shipley J., Raynaud F. I., Westaway S. M., Prinjha R. K., Fedorov O., Burke R., Schofield C. J., Westwood I. M., Bountra C., Müller S., van Montfort R. L. M., Brennan P. E., Blagg J., 8-substituted pyrido[3,4-d]pyrimidin-4(3H)-one derivatives as potent, cell permeable, KDM4 (JMJD2) and KDM5 (JARID1) histone lysine demethylase inhibitors. J. Med. Chem. 59, 1388–1409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao J., Kessler M., Helmling S., O’Connor J. P., Moore C., Pta1, a component of yeast CF II, is required for both cleavage and poly(A) addition of mRNA precursor. Mol. Cell. Biol. 19, 7733–7740 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pearson E., Moore C., The evolutionarily conserved Pol II flap loop contributes to proper transcription termination on short yeast genes. Cell Rep. 9, 821–828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collart M. A., Oliviero S., Preparation of yeast RNA. Curr. Protoc. Mol. Biol. Chapter 13, Unit13.12 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zang C., Schones D. E., Zeng C., Cui K., Zhao K., Peng W., A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S, Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Selth L. A., Gilbert C., Svejstrup J. Q., RNA immunoprecipitation to determine RNA-protein associations in vivo. Cold Spring Harb. Protoc. pdb.prot5234 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1501662/DC1
fig. S1. Mass spectrometry results.
fig. S2. Western blot analysis of TAP-tagged proteins.
fig. S3. Materials for in vitro RNA binding experiments.
fig. S4. Loss of Jhd2 does not affect processing or polyA-dependent termination in vitro.
fig. S5. RT-qPCR and Western blot analysis for Jhd2 mRNA and protein used in PMA1 studies.
fig. S6. RT-qPCR and Western blot analysis for Jhd2 mRNA and protein used in KlCYC1 studies.
fig. S7. Metagene plots of H3K4me3 levels.
table S1. Jhd2 bound peaks identified using SICER.
table S2. Pcf11 bound peaks identified using SICER.
table S3. List of genes with APA after Jhd2 deletion.
table S4. Plasmids used in this study.
table S5. Yeast strains used in this study.
table S6. Primers used in this study.