

Mammalian heart regeneration after acute heart attacks can be induced by decellularized zebrafish cardiac extracellular matrix.
Keywords: Extracellular matrix, decellularization, zebrafish, myocardial infarction, heart regeneration, ischemic heart disease, cardiac repair, cardiomyogenesis
Abstract
Heart attack is a global health problem that leads to significant morbidity, mortality, and health care burden. Adult human hearts have very limited regenerative capability after injury. However, evolutionarily primitive species generally have higher regenerative capacity than mammals. The extracellular matrix (ECM) may contribute to this difference. Mammalian cardiac ECM may not be optimally inductive for cardiac regeneration because of the fibrotic, instead of regenerative, responses in injured adult mammalian hearts. Given the high regenerative capacity of adult zebrafish hearts, we hypothesize that decellularized zebrafish cardiac ECM (zECM) made from normal or healing hearts can induce mammalian heart regeneration. Using zebrafish and mice as representative species of lower vertebrates and mammals, we show that a single administration of zECM, particularly the healing variety, enables cardiac functional recovery and regeneration of adult mouse heart tissues after acute myocardial infarction. zECM-treated groups exhibit proliferation of the remaining cardiomyocytes and multiple cardiac precursor cell populations and reactivation of ErbB2 expression in cardiomyocytes. Furthermore, zECM exhibits pro-proliferative and chemotactic effects on human cardiac precursor cell populations in vitro. These contribute to the structural preservation and correlate with significantly higher cardiac contractile function, notably less left ventricular dilatation, and substantially more elastic myocardium in zECM-treated hearts than control animals treated with saline or decellularized adult mouse cardiac ECM. Inhibition of ErbB2 activity abrogates beneficial effects of zECM administration, indicating the possible involvement of ErbB2 signaling in zECM-mediated regeneration. This study departs from conventional focuses on mammalian ECM and introduces a new approach for cardiac tissue regeneration.
INTRODUCTION
The extracellular matrix (ECM) is the architectural foundation of organ morphogenesis, development, homeostasis, and regeneration across the animal kingdom (1–5). The ECM is involved in nearly all cellular activities, such as cell adhesion, migration, survival, proliferation, and differentiation (2, 6, 7). During tissue development and regeneration, the ECM not only serves as a scaffold to support proliferating and migrating cells but also provides a wide variety of biochemical and physical signaling to facilitate the processes (8–10).
Evolutionarily primitive species generally have greater regenerative capabilities than mammals (11–14). An example is the major difference in the regenerative capacity between adult zebrafish and mammalian hearts. An adult zebrafish heart can fully regenerate after up to 20% volumetric loss by ventricular amputation, whereas a mammalian counterpart cannot sustain such an injury (15). On the other hand, embryonic mammals also have a robust regenerative capability that is rapidly lost during postnatal development (16–18). For example, a neonatal mouse can regenerate up to 10% of its heart apex after ventricular resection, an ability that disappears within 1 week after birth (19). More specifically, although mammalian cardiomyocytes proliferate rapidly during the fetal period, their proliferative capacity quickly ceases after birth (19, 20), with the exception of a brief proliferative burst at preadolescence (21). Despite a low turnover rate throughout adulthood, most adult mammalian cardiomyocytes remain mitotically quiescent and cannot spontaneously regenerate after severe injuries such as myocardial infarction (MI) (8, 22, 23). Consequently, how the regenerative capacity lost in evolution can be effectively restored or reactivated in adult mammalian hearts remains to be explored. The ECM may contribute to this evolutionary difference in cardiac regeneration (4, 24). We hypothesize that ECM in the zebrafish heart contributes to its regenerative capability and may be used to induce mammalian heart regeneration.
Decellularized mammalian ECM has been extensively investigated in tissue engineering and regenerative medicine (25, 26). Mammalian cardiac ECM has reportedly been beneficial when administered to the ischemic heart (27, 28). However, instead of regeneration, fibrosis typically occurs in adult mammalian hearts after severe ischemic insults, which, in turn, impedes further structural or functional recovery (14). We believe that the ECM from a regenerative tissue is more likely to revitalize mammalian hearts after MI than that from a fibrosis-prone tissue. Besides, mammalian organs are relatively thick and thus typically require the involvement of detergents or other potent chemicals for their decellularization (29, 30). This may reduce the availability and activity of trophic molecules in the decellularized ECM and alter their composition and/or three-dimensional configuration (31, 32). In sharp contrast, the zebrafish ventricular wall consists of only four to five layers of cardiomyocytes in a compact myocardium and is <20 μm thick (33). This enables a physical decellularization protocol minimizing chemical perturbations to the native ECM composition and structure. In addition, we use a mechanical process to dissociate the decellularized ECM into fine powders for injection without any chemical or biological reagent to further preserve the integrity of the zebrafish heart ECM. Zebrafish are small, easy to breed, and routinely raised in large populations, allowing researchers to pool zebrafish hearts for decellularization.
Here, we established a protocol to physically decellularize zebrafish cardiac ECM (zECM) from normal and healing [3 days post-amputation (dpa)] zebrafish hearts. We characterized zECM, examined the bioactivity of zECM on human cardiac precursor cell populations, and investigated the regenerative capability of zECM in vivo, using acutely infarcted adult mouse heart as a model organ and decellularized adult mouse cardiac ECM (mECM) as a control. As a starting point to understand the working mechanism of zECM on mammalian heart regeneration, we also investigated the role of the epidermal growth factor receptor-2 (EGFR-2) receptor tyrosine kinase (also known as ErbB2, Neu, or HER2) in zECM-induced effects in ischemic hearts. This proof-of-concept study explores the potential of zECM as a new biological material for cardiac tissue regeneration, departing from conventional focuses on mammalian cardiac ECM.
RESULTS
Decellularized zECM is produced and characterized
We developed a physical decellularization protocol with mechanical dissociation to minimize the use of chemical or biological reagents that may disturb the native ECM composition, taking advantage of the very thin zebrafish ventricular wall. To compare the difference in the regenerative capacity of cardiac ECM between healthy and regenerating zebrafish hearts, we harvested normal and healing (3 dpa) adult zebrafish ventricles (fig. S1A) and thoroughly washed them in antibiotic/antifungal solutions. Typically, 50 to 60 zebrafish ventricles were collected and pooled per batch. Healthy adult mouse ventricles were used as a treatment control and minced into very small fragments (<1 mm3) for physical decellularization. Briefly, all three groups were decellularized by repeated freeze-thaw cycles with the removal of red blood cells and DNA/RNA by the erythrolysis buffer and deoxyribonuclease/ribonuclease, respectively (fig. S1B). After lyophilization, ECM was mechanically ground into fine powders in a liquid nitrogen–chilled container and stored at −80°C.
Scanning electron microscopy (SEM) was used to observe the morphological change following decellularization. SEM images revealed the differences of fresh and decellularized zebrafish ventricular tissues and ECM particles (Fig. 1A). Dynamic light scattering showed that the decellularized normal zECM (nzECM) and healing zECM (hzECM) were micro- to nanoparticles with an average radius of approximately 306.3 nm (Fig. 1B). The final yield of lyophilized zECM powders was approximately 6 to 8% of the original wet weight. zECM contained approximately 1.6 to 1.9% residual DNA compared with the native tissue (fig. S1C). mECM generated with the same protocol had roughly 4.5% residual DNA (fig. S1C). Analyses of cardiac ECM composition revealed that mECM contains 408.80 ± 25.81 μg of collagen, 45.70 ± 9.51 μg of elastin, and 2.35 ± 0.58 μg of glycosaminoglycans (GAGs) per milligram of material (Fig. 1C). nzECM contained 299.10 ± 32.05 μg of collagen (73.2%, P < 0.01), 62.67 ± 9.07 μg of elastin (137.1%, P < 0.05), and 3.34 ± 0.61 μg of GAGs (142.1%, P < 0.05) per milligram of material, exhibiting significant differences in major ECM structural components when compared with mECM (Fig. 1C).
Fig. 1. Characterization of decellularized zECM.
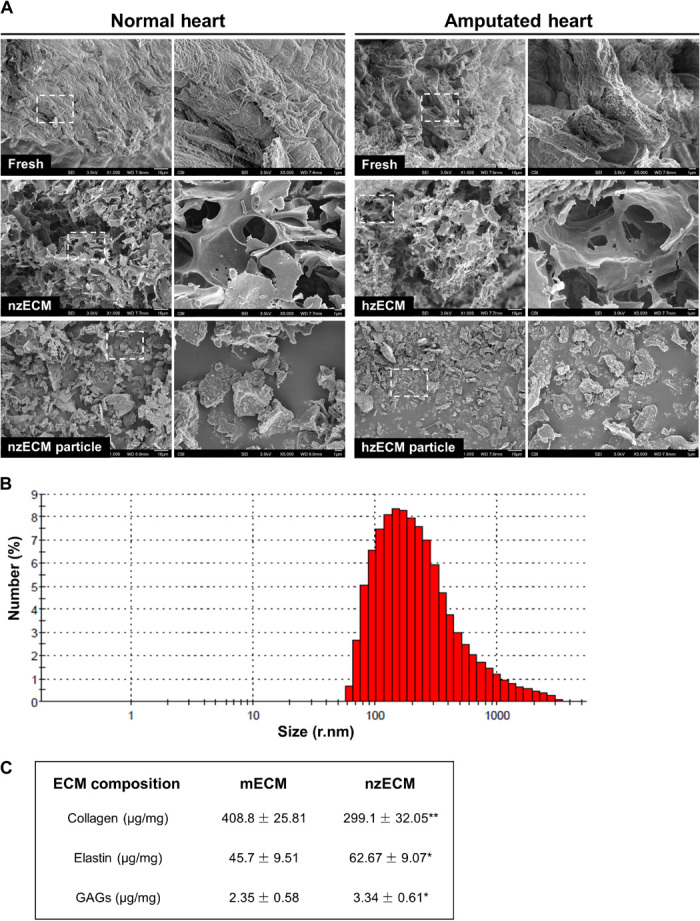
(A) SEM images of fresh, decellularized, and ground decellularized normal and healing (3 dpa) zebrafish ventricular tissues. Scale bars, 10 μm (magnification, ×1000) and 1 μm (magnification, ×5000). (B) Particle size analysis of ground zECM by dynamic light scattering (n = 3). (C) Composition analyses of nzECM and adult mECM showing the amount of collagen, elastin, and GAGs in each group, respectively (n = 3 per group; data are means ± SD; *P < 0.05, **P < 0.01).
Mass spectrometry (MS) was used to characterize types of proteins present in hzECM, nzECM, and mECM. ECM extracts were loaded on an SDS–polyacrylamide gel electrophoresis (PAGE) gel to separate protein from low–molecular weight analytes and buffers. Tryptic digestion of a 1-cm band that contains unresolved protein resulted in the detection of many cellular and structural proteins by liquid chromatography (LC)–MS/MS from 3 μg of hzECM, nzECM, or mECM extract. Results were summarized in tables S1 to S3. For example, natriuretic peptide and fibrinogen β and γ polypeptides were detected in hzECM (table S3). Overall, these results suggest that the decellularized cardiac ECM extracts are amenable to the LC-MS/MS analysis and that quantitative measurements may offer new insights into compositional and relative abundance differences.
Decellularized zECM exhibits bioactivity in vitro
As a first step to investigate the cardiac regenerative potential of zECM for mammalian hearts, we examined the bioactivity of decellularized zECM on the proliferation and migration of human cardiac precursor cell populations in vitro. To simulate the harsh microenvironment in the ischemic myocardium, we applied stressed growth conditions, including nutrient deprivation and dual hypoxia/nutrient deprivation, to cell cultures. Both hzECM and nzECM displayed pro-proliferative effects on human cardiac stem cells (hCSCs) and human heart perivascular mesenchymal stem/stromal cell (MSC)–like precursors (hHPs) under each stressed culture condition. When deprived of nutrition [hCSC: 25% complete culture medium; hHP: 2.5% fetal bovine serum (FBS)] for 4 days, hzECM-treated hCSCs and hHPs both exhibited significantly higher proliferation rates when compared with mECM- or nontreated controls (hCSC, both P < 0.005; hHP, both P < 0.05), whereas nzECM- and mECM-treated cells showed notably faster growth than control cells did (all P < 0.05) (Fig. 2, A and B). However, under dual hypoxic (2.5% O2) and nutritional deprivation, hzECM markedly enhanced hCSC and hHP proliferation when compared with nzECM, mECM, and control groups (all P < 0.01). nzECM significantly promoted hCSC growth (when compared with mECM- and nontreated control cells, both P < 0.001) and hHP growth (when compared with nontreated control cells, P = 0.01). In sharp contrast, mECM showed no notable stimulatory effect under the dual stress culture condition (Fig. 2, C and D).
Fig. 2. Bioactivity of zECM on human cardiac precursor cell proliferation and migration.

(A to D) Relative proliferation rates of hCSCs and hHPs under stressed culture conditions following different cardiac ECM treatments. Addition of hzECM, nzECM, or mECM in the culture medium partially rescued the proliferation of (A) hCSCs and (B) hHPs under nutrient-deprived culture conditions. Addition of hzECM or nzECM, but not mECM, in the culture medium partially rescued the proliferation of (C) hCSCs and (D) hHPs under dual hypoxic (2.5% O2) and nutrient-deprived culture conditions. *P ≤ 0.05, †P ≤ 0.01, §P ≤ 0.005, #P ≤ 0.001 compared to nutrient-deprived controls in all graphs. (E to H) Transwell chemotaxis assays with different cardiac ECM showing the migration of hCSCs and hHPs under nutrient-deprived culture conditions. hzECM and nzECM, but not mECM, induced prominent migration of (E) hCSCs and (G) hHPs (cells stained in green; scale bars, 50 μm). Significantly more (F) hCSCs and (H) hHPs migrated in hzECM- and nzECM-induced groups than in the mECM-induced group and saline control (n = 4 per group; data normalized to the respective saline control; **P < 0.01, ***P < 0.001 compared to mECM and saline; #P < 0.05 hzECM versus nzECM). Quantitative data represent means ± SD.
To simulate cell migration in the ischemic myocardium, we applied nutrient-deprived culture conditions in the Transwell chemotaxis assay with induction of different decellularized cardiac ECM. The results showed that hzECM and nzECM, but not mECM, induced prominent migration of hCSCs (Fig. 2E) and hHPs (Fig. 2G). Quantification data indicate that significantly more hCSCs (both P < 0.01; Fig. 2F) and hHPs (hzECM, P < 0.001; nzECM, P < 0.01; Fig. 2H) migrated under the hzECM and nzECM inductions than under the mECM induction and saline control. hzECM induced notably higher hHP migration than nzECM under stress (P < 0.05; Fig. 2H). Together, these data suggest the preservation of zECM bioactivity after decellularization and pro-proliferative and chemotactic effects of zECM on two human cardiac precursor cell populations under stressed microenvironments.
Intramyocardial administration of zECM improves cardiac function
The efficacy of decellularized zECM on cardiac function was investigated in an adult mouse acute MI (AMI) model (Fig. 3A). There was minimal mortality (zero to one animal per group) during surgery. No major difference in the mortality rate was observed between treatment groups. For the functional studies, all mice that survived the MI induction surgery lived through the 6-week experimental duration. Long-term cardiac function was repeatedly assessed by M- and B-mode echocardiography before (baseline) and after surgery at 5, 14, and 42 days (fig. S2). The results showed that hzECM exhibited substantially higher overall treatment efficacy in left ventricular (LV) fractional area change (LVFAC) (Fig. 3B) and LV ejection fraction (LVEF) (Fig. 3C) than all other groups (all P < 0.001) did, whereas nzECM was significantly higher than mECM and saline control (all P < 0.005). mECM was less effective but exhibited notable beneficial effects when compared with saline (both P < 0.05). These data indicate significant preservation of LV contractility after AMI, following zECM treatment. Moreover, both hzECM and nzECM had markedly reduced LV end-diastolic area (EDA) (Fig. 3D) and end-systolic area (ESA) (Fig. 3E) compared to mECM and saline (all P < 0.005), suggesting amelioration of progressive LV dilatation with zECM treatment. All echocardiographic measurements are listed in table S4.
Fig. 3. Echocardiographic analyses of cardiac function.
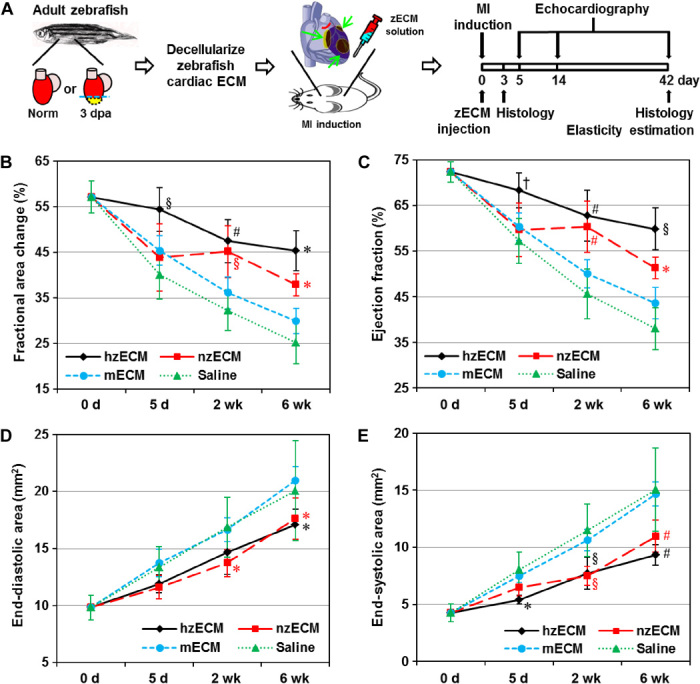
(A) Schematic representation of the work flow using zECM (0.5 mg of suspension) for mammalian heart regeneration after AMI. Cardiac contractile function is indicated by (B) fractional area change and (C) ejection fraction; LV dimension is indicated by (D) EDA and (E) ESA (n = 7 per group; data analyzed by two-way repeated-measures analysis of variance (ANOVA); *P ≤ 0.05, †P ≤ 0.01, §P ≤ 0.005, #P ≤ 0.001 compared to saline controls in all graphs).
zECM amends elasticity of the infarcted myocardium
To understand the effect of zECM on the ventricular elasticity in the ischemic heart, we performed myocardial strain analysis at 6 weeks after MI. The data revealed that zECM-treated myocardium had similar intercardiac strain [defined as Δ(end-diastole and end-systole) × 100%] compared to the noninfarcted counterparts, whereas mECM- and saline-treated ones were substantially stiffer. The strain of the infarcted myocardium was estimated by normalizing the spatially averaged axial strain in the infarcted area (A) to that of four noninfarct areas (B, C, D, and E) in LV walls during a cardiac cycle (Fig. 4A), using VevoStrain cardiac strain analysis (Fig. 4B). Both hzECM (all P < 0.001) and nzECM (all P < 0.001) had substantially greater normalized radial (Fig. 4C) and circumferential (Fig. 4D) strains than mECM and saline. mECM showed moderately increased radial and circumferential strains (both P < 0.01) compared to saline. No significant difference was observed between hzECM and nzECM (both P > 0.05). hzECM exhibited the highest strains in both directions among all groups with no statistical difference from normal hearts (both P > 0.05). These data suggest the effectiveness of zECM, but not of mECM, in preserving or restoring LV myocardial elasticity after MI.
Fig. 4. Ultrasonic myocardial strain analysis.
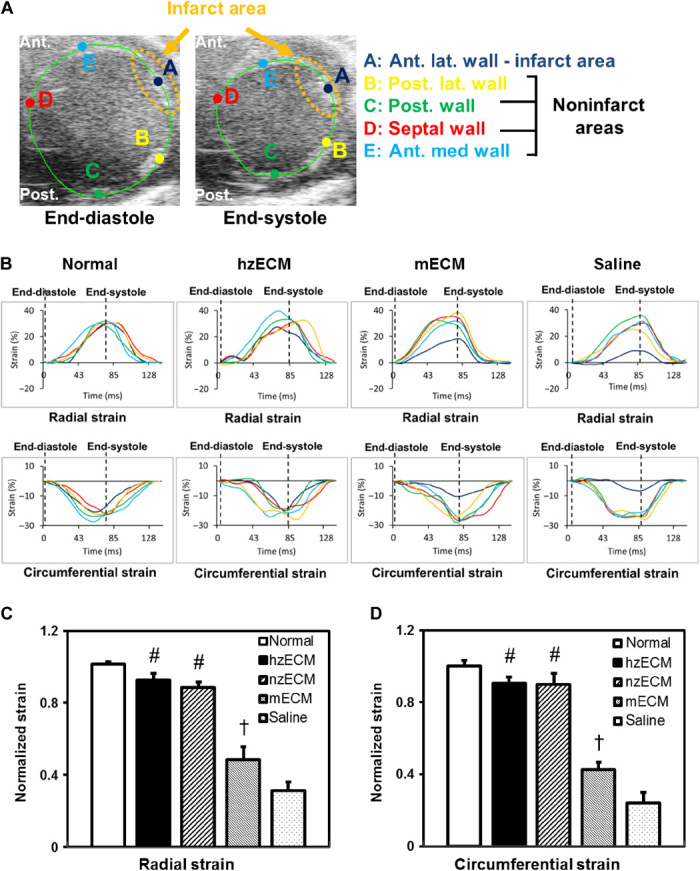
(A) Representative B-mode images showing ROI selection at end-diastole and end-systole for myocardial strain analysis. Ant., anterior; Post., posterior; lat., lateral. (B) Representative graphs showing radial (upper panels) and circumferential (lower panels) strain estimation during a cardiac cycle: Strain of the infarcted area (dark blue) is closer to the noninfarcted area (yellow, green, red, and cyan) and normal heart in hzECM-treated group than in mECM- or saline-treated groups. Quantification of (C) radial and (D) circumferential strain (n = 3 per group; †P ≤ 0.01, #P ≤ 0.001 for hzECM and nzECM compared to mECM and saline; mECM compared to saline in all graphs).
zECM promotes structural preservation in the infarcted heart
Hematoxylin and eosin (H&E) staining on serially sectioned mouse hearts revealed that zECM treatment alters pathological remodeling after MI (fig. S3A). LV chambers in both zECM-treated groups are notably smaller with less infarct area (fig. S3B) and thicker ventricular walls (fig. S3C) than those in mECM and saline groups. In zECM-treated groups, Masson’s trichrome staining and anti-mouse CD68 staining at 6 weeks after MI showed no increase in fibrotic scarring (fig. S4A) and chronic inflammation (fig. S4B), respectively, because of species difference. Consistent with the reduction of the infarct size, elastin is preserved locally at the infarct area in zECM-treated groups (fig. S5). We detected a significantly higher level of elastin in both zECM groups, particularly hzECM, than in mECM and saline groups. hzECM-treated hearts had approximately 60% of the elastin content detected in healthy mouse hearts (approximately 26% by area size), suggesting improved elastin retention after MI (fig. S5). Immunodetection of apoptotic cardiomyocytes with cleaved caspase-3 and cardiac troponin T (cTnT) at 3 days after MI showed that hzECM treatment appears to reduce apoptosis (fig. S6).
zECM increases proliferation of mammalian cardiac precursor cell populations in vivo
We investigated whether the differences in functional outcomes of zECM and mECM treatment correlate with distinctive regenerative responses in the ischemic myocardium. Immunohistochemistry at 6 weeks after MI showed that all zECM-treated hearts had significantly higher numbers of c-kit+/Ki67+ proliferating murine cardiac stem cells (mCSCs) than mECM- and saline-treated ones at the infarct and peri-infarct areas (c-kit: stem cell growth factor receptor or CD117; Ki67: a cellular proliferation marker) (Fig. 5, A to C). hzECM- and nzECM-treated hearts exhibited markedly more platelet-derived growth factor receptor-β (PDGFRβ)+/Ki67+ proliferating cardiac MSCs (cMSCs) than mECM- and saline-treated hearts did at the infarct and peri-infarct areas (Fig. 5, D to F). Activation of Wilms tumor protein (Wt) 1+/Ki67+ proliferating epicardium-derived progenitor cells (EPDCs) was observed only at the epicardium of the infarct (Fig. 5G). The hzECM-treated group had substantially more proliferating EPDCs than all other groups; nzECM had notably more proliferating EPDCs than mECM and saline (Fig. 5H).
Fig. 5. Proliferation of cardiac precursor cell populations.

Dual immunofluorescence detection and quantification of (A to C) c-kit+/Ki67+ proliferating cardiac stem cells, (D to F) PDGFRβ+/Ki67+ proliferating cardiac mesenchymal stromal cells, and (G and H) Wt1+/Ki67+ proliferating EPDCs at 6 weeks after MI at the mid-infarct level of mouse left ventricles. Arrows indicate doubly positive cells. All image analyses are performed within 20 × 10–μm areas in five images of each heart (n = 4 per group). All quantitative data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 compared to saline and mECM; #P < 0.05, ##P < 0.01 for hzECM versus nzECM. Scale bars, 50 μm.
zECM augments adult mammalian cardiomyocyte proliferation in the ischemic myocardium
We examined whether the administration of zECM increases the host cardiomyocyte proliferation after the ischemic insult. Dual detection of cTnT and Ki67 at 3 days after treatment revealed the proliferation of adult mouse cardiomyocytes in zECM-treated animals (Fig. 6A). Image analyses showed that both zECM groups have notably higher numbers of cTnT+/Ki67+ proliferating cardiomyocytes than control groups at the infarct and peri-infarct areas (Fig. 6, B and C). hzECM had the highest cTnT+/Ki67+ cell number among all groups, especially at the infarct area (Fig. 6, B and C). In sharp contrast, cardiomyocyte proliferation is very limited in mECM and saline controls (Fig. 6, A to C), which is consistent with previous reports (34, 35). Nevertheless, there are few cTnT+/Ki67+ cardiomyocytes in all four groups at 6 weeks after MI (all P > 0.05; fig. S7), suggesting a limited duration of zECM treatment. This limited window may reduce the chance of hypertrophy and tumor development due to prolonged cell proliferation. On the other hand, healthy adult mouse hearts have little ErbB2 expression, which plays a significant role in mammalian heart regeneration (36). However, at 3 days after treatment, we observed the presence of ErbB2+/cTnT+ cardiomyocytes in both zECM-treated groups (Fig. 6D). hzECM and nzECM groups exhibited markedly larger presences of ErbB2+/cTnT+ cardiomyocytes than mECM and saline controls at the infarct and peri-infarct areas (Fig. 6, E and F), suggesting that zECM reactivates ErbB2 expression in adult mammalian cardiomyocytes after MI.
Fig. 6. Cardiomyocyte proliferation and ErbB2 expression.

(A to C) Dual immunofluorescence detection and quantification of cTnT+/Ki67+ proliferating cardiomyocytes at 3 days after MI at the mid-infarct level of mouse left ventricles. Arrows indicate doubly positive cells. (D to F) Dual immunofluorescence detection and quantification of ErbB2+/cTnT+ cardiomyocytes at 3 days after MI at the mid-infarct level suggest the involvement of NRG1 signaling in zECM-treated groups. All image analyses were performed using 20 × 10–μm areas in five images of each heart (n = 4 per group). All quantitative data represent means ± SD. ***P < 0.001 compared to mECM and saline; #P < 0.05 for hzECM versus nzECM. Scale bars, 50 μm.
Neuregulin-1, a mitogen of cardiomyocytes, is present in zECM
Neuregulin-1 (NRG1) is a mitogen capable of stimulating cardiomyocyte proliferation in regenerating zebrafish myocardium (37, 38). Immunohistochemical analysis showed that zebrafish hearts, especially the actively healing ones, express NRG1 (Fig. 7, A and B) (37), which is absent in adult mouse hearts (Fig. 7B). Western blot analysis of NRG1 in all three ECM groups revealed the presence of NRG1 protein in both hzECM and nzECM (Fig. 7C). Quantification of Western blotting showed that hzECM and nzECM contain approximately 6.5 and 5 times more NRG1 protein, respectively, when compared with mECM (both P < 0.001; Fig. 7D).
Fig. 7. Detection of NRG1 in zECM.

(A) Immunohistochemical detection of NRG1 (brown arrowheads) in normal and healing (3 dpa) zebrafish hearts. (B) Positive immunofluorescence detection of NRG1 (green) at the ventricular apex of normal zebrafish heart (nzH) and healing zebrafish heart (hzH) but not in the adult mouse heart (mH). (C) Consistent with the in situ NRG1 staining, Western blotting showed that both nzECM and hzECM ECM contain NRG1 protein. (D) Quantification data indicate hzECM and nzECM contain approximately 6.5 and 5 times more NRG1 than normal mECM, respectively. Data represent means ± SD. ***P < 0.001 versus mECM. Scale bars, 50 μm.
Inhibiting ErbB2 signaling obliterates major beneficial effects of zECM treatment
We hypothesize that the ErbB2 signaling pathway plays a role in the cardiac repair and regeneration observed in zECM-treated groups. To evaluate the importance of ErbB2 signaling in zECM-mediated therapeutic effects, we performed a loss-of-function study. We injected zECM intramyocardially after AMI while selectively inhibiting ErbB2 receptor kinase activity with a tyrphostin, AG825, dissolved in dimethyl sulfoxide (DMSO) via intraperitoneal administration (39–41). Echocardiographic analyses showed that blocking ErbB2 signaling during zECM treatment renders zECM ineffective: It abolished the improvement of LV contractility (both P > 0.05; Fig. 8, A and B) and eliminated the prevention of LV dilatation (both P > 0.05; Fig. 8, C and D). All echocardiographic measurements are listed in table S5. Control mice receiving intramyocardial saline injections with intraperitoneal sham or DMSO administration with or without AG825 showed no significant difference in cardiac functional outputs (all P > 0.05; fig. S8). Dual immunofluorescence detection and quantification of c-kit+/Ki67+ proliferating mCSCs at 3 days after treatment exhibited no notable difference between all four groups (P > 0.05; Fig. 8, E and F). Similarly, no significant difference in the number of ErbB2+/cTnT+ cardiomyocytes between all groups was observed following AG825 treatment (P > 0.05; Fig. 8, G and H). These results suggest that ErbB2 is involved in the mediation of zECM effects in the mammalian cardiac tissue after AMI.
Fig. 8. Inhibition of ErbB2 activity with decellularized cardiac ECM treatment.

To inhibit ErbB2 activity in vivo, the ErbB2 inhibitor AG825 was intraperitoneally injected once (5 mg/kg) immediately after the administration of decellularized cardiac ECM. Cardiac contractile function is indicated by (A) fractional area change and (B) ejection fraction; LV dimension is indicated by (C) EDA and (D) ESA. No significant difference is observed between all groups at all time points (n = 7 per group; all P > 0.05; data analyzed by two-way repeated-measures ANOVA). Dual immunofluorescence detection and quantification of (E and F) c-kit+/Ki67+ proliferating cardiac stem cells and (G and H) ErbB2+/cTnT+ cardiomyocytes. No significant difference is observed between all groups (n = 4 per group, all P > 0.05). Scale bars, 50 μm.
DISCUSSION
Although mammalian cardiomyocytes spontaneously proliferate at a very low rate throughout adulthood (42), unlike the evolutionarily primitive zebrafish, adult mammalian hearts have very limited regenerative capacity after MI or other injuries involving massive loss of cardiomyocytes (43). We suspect that ECM contributes to this genus difference and therefore hypothesize that ECM from a regenerable tissue, such as the zebrafish heart, can induce and/or facilitate adult mammalian heart regeneration after injury. To our knowledge, decellularized cardiac ECM from nonmammalian vertebrates has not been tested in a mammalian MI model despite favorable results of mammalian ECM for cardiac repair and regeneration (27, 44). Here, we used zebrafish and mice as representative species for lower vertebrates and mammals, respectively. We found that the composition of nzECM is significantly different from mECM, with more elastin and GAGs and less collagen. Both hzECM and nzECM had significant pro-proliferative and chemotactic effects on human cardiac precursor cells, including hCSCs and hHPs, under stress in vitro, whereas the efficacy of mECM was rather limited. The differences in the cellular proliferation and migratory rates suggest differential rescue and/or inductive capacities with each ECM treatment under deprived growth conditions.
Intramyocardial administration of decellularized cardiac ECM suspension showed that a single treatment of zECM, particularly hzECM, enables endogenous regeneration of murine heart tissue after AMI. We observed notably increased proliferation of multiple resident cardiac precursor cell populations, including mCSCs, cMSCs, and EPDCs, in both zECM groups. c-kit+ mCSCs did not express tryptase, a mast cell marker, in zECM and control groups (fig. S9). Furthermore, a fraction of the remaining cardiomyocytes not only reexpressed ErbB2 but also proliferated after zECM treatment. These cellular regenerative responses following zECM induction contribute to the overall architectural preservation and structural regeneration, which correlates with approximately 61% recovery of cardiac ejection fraction {defined as [Δ(treatment-saline)/Δ(healthy-saline)] × 100%}. Under identical conditions, mECM yields only 17% functional recovery with limited improvement of myocardial elasticity and minimal proliferation of cardiac precursor cells and cardiomyocytes, consistent with literature reports on mammalian ECM in MI treatment (45, 46). Our data indicate that a single intervention with zECM is sufficient to lead to significant improvement in cardiac output and remodeling as well as near-normal LV wall motion. Together, these results suggest the efficacy of zECM, especially hzECM, in the preservation and/or recovery of global cardiac milieu after MI.
zECM likely exerts its activities via multiple mechanisms in the ischemic myocardium, for example, the alteration of local ECM composition with improved elastin preservation and the release of incorporated inductive factor(s). The higher elastin content is consistent with greater myocardial strain and may augment cardiac stem/progenitor cell proliferation (47). In addition, it is possible that the altered ECM composition in the healing phase of amputated zebrafish heart partly contributes to the highest regenerative efficacy of hzECM for ischemic mouse hearts (48). On the other hand, we detected the presence of NRG1, a mitogen of cardiomyocytes and a ligand of ErbB2/ErbB4 complex, in both hzECM and nzECM but only minimally in mECM. NRG1 induces cardiomyocyte proliferation and myocardial regeneration in injured mammalian hearts (49), especially in neonates (36, 50), largely via ErbB2/ErbB4 signaling pathways (36, 49, 50). The presence of NRG1 in zECM and the reactivation of its receptor ErbB2 in zECM-treated hearts are consistent with the observed proliferation of cardiomyocytes and improvement of cardiac function. Moreover, one intramyocardial zECM treatment yields significant functional recovery and structural change in infarcted hearts, in contrast to similar outcomes yielded by daily systemic injections of recombinant NRG1 protein for 12 weeks (49). These results imply a role of NRG1 in zECM-induced cardiomyocyte proliferation after MI. However, because of the lethality of homozygous nrg1 deficiency in developing zebrafish, we cannot obtain a sufficient amount of NRG1-depleted zECM for testing. Thus, we directly probed ErbB2 signaling in the context of zECM administration by inhibiting ErbB2 activity.
ErbB2 signaling is essential to the survival, repair, growth, and regeneration of postnatal mammalian cardiomyocytes (51, 52). Ventricular-restricted ErbB2-deficient mice exhibit phenotypes of dilated cardiomyopathy in their adulthood, indicated by decreased contractility, wall thinning, and chamber dilation (53). ErbB2 activation promotes dedifferentiation, proliferation, and hypertrophy of cardiomyocytes (36). To block ErbB2 signal transduction, we administered AG825, a small-molecule tyrphostin that selectively inhibits ErbB2 autophosphorylation nearly 60-fold more potently than ErbB1 (EGFR), immediately following zECM treatment (39). Surprisingly, early ErbB2 inhibition not only obliterated the improvement of cardiac function but also eliminated the reduction of LV dilatation in both hzECM and nzECM groups after MI. These results agree with previous findings showing notable deterioration in cardiac contractility of normal or diabetic murine hearts subjected to ischemia-reperfusion injury ex vivo after repeated intraperitoneal administration of AG825 (41). Moreover, early ErbB2 inhibition fully prevented mCSC proliferation and ErbB2 reactivation in cardiomyocytes after zECM treatment. These data indicate the participation of ErbB2 signaling in zECM bioactivity after MI. Future studies using zECM from heterozygous nrg1 mutant zebrafish may provide more insight into the impact of altered NRG1 and/or ErbB2 signaling in zECM-mediated cardiotrophic efficacy.
The substantially more proliferating cardiomyocytes in both zECM groups at 3 days after MI, but not at 6 weeks after MI, suggest a limited duration of zECM activities and possibly a finite therapeutic window for zECM-based intervention. This may reduce the risk of cardiac hypertrophy associated with unrestrained activation of NRG1/ErbB2 signaling (36, 37). Nevertheless, depending on the length of the therapeutic window, whether zECM is capable of inducing similar benefits in hearts that suffer from subacute or chronic MI needs further investigation. Local injections of zECM limit potential oncogenic risks associated with systemic growth factor administrations such as NRG1 treatment (38, 54). In addition to local injections, the nano- to microsize zECM particulate might also be administered through an intracoronary route, making it more clinically applicable and less invasive. The proliferation of multiple cardiac progenitors further indicates the broader biological activities of zECM beyond NRG1 (49). Moreover, it is possible that the significant functional improvement, especially in the hzECM group, can be attributed to other cellular mechanisms such as the increase of cardiomyocyte survival or alteration of cell metabolism, potentially mediated via signaling molecules present in zECM.
Previous studies have shown that genes of a number of secretory trophic molecules, such as apoEb, vegf-c, granulin-A, thymosin β4, and pdgf-b, are up-regulated in regenerating zebrafish hearts at 3 dpa (55). Identification of bioactive molecules, other than NRG1, within zECM with proteomic analysis could reveal additional pathways that are important to mammalian cardiac tissue regeneration. Consequently, we have performed MS to characterize the types of proteins that can be detected in hzECM, nzECM, and mECM (tables S1 to S3). These data suggest that decellularized cardiac ECM extracts are amenable to a qualitative LC-MS/MS analysis. The inability to detect the presence of NRG1 or other known trophic factors of zebrafish heart regeneration, such as PDGF-BB, by the MS analysis in cardiac ECM may be attributed to multiple technical limitations: (i) Signals of abundant proteins, such as collagen or cellular structural proteins, suppress and mask signals of low-abundance proteins, particularly in the LC-MS/MS–based analysis, because of the wide protein concentration range in ECM extracts (56); (ii) gel-based and LC-MS/MS techniques were reported to have limited sensitivity to molecules in low concentrations (10 to 20 fmol) (57); and (iii) the MS technique used in this study is for qualitative detection of proteins, not for quantitative measurement, and cannot exclude the presence of nondetectable proteins within the sample. We are attempting to use more advanced sample preparation techniques in a follow-up study that may lead to a more comprehensive analysis of trophic factors in zECM (56, 58). Although our current data do not provide a definitive answer to how the protein composition affects the relative therapeutic outcome, the results provide a foundation for ongoing quantitative proteomic studies that aim to (i) identify and measure the relative abundance of large numbers of proteins present in zECM, including many uncharacterized proteins, (ii) precisely compare compositional differences in decellularized zECM and mECM, and (iii) reveal protein(s) correlated with the observed therapeutic outcome as potential target(s) for intervention.
The mammalian developmental process roughly restates the phylogeny of the animal kingdom, especially from the perspective of cardiac regenerative capacity (15, 19, 59). This is reflected by the higher regenerative capability of mammals at the embryonic and fetal stages, which subsides during postnatal development in a tissue-dependent manner. This “regenerative phylogeny” is also recapitulated by the differential bioinductive potency of decellularized ECM derived from different mammalian developmental stages. For example, fetal, neonatal, and adult rat decellularized cardiac ECM has been shown to differentially promote the proliferation of neonatal rat ventricular cardiomyocytes (NRVMs) in culture, with the fetal ECM being the most potent (31). The same group further reports that, by adjusting the pepsin digestion time, the partially digested decellularized adult cardiac ECM can release more cues for NRVM proliferation (60). These results suggest a correlation between fetal and adult murine cardiac ECM in their relative cardioinductive efficacy as well as the possible developmental conservation of certain cardiotrophic substances. In addition, decellularized ECM from fetal porcine brain has been demonstrated to induce neural network formation in a bioengineered brain tissue more effectively than its adult counterpart (61). Therefore, it is possible that decellularized ECM from other primitive life-forms can also promote the regeneration of mammalian heart or other organs with limited regenerative capabilities.
In conclusion, the current study demonstrated that decellularized cardiac ECM from zebrafish induces the proliferation of murine and human cardiac precursor cell populations and murine adult cardiomyocytes under stress and significantly augments cardiac function and myocardial elasticity after MI. Decellularized adult mECM only slightly promoted cardiac progenitor cell and cardiomyocyte proliferation under stress and marginally improved functional recovery and myocardial elasticity, as typically seen in previous approaches using mammalian ECM. Through the selective inhibition of ErbB2 activity, we found that ErbB2 signaling is involved in zECM-mediated anatomical and functional improvement. Overall, our data demonstrated the potential of zECM as a new candidate to induce cardiac regeneration. Tests of zECM efficacy in large animal models of MI using a less invasive delivery method are under investigation.
MATERIALS AND METHODS
Animals
The Institutional Animal Care and Use Committee at the University of Pittsburgh approved the animal usage and surgical procedures performed in this study. Adult (6 to 12 months) wild-type AB* zebrafish (mixed male and female) were maintained at 28°C. Approximately 850 zebrafish were used for this study. A total of 115 male BALB/cJ mice at 9 to 12 weeks old (The Jackson Laboratory) were used for this study.
Ventricular amputation and procurement of zebrafish hearts
Healing zebrafish hearts were obtained after ventricular resection surgeries, as described by Poss et al. (15). The regenerating ventricle (lower two-thirds, including the apex) was collected for the decellularization. Approximately 50 to 60 ventricles were collected per batch and immediately washed in antibiotic-antimycotic solutions. Details are described in the Supplementary Materials.
Procurement of adult mouse ventricular tissues
Mouse left ventricles were dissected out and finely chopped to <1 mm3. Mouse ventricular pieces were washed in antibiotic-antimycotic solutions before the decellularization process. Details are described in the Supplementary Materials.
Decellularization of cardiac ECM
Zebrafish ventricles and mouse ventricular pieces were weighed in sterile microcentrifuge tubes and subjected to three freeze-thaw cycles: submerging in liquid nitrogen for 10 min and completely thawing at 37°C in a water bath. Erythrocyte lysis, an additional freeze-thaw cycle, and DNA/RNA removal were subsequently performed twice. Samples were then washed, centrifuged, and subjected to lyophilization for 72 hours. Lyophilized samples were weighed, finely ground into fine powders in a cooled mini mortar, and stored at −80°C for future use. Details are described in the Supplementary Materials.
Preparation of cardiac ECM suspension and particle size measurement
Lyophilized cardiac ECM powders were weighed, resuspended in 0.9% normal saline, and sonicated in cool water for 15 min. Large particles were removed by centrifugation. Particle size was measured by a Zetasizer Nano ZS90 (Malvern). For in vivo administration, 0.5 mg of lyophilized powders was resuspended in 30 μl of normal saline. Details are described in the Supplementary Materials.
ECM composition analysis
The amount of collagen in cardiac ECM was measured with a Sircol collagen assay kit (Biocolor Ltd.) following the manufacturer’s manual. Briefly, collagen was extracted by a fragmentation reagent provided with the kit. Samples were incubated at 65°C for 2 to 3 hours and precipitated out by adding the Sircol dye reagent. An ice-cold acid-salt wash reagent was gently layered onto the collagen-dye pellet to remove unbound dye. Samples were centrifuged at 12,000 rpm for 10 min. Precipitates were further recovered by the addition of an alkali reagent. Absorbance was measured at 550 nm with a SynergyMX plate reader (BioTek). The amount of elastin in cardiac ECM was measured with a Fastin elastin assay kit (Biocolor Ltd.). Briefly, elastin was extracted from tissue samples by digesting with oxalic acid at 100°C and precipitated out using the supplied elastin-precipitating reagent. Precipitates were then incubated with the elastin dye reagent followed by dissociation in the dye dissociation reagent. Absorbance was measured at 513 nm. The amount of GAGs was measured using the Blyscan Sulfated Glycosaminoglycan Kit (Biocolor Ltd.). Briefly, tissue samples were digested with the supplied papain extraction reagent at 65°C. Extracted GAGs were incubated with the Blyscan dye reagent to form precipitates of sulfated glycosaminoglycan-dye complex. Precipitates were further dissociated by the dissociation reagent. Absorbance was measured at 656 nm.
DNA extraction
Decellularized ECM was aliquoted and weighed in a preweighed microcentrifuge tube. DNA was extracted from decellularized ECM and normal zebrafish hearts with a DNeasy Blood & Tissue Kit (69504, Qiagen) following the manufacturer’s instructions. DNA concentration was measured with a NanoDrop 2000 spectrophotometer (ND-2000, Thermo Fisher Scientific). The amount of residual DNA in decellularized ECM per unit dry weight was calculated using the following formula: multiplying the DNA concentration by the total volume of the elution buffer and then dividing by the amount of decellularized ECM (in micrograms) used for DNA extraction. The results were normalized to DNA content in normal zebrafish hearts by unit dry weight.
MI model
The induction of MI and intramyocardial injections were performed, as we previously reported (62, 63). After MI induction, mice were randomly assigned to one of the four groups: hzECM, nzECM, mECM, or saline control. For ErbB2 inhibition in vivo, the ErbB2 inhibitor AG825 (sc-202045A, Santa Cruz Biotechnology) dissolved in DMSO was intraperitoneally injected once at 5 mg/kg immediately after the cardiac ECM administration (40, 41). The investigator who induced MI and performed injections was blinded to the content of the injectant. Details are described in the Supplementary Materials.
Echocardiography
Echocardiographic studies were performed by a blinded investigator repeatedly before surgery and at 5 days, 2 weeks, and 6 weeks after surgery, as previously described (63, 64). Echocardiographic parameters were measured using a high-frequency linear probe (MS400, 30 MHz) connected to a high-resolution ultrasound imaging system (Vevo 2100; FUJIFILM VisualSonics). ESA and EDA were measured from short-axis images of the left ventricle using B-mode scan. Functional parameters, including the LV fractional shortening (LVFS), LVFAC, and LVEF, were determined, as previously described (65, 66). Mice that died or displayed morphological/behavioral abnormality before the final time point were excluded. Details are described in the Supplementary Materials.
Ultrasonic strain analysis
The ultrasound B-mode frames of LV short-axis view acquired at 6 weeks after MI were analyzed using a cardiac strain analysis software package (VevoStrain, Vevo 2100; FUJIFILM VisualSonics) by a blinded investigator (n = 3 per group) (67, 68). Five regions of interest (ROIs) were selected along the LV midwall including one ROI in the anterior lateral (infarcted area) and four ROIs in the anterior medial, posterior lateral, posterior, and posterior medial (noninfarct areas) walls of the LV, as shown in Fig. 3. Radial and circumferential intercardiac strain (that is, change in strain from end-diastole to end-systole) in all five ROIs were obtained. The end-diastole and end-systole were determined according to EDA and ESA, respectively, as described above. The estimated intercardiac strain in the infarcted area was normalized to the average intercardiac strain of the four ROIs in unaffected LV walls.
Primary human cell culture
Primary hCSCs were purchased from Celprogen Inc. (36099-26) and expanded with hCSC complete medium with serum (hCSC-CM, M36099-26S; Celprogen Inc.) and ECM precoated T75 flasks (E36099-26; Celprogen Inc.), according to the manufacturer’s protocol. Primary hHPs were isolated from ventricular myocardium and purified by flow cytometry, as previously reported (69), in compliance with Institutional Review Board protocol number 0506176 at the University of Pittsburgh. hHPs were expanded in complete growth medium (hHP-CM) containing high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (11965-118, without sodium pyruvate), 20% FBS, and 1% penicillin/streptomycin (all from Life Technologies). hCSCs at passages 2 to 4 and hHPs at passages 6 to 8 were used in subsequent assays.
Measurement of cell proliferation in vitro
To investigate the effect of zECM on the proliferation of hCSCs and hHPs under stress, we used two stressed culture conditions. For the nutrient deprivation stress assay, we used 25% complete culture medium for hCSCs (25% hCSC-CM) and DMEM supplemented with 2.5% FBS and 1% penicillin/streptomycin for hHPs (2.5% FBS-CM). For the ischemic stress assay, we cultured cells under 2.5% O2 concentration (with 5% CO2 and 92.5% N2) in addition to nutrient deprivation. Briefly, hCSC-CM and hHP-CM were diluted at 1:3 and 1:7 ratios, respectively, with high-glucose DMEM containing 1% penicillin/streptomycin before supplementing with 25 μg of hzECM, nzECM, or mECM per 96-well plate. Full CM and diluted CM without ECM served as positive and negative controls, respectively. hCSCs were first plated in ECM precoated 96-well plates (E36099-26, Celprogen Inc.) in triplicate (103 cells per well) overnight with 100 μl of 25% hCSC-CM. hHPs were first plated in regular 96-well culture plates (Corning) in triplicate (103 cells per well) overnight with 100 μl of hHP-CM. After washing, media were replaced with 200 μl of 25% hCSC-CM and 2.5% FBS-CM, with or without ECM, for hCSCs and hHPs, respectively. Distinct medium dilutions were applied for different cell types because of their differential cell doubling time and endurance under stress. All plates were subsequently incubated under ambient or 2.5% O2 conditions for 4 days. At respective time points, after washing, 20 μl of CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) reagent (Promega) in DMEM was added to all wells containing 100 μl of fresh medium. The plate was then incubated at 37°C for 2.5 hours, at which point the absorbance at 490 nm (with reference at 650 nm) was read with an Infinite 200 PRO plate reader (Tecan). All experiments were independently repeated three times. Results were individually normalized to each experimental control and then averaged.
Measurement of cell migration in vitro
A Transwell chemotaxis assay was used to investigate the effect of zECM on the migration of hCSCs and hHPs under nutrient deprivation stress (with 25% hCSC-CM for hCSCs and 2.5% FBS-CM for hHPs, as described above). Media supplemented with 125 μg of hzECM, nzECM, mECM, or saline (negative control) were loaded into bottom wells. hCSCs and hHPs were seeded in Transwell inserts with an 8-μm pore size (Millipore) at a density of 10,000 cells per cm2. After incubation at 37°C for 6 hours, nonmigrated cells were removed with cotton swabs. Migrated cells were fixed with methanol for 10 min and stained with Quant-iT PicoGreen dsDNA reagent (P7581; Thermo Fisher Scientific). Fluorescent images were captured using a Nikon Eclipse Ti fluorescence microscope equipped with NIS-Elements AR imaging software (both from Nikon). The number of migrated cells was quantified and averaged from three independent images taken in three different areas per sample per group (n = 4). The cell number of each group was individually normalized to the average number of cells in the saline control group.
Histological and immunohistochemical analyses
Animals were sacrificed at 3 days and 6 weeks after surgery. Hearts were arrested in diastole by intraventricular injection of 1 M potassium chloride (KCl) and processed as previously described (62). For histology and immunohistochemistry, harvested hearts were flash-frozen in 2-methylbutane (M32631, Sigma-Aldrich), precooled in liquid nitrogen, and serially cryosectioned (8 μm thick) from the apex to the ligation level (approximately 0.5 mm in length). Each series contained 18 to 21 heart sections, which were roughly 200 μm apart natively and collected on one glass slide. Sections were fixed in a precooled (−20°C) mixture of methanol (322415, Sigma-Aldrich) and acetone (320110, Sigma-Aldrich) (1:1) for 5 min or in 4% paraformaldehyde (P6148, Sigma-Aldrich) for 8 min at room temperature (RT) immediately before staining. H&E staining was performed following the standard protocol. Masson’s trichrome staining kit (IMEB) was used to reveal collagen deposition on heart serial cross sections, following the manufacturer’s instruction.
For immunohistochemistry, nonspecific antibody binding was blocked with 10% donkey or goat serum at RT for 1 to 2 hours and, if necessary, with the Mouse on Mouse Antibody Staining Kit (Vector Laboratories). For evaluation of chronic inflammation, sections were incubated overnight at 4°C with rat anti-mouse CD68 primary antibody (1:200; Ab53444, Abcam), followed by goat anti-rat Alexa488 immunoglobulin G (IgG) (1:400; A-11006, Life Technologies). To examine mCSCs, cMSCs, and EPDCs, sections were first incubated overnight at 4°C with rat anti-mouse CD117 (1:50; CL8936AP, Cedarlane Laboratories), goat anti-mouse PDGFRβ (1:100; AF1042, R&D Systems), or rabbit anti–Wilms tumor (1:100; E3990, Spring Bioscience) antibody, respectively, followed by donkey anti-rat Alexa594 IgG (1:400; A-21209, Life Technologies), donkey anti-goat Alexa594 IgG (1:400; A-11058, Life Technologies), or donkey anti-rabbit Cy3 IgG (1:300; 711-165-152, Jackson ImmunoResearch Laboratories), respectively, at RT for 1 hour. For the detection of cardiomyocytes, sections were incubated overnight at 4°C with mouse anti-cTnT primary antibody (1:200; ab10214, Abcam), followed by goat anti-mouse Alexa488 IgG (1:400; A-11001, Life Technologies) at RT for 1 hour. To detect proliferating cells, after the first staining for one of the cell lineage markers above, a second overnight incubation was performed at 4°C with rabbit anti-mammalian Ki67 primary antibody (1:200; ab15580, Abcam), followed by donkey anti-rabbit Alexa488 IgG (1:400; A-21206, Life Technologies) at RT for 1 hour. For the detection of cells expressing ErbB2, after the first staining of cTnT, a second overnight incubation was performed at 4°C with chicken anti-ErbB2 primary antibody (1:100; ab14027, Abcam), followed by goat anti-chicken IgY (H+L) Alexa594 (1:400; A-11042, Life Technologies) at RT for 1 hour. To detect elastin, sections were incubated overnight at 4°C with mouse anti-elastin primary antibody (1:100; sc-374638, Santa Cruz Biotechnology), followed by goat anti-mouse Alexa488 IgG at RT for 1 hour. The elastin content at the infarct zone was quantified by the area size of the positive fluorescent signal. To ensure that c-kit+ cells were not mast cells, sections were first stained with anti-CD117, as described above, followed by a second overnight incubation with mouse anti-tryptase primary antibody (1:100; ab2378, Abcam) at 4°C and then with goat anti-mouse Alexa488 IgG at RT for 1 hour. To determine the effect of zECM treatments on apoptosis of cardiomyocytes, sections were first stained with anti-cTnT, as described above, followed by incubation with rabbit anti-cleaved caspase-3 primary antibody (1:100; 9661, Cell Signaling Technology) at RT for 1 hour and then with goat anti-rabbit Alexa594 at RT for 1 hour. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) at RT for 5 min. Immunofluorescence images were taken using a Nikon Eclipse Ti fluorescence microscope equipped with NIS-Elements AR imaging software (both from Nikon). Images were quantified using ImageJ software (National Institutes of Health).
For the histological analysis of the zebrafish heart, zebrafish were euthanized using tricaine (0.168 g/liter) for 3 to 5 min. Zebrafish hearts were collected in cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 2 hours. After two washes in PBS, hearts were cryopreserved overnight with 30% sucrose in PBS. Samples were embedded in tissue freezing medium (39475237, Leica Biosystems). Tissue blocks were cryosectioned at 14 μm thickness. Sections were allowed to dry at 37°C for 2 hours before storing them at −20°C. Acid Fuchsin Orange-G staining was performed as previously described (1). To detect NRG1 in situ, sections were incubated with rabbit anti-NRG1 antibody (1:100; ab27303, Abcam) overnight at 4°C, followed by goat anti-rabbit horseradish peroxidase–conjugated IgG (1:1000; G-21234, Life Technologies) at RT for 1 hour. Images were captured with a Leica MZ 16 microscope with a QImaging Retiga 1300 camera (Leica Biosystems). To fluorescently detect NRG1, mouse and zebrafish ventricular tissue sections were incubated with rabbit anti-NRG1 antibody (1:100; ab27303, Abcam) overnight at 4°C, followed by donkey anti-rabbit Alexa488 IgG (1:400; A-21206, Life Technologies) at RT for 1 hour. Nuclei were stained with DAPI at RT for 5 min. Immunofluorescence images were taken by a Nikon Eclipse Ti fluorescence microscope equipped with a QImaging Retiga 1300 camera. Images were quantified using ImageJ (National Institutes of Health).
Scanning electron microscopy
Fresh and decellularized normal and healing zebrafish hearts and respective lyophilized powders were processed for SEM. Briefly, fresh whole hearts were fixed in 2.5% glutaraldehyde (G5882, Sigma-Aldrich) in 0.1 M PBS (pH 7.4) for at least 10 min. Ventricles were dissected out under a dissection microscope, and fixation was then continued for no more than 50 min. Tissues were washed thoroughly in three changes of 0.1 M PBS for 15 min each. Tissues were fixed in 1% OsO4 in 0.1 M PBS for 60 min and then washed thoroughly in three changes of 0.1 M PBS for 15 min each. Tissues were subsequently dehydrated in graded series of ethanol (in PBS) for 15 min each: 30%, 50%, 70%, 90%, and 3 × 100%. All specimens were then subjected to critical point drying, mounted on studs, and sputter-coated with 3.5 nm of gold/palladium alloy using a Cressington 108auto sputter coater (Cressington Scientific Instruments). Samples were then imaged on a JSM-6335F SEM (JEOL USA). The samples were stored in a desiccator for future imaging.
Western blot
Total proteins were extracted and purified using protein extraction reagents (RIPA Lysis Buffer, 040-483; ProteinSimple) for zebrafish and mouse ventricular tissues (n = 4 per group). The equivalent of 50 μg of protein from each sample was separated by 11% gel [for 10 ml of gel: 3.85 ml of deionized water, 3.53 ml of 30% acrylamide, 2.6 ml of tris-HCl (pH 8.8), 50 μl of ammonium persulfate, and 10 μl of tetramethylethylenediamine; all from Bio-Rad Laboratories] and then transferred onto a polyvinylidene fluoride (PVDF) membrane (BioTrace PVDF Transfer Membrane, 66594). After blocking with 5% bovine serum albumin (OmniPur BSA, Fraction V, 2930-100GM; EMD Millipore), membranes were incubated with the anti-NRG1 antibody (1:300; ab27303, Abcam) at 4°C with shaking at 90 rpm overnight. Membranes were then washed with tris-buffered saline and treated with a goat anti-rabbit secondary antibody (1:1000; sc-2004, Santa Cruz Biotechnology) at RT for 2 hours. The signals were visualized with the ChemiDoc XRS+ Imaging System (Bio-Rad Laboratories). The band densities were quantified using ImageJ (National Institutes of Health).
Mass spectrometry
ECM (2 mg) was boiled in 0.2 ml of RIPA buffer (with 0.1% SDS, 1% sodium deoxycholate, and 1% NP-40) for 10 min. Solubilized proteins were quantified with the bicinchoninic acid assay, and 3 μg of protein was resolved in a gel (4% stacking and 7.5% SDS-PAGE running gel) at 150 V for 12 min and then stained with SimplyBlue SafeStain (LC6060, Thermo Fisher Scientific). In-gel trypsin digestion was carried out as previously described (70). Excised gel bands were washed with high-performance liquid chromatography (HPLC) water and destained with 50% acetonitrile (ACN)/25 mM ammonium bicarbonate until no visible staining. Gel pieces were dehydrated with 100% ACN and reduced with 10 mM dithiothreitol (DTT) at 56°C for 1 hour, followed by alkylation with 55 mM iodoacetamide (IAA) at RT for 45 min in the dark. Gel pieces were then again dehydrated with 100% ACN to remove excess DTT and IAA and rehydrated with trypsin (20 ng/μl)/25 mM ammonium bicarbonate and digested overnight at 37°C. The resultant tryptic peptides were extracted with 70% ACN/5% formic acid, vacuum-dried, and reconstituted in 18 μl of 0.1% formic acid. Proteolytic peptides from in-gel trypsin digestion were analyzed by a nanoflow reversed-phase LC-MS/MS. Tryptic peptides were loaded onto a C18 column (PicoChip column packed with 10.5 cm Reprosil C18 3-μm 120 Å chromatography media with a 75-μm inner diameter column and a 15-μm tip; New Objective Inc.) using a Dionex HPLC system (Dionex Ultimate 3000, Thermo Fisher Scientific) operated with a double-split system (personal communication with S. Gygi from the Department of Cell Biology, Harvard Medical School) to provide an in-column nanoflow rate (~300 nl/min). Mobile phases used were 0.1% formic acid for A and 0.1% formic acid in acetonitrile for B. Peptides were eluted off the column using a 52-min gradient (2 to 40% B in 42 min, 40 to 95% B in 1 min, 95% B for 1 min, and 2% B for 8 min) and injected into a linear ion trap MS (LTQ XL, Thermo Fisher Scientific) through electrospray. The LTQ XL was operated in a date-dependent MS/MS mode in which each full MS spectrum [acquired at 30,000 automatic gain control (AGC) target, 50 ms maximum ion accumulation time, precursor ion selection range of mass/charge ratio (m/z) of 300 to 1800] was followed by MS/MS scans of the five most abundant molecular ions determined from full MS scan (acquired based on the setting of 1000 signal threshold, 10,000 AGC target, maximum accumulation time of 100 ms, isolation width of 2.0 Da, activation time of 30 ms, and 35% normalized collision energy). Dynamic exclusion was enabled to minimize redundant selection of peptides previously selected for collision-induced dissociation. MS/MS spectra were searched using MASCOT search engine (version 2.4.0, Matrix Science Ltd.) against the UniProt zebrafish and mouse proteome database. The following modifications were used: static modification of cysteine (carboxyamidomethylation, +57.05 Da), variable modification of methionine (oxidation, +15.99 Da). The mass tolerance was set at 1.4 Da for the precursor ions and 0.8 Da for the fragment ions. Peptide identifications were filtered using PeptideProphet and ProteinProphet algorithms with a protein threshold cutoff of 99% and peptide threshold cutoff of 90% implemented in Scaffold 4 software (Proteome Software).
Statistical analysis
All measured data were presented as means ± SD. Statistical differences between groups were analyzed by Student’s t test (two groups), one-way ANOVA (multiple groups), or two-way repeated ANOVA (repeated echocardiographic measurements) with a 95% confidence interval. Statistical significance was set at P ≤ 0.05. Bonferroni multiple comparison test was performed for ANOVA post hoc analysis. Statistical analyses were performed with SigmaStat 3.5 (Systat Software), GraphPad Prism 5.0 (GraphPad Software), and SPSS21 (IBM) statistics software.
Supplementary Material
Acknowledgments
We thank M. Tsang for expert advice on zebrafish biology and J. Franks and D. B. Stolz for assistance and advice with SEM. Funding: This work was supported by American Heart Association Established Investigator Award 12EIA9020016 (to Y.W.). In vivo animal imaging was performed using an ultrasound scanner supported by NIH shared instrument grant NIH1S10RR027383-01 (to K.K.). This project used the University of Pittsburgh Cancer Institute Cancer Proteomics Facility that is supported, in part, by NIH award P30CA047904. Author contributions: W.C.W.C. prepared materials, designed and performed research, analyzed the data, and wrote the manuscript; Z.W. prepared the materials, designed the experiments, performed the research, and analyzed the data; M.A.M. prepared the materials and performed the research; D.W.P. performed the research and analyzed the data; D.W.L. and H.-J.L. prepared the samples, performed the experiments, and analyzed the data; X.Z. and N.A.Y. performed the experiments and analyzed the data; K.K. analyzed the data, provided the laboratory space and equipment, and edited the manuscript; Y.W. conceptualized and designed the research, provided the equipment and funding, and outlined and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1600844/DC1
Supplementary Materials and Methods
fig. S1. Amputated zebrafish hearts and decellularization processes.
fig. S2. Representative LV images of M- and B-mode echocardiography.
fig. S3. Histological analysis of mouse hearts after zECM treatment.
fig. S4. Fibrosis and chronic inflammation.
fig. S5. Analysis of elastin within the infarct zone in zECM-treated hearts.
fig. S6. Cardiomyocyte apoptosis.
fig. S7. Cardiomyocyte proliferation at 6 weeks after MI.
fig. S8. Echocardiographic analysis of cardiac function in control mice receiving myocardial saline injections and an intraperitoneal sham or DMSO injection with or without the ErbB2 inhibitor AG825 after AMI.
fig. S9. c-kit+ cells do not express mast cell marker tryptase.
table S1. zECM Protein Identification Probability.
table S2. mECM Protein Identification Probability.
table S3. Summary of detected proteins in zECM and mECM.
table S4. Echocardiographic parameters of infracted mouse hearts following ECM treatment.
table S5. Echocardiographic parameters of infracted mouse hearts following ECM treatment and ErbB2 inhibitor administration.
REFERENCES AND NOTES
- 1.Li D. Y., Brooke B., Davis E. C., Mecham R. P., Sorensen L. K., Boak B. B., Eichwald E., Keating M. T., Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Daley W. P., Yamada K. M., ECM-modulated cellular dynamics as a driving force for tissue morphogenesis. Curr. Opin. Genet. Dev. 23, 408–414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clause K. C., Barker T. H., Extracellular matrix signaling in morphogenesis and repair. Curr. Opin. Biotechnol. 24, 830–833 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godwin J., Kuraitis D., Rosenthal N., Extracellular matrix considerations for scar-free repair and regeneration: Insights from regenerative diversity among vertebrates. Int. J. Biochem. Cell Biol. 56, 47–55 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Bowers S. L. K., Banerjee I., Baudino T. A., The extracellular matrix: At the center of it all. J. Mol. Cell. Cardiol. 48, 474–482 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valiente-Alandi I., Schafer A. E., Blaxall B. C., Extracellular matrix-mediated cellular communication in the heart. J. Mol. Cell. Cardiol. 91, 228–237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watt F. M., Huck W. T. S., Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Biol. 14, 467–473 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Zacchigna S., Giacca M., Extra- and intracellular factors regulating cardiomyocyte proliferation in postnatal life. Cardiovasc. Res. 102, 312–320 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Janson I. A., Putnam A. J., Extracellular matrix elasticity and topography: Material-based cues that affect cell function via conserved mechanisms. J. Biomed. Mater. Res. A 103, 1246–1258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handorf A. M., Zhou Y., Halanski M. A., Li W.-J., Tissue stiffness dictates development, homeostasis, and disease progression. Organogenesis 11, 1–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oberpriller J., Oberpriller J. C., Mitosis in adult newt ventricle. J. Cell Biol. 49, 560–563 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brockes J. P., Kumar A., Comparative aspects of animal regeneration. Annu. Rev. Cell Dev. Biol. 24, 525–549 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Bonfanti L., From hydra regeneration to human brain structural plasticity: A long trip through narrowing roads. Scientific World Journal 11, 1270–1299 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kikuchi K., Poss K. D., Cardiac regenerative capacity and mechanisms. Annu. Rev. Cell Dev. Biol. 28, 719–741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poss K. D., Wilson L. G., Keating M. T., Heart regeneration in zebrafish. Science 298, 2188–2190 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Drenckhahn J.-D., Schwarz Q. P., Gray S., Laskowski A., Kiriazis H., Ming Z., Harvey R. P., Du X.-J., Thorburn D. R., Cox T. C., Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev. Cell 15, 521–533 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Sturzu A. C., Rajarajan K., Passer D., Plonowska K., Riley A., Tan T. C., Sharma A., Xu A. F., Engels M. C., Feistritzer R., Li G., Selig M. K., Geissler R., Robertson K. D., Scherrer-Crosbie M., Domian I. J., Wu S. M., Fetal mammalian heart generates a robust compensatory response to cell loss. Circulation 132, 109–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yates C. C., Hebda P., Wells A., Skin wound healing and scarring: Fetal wounds and regenerative restitution. Birth Defects Res. C Embryo Today 96, 325–333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porrello E. R., Mahmoud A. I., Simpson E., Hill J. A., Richardson J. A., Olson E. N., Sadek H. A., Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh S., Pontén A., Fleischmann B. K., Jovinge S., Cardiomyocyte cell cycle control and growth estimation in vivo—An analysis based on cardiomyocyte nuclei. Cardiovasc. Res. 86, 365–373 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Naqvi N., Li M., Calvert J. W., Tejada T., Lambert J. P., Wu J., Kesteven S. H., Holman S. R., Matsuda T., Lovelock J. D., Howard W. W., Iismaa S. E., Chan A. Y., Crawford B. H., Wagner M. B., Martin D. I. K., Lefer D. J., Graham R. M., Husain A., A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157, 795–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabé-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H., Jovinge S., Frisén J., Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mollova M., Bersell K., Walsh S., Savla J., Tanmoy Das L., Park S.-Y., Silberstein L. E., dos Remedios C. G., Graham D., Colan S., Kühn B., Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. U.S.A. 110, 1446–1451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarras M. P., Jr, Components, structure, biogenesis and function of the Hydra extracellular matrix in regeneration, pattern formation and cell differentiation. Int. J. Dev. Biol. 56, 567–576 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Hoshiba T., Lu H., Kawazoe N., Chen G., Decellularized matrices for tissue engineering. Expert Opin. Biol. Ther. 10, 1717–1728 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Badylak S., Decellularized allogeneic and xenogeneic tissue as a bioscaffold for regenerative medicine: Factors that influence the host response. Ann. Biomed. Eng. 42, 1517–1527 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Seif-Naraghi S. B., Singelyn J. M., Salvatore M. A., Osborn K. G., Wang J. J., Sampat U., Kwan O. L., Monet Strachan G., Wong J., Schup-Magoffin P. J., Braden R. L., Bartels K., DeQuach J. A., Preul M., Kinsey A. M., DeMaria A. N., Dib N., Christman K. L., Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction. Sci. Transl. Med. 5, 173ra25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wassenaar J. W., Gaetani R., Garcia J. J., Braden R. L., Luo C. G., Huang D., DeMaria A. N., Omens J. H., Christman K. L., Evidence for mechanisms underlying the functional benefits of a myocardial matrix hydrogel for post-MI treatment. J. Am. Coll. Cardiol. 67, 1074–1086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He M., Callanan A., Comparison of methods for whole-organ decellularization in tissue engineering of bioartificial organs. Tissue Eng. Part B Rev. 19, 194–208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert T. W., Strategies for tissue and organ decellularization. J. Cell. Biochem. 113, 2217–2222 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Williams C., Quinn K. P., Georgakoudi I., Black L. D. III, Young developmental age cardiac extracellular matrix promotes the expansion of neonatal cardiomyocytes in vitro. Acta Biomater. 10, 194–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roosens A., Somers P., De Somer F., Carriel V., Van Nooten G., Cornelissen R., Impact of detergent-based decellularization methods on porcine tissues for heart valve engineering. Ann. Biomed. Eng. 44, 2827–2839 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Lafontant P. J., Behzad A. R., Brown E., Landry P., Hu N., Burns A. R., Cardiac myocyte diversity and a fibroblast network in the junctional region of the zebrafish heart revealed by transmission and serial block-face scanning electron microscopy. PLOS ONE 8, e72388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senyo S. E., Steinhauser M. L., Pizzimenti C. L., Yang V. K., Cai L., Wang M., Wu T.-D., Guerquin-Kern J.-L., Lechene C. P., Lee R. T., Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493, 433–436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ali S. R., Hippenmeyer S., Saadat L. V., Luo L., Weissman I. L., Ardehali R., Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. U.S.A. 111, 8850–8855 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Uva G., Aharonov A., Lauriola M., Kain D., Yahalom-Ronen Y., Carvalho S., Weisinger K., Bassat E., Rajchman D., Yifa O., Lysenko M., Konfino T., Hegesh J., Brenner O., Neeman M., Yarden Y., Leor J., Sarig R., Harvey R. P., Tzahor E., ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 17, 627–638 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Gemberling M., Karra R., Dickson A. L., Poss K. D., Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife 4, e05871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yutzey K. E., Regenerative biology: Neuregulin 1 makes heart muscle. Nature 520, 445–446 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Osherov N., Gazit A., Gilon C., Levitzki A., Selective inhibition of the epidermal growth factor and HER2/neu receptors by tyrphostins. J. Biol. Chem. 268, 11134–11142 (1993). [PubMed] [Google Scholar]
- 40.Miyagawa S., Katsu Y., Watanabe H., Iguchi T., Estrogen-independent activation of erbBs signaling and estrogen receptor α in the mouse vagina exposed neonatally to diethylstilbestrol. Oncogene 23, 340–349 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Akhtar S., Yousif M. H. M., Chandrasekhar B., Benter I. F., Activation of EGFR/ERBB2 via pathways involving ERK1/2, P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from ischemia-reperfusion injury. PLOS ONE 7, e39066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foglia M. J., Poss K. D., Building and re-building the heart by cardiomyocyte proliferation. Development 143, 729–740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uygur A., Lee R. T., Mechanisms of cardiac regeneration. Dev. Cell 36, 362–374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singelyn J. M., Christman K. L., Injectable materials for the treatment of myocardial infarction and heart failure: The promise of decellularized matrices. J. Cardiovasc. Transl. Res. 3, 478–486 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okada M., Payne T. R., Oshima H., Momoi N., Tobita K., Huard J., Differential efficacy of gels derived from small intestinal submucosa as an injectable biomaterial for myocardial infarct repair. Biomaterials 31, 7678–7683 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Sonnenberg S. B., Rane A. A., Liu C. J., Rao N., Agmon G., Suarez S., Wang R., Munoz A., Bajaj V., Zhang S., Braden R., Schup-Magoffin P. J., Kwan O. L., DeMaria A. N., Cochran J. R., Christman K. L., Delivery of an engineered HGF fragment in an extracellular matrix-derived hydrogel prevents negative LV remodeling post-myocardial infarction. Biomaterials 45, 56–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu Y., Bayomy A. F., Gomez M. V., Bauer M., Du P., Yang Y., Zhang X., Liao R., A role for matrix stiffness in the regulation of cardiac side population cell function. Am. J. Physiol. Heart Circ. Physiol. 308, H990–H997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Missinato M. A., Tobita K., Romano N., Carroll J. A., Tsang M., Extracellular component hyaluronic acid and its receptor Hmmr are required for epicardial EMT during heart regeneration. Cardiovasc. Res. 107, 487–498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bersell K., Arab S., Haring B., Kühn B., Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Polizzotti B. D., Ganapathy B., Walsh S., Choudhury S., Ammanamanchi N., Bennett D. G., dos Remedios C. G., Haubner B. J., Penninger J. M., Kühn B., Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci. Transl. Med. 7, 281ra45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Force T., Krause D. S., Van Etten R. A., Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 7, 332–344 (2007). [DOI] [PubMed] [Google Scholar]
- 52.D’Uva G., Tzahor E., The key roles of ERBB2 in cardiac regeneration. Cell Cycle 14, 2383–2384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crone S. A., Zhao Y.-Y., Fan L., Gu Y., Minamisawa S., Liu Y., Peterson K. L., Chen J., Kahn R., Condorelli G., Ross J. Jr, Chien K. R., Lee K.-F., ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 8, 459–465 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Cote G. M., Sawyer D. B., Chabner B. A., ERBB2 inhibition and heart failure. N. Engl. J. Med. 367, 2150–2153 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Lien C.-L., Schebesta M., Makino S., Weber G. J., Keating M. T., Gene expression analysis of zebrafish heart regeneration. PLOS Biol. 4, e260 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Such-Sanmartín G., Bache N., Callesen A. K., Rogowska-Wrzesinska A., Jensen O. N., Targeted mass spectrometry analysis of the proteins IGF1, IGF2, IBP2, IBP3 and A2GL by blood protein precipitation. J. Proteomics 113, 29–37 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Ranganath S. H., Levy O., Inamdar M. S., Karp J. M., Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 10, 244–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn S.-B., Khan A., Detection and quantitation of twenty-seven cytokines, chemokines and growth factors pre- and post-high abundance protein depletion in human plasma. EuPA Open Proteom. 3, 78–84 (2014). [Google Scholar]
- 59.Becker R. O., Chapin S., Sherry R., Regeneration of the ventricular myocardium in amphibians. Nature 248, 145–147 (1974). [DOI] [PubMed] [Google Scholar]
- 60.Williams C., Sullivan K., Black L. D. III, Partially digested adult cardiac extracellular matrix promotes cardiomyocyte proliferation in vitro. Adv. Healthc. Mater. 4, 1545–1554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sood D., Chwalek Karolina, Stuntz Emily, Pouli Dimitra, Du Chuang, Tang-Schomer Min, Georgakoudi Irene, Black Lauren D. III, Kaplan David L., Fetal brain extracellular matrix boosts neuronal network formation in 3d bioengineered model of cortical brain tissue. ACS Biomater. Sci. Eng. 2, 131–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen C.-W., Okada M., Proto J. D., Gao X., Sekiya N., Beckman S. A., Corselli M., Crisan M., Saparov A., Tobita K., Péault B., Huard I., Human pericytes for ischemic heart repair. Stem Cells 31, 305–316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen W. C. W., Lee Brandon G., Park Dae Woo, Kim Kyobum, Chu Hunghao, Kim Kang, Huard Johnny, Wang Yadong, Controlled dual delivery of fibroblast growth factor-2 and Interleukin-10 by heparin-based coacervate synergistically enhances ischemic heart repair. Biomaterials 72, 138–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chu H., Chen C.-W., Huard J., Wang Y., The effect of a heparin-based coacervate of fibroblast growth factor-2 on scarring in the infarcted myocardium. Biomaterials 34, 1747–1756 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Pollick C., Hale S. L., Kloner R. A., Echocardiographic and cardiac Doppler assessment of mice. J. Am. Soc. Echocardiogr. 8, 602–610 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Wandt B., Bojö L., Tolagen K., Wranne B., Echocardiographic assessment of ejection fraction in left ventricular hypertrophy. Heart 82, 192–198 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Donnell M., Skovoroda A. R., Shapo B. M., Emelianov S. Y., Internal displacement and strain imaging using ultrasonic speckle tracking. IEEE Trans. Ultrason. Ferroelect. Freq. Contr. 41, 314–325 (1994). [Google Scholar]
- 68.Lubinski M. A., Emelianov S. Y., O’Donnell M., Speckle tracking methods for ultrasonic elasticity imaging using short-time correlation. IEEE Trans. Ultrason. Ferroelect. Freq. Contr. 46, 82–96 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Chen W. C. W., Baily J. E., Corselli M., Díaz M. E., Sun B., Xiang G., Gray G. A., Huard J., Péault B., Human myocardial pericytes: Multipotent mesodermal precursors exhibiting cardiac specificity. Stem Cells 33, 557–573 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M., In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1600844/DC1
Supplementary Materials and Methods
fig. S1. Amputated zebrafish hearts and decellularization processes.
fig. S2. Representative LV images of M- and B-mode echocardiography.
fig. S3. Histological analysis of mouse hearts after zECM treatment.
fig. S4. Fibrosis and chronic inflammation.
fig. S5. Analysis of elastin within the infarct zone in zECM-treated hearts.
fig. S6. Cardiomyocyte apoptosis.
fig. S7. Cardiomyocyte proliferation at 6 weeks after MI.
fig. S8. Echocardiographic analysis of cardiac function in control mice receiving myocardial saline injections and an intraperitoneal sham or DMSO injection with or without the ErbB2 inhibitor AG825 after AMI.
fig. S9. c-kit+ cells do not express mast cell marker tryptase.
table S1. zECM Protein Identification Probability.
table S2. mECM Protein Identification Probability.
table S3. Summary of detected proteins in zECM and mECM.
table S4. Echocardiographic parameters of infracted mouse hearts following ECM treatment.
table S5. Echocardiographic parameters of infracted mouse hearts following ECM treatment and ErbB2 inhibitor administration.