

Summary
Fringe proteins are β3-N-acetylglucosaminyltransferases that modulate Notch activity by modifying O-fucose residues on Epidermal Growth Factor-like (EGF) repeats of Notch. Mammals have three Fringes: Lunatic, Manic, and Radical. While Lunatic and Manic Fringe inhibit Notch1 activation from Jagged1 and enhance activation from Delta-like 1, Radical Fringe enhances signaling from both. We used a mass spectral approach to determine whether the variable effects of Fringes on Notch1 result from generation of unique glycosylation patterns on Notch1. We found that Lunatic and Manic Fringe modified similar sites on Notch1, while Radical Fringe modified a subset. Fringe modifications at EGF8 and 12 enhanced Notch1 binding to and activation from Delta-like 1, while modifications at EGF6 and 36 (added by Manic and Lunatic but not Radical) inhibited Notch1 activation from Jagged1. Combined, these results suggest that Fringe modifications “mark” different regions in the Notch1 extracellular domain for activation or inhibition.
Keywords: Notch, Fringe, O-fucose, EGF repeats, glycosylation, signal transduction, development
eTOC Blurb
Kakuda and Haltiwanger examine how Fringes regulate Notch1. They show that Radical Fringe modifies a subset of EGF repeats modified by Lunatic and Manic Fringe. Modifications at distinct EGF repeats “mark” the Notch1 extracellular domain, either enhancing or inhibiting activity.
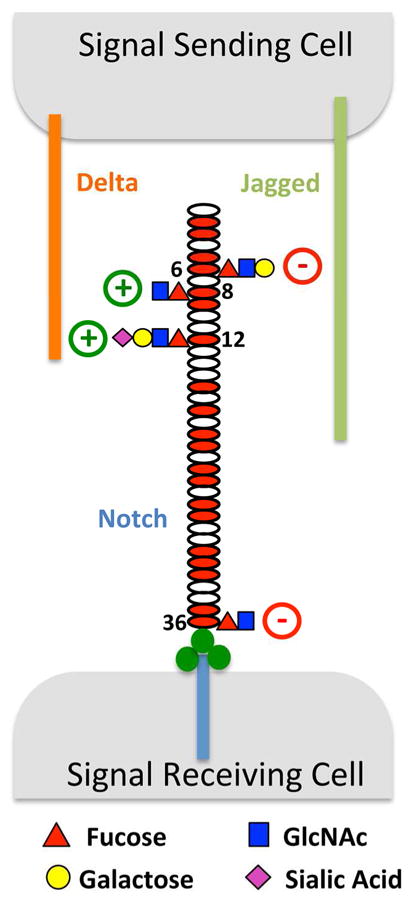
Introduction
Fringe was originally described in Drosophila, where loss of its expression caused a “notching” phenotype in the wing, suggesting involvement of the Notch pathway (Irvine and Wieschaus, 1994). Subsequent studies revealed that Fringe is a cell-autonomous modulator of Notch activity, potentiating activation from one Notch ligand, Delta, while inhibiting activation from the other, Serrate (Panin et al., 1997) (Fig. 1A). Three mammalian orthologs have been identified, Lunatic Fringe (Lfng), Manic Fringe (Mfng), and Radical Fringe (Rfng), which are non-redundant in mice (Moran et al., 2009) where they are expressed in both a tissue-specific and developmentally regulated manner (Johnston et al., 1997). The mammalian Fringes also modulate Notch1 activity, typically activating signaling from Delta family ligands (i.e. Delta-like (DLL) 1 and 4) and inhibiting signaling from Serrate family ligands (i.e. Jagged1, J1). However, some differences exist; for example Rfng can activate signaling from both DLL1 and J1 (LeBon et al., 2014; Yang et al., 2005).
Fig. 1. Lfng, Mfng, and Rfng elicit distinct responses from DLL1 and J1 ligands.

(A) Fringes modulate Notch activity by elongation of O-fucose residues on Notch, resulting in enhanced activation from Delta-family ligands but reduced activation from Serrate/Jagged ligands. (B) Structure of EGF12 from N1 modified with an O-fucose tetrasaccharide (colors for monosaccharides are the same as in the key in panel A). The galactose and sialic acid were modeled onto the structure from (Taylor et al., 2014). (C) Domain map of mouse N1 ECD. Each oval represents an EGF repeat. EGFs containing the consensus sequence for O-fucosylation, C2xxxx(S/T)C3, where C2 and C3 are the second and third conserved Cysteine in the EGF repeat (Rana and Haltiwanger, 2011) are colored red. (D) Cell-based co-culture N1 activation assays as described in Experimental Procedures. Cells were co-transfected with plasmids encoding N1 and one of the three Fringes or an empty vector (-Fng) control. Relative Luciferase units (RLU) compared to controls (-Fng) (normalized to 1 for each ligand) were calculated. Statistical significance of controls versus plus Fringe was determined using one-way ANOVA. Bar graph shows mean +/− SD; two independent experiments n = 6 were analyzed. ***, p < 0.0001; **, p < 0.001; *, p < 0.01. (E) Cell-based N1-ligand binding assays were performed by flow cytometric analysis. Cells were co-transfected with plasmids encoding N1 and one of the Fringes or an empty vector control (- Fng) and incubated with increasing concentrations of soluble DLL1-Fc or J1-Fc. Binding curves were generated by determining the MFI at each ligand concentration.
Fringes play important and distinct biological roles in modulating Notch activity in mammals as well as in flies (Rana and Haltiwanger, 2011; Takeuchi and Haltiwanger, 2014). Elimination of Lfng in mice results in a severe defect in somitogenesis (Evrard et al., 1998; Zhang and Gridley, 1998) and Lfng is a component of the somitogenesis “clock”, regulating mouse Notch1 (N1) activity in this context (Wahi et al., 2016). Mutations in Lfng are also known to cause Spondylocostal Dysostosis 3 (MIM #609813) in humans which results in severe congenital vertebral abnormalities, similar to those seen in Lfng null mice (Sparrow et al., 2006). Lfng has also been implicated as a regulator of Notch activity during angiogenesis (Benedito et al., 2009), hematopoiesis (Stanley and Guidos, 2009), and kidney development (Liu et al., 2013). Elimination of Mfng or Rfng in mice has no obvious developmental phenotype (Moran et al., 2009) but both have been implicated in a variety of Notch-dependent processes such as bile duct remodeling (D'Amato et al., 2016; Ryan et al., 2008). All three Fringes cooperate to modulate Notch activity during both B and T cell differentiation (Song et al., 2016) and have been linked to breast cancer (Xu et al., 2012; Zhang et al., 2015).
All three Fringes are β3-N-acetlyglucosaminyltransferases that transfer a GlcNAc to O-fucose residues on the Epidermal Growth Factor-like (EGF) repeats in the Notch extracellular domain (ECD) (Bruckner et al., 2000; Moloney et al., 2000a; Rampal et al., 2005b). In mammals, the resulting GlcNAcβ1-3Fucose disaccharide can be further elongated to a tetrasaccharide: Siaα2-6Galβ1-4GlcNAcβ1-3Fuc (Moloney et al., 2000a; Moloney et al., 2000b) (Fig. 1A, B). Elimination of the catalytic activity of Fringe prevents it from modulating Notch function, indicating that Notch activity is regulated by changes in O-fucose glycan structure (Bruckner et al., 2000; Moloney et al., 2000a). Several in vitro studies demonstrated that Fringe modifications alter Notch-ligand binding (Hou et al., 2012; Taylor et al., 2014; Xu et al., 2007; Yang et al., 2005). The ability of Fringes to enhance binding of Notch to Delta-family ligands has been consistent, but Fringes sometimes inhibit, have no effect, or enhance binding to Jagged-family ligands (Hicks et al., 2000; Stahl et al., 2008; Taylor et al., 2014; Yang et al., 2005). Recent structural studies have shown that the O-fucose on EGF12 of N1 is in direct contact with residues in DLL4 and therefore plays an essential role in ligand binding (Luca et al., 2015). Additional studies revealed that Fringe modification of the O-fucose at this site enhances binding to DLL1, DLL4 and J1 (Taylor et al., 2014). These results provide at least a partial molecular explanation for how Fringe modifications enhance signaling from Delta-family ligands, but not for how they inhibit signaling from Serrate or J1. In fact, the observed enhancement of J1 binding by Fringe modification at EGF12 was counterintuitive.
A number of questions remain regarding the mechanism of Fringe action. The studies described above demonstrate that Fringe modification of O-fucose on EGF12 plays an important role enhancing binding to Delta-family ligands. However, elimination of the O-fucose site on EGF12 in endogenous mouse N1 results in a hypomorphic allele with no obvious defect in somitogenesis where DLL1 is the predominant ligand (Ge and Stanley, 2008). This result suggests that Lfng modulates N1 activity during somitogenesis by modifying O-fucose sites other than EGF12. N1 contains 36 EGF repeats in its ECD, 20 of which contain a consensus sequence for O-fucosylation (Fig. 1C). We hypothesize that Fringe modifications at sites other than EGF12 affect N1 function. In addition, the differences observed in the ability of the three mammalian Fringes to modulate N1 activity ((LeBon et al., 2014; Yang et al., 2005) and Fig. 1D) suggest that they may modify O-fucose on different sets of EGF repeats. Using mouse N1 as a model system, here we determined which predicted O-fucosylation sites are modified with O-fucose, which are elongated by Lfng, Mfng, or Rfng, and which are required for these Fringes to modulate N1 activation by DLL1 or J1.
Results
Radical Fringe provides a clue to sites involved in activation of N1
As an initial step towards determining which O-fucose sites are necessary for Fringe to modulate N1 activity, we compared the activity of the three Fringes in a cell-based N1 signaling assay (Fig. 1D). Both Lfng and Mfng showed a classic Fringe effect, enhancing signaling from DLL1 but inhibiting signaling from J1. In contrast, Rfng activated signaling from both ligands, suggesting that sites modified by Rfng activate N1 from both DLL1 and J1. To demonstrate that this pattern of Fringe modulation is not unique to the NIH3T3 cells used for this assay, we showed a similar pattern using a human Notch1-Gal4 construct in HEK293T cells (Fig. S1A). The same pattern has also been observed previously in CHO cells (LeBon et al., 2014). Based on these results, we hypothesized that the difference in the ability of Rfng to modulate N1 activity was due to a difference in the O-fucose sites it modifies compared to those modified by either Lfng or Mfng.
Consistent with the Notch activation data, all three Fringes enhanced binding of DLL1 to N1 in a cell-based binding assay (Fig. 1E and Fig. S1B). Despite the negative effects of Lfng and Mfng on J1 (Fig. 1D), all three Fringes also enhanced binding of J1 to N1 (Fig. 1E and Fig. S1B). Enhanced ligand binding was not due to a Fringe-mediated increase in cell-surface expression level of N1 (Fig. S1C). Similar results were obtained using purified proteins in a pull-down assay, demonstrating that the enhanced binding occurs even in the absence of other cellular components (Fig. S1D). The increased J1 binding is in contrast to the inhibition of N1 activity by Lfng and Mfng in cell-based assays (Fig. 1D), but consistent with what we observed previously using the ligand-binding domain only (Taylor et al., 2014) and with other reports (Yang et al., 2005). These results suggest that inhibition of J1-mediated N1 activation by Lfng or Mfng occurs after initial ligand binding.
Predicted O-fucose sites on N1 are modified at high stoichiometries, and Rfng elongates O-fucose on a subset of sites modified by Lfng and Mfng
We used mass spectral glycoproteomic methods to identify which EGF repeats with predicted O-fucosylation sites are modified with O-fucose (Fig. 1C) and which of these are further elongated by a Fringe. A typical example is shown in Fig. 2A, where selection and fragmentation of a peptide from EGF35 containing an O-fucose consensus sequence is shown. Searching the MS data in the chromatogram for the ion representing this glycopeptide (m/z 720.0, Fig. 2A) resulted in the Extracted Ion Chromatogram (EIC) on the right. Searching for other glycoforms of this peptide (unmodified, or O-fucose di-, tri-, or tetrasaccahride) revealed that only the monosaccharide form was present. These results suggested that this site was modified with O-fucose at high stoichiometry (i.e. no unmodified peptide detected) and that the fucose was not elongated by any Fringe, consistent with the absence of Fringe activity in HEK293T cells.
Fig. 2. Lfng, Mfng, and Rfng generate unique patterns of O-fucose glycoforms on N1 EGF1-36.

Peptides derived from mouse N1 EGF1-36 produced in the absence (A) or presence (B) of Lfng were analyzed by mass spectrometry. The top panels show an MS spectrum at 4.5 min, and the bottom panels are the MS/MS spectrum of the indicated glycopeptide from EGF35, confirming the assignment. An Extracted Ion Chromatogram (EIC) of the MS data following the relative amounts of the ions corresponding to the potential glycoforms of this peptide (unmodified, mono-, di-, tri- and tetra-saccharide glycoforms, see key) is shown on the right. (C) EICs were generated as in (A) and (B) to evaluate the relative amounts of the O-fucose glycoforms (see key on right) on each O-fucosylated peptide. Spectra supporting the assignments of the ions used to generate the EICs for each site can be found in Data S1. Several of the peptides have additional modifications (see footnotes). Key: black bar, peptide; red triangle, fucose; blue square, GlcNAc; yellow circle, galactose; purple diamond, sialic acid.
To evaluate which sites were modified by Fringe family members, we examined the peptide glycoforms after co-transfecting plasmids encoding Lfng, Mfng, or Rfng. Fig. 2B shows analysis of the same peptide from EGF35 in a sample where Lfng was expressed. In addition to the monosaccharide glycoform of the peptide, ions corresponding to the di- and tri-saccharide glycoforms also appeared in the MS spectra (m/z 821.0, and 902.0, respectively), showing that Lfng elongates O-fucose on EGF35. EIC searches of this data revealed that the major form was the tri-saccharide, with roughly equivalent amounts of mono- and di-saccharide.
We performed a similar analysis for peptides from all EGF repeats containing O-fucose consensus sequences (Fig. 2C, Table S1 and Data S1). In the absence of any Fringe, most sites were O-fucosylated at high stoichiometry except for those at EGF23, 24, 26 and 32. These results demonstrate that O-fucosylation is a highly efficient process, as we previously observed for EGF12 (Rana et al., 2011), and suggested that some unique feature at EGF23, 24, 26 and 32 reduced or blocked O-fucosylation.
In contrast to the high stoichiometry of O-fucosylation at most sites, elongation by the Fringes was selective. Only two sites were modified by all three Fringes (EGF 8 and 26). Surprisingly, EGF12 was modified by Lfng and Rfng but poorly by Mfng. Six sites were modified by Lfng and Mfng, but poorly or not at all by Rfng (EGF 6, 9, 27, 30, 35, and 36, Fig. 2C). Other sites (EGF2, 3, 5, 16, 18, 20, 21, 24, and 31) were either poorly modified or unmodified by any Fringe (Fig. 2C). Control analyses confirmed that differences in glycan structures at individual sites were not due to differences in ionization efficiencies in different glycoforms of the peptides (Fig. S2A). The relative amount of O-fucose elongation was also responsive to the level of Fringe plasmid transfected into cells (Fig. S2B), providing further evidence that the EICs can be used to monitor the extent of Fringe-mediated elongation at each site. Similar mass spectral results were obtained using full-length N1, indicating that elongation by Fringe is not significantly affected by whether the N1 is tethered to the membrane or not (Fig. S2C). A summary of the results from Fig. 2C is shown in Fig. 4. These results are consistent with our hypothesis that Rfng modifies different sites than Mfng and/or Lfng and suggest that Fringe modifications at EGF8, 12, and/or 26 (those added by Rfng) are important for Notch activation regardless of ligand.
Fig. 4. The Fringe-mediated Notch Code.

A summary of the data from Fig. 2C. The most abundant O-fucose glycan found at each site is diagrammed. Fringe modification at EGF12 enhances binding to and activation of N1 by both ligands (green arrow), while modification at EGF8 enhances binding and activation from DLL1 (blue arrow). Fringe modifications at EGF6 and 36 inhibit N1 activation from J1 even though binding is increased due to modification at EGF12 (red arrows).
O-Fucose on EGF8 is important for DLL1 and J1 to bind and activate mouse Notch1, and O-fucose on EGF12 is more important for DLL1 than J1
We first examined the effects of O-fucose on N1 activity in the absence of Fringe. We performed site-directed mutation of all Fringe modified O-fucose sites by replacing the modified threonine (T) with valine (V), a change that removes only the hydroxyl group necessary for fucosylation. The EGF8V mutant had the most dramatic effect on the ability of both DLL1 and J1 to activate N1 in cell-based assays in the absence of any Fringe (Fig. S3A). EGF12V significantly reduced activation of N1 by DLL1 with almost no effect on J1, but the EGF8V12V double mutant markedly decreased N1 activation from both ligands. A few other sites also had effects: EGF9V and EGF27V reduced activation of N1 by J1 with little effect on DLL1, and EGF26V reduced activation from DLL1 but not J1 (Fig. S3A). Several other sites had smaller or no significant effect on N1 activation by either ligand (EGFs 6, 30, 35, 36) (Fig. S3A). Consistent with the activity assays, cell-based binding assays showed that EGF8V significantly reduced binding of DLL1 and J1, EGF12V reduced DLL1 binding but not J1, and the EGF8V12V double mutant nearly eliminated binding of both (Fig. S3B). None of these mutations caused reduced cell-surface expression of N1 (Fig. S3C). Thus, O-fucose on EGF8 affected binding to and activation of N1 by both ligands, while O-fucose on EGF12 affected DLL1 more than J1.
Fringe modification of EGF8 and 12 enhances Notch1 activation from DLL1 by enhancing binding
To evaluate which O-fucosylation sites are needed for any of the Fringes to enhance N1 activation from DLL1, we mutated all of the sites elongated by any Fringe and repeated the cell-based N1 activation assays shown in Fig. 1D. Since Rfng enhanced activation from DLL1 but only modified EGF8, 12, and 26 (Fig. 2C), mutation of those sites provided significant information regarding their importance. Compared to WT N1, both EGF8V and EGF12V showed smaller increases in N1 activation by Rfng, and the double mutant EGF8V12V could no longer be enhanced by Rfng, indicating the importance of EGF8 and 12 for Fringe-mediated enhancement of N1 activation by DLL1 (Fig. 3A). Like Rfng, the effects of Lfng were eliminated completely with the EGF8V12V. EGF8V and EGF9V almost completely eliminated the ability of Mfng to enhance DLL1 activation of N1. These results are consistent with the observation that Lfng and Rfng modified both EGF8 and 12 while Mfng modified EGF8 and EGF9, but not EGF12 (Fig. 2C). This indicates that modification of EGF9 by Mfng may partially compensate for lack of modification at EGF12. Unexpectedly, EGF26V had little effect on whether any of Fringes could enhance N1 activity. Other O-fucose site mutants had no significant effects (EGF6, 27, 30, 35, and 36, Fig. S3D).
Fig. 3. Fringe-mediated elongation of O-fucose at specific EGF repeats modulates N1 activation from DLL1 or J1.

(A) WT N1 or O-fucosylation site mutants (or Empty Vector, EV) were tested in cell-based N1 activation assays using DLL1 as activating ligand. Assays were performed as indicated in the absence (blue) or presence of Lfng (red), Mfng (green) or Rfng (purple). Analysis of additional mutants is shown in Fig. S3D. Bar graph shows mean +/− SD; Statistical significance of the enhancement of activation relative to –Fng for each mutant was determined using one-way ANOVA. Three independent experiments n = 9 were analyzed. ***, p < 0.0001; **, p < 0.001; *, P < 0.01. (B) Cell-based N1-DLL1 binding assays at a fixed concentration of DLL1 were performed. HEK293T cells were co-transfected with WT or mutant N1 (or EV) and GFP along with increasing amounts of Lfng (left), Mfng (middle), or Rfng (right). Cells were incubated with 3.6 nM DLL1-Fc pre-incubated with PE-Anti-mouse IgG as described in Supplemental Experimental Procedures. (C). N1 activation assays were done as in panel A but J1 was used as activating ligand. Analysis of additional mutants is shown in Fig. S3E. (D). N1-J1 binding assays at a fixed concentration of J1 were performed as in panel B. Cells were incubated with 2.1 nM J1-Fc pre-incubated with PE-Anti-human IgG as described in Supplemental Experimental Procedures.
To examine whether the Fringe-mediated enhancement in DLL1-N1 activation correlates with increases in DLL1-N1 binding, cell-based binding assays were performed where the ratio of Fringe to N1 was varied (Fig. 3B). Single mutants at EGF8V or EGF12V reduced the ability of Lfng and Rfng to enhance DLL1 binding to N1, and the EGF8V12V double mutant showed no enhancement of DLL1 binding by Lfng or Rfng even at the highest Fringe/N1 ratio, consistent with the observation that these mutations eliminated the ability of Lfng and Rfng to enhance N1 activation (Fig. 3A). Taken together these results indicate that Fringe modifications at EGF8 and 12 are essential for enhancement of DLL1-N1 binding, leading to enhanced activation. EGF8V and EGF9V had a similar effect on Mfng-induced binding, consistent with the Mfng N1 activation data in Fig. 3A.
Fringe modification at EGF6 or 36 inhibits J1-induced N1 activation
In contrast to DLL1, Lfng or Mfng significantly inhibit the activation of N1 by J1 (Fig. 3C). This inhibitory effect is reduced in the EGF6V and EGF36V single mutants. In the presence of Lfng, the EGF6V single and EGF6V36V double mutant caused a statistically significant enhancement of J1 activation of N1, similar to the effect of Rfng on N1 (which does not modify EGF6 or 36, Fig. 2C). EGF6V and EGF36V had no effect on the ability of Rfng to enhance N1 activity from J1 (Fig. 3C) or DLL1 (Fig. S3D). In contrast, EGF12V eliminated the Rfng-mediated enhancement (Fig. 3C). The EGF8V mutant reduced the ability of Lfng to inhibit N1 activation from J1, although the overall loss of N1 activity with this mutation makes it more difficult to accurately measure the level of inhibition at this site (Fig. 3C). Elimination of other Fringe modified sites had little or no effect (Fig. S3E).
Cell-based J1-N1 binding assays with increasing amounts of Lfng, Mfng and Rfng relative to N1 showed increased binding to WT N1 in all cases as expected (Fig. 3D). None of the mutations affecting Lfng or Mfng-mediated inhibition of N1 activation (6V, 36V, 6V36V) altered J1-N1 binding, providing additional support for the hypothesis that Fringe-mediated inhibition of J1-induced N1 activation results from a process which occurs after initial binding. Consistent with the N1 activation data shown in Fig. 3C, Rfng did not enhance binding of J1 to EGF12V. Thus, EGF12 appears to be the major site for Rfng to enhance binding and activation of N1 by J1. Lfng did not enhance binding of J1 to EGF12V either, supporting the importance of EGF12 as the major sites for Fringe-mediated increases in N1-J1 binding.
Discussion
The results presented here suggest that Fringes “mark” different regions of the N1 ECD resulting in activation or inhibition of N1 activity (Fig. 4), much like marks on histones regulate transcriptional activity (Jenuwein and Allis, 2001). Modification of O-fucose at EGF12 by Rfng in the ligand-binding region (EGF8-12 as recently defined (Andrawes et al., 2013; Yamamoto et al., 2012)) enhanced N1 binding to and activation by either ligand. Modification of EGF8 and 12 by any Fringe enhanced DLL1 binding and activation. This result helps to explain why mutation of EGF12 in endogenous N1 in mice resulted in a hypomorphic allele with no defects in somitogenesis as would be expected if EGF12 were the only biologically important O-fucosylation site for Lfng (Ge and Stanley, 2008). In contrast, modification at EGF6 and 36 by Lfng or Mfng inhibited J1-induced N1 activation in a dominant fashion, overcoming the enhanced binding of J1 resulting from modification at EGF 12. Rfng did not modify EGF6 or 36, explaining why it activated signaling from both DLL1 and J1 while Lfng and Mfng inhibited signaling from J1. Biological roles for EGF6 or 36 of the Notch ECD have not been previously described and they are not known to physically interact with ligand.
Our data shows that Fringe modifications at EGF8 and 12 enhance N1 activation in response to DLL1 directly by increasing binding. Recent structural and biophysical studies using small fragments of N1 support this idea. Taylor and co-workers showed that addition of fucose to EGF12 enhances binding of DLL1, J1 and DLL4 to N1 EGF11-13 (Taylor et al., 2014). Fringe modification of the O-fucose on EGF12 significantly increased the binding to DLL1 and J1, with a smaller increase in binding to DLL4. Structural analysis of the different glycoforms of EGF11-13 revealed that sugars do not alter the structure of the protein but exist in a highly ordered state on the surface of EGF12 with specific interactions to underlying amino acids, suggesting that they serve as an extension of the ligand binding site. This proposal was confirmed in a recent co-crystal of EGF11-13 and DLL4 showing the fucose on EGF12 in a highly ordered state in the interface between the two proteins mediating specific interactions with both (Luca et al., 2015). Modeling in the GlcNAc added by Fringe suggested additional interactions, supporting the model proposed by Taylor et al. (Taylor et al., 2014). Although these results nicely explain the importance of O-fucose and Fringe modifications at EGF12, they do not explain how the O-fucose glycans on EGF8 enhance ligand binding. Our results suggest that the O-fucose glycans on EGF8 are directly involved in ligand binding like those on EGF12. Work on mouse Notch2 suggests that O-fucose on EGF8 is not required for DLL1 or J1-induced Notch2 activation in cell-based co-culture assays (Yamamoto et al., 2012), indicating that differences in the roles of O-fucosylation on individual EGF repeats between Notch orthologues exist. Hou and coworkers recently demonstrated that addition of galactose to O-fucose glycans (required for extension to a tri- or tetra-saccharide form of O-fucose) increases the ability of Lfng to enhance DLL1-N1 activation but has the opposite effect with Mfng (Hou et al., 2012). Addition of galactose to O-fucose on EGF12 of EGF11-13 has little or no effect on DLL1 or J1 binding (Taylor et al., 2014), suggesting that galactose on other sites (e.g. 6, 9, 26, 27, 30, 35, all modified by galactose, Fig. 4) may account for these differences.
Inhibition of J1 signaling appears not to be mediated by a corresponding decrease in J1-N1 binding but through some other mechanism, possibly a structural change that is required to link ligand binding to the conformational changes required to allow proteolytic activation of Notch. The inhibition appears to be downstream of the increased J1 binding resulting from the Fringe modification at EGF12 since binding increases but activation is inhibited. Interestingly, our data showing that efficiency of O-fucosylation at EGF26 increases in the presence of Fringe (Fig. 2C) strongly suggests that Fringe modifications induce a conformational change in N1, leading to exposure of the site in EGF26, permitting more efficient modification by POFUT1. Thus, the site at EGF26 may be partially sterically blocked in the absence of Fringe, reducing efficiency of modification. Since POFUT1 acts in the ER (Luo and Haltiwanger, 2005) and Fringe is localized to the Golgi (Shifley and Cole, 2008), N1 may be undergoing retrograde transport to the ER after Fringe modification. Similar conformational models have been proposed for how O-glucose glycans affect Notch activity, which also appear to affect a step between ligand binding and Notch proteolytic activation (Jafar-Nejad et al., 2010; Takeuchi and Haltiwanger, 2014). Chen and co-workers showed that the trisaccharide form of O-fucose (addition of galactose) is required for Lfng or Mfng to inhibit N1 activation by J1 (Chen et al., 2001). Since both Mfng and Lfng elongate O-fucose on EGF6 (but not EGF36) to the trisaccharide (Fig. 4), the proposed conformational changes required for inhibition may require addition of the galactose to EGF6.
The variability in O-fucose residues being elongated by Fringes supports the concept that Fringe enzymes recognize specific features of individual EGF repeats, either enhancing or reducing modification, consistent with our previous studies showing that EGF repeats from human factor VII and IX encode specificity determinants for Fringe-mediated O-fucose elongation (Rampal et al., 2005b; Shao et al., 2003). Our prior studies showed the Lfng has the highest in vitro catalytic activity of the three Fringes (Rampal et al., 2005b), which may explain why it modifies sites more fully than either Mfng or Rfng (Fig. 4). It may also explain why more complete elongation to tetrasaccharide occurs with Lfng. More rapid modification of a site while N1 is moving through the Golgi apparatus would provide more time for β4galactosyltransferase and α6sialyltransferase to complete the tetrasaccharide modification. The fact that Rfng modifies fewer sites than either Lfng or Mfng suggests that something in those EGF repeats inhibits Rfng modification, or that something in those EGF repeats enhances modification by Lfng or Mfng.
Our prior studies showed that a EGF12A mutant (threonine mutated to alanine) caused a loss of N1 activation by either ligand, while EGF26A enhanced N1 activity (Rampal et al., 2005a). Here we showed that EGF12V reduced binding to and activation from DLL1 but had almost no effect on J1 (Fig. S3A, B). This result is consistent with our recent studies showing that the T to V mutation in EGF12 also significantly rescued binding of J1 to EGF11-13 relative to the T to A mutant (Taylor et al., 2014). We also did not observe an increase in N1 activity with either EGF26A or EGF26V mutants (Fig. S3A and data not shown). In the prior study we used COS-7 cells in the N1 activation assays, which express functional, endogenous Fringes based on elongation of O-fucose at EGF12, 26 and 27 (Rampal et al., 2005a). Here we used NIH3T3 cells for these assays, which do not express functional, endogenous Fringes based on lack of O-fucose elongation at these sites (Fig. S2D). It was for this reason that we chose NIH3T3 cells in this study. These results raise the intriguing possibility that further regulation of N1 activity could occur depending on the complement of Fringes expressed in a cell and the extent of Fringe modification at various sites.
Experimental Procedures
Glycoproteomic analysis of EGF1-36 and smaller fragments of N1 ECD
Purified N1 proteins (EGF1-36 or EGF fragment) were reduced, alkylated and subjected to in-gel digestion with trypsin, chymotrypsin or V8 as described (Kakuda and Haltiwanger, 2014; Rana et al., 2011). The resulting peptides were analyzed by nanoLC-MS/MS using Agilent nano HPLC-CHIP system coupled to a model 6430 Ion Trap mass spectrometer as described (Kakuda and Haltiwanger, 2014; Rana et al., 2011). O-Fucosylated peptides were identified by neutral loss searches, and semi-quantitative Extracted Ion Chromatograms (EIC) of selected ions were generated to compare relative amounts of O-fucosylated and unfucosylated forms of each peptide. The most abundant ion for each peptide from an EGF repeat was chosen for generation of EICs. Raw data for the EIC chromatograms was smoothed using a gauss algorithm.
Cell-based co-culture N1 activation assay
NIH3T3 cells (0.5 × 105) were seeded in a 24-well tissue culture plate and co-transfected with 0.1 μg of WT or mutant pcDNA1-N1-myc, and either 0.05 μg of SEAP (EV), LfngAP (Lfng), MfngAP (Mfng) or RfngAP (Rfng) plasmid, along with 0.12 μg of TP1-1 luciferase reporter construct and 0.06 μg of gWIZ β-galactosidase construct for transfection efficiency normalization using Lipofectamine 2000 (Invitrogen), according to the manufacture’s instruction. After 4 h, L cells stably expressing J1 or DLL1 were overlaid on the transfected NIH3T3 cells at a density of 1.5 × 105 cells/well for another 24 h. Cells were lysed and luciferase assays were performed based on the manufacture’s instructions (Luciferase Assay System, Promega) as described previously (Rana et al., 2011; Yamamoto et al., 2012).
Supplementary Material
Highlights.
Fringe modulates Notch activity by addition of GlcNAc to O-fucose on EGF repeats.
O-Fucose was found on 17 EGF repeats, but only a few are modified by any Fringe.
Fringe modifications at EGF8 and 12 enhance activation of Notch1 by Delta-like 1.
Fringe modifications at EGF6 and 36 inhibit Notch1 activation by Jagged1.
Acknowledgments
The authors would like to thank Dr. Vincent Luca for providing the EGF12 structure in Fig. 1B, and Drs. Bernadette Holdener, Chris Garcia, Vincent Luca, and members of the Haltiwanger laboratory for discussions and comments on this manuscript. This work was supported by NIH grant GM061126.
Footnotes
Author contributions
SK and RSH designed experiments, and SK performed experiments. SK and RSH interpreted results. RSH wrote the first draft of the manuscript, and SK and RSH edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrawes MB, Xu X, Liu H, Ficarro SB, Marto JA, Aster JC, Blacklow SC. Intrinsic Selectivity of Notch 1 for Delta-like 4 over Delta-like 1. J Biol Chem. 2013;288:25477–25489. doi: 10.1074/jbc.M113.454850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature. 2000;406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- Chen J, Moloney DJ, Stanley P. Fringe modulation of Jagged1-induced Notch signaling requires the action of ß4Galactosyltransferase-1. Proc Natl Acad Sci USA. 2001;98:13716–13721. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amato G, Luxan G, Del Monte-Nieto G, Martinez-Poveda B, Torroja C, Walter W, Bochter MS, Benedito R, Cole S, Martinez F, et al. Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol. 2016;18:7–20. doi: 10.1038/ncb3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrard YA, Lun Y, Aulehla A, Gan L, Johnson RL. Lunatic fringe is an essential mediator of somite segmentation and patterning. Nature. 1998;394:377–381. doi: 10.1038/28632. [DOI] [PubMed] [Google Scholar]
- Ge C, Stanley P. The O-fucose glycan in the ligand-binding domain of Notch1 regulates embryogenesis and T cell development. Proc Natl Acad Sci U S A. 2008;105:1539–1544. doi: 10.1073/pnas.0702846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks C, Johnston SH, DiSibio G, Collazo A, Vogt TF, Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nature Cell Biology. 2000;2:515–520. doi: 10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- Hou X, Tashima Y, Stanley P. Galactose differentially modulates lunatic and manic fringe effects on Delta1-induced NOTCH signaling. J Biol Chem. 2012;287:474–483. doi: 10.1074/jbc.M111.317578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine KD, Wieschaus E. fringe, a Boundary-specific signaling molecule, mediates interactions between dorsal and ventral cells during Drosophila wing development. Cell. 1994;79:595–606. doi: 10.1016/0092-8674(94)90545-2. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H, Leonardi J, Fernandez-Valdivia R. Role of glycans and glycosyltransferases in the regulation of Notch signaling. Glycobiology. 2010;20:931–949. doi: 10.1093/glycob/cwq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Johnston SH, Rauskolb C, Wilson R, Prabhakaran B, Irvine KD, Vogt TF. A family of mammalian Fringe genes implicated in boundary determination and the Notch pathway. Development. 1997;124:2245–2254. doi: 10.1242/dev.124.11.2245. [DOI] [PubMed] [Google Scholar]
- Kakuda S, Haltiwanger RS. Analyzing the posttranslational modification status of Notch using mass spectrometry. Methods Mol Biol. 2014;1187:209–221. doi: 10.1007/978-1-4939-1139-4_16. [DOI] [PubMed] [Google Scholar]
- LeBon L, Lee TV, Sprinzak D, Jafar-Nejad H, Elowitz MB. Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states. eLife. 2014;3:e02950. doi: 10.7554/eLife.02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen S, Boyle S, Zhu Y, Zhang A, Piwnica-Worms DR, Ilagan MX, Kopan R. The extracellular domain of Notch2 increases its cell-surface abundance and ligand responsiveness during kidney development. Dev Cell. 2013;25:585–598. doi: 10.1016/j.devcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science. 2015;347:847–853. doi: 10.1126/science.1261093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Haltiwanger RS. O-fucosylation of Notch occurs in the endoplasmic reticulum. J Biol Chem. 2005;280:11289–11294. doi: 10.1074/jbc.M414574200. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, et al. Fringe is a Glycosyltransferase that modifies Notch. Nature. 2000a;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Shair L, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on Epidermal Growth Factor-like modules. J Biol Chem. 2000b;275:9604–9611. doi: 10.1074/jbc.275.13.9604. [DOI] [PubMed] [Google Scholar]
- Moran JL, Shifley ET, Levorse JM, Mani S, Ostmann K, Perez-Balaguer A, Walker DM, Vogt TF, Cole SE. Manic fringe is not required for embryonic development, and fringe family members do not exhibit redundant functions in the axial skeleton, limb, or hindbrain. Dev Dyn. 2009;238:1803–1812. doi: 10.1002/dvdy.21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Fringe modulates notch ligand interactions. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- Rampal R, Arboleda-Velasquez JF, Nita-Lazar A, Kosik KS, Haltiwanger RS. Highly conserved O-fucose sites have distinct effects on Notch1 function. J Biol Chem. 2005a;280:32133–32140. doi: 10.1074/jbc.M506104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal R, Li AS, Moloney DJ, Georgiou SA, Luther KB, Nita-Lazar A, Haltiwanger RS. Lunatic Fringe, Manic Fringe, and Radical Fringe Recognize Similar Specificity Determinants in O-Fucosylated Epidermal Growth Factor-like Repeats. J Biol Chem. 2005b;280:42454–42463. doi: 10.1074/jbc.M509552200. [DOI] [PubMed] [Google Scholar]
- Rana NA, Haltiwanger RS. Fringe benefits: functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Curr Opin Struct Biol. 2011;21:583–589. doi: 10.1016/j.sbi.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS. O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J Biol Chem. 2011;286:31623–31637. doi: 10.1074/jbc.M111.268243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MJ, Bales C, Nelson A, Gonzalez DM, Underkoffler L, Segalov M, Wilson-Rawls J, Cole SE, Moran JL, Russo P, et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology. 2008;48:1989–1997. doi: 10.1002/hep.22538. [DOI] [PubMed] [Google Scholar]
- Shao L, Moloney DJ, Haltiwanger RS. Fringe Modifies O-Fucose on Mouse Notch1 at Epidermal Growth Factor-like Repeats within the Ligand-binding Site and the Abruptex Region. J Biol Chem. 2003;278:7775–7782. doi: 10.1074/jbc.M212221200. [DOI] [PubMed] [Google Scholar]
- Shifley ET, Cole SE. Lunatic fringe protein processing by proprotein convertases may contribute to the short protein half-life in the segmentation clock. Biochimica et biophysica acta. 2008;1783:2384–2390. doi: 10.1016/j.bbamcr.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Song Y, Kumar V, Wei HX, Qiu J, Stanley P. Lunatic, Manic, and Radical Fringe Each Promote T and B Cell Development. J Immunol. 2016;196:232–243. doi: 10.4049/jimmunol.1402421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, Fatkin D, Turnpenny PD, Kusumi K, Sillence D, Dunwoodie SL. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 2006;78:28–37. doi: 10.1086/498879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J Biol Chem. 2008;283:13638–13651. doi: 10.1074/jbc.M802027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Guidos CJ. Regulation of Notch signaling during T- and B-cell development by O-fucose glycans. Immunol Rev. 2009;230:201–215. doi: 10.1111/j.1600-065X.2009.00791.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Haltiwanger RS. Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun. 2014;453:235–242. doi: 10.1016/j.bbrc.2014.05.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor P, Takeuchi H, Sheppard D, Chillakuri C, Lea SM, Haltiwanger RS, Handford PA. Fringe-mediated extension of O-linked fucose in the ligandbinding region of Notch1 increases binding to mammalian Notch ligands. Proc Natl Acad Sci U S A. 2014;111:7290–7295. doi: 10.1073/pnas.1319683111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahi K, Bochter MS, Cole SE. The many roles of Notch signaling during vertebrate somitogenesis. Semin Cell Dev Biol. 2016;49:68–75. doi: 10.1016/j.semcdb.2014.11.010. [DOI] [PubMed] [Google Scholar]
- Xu A, Haines N, Dlugosz M, Rana NA, Takeuchi H, Haltiwanger RS, Irvine KD. In vitro reconstitution of the modulation of Drosophila notch-ligand binding by fringe. J Biol Chem. 2007;282:35153–35162. doi: 10.1074/jbc.M707040200. [DOI] [PubMed] [Google Scholar]
- Xu K, Usary J, Kousis PC, Prat A, Wang DY, Adams JR, Wang W, Loch AJ, Deng T, Zhao W, et al. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer cell. 2012;21:626–641. doi: 10.1016/j.ccr.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Charng WL, Rana NA, Kakuda S, Jaiswal M, Bayat V, Xiong B, Zhang K, Sandoval H, David G, et al. A mutation in EGF repeat-8 of Notch discriminates between Serrate/Jagged and Delta family ligands. Science. 2012;338:1229–1232. doi: 10.1126/science.1228745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell. 2005;16:927–942. doi: 10.1091/mbc.E04-07-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Gridley T. Defects in somite formation in lunatic fringe-deficient mice. Nature. 1998;394:374–377. doi: 10.1038/28625. [DOI] [PubMed] [Google Scholar]
- Zhang S, Chung WC, Wu G, Egan SE, Miele L, Xu K. Manic Fringe promotes a claudin-low breast cancer phenotype through Notch-mediated PIK3CG induction. Cancer Res. 2015 doi: 10.1158/0008-5472.CAN-14-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.