

Abstract
Background
Bruton’s tyrosine kinase (Btk) is a central player in multiple signaling pathways of lymphoid and myeloid cells. Myeloid cells are crucial early effectors in organ ischemia/reperfusion injury (IRI). BTKB66 is a selective, irreversible inhibitor of Btk. In this study, we hypothesized that Btk inhibition would reduce hepatocellular injury in a murine model of liver warm hepatic ischemia and reperfusion.
Methods
First, BTKB66 was tested in in vitro models of LPS-mediated neutrophil and macrophage activation. Then, to assess its efficacy in vivo, BTKB66 was administered orally to mice for seven days prior to subjecting them to 90 minutes of warm hepatic ischemia followed by reperfusion for 6 or 24 hours. Clinical and pathologic features in the livers, including AST, ALT, and a panel of cytokines and chemokines, were examined.
Results
BTKB66 potently inhibited LPS-mediated activation of bone-marrow derived neutrophils and macrophages in vitro. It also reduced the severity of IRI as determined by AST and ALT levels, as well as immune cell infiltrates. BTKB66 significantly decreased hepatic markers of sterile inflammation, such as CXCL1, CXCL2, and CXCL10, in parallel with depression of serum markers of the myeloid cell activation, such as CCL5, CCL11, and CXCL5.
Conclusion
BTKB66 treatment ameliorated hepatocellular injury in a well-established model of liver partial warm ischemia and in situ reperfusion. These findings confirm that neutrophil recruitment and activation play an essential role in IR-stress, and that targeting Btk activity may provide a useful approach for preventing hepatocellular damage and improving outcomes in liver transplantation.
INTRODUCTION
Reperfusion of ischemic tissues leads to a complex inflammatory response occurring without exogenous antigens. This inflammatory injury continues to affect clinical outcomes in transplantation, trauma, myocardial infarction, and stroke. Ischemia/reperfusion injury (IRI) involves leukocyte recruitment and activation; the production of reactive oxygen species; secretion of pro-inflammatory cytokines and chemokines; and activation of complement. 1 The cellular components include Kupffer cells, dendritic cells, neutrophils, T cells, and natural killer cells. The pathogenesis of IRI has been divided into two distinct, but overlapping, phases. In the first phase of hepatic IRI, liver resident Kupffer cells and dendritic cells are activated by “danger” molecules, thus facilitating a pro-inflammatory milieu and potentiating the inflammatory response. The next phase involves circulating monocytes and neutrophils, which become activated and recruited to the target organ, perpetuating and amplifying the local immune response and tissue destruction. 1 The resulting tissue damage initiates a series of responses dominated by innate immune activation, leading to local inflammation, cell death, and possibly even organ loss.
The multiplicity of effector cells and effector molecules has meant that signaling pathways as diverse as NF-κB 2 and PI3K 3 have been targeted for the development of novel anti-IRI therapies. Neutrophil activation in particular plays an essential role in liver IRI. 4 Rolling, adhesion, and migration of leukocytes into tissues have been established in the model for leukocyte activation. Our group has also shown the importance of neutrophil elastase, 5 selectin-mediated neutrophil recruitment, 6 and the JAK/STAT pathway in IRI. 7 Other groups have reported on the importance of neutrophil activation, specifically the formation of neutrophil extracellular traps, during liver IRI. 8 There is strong evidence for the importance of other myeloid cells such as macrophages in the inflammatory IRI cascade, likely through the TIM-4 and/or PTEN pathways. 9,10
Bruton’s tyrosine kinase (Btk) is a nonreceptor tyrosine kinase in the Tec family initially implicated in X-linked agammaglobulinema, a disorder of B cell development. The clinical significance of Btk has centered on its role in B cell activation following the engagement of B cell antigen receptors 11,12 inhibitors of Btk have been used successfully to treat chronic lymphocytic leukemia. 13 However, Btk’s role is not limited to B cell development, as it has been found in hematopoietic stem cells, myeloid cells, erythrocytes, and platelets. It is found in B cells, but not plasma cells; it is also absent in T lymphocytes. 14–16 Btk also has a significant role in Fcγ receptor-induced signaling and phagocytosis in monocytes/macrophages and neutrophils 17, and histamine release, FcεRI-dependent degranulation, and cytokine production in mast cell. 18 Several groups have also implicated Btk in the development, recruitment, and activation of neutrophils in inflammation. 19 Specifically, Btk mediates signaling via the TLR4 receptor and G-protein coupled receptors in neutrophils. 20 There are also indications that the role of Btk in inflammation is not limited to neutrophil but rather that it is involved at multiple points in the development and activation of myeloid cells. 21 Upon activation, Btk directly binds TLR4 and several other signaling proteins, resulting in the induction of pro-inflammatory cytokines, which then trigger and potentiate IRI. 22
Given the evidence that TLR4 signaling triggers local sterile inflammatory responses, 1 we hypothesized that inhibiting Btk would attenuate the pathogenesis of liver IRI. To the best of our knowledge, this is the first study to document that Btk signaling is critical in the mechanism of liver IRI and that its selective inhibition blocks IR-induced hepatocellular damage and protects livers from severe inflammation. Our findings provide a further rationale for the ongoing development of selective Btk inhibitors, which have recently come into clinical use, and demonstrate their potential utility in mitigating IRI in clinical practice.
METHODS
Animals
Male C57BL/6 mice (8–12 weeks old; designated WT) were purchased from Jackson Laboratory, (Bar Harbor, ME), and housed in the University of California, Los Angeles, facility under specific pathogen-free conditions and received humane care according to criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23 revised 1985).
Mouse Warm Hepatic Ischemia/Reperfusion Injury Model
Warm hepatic IRI operations in mice were performed as previously described.5 Briefly, animals (n=5–6 per group) were injected with heparin (100 U/kg) and an atraumatic clip was used to interrupt the arterial-portal venous blood supply to the cephalad liver lobes. After 90 min of partial hepatic ischemia, the clip was removed, initiating reperfusion. Animals were sacrificed at 6 hours or 24 hours; liver tissue and blood samples were obtained for analysis. Sham controls underwent the same procedure without vascular occlusion. The extent and severity of hepatic IRI were assessed in each group.
Treatment Protocol
Water for the mice was replaced either with a solution of vehicle alone (0.16M citric acid in DI water) or Btk inhibitor BTKB66 dissolved in the vehicle (0.32 mg/mL in 0.16 M citric acid in DI water). The mice were given this solution for seven days before the IRI operation. All mice consumed 4–6 mL of the solutions per day in place of water. The dose was chosen according to previously established concentrations needed for significant inhibition of Btk activity using its structural analogue ibrutinib. 23
Hepatocellular Injury Assay
Serum glutamic-oxaloacetic transaminase (SGOT, AST) and glutamic-pyruvic transaminase (SGPT, ALT) levels, indicators of hepatocyte injury, were measured in blood samples obtained. The measurements were performed using an autoanalyzer by IDEXX Laboratories (Irvine, CA).
Immunohistochemical Analysis
Liver tissue samples were embedded in Tissue Tec OCT compound (Miles, Elkhart, IN), snap-frozen in liquid nitrogen, and stored at −80°C. Cryostat sections (5 μm) were fixed in acetone and then endogenous peroxidase activity was inhibited with 0.3% H2O2 in phosphate-buffered saline. Primary rat antibody against mouse CD3, Ly-6G, and F4/80 (BD Biosciences Pharmingen, San Diego, CA) were used to stain for T cells, neutrophils, and macrophages. Bound primary antibody was detected using biotinylated anti-rat IgG and streptavidin peroxidase-conjugated complexes (DAKO, Carpinteria, CA). Slides were examined in a blinded fashion by counting labeled cells in triplicate within 10 high-power fields (HPF) per section.
Apoptosis Assay
Apoptosis in 5 μm cryostat liver sections was detected by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) method with an in situ cell death detection kit (Roche) according to the manufacturer’s protocol. The peroxidase activity was visualized with a diaminobenzidine substrate, which yielded a brown oxidation product; methyl green was used for counterstaining. TUNEL-positive cells were counted in 10 HPFs per section under light microscopy (×400), and the numbers of cells per HPF (mean ± SD) are shown.
Isolation of Bone Marrow Derived Mouse Neutrophils and Macrophages
To obtain the bone marrow cell suspensions, the femurs of a mouse were flushed with PBS containing 2% of fetal bovine serum (FBS). To isolate neutrophils, after dispersing cell clumps, the cell suspension was centrifuged (1200 rpm for 10 min at 4 °C). The cell pellets were resuspended in RPMI 1640/2% FBS and then incubated with primary antibodies specific for cell surface markers CD117, F4/80, CD4, CD45R, CD5, and TER119 for 30 min at 4 °C. After a 30 min incubation, anti-biotin MACSiMBeads were added according to the manufacturer’s instructions (Miltenyi Biotec Inc., San Diego, CA), and the cells were incubated for 15 min at 4 °C. The entire cell suspension was then placed into the magnetic field of an EasySep Magnet separator (StemCell Technologies, Vancouver, BC). The MACSiBead-labeled cells were allowed to adhere to the wall of the tube for 5 min. Viability, as determined by trypan blue exclusion, was consistently > 98%. Bone marrow cells were stained with PE conjugated anti-Ly-6G antibody, FITC-conjugated anti-CD11b antibody. All antibodies were purchased from BD Biosciences (San Jose, CA) or eBiosciences (San Diego, CA). Dead cells were excluded with (4′, 6-diamidino-2-phenylindole) DAPI staining dye (ThermoFisher, Waltham, MA). Cells were analyzed on LSRII or FACSVersa with FlowJo (Ashland, OR).
To isolate bone marrow-derived macrophages, the marrow tissue of mouse femurs was flushed by irrigation with media. The marrow plugs were dispersed by passing through a 25-gauge needle, and the cells were suspended by vigorous pipetting and washed by centrifugation. The cells were cultured in plastic tissue-culture dishes (150 mm) in 40 mL DMEM containing 20% FBS (Omega, Tarzana, CA), 5% M-CSF conditioned media, as a source of macrophage colony-stimulating factor (M-CSF), 1% pen/strep, 1% glutamine, and 0.5% sodium pyruvate (ThermoFisher, Waltham, MA) for 7–9 days prior to experimental use.
Macrophage and Neutrophil Activation Assays
Bone marrow derived neutrophils or macrophages (1 × 106/ml) were resuspended in PBS containing 1mM each CaCl2 and MgCl2 and incubated for 1 hour by shaking at room temperature with or without BTKB66 (1μM unless otherwise indicated) prior to lipopolysaccharide (LPS) stimulation (10 ng/ml) (Sigma, St. Louis, MO). After LPS stimulation for the indicated period of time, the cells were collected and protein was extracted.
Protein Isolation and Immunoblotting
Western blot of cell lysates was performed following standard procedures. Cells were lysed using RIPA buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA) supplemented with protease inhibitors (Roche, Basel, Switzerland). Protein concentrations were determined by BCA assays (ThermoFisher, Waltham, MA). Samples were boiled in SDS-PAGE sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromophenol blue) with β-ME and analyzed by SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA), blocked in 5% nonfat milk in 0.1% Tween in phosphate-buffered saline, and immunoblotted with the indicated primary antibodies and appropriate horseradish peroxidase conjugated secondary antibodies. Phospho-p38 MAPK (Thr180/Tyr182) (clone #9211), p38 MAPK Antibody (#9212), p-ERK1/2(Thr202/Tyr204)(#9101), ERK(#9102), p-JNK(Thr183/Tyr185)(#9251), JNK(#9252) were purchased from Cell Signaling (Danvers, MA). Immunoblots were developed by chemiluminescence (ECL System; Amersham Pharmacia Biotech, Piscataway, NJ).
qRT-PCR
RNA was extracted from liver tissue or cultured cells using Trizol (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. Total RNA was reverse transcribed to form cDNA, which was then subjected to real time PCR with gene specific primers using IQ SYBR Green Supermix on a CFX384 detection system both from Biorad (Hercules, CA). The primers used to amplify specific mouse gene fragments were as follow: CXCL10, 5′-ccaagtgctgccgtcattttc-3′ (sense) and 5′-ggctcgcagggatgatttcaa-3′ (antisense); CXCL1, 5′-ctgggattcacctcaagaacatc-3′ (sense) and 5′-cagggtcaaggcaagcctc-3′ (antisense); CXCL2, 5′-aatcatccaaaagatactgaacaaag-3′ (sense) and 5′-ttctctttggttcttccgttg-3′ (antisense); IL-6, 5′-agttgccttcttgggactga-3′ (sense) and 5′-tccacgatttcccagagaac-3′ (antisense); 36B4, 5′-ctgtgccagctcagaacactg-3′ (sense) and 5′-tgatcagcccgaaggagaag-3′ (antisense). Relative expression values are normalized to a control gene (36B4).
Myeloperoxidase assay
Myeloperoxidase (MPO) activity was measured using a Myeloperoxidase Colorimetric Activity Assay Kit (Sigma, St. Louis, MO) according to manufacturer instructions. MPO activity was reported as nmole/min/mL.
Serum Protein Quantification
Mouse cytokine/chemokines were quantitated in sera using the Luminex xMAP Immunoassay (Luminex Corp. Austin, TX).
Kinase Assays
In vitro kinase IC50s were measured using fluorescence binding assays after 1 h incubation of kinase, ATP, inhibitor, and substrate. Assays were performed by ThermoFisher Scientific SelectScreen™ Biochemical Kinase Profiling Service (Madison, WI). Kinase specific assay conditions and analyses are available online (https://www.thermofisher.com/us/en/home/products-and-services/services/custom-services/screening-and-profiling-services/selectscreen-profiling-service.html). All tests were run in duplicate.
Statistical Analysis
All data are expressed as mean ± SD. Statistical comparisons between groups were analyzed by two-sided Student’s t test. All differences were considered statistically significant at a value of P<0.05.
RESULTS
Inhibition of Btk Signaling Reduces IR-Triggered Hepatocellular Damage
BTKB66 is a highly selective, irreversible, heteroaromatic inhibitor of Btk with an IC50 13 nM. (Fig. 1) The IC50 of this compound for a panel of kinases selected to broadly evaluate selectivity is shown in Table 1. In order to investigate the role of Btk in liver IRI, mice were treated with BTKB66 or vehicle in the drinking water for seven days. They then underwent 90 minutes of partial hepatic warm ischemia by vascular clamping, followed by 6 or 24 hours of reperfusion. Livers in mice pre-treated with BTKB66 were resistant to IR-induced hepatocellular injury at 6 hours, as indicated by serum ALT and AST levels (IU/L), when compared with vehicle-treated controls (Fig. 2A; ALT: 722±189 versus 2814±617 IU/L, P = 0.009; AST: 296±88 versus 1287±294 IU/L, P = 0.009). This IR-protection persisted up to at least 24 hours (Fig. 2B; ALT: 393±134 versus 779±115 IU/L, P = 0.046; AST: 274±129 versus 710±148 IU/L, P = 0.048). BTKB66 treatment led to preserved lobular liver architecture with significantly decreased hepatocyte necrosis. In contrast, livers from vehicle-treated control mice had severe sinusoidal and vascular congestion, along with marked vacuolization and necrosis (Fig. 3). Neither treatment at the time of ischemia (intraperitoneal or by gavage) nor treatment for 24 hours prior to IRI significantly reduced hepatocellular damage. That pretreatment for seven days is required for efficacy is likely due to a loading effect, similar to that seen using its structural analogue ibrutinib. 23–25
Figure 1.
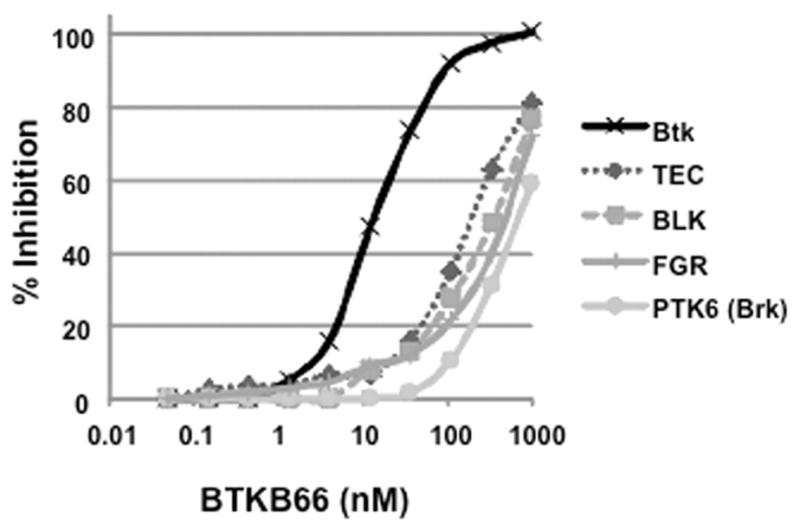
BTKB66 potently and selectively inhibits the kinase activity of Btk. In vitro kinase IC50s were measured using fluorescence resonance energy transfer binding based assays after 1 h incubation of kinase, ATP, inhibitor, and substrate. BTKB66 inhibits substrate phosphorylation with an IC50 of 13.3 nM, whereas TEC is inhibited with an IC50 of 195 nM.
Table 1.
IC50 values for inhibition of kinase activity by BTKB66
| Kinase | IC50, nM | ||
|---|---|---|---|
| Btk | 13.3 | ||
|
| |||
| TEC | 195 | ||
|
| |||
| BLK | 325 | ||
|
| |||
| FGR | 469 | ||
|
| |||
| PTK6 (Brk) | 493 | ||
|
| |||
| ABL1 | FRK (PTK5) | PAK1 | >1000 |
| ACVR1B (ALK4) | FYN | PAK2 (PAK65) | |
| ADCK3 | GSK3B | PAK4 | |
| AKT1 (PKB alpha) | HCK | PDGFRA | |
| AKT2 (PKB beta) | IGF1R | PDGFRB | |
| ALK | IKBKB | PDK1 Direct | |
| AURKA (Aurora A) | INSR | PIK3C2B | |
| AURKB (Aurora B) | ITK | PIK3CA/PIK3R1 | |
| AXL | JAK1 | PIK3CG | |
| BMPR2 | JAK2 | PIM1 | |
| BRAF | JAK3 | PIM2 | |
| CDK11 | KDR (VEGFR2) | PIM3 | |
| CDK16 | KIT | PLK1 | |
| CDK2/cyclin A | LCK | PRKACA (PKA) | |
| CDK3/cyclin E1 | LYN A | PRKCE | |
| CDK7/cyclin H | MAP2K1 (MEK1) | PTK2 (FAK) | |
| CDK9/cyclin T1 | MAP2K2 (MEK2) | RAF1 (cRAF) | |
| CHEK1 (CHK1) | MAP3K9 (MLK1) | RET | |
| CHUK (IKK alpha) | MAPK10 (JNK3) | RIPK2 | |
| CSF1R (FMS) | MAPK14 (p38 alpha) | ROCK2 | |
| CSK | MAPK3 (ERK1) | RPS6KA1 (RSK1) | |
| CSNK1D | MAPK8 (JNK1) | SRC | |
| CSNK1G2 | MAPK9 (JNK2) | STK22D (TSSK1) | |
| DCAMKL1 | MAPKAPK2 | STK32C (YANK3) | |
| DYRK1B | MARK3 | SYK | |
| EGFR (ErbB1) | MET (cMet) | TEK (Tie2) | |
| EPHA2 | MKNK1 | TGFBR1 (ALK5) | |
| ERBB2 (HER2) | MKNK2 | TYK2 | |
| FER | NEK2 | ULK2 | |
| FGFR2 | NTRK1 (TRKA) | YES1 | |
| FGFR3 | NUAK2 | ZAP70 | |
| FLT3 | |||
Figure 2.
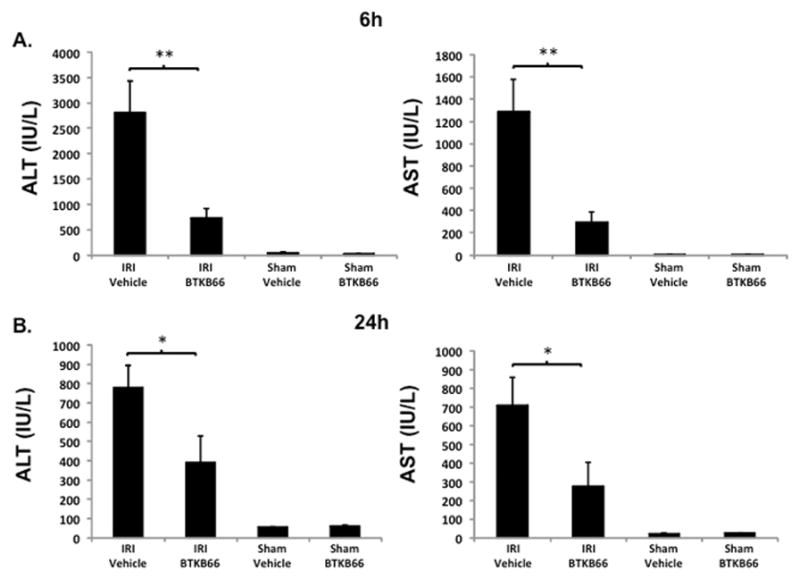
Inhibition of Btk Signaling Reduces IR-Triggered Hepatocellular Damage. Serum ALT and AST levels after liver warm ischemia (90 minutes) followed by reperfusion (6 hours [A] and 24 hours [B]) were lower in the BTKB66 treatment group in comparison with vehicle treated group. Sham operation groups are represented for comparison. * P < 0.05, ** P < 0.01; n = 4–6/group. Means and standard deviation are shown.
Figure 3.
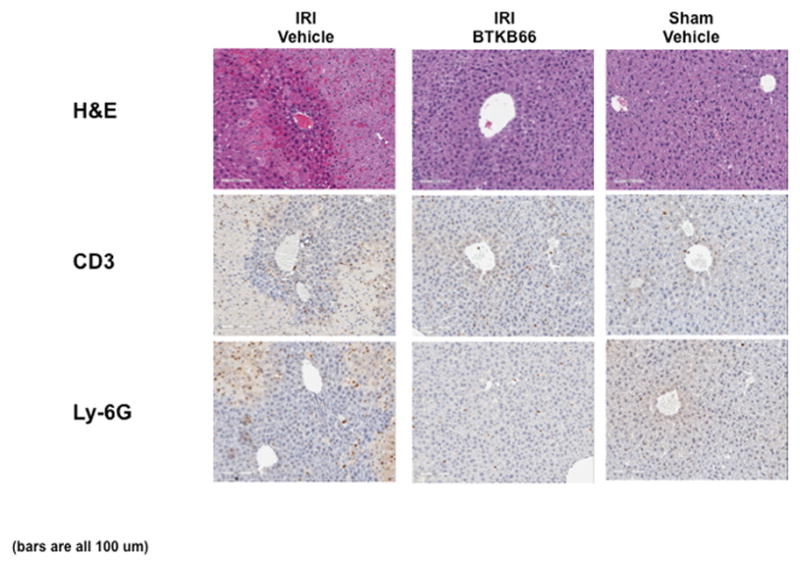
Inhibition of Btk Signaling Reduces IR-Triggered Hepatocyte Necrosis. Representative liver histology (hematoxylin-eosin staining; magnification 100x) of ischemic (90 minutes) liver lobes harvested after 6 hours of reperfusion. BTKB66 treatment leads to preservation of lobular architecture and minimal signs of hepatocyte necrosis after warm hepatic IRI. Untreated livers have severe sinusoidal and vascular congestion, vacuolization, and necrosis. Bars are 100 μm.
Inhibition of Btk Signaling Reduces Liver Neutrophil and Lymphocyte Infiltration and Apoptosis
We performed immunohistochemical staining for neutrophils, lymphocytes, and macrophages in livers after 6 hours of reperfusion following 90 minutes of warm ischemia (Fig. 3). Disruption of Btk signaling diminished the numbers of liver-infiltrating CD3-positive lymphocytes (positive cells/HPF: 15±1.8 versus 48±3.5, P < 0.001) and Ly-6G positive neutrophils (positive cells/HPF: 12±0.9 versus 116±2.3, P < 0.001) in comparison with controls (Fig. 4A). The frequency of liver-infiltrating macrophages (F4/80) was comparable in untreated and BTKB66 treated mice (data not shown). The decrease in neutrophil activity was confirmed by MPO assay, which was decreased in the BTKB66 treated group after 6 hours (nmol/min/mL: 20±18 versus 123±64, P = 0.048) compared to the vehicle (Fig. 4B). TUNEL staining was performed to evaluate hepatocyte apoptosis. At both 6 hours and 24 hours, hepatocyte apoptosis (TUNEL-positive cells/field) was notably increased in the untreated IR-stressed livers as compared to those treated with BTKB66 (positive cells/HPF: 28.3±8.1 versus 1.3±1.5, P < 0.001 at 6 h) (Fig. 5).
Figure 4.
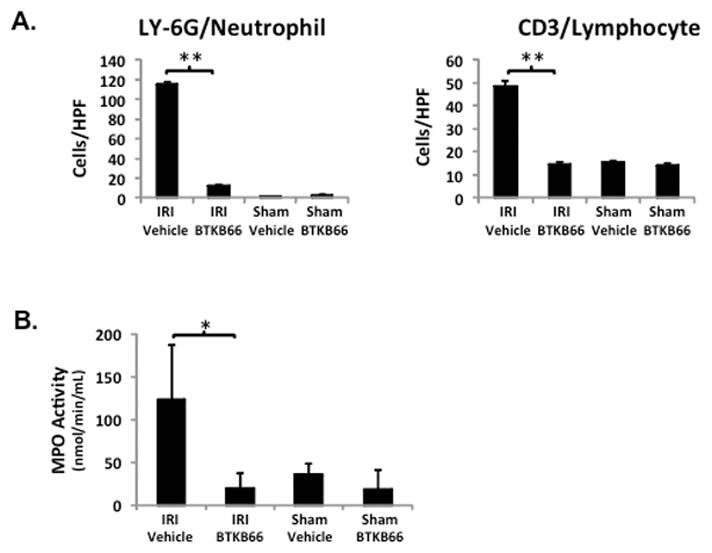
Inhibition of Btk Signaling Reduces IR-Triggered Liver Neutrophil and Lymphocyte Infiltration. (A) Histopathological analysis of liver tissues after 6 hours of IRI. Liver sections were evaluated in a blinded fashion by counting labeled cells within 10 high-power fields per section. BTKB66 led to less neutrophil and T cell infiltration. (B) Myeloperoxidase activity in liver tissue. * P<0.05, ** P<0.001; n = 3/group. Means and standard deviation are shown.
Figure 5.
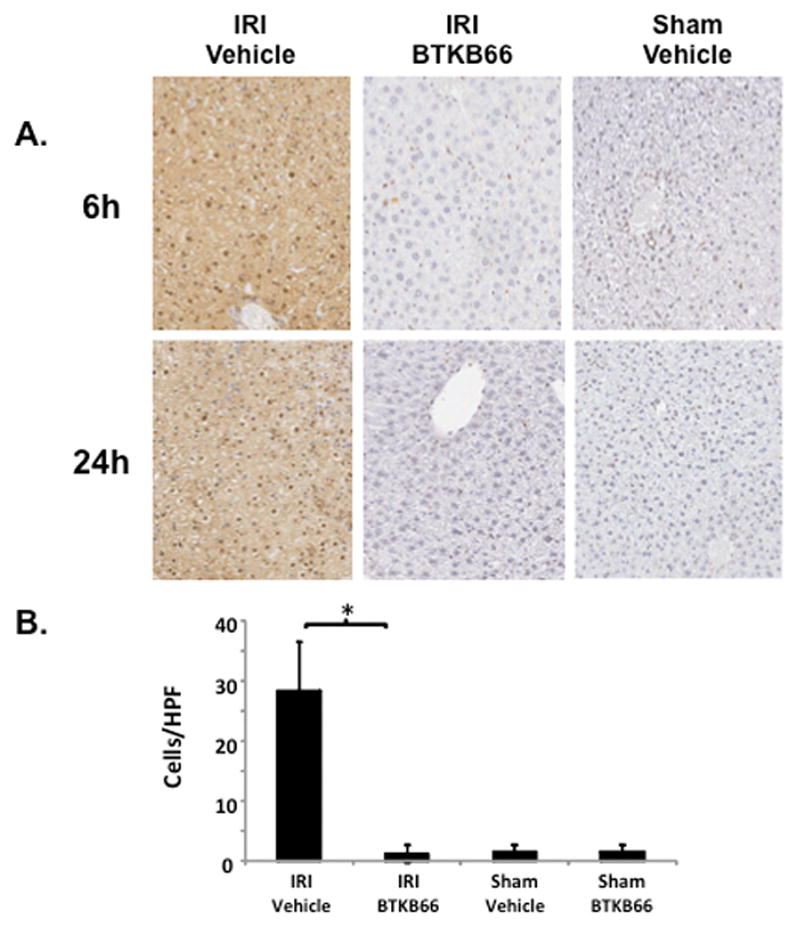
Inhibition of Btk Signaling Reduces IR-Triggered Hepatocyte Apoptosis. (A) TUNEL assay of liver tissues after 6 and 24 hours of IRI. (B) Histopathological analysis of liver tissues after 6 hours of IRI. Liver sections were evaluated in a blinded fashion by counting labeled cells within 10 high-power fields per section. * P<0.001; n = 3/group. Means and standard deviation are shown.
Inhibition of Btk Signaling Diminishes IR-Triggered Liver Chemokine Programs
Using RT-PCR we measured the relative level of mRNA coding for various cytokine and chemokine programs in the liver (Fig. 6A). Compared to controls, BTKB66 treatment diminished the expression of pro-inflammatory mediators, including CXCL1 (CXCL1/36B4 mRNA: 288.2±391.0 versus 32.4±32.1, P = 0.045), CXCL2 (CXCL2/36B4 mRNA: 76.2±80.1 versus 10.2±10.9, P = 0.02), and CXCL-10 (CXCL-10/HPRT mRNA: 21.2±24.8 versus 0.8±0.5, P = 0.02) after 6 hours of reperfusion.
Figure 6.
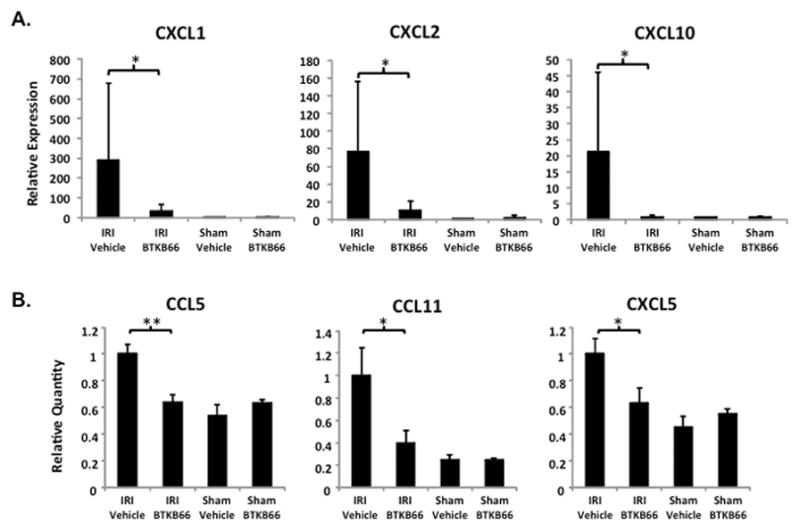
Inhibition of Btk Signaling Diminishes IR-Triggered Chemokine Programs. (A) qRT-PCR analysis of liver tissues after 6 hours of IRI. Liver tissues were evaluated for mRNA levels of selected chemokines. BTKB66 led to less expression of proinflammatory mediators CXCL1, CXCL2, and CXCL-10. * P<0.05. (B) Quantitative analysis of serum after 6 hours of IRI by multi-plex ELISA. Mouse sera were evaluated for protein levels of cytokines and chemokines. BTKB66 led to less pro-inflammatory mediators. * P<0.05, ** P < 0.005; n = 4–6/group. Means and standard deviation are shown.
Inhibition of Btk Signaling Diminishes IR-Triggered Systemic Chemokine Programs
Next we performed multi-plex cytokine analysis on serum 6 hours post-IR (Fig. 6B). Compared to vehicle controls, BTKB66 treatment decreased the expression of the pro-inflammatory chemokines, CCL5 (3217±240 versus 2050±181, P = 0.003), CCL11 (4368±1084 versus 1721±503, P = 0.03), and CXCL5 (38±4 versus 24±4, P = 0.046).
Btk Blockade Reduces p38, ERK1/2, and JNK Phosphorylation in LPS-Stimulated Bone Marrow Neutrophil/Macrophage Cultures
We hypothesized that the possible targets of BTKB66 were the activation of neutrophils and macrophages. To test this hypothesis in controlled cell culture experiments designed to mimic in vivo liver IRI, we used the model of LPS-induced (10 ng/mL) neutrophil and macrophage activation. While the use of many stimulating factors such as fMLP, hydrogen peroxide, PMA, or other DAMPS such as necrotic media from hepatocytes have been documented in the literature, 8 LPS is an accepted standard by which TLR4 stimulation of Btk is initiated. 22 It is also a well-documented major mediator of neutrophil activation leading to early down-regulation of L-selectin and up-regulation of CD1lb/CD18. 26 This myeloid cell stimulation was examined by Western blot analysis. As shown in Figure 7, the addition of LPS triggered robust phosphorylation of p38, ERK1/2, and JNK. However, cells that were treated with BTKB66 (1μM) for 60 minutes prior to LPS stimulation had markedly diminished phospho-p38, phospho-ERK1/2, and phospho-JNK levels. We observed that this effect was consistent from 30 minutes up to 4 hours of LPS stimulation.
Figure 7.
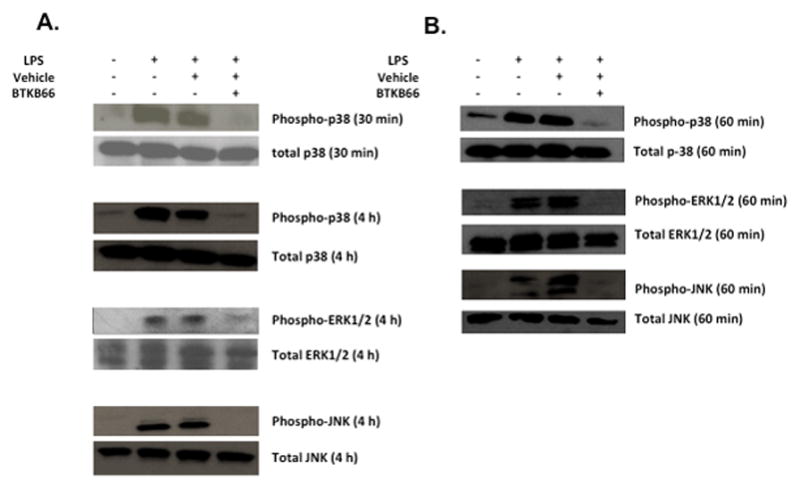
Btk Blockade Reduces p38, ERK1/2, and JNK Phosphorylation in LPS-Stimulated Bone Marrow Neutrophils and Macrophages. Western blot analysis of bone marrow derived (A) neutrophils and (B) macrophage after LPS activation for the indicated amount of time. BTKB66 led to inhibition of p38, ERK1/2, and JNK phosphorylation in response to LPS.
Btk Inhibition Decreases the Expression of Cytokines in LPS-Stimulated Bone Marrow Neutrophil Cultures
We stimulated bone marrow derived neutrophils with LPS (10 ng/mL) and assessed the gene expression of pro-inflammatory cytokines and chemokines. As shown in Figure 8, addition of LPS triggered the expression of a classic liver IRI pro-inflammatory gene signature [IL-6 (P < 0.001), IL1-β (P < 0.001), CXCL-2 (P < 0.001), and CXCL-10 (P < 0.001)]. In contrast, pretreatment of neutrophils with serial dilutions of BTKB66 (10 nM to 1 μM) prior to LPS stimulation markedly reduced the induction of these inflammatory genes in a dose dependent manner.
Figure 8.
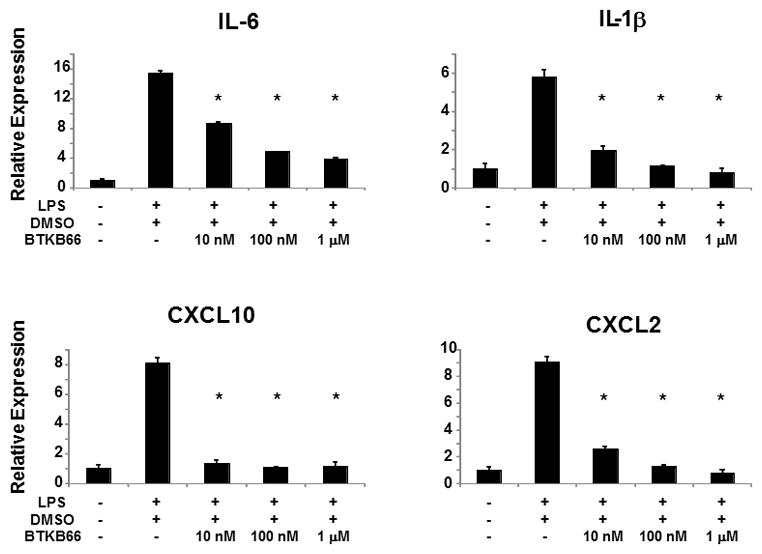
Btk Inhibition Decreases the Expression of Cytokines in LPS-Stimulated Bone Marrow Neutrophils. qRT-PCR analysis of bone marrow derived neutrophils after 30 minutes of LPS activation. BTKB66 led to less proinflammatory mediators IL-1β, IL-6, CXCL2, and CXCL-10 in a dose dependent manner. *P<0.001; n = 3/group. Means and standard deviation are shown.
DISCUSSION
Herein we examined the impact of inhibiting the Btk signaling pathway on the pathogenesis of liver ischemia reperfusion injury (IRI). Using BTKB66, a highly selective, orally available, irreversible showed the critical role of Btk signaling in IR-triggered, innate immunity dominated liver damage. Mice pretreated for a week with BTKB66 had significant improvement in both liver function and histology, compared to controls treated with vehicle. Consistent with these improvements were the associated reduction of lymphocyte and neutrophil recruitment to or localization in IR-stressed liver, as well as down-regulation of pro-inflammatory cytokine/chemokine gene programs. The in vitro studies support our in vivo findings, strongly indicating that liver IRI proceeds via Btk-mediated activation of neutrophils and macrophages. This is the first study, to the best of our knowledge, which documents that Btk inhibition mitigates local sterile inflammation and provides hepatoprotection against IR stress. This study, therefore, implicates Btk as a putative target for the generation of much needed clinically applicable inhibitors against liver IRI.
Our previous observations that selectin-mediated neutrophil activation and recruitment is required to initiate the disease process stimulated an interest in dissecting the complex role of neutrophils in liver IRI. 27 Multiple studies support the hypothesis that the activation of innate immunity and myeloid cells in particular can serve as a central point in the signaling cascades during IR-induced hepatocellular damage. Although earlier studies have shown that neutrophil depletion is a potent means to inhibit hepatic warm IRI, 28 not much is known about how Btk can influence IR-mediated organ damage. However, it is known that Btk is involved in signaling pathways that regulate leukocyte function and recruitment. 19 The recruitment of neutrophils to sites of inflammation is a tightly regulated multistep process. An overlapping expression cascade of selectins increases and enforces leukocyte recruitment during inflammation, with neutrophils expressing various binding ligand partners. Among the various signaling proteins involved in this process is Btk, which is expressed on a variety of hematopoietic cells including neutrophils. 29 By directly binding TLR4 upon activation, Btk activates multiple signaling pathways, including NF-κB, p38 MAPK, and iNOS, whereas Btk inhibition has been shown to protect against sepsis-induced acute lung injury. 30 Previous studies have also shown that inhibition of Btk can lead to reduced fMLP-mediated tyrosine phosphorylation, neutrophil adhesion, and chemotaxis. 31
These findings imply that Btk signaling can mediate local infiltration by inflammatory cells. The protective effects of Btk inhibition have previously been demonstrated in an acute lung injury model. 32 In IR-mediated inflammation, the initial activation of neutrophils results in the release of IL-1β, IL-6, and CXCL-10, a signature of liver IRI. 33 To confirm the effect of Btk signaling on cytokine expression, we used an in vitro assay with bone marrow derived neutrophils. Pretreatment with BTKB66 efficiently diminished LPS-triggered induction of numerous neutrophil mediators, including IL-1β, IL-6, CXCL2, and CXCL10. BTKB66 pretreatment also significantly decreased both the number of neutrophils sequestered in the liver and the resulting expression of pro-inflammatory gene programs, compared to controls. There was also a significant increase in Ly-6G neutrophil infiltration in vehicle-treated livers, compared to sham-operated mouse livers. Furthermore, as opposed those in the vehicle-treated group, livers in BTKB66 pretreated mice were characterized by decreased neutrophil sequestration and lower expression of CXCL2 (MIP-2; macrophage inflammatory protein-2), a chemokine responsible for attracting other granulocytes to sites of inflammation. 34
It is important to note that, while we did not observe a difference in the frequency of macrophages infiltrating IR-stressed liver, Btk inhibition does appear to block macrophage activation in vitro. The lack of an observed difference in hepatic macrophage sequestration may be due to distinct infiltration kinetics to sites of sterile inflammation. 10 There is also evidence that Btk-mutant mice have impaired macrophage function, including TNFα production. 21,35,36 Nevertheless, the recruitment and activation of neutrophils occurs early in the IRI process and this timeframe is more in line with the observed decrease in IR-induced hepatocellular injury. BTKB66 clearly inhibits both of these functions. We have also tested hepatocytes directly and seen no activity. While the effect of Btk inhibition on IRI may be due to its pleotropic effects, our results lead us to favor the hypothesis that the beneficial biological effects of Btk inhibition upon IRI pathology are largely due to inhibition of neutrophil activation and function.
The possibility that Btk inhibition decreases IRI by inhibiting B cells, while not excluded, is also unlikely given the timeframe of therapeutic manipulation and the dynamics of the disease state. Although studies examining the effect of PI3K-p110γ do implicate B cell migration and lymphocytic infiltration, these are seen at 24 hours and not at 6 hours, as in our model. 3 Similarly, studies examining the role of T cells through NF-κB inhibition in renal IRI show an effect not earlier than after 24 hours. 2
In conclusion, our present data implicate the Btk signaling pathway in the activation of myeloid cells, specifically neutrophils and macrophages, and the resultant sterile inflammation that leads to IR-induced liver damage. Our findings are the first to demonstrate that targeting Btk can ameliorate hepatocellular damage due to warm liver ischemia-reperfusion. These results should further encourage the development of inhibitors that specifically target the Btk pathway as a means to combat tissue IRI and improving outcomes in organ transplantation.
Acknowledgments
We would like to thank Azar Ghomian and Nakul Datta for their assistance in the performing the experiments described above. We would like to thank Betty Y. Chang for her input and fruitful discussions.
ABBREVIATIONS
- Btk
Bruton’s tyrosine kinase
- CXCL1
C-X-C motif chemokine 1
- CXCL2
C-X-C motif chemokine 2
- CXCL10
C-X-C motif chemokine 10
- ERK1/2
extracellular signal-regulated kinases 1/2
- FBS
fetal bovine serum
- IL-1β
interleukin 1 beta
- IL-6
interleukin 6
- IRI
ischemia/reperfusion injury
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MPO
myeloperoxidase
- qRT-PCR
quantitative real time PCR
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
Footnotes
DISCLOSURES
This work was supported by Pharmacyclics LLC, Sunnyvale, CA. The authors report no other proprietary or commercial interest in any product mentioned or concept discussed in this article.
AZ and JWK conceived and designed the study. AZ, TP, CL, YK, and KN acquired the data, and analyzed and interpreted them. AZ and TP drafted the article. KN, CL, SJB, RWB, and JWK revised it critically for important intellectual content. All authors provided final approval of the version to be submitted.
References
- 1.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11(8):1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue C, Liu Y, Li C, et al. Powerful protection against renal ischemia reperfusion injury by T cell-specific NF-kappaB inhibition. Transplantation. 2014;97(4):391–396. doi: 10.1097/01.TP.0000438622.89310.95. [DOI] [PubMed] [Google Scholar]
- 3.Kim N, Woo DC, Joo SJ, et al. Reduction in Renal Ischemia-Reperfusion Injury in Mice by a Phosphoinositide 3-Kinase p110gamma-Specific Inhibitor. Transplantation. 2015;99(10):2070–2076. doi: 10.1097/TP.0000000000000742. [DOI] [PubMed] [Google Scholar]
- 4.Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006;26(8):912–919. doi: 10.1111/j.1478-3231.2006.01327.x. [DOI] [PubMed] [Google Scholar]
- 5.Uchida Y, Freitas MC, Zhao D, Busuttil RW, Kupiec-Weglinski JW. The protective function of neutrophil elastase inhibitor in liver ischemia/reperfusion injury. Transplantation. 2010;89(9):1050–1056. doi: 10.1097/TP.0b013e3181d45a98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dulkanchainun TS, Goss JA, Imagawa DK, et al. Reduction of hepatic ischemia/reperfusion injury by a soluble P-selectin glycoprotein ligand-1. Ann Surg. 1998;227(6):832–840. doi: 10.1097/00000658-199806000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freitas MC, Uchida Y, Zhao D, Ke B, Busuttil RW, Kupiec-Weglinski JW. Blockade of Janus kinase-2 signaling ameliorates mouse liver damage due to ischemia and reperfusion. Liver Tranpl. 2010;16(5):600–610. doi: 10.1002/lt.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H, Tohme S, Al-Khafaji AB, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–614. doi: 10.1002/hep.27841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji H, Liu Y, Zhang Y, et al. T-cell immunoglobulin and mucin domain 4 (TIM-4) signaling in innate immune-mediated liver ischemia-reperfusion injury. Hepatology. 2014;60(6):2052–2064. doi: 10.1002/hep.27334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue S, Rao J, Zhu J, et al. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol. 2014;192(11):5343–5353. doi: 10.4049/jimmunol.1400280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan WN. Regulation of B lymphocyte development and activation by Bruton’s tyrosine kinase. Immunol Res. 2001;23(2–3):147–156. doi: 10.1385/IR:23:2-3:147. [DOI] [PubMed] [Google Scholar]
- 12.Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72(2):279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 13.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benson MJ, Rodriguez V, von Schack D, et al. Modeling the clinical phenotype of BTK inhibition in the mature murine immune system. J Immunol. 2014;193(1):185–197. doi: 10.4049/jimmunol.1302570. [DOI] [PubMed] [Google Scholar]
- 15.Smith CI, Baskin B, Humire-Greiff P, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994;152(2):557–565. [PubMed] [Google Scholar]
- 16.Vargas L, Hamasy A, Nore BF, Smith CI. Inhibitors of BTK and ITK: state of the new drugs for cancer, autoimmunity and inflammatory diseases. Scand J Immunol. 2013;78(2):130–139. doi: 10.1111/sji.12069. [DOI] [PubMed] [Google Scholar]
- 17.Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. 2008;181(1):288–298. doi: 10.4049/jimmunol.181.1.288. [DOI] [PubMed] [Google Scholar]
- 18.Hata D, Kawakami Y, Inagaki N, et al. Involvement of Bruton’s tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. The Journal of experimental medicine. 1998;187(8):1235–1247. doi: 10.1084/jem.187.8.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Block H, Zarbock A. The role of the tec kinase Bruton’s tyrosine kinase (Btk) in leukocyte recruitment. Int Rev Immunol. 2012;31(2):104–118. doi: 10.3109/08830185.2012.668982. [DOI] [PubMed] [Google Scholar]
- 20.Zemans RL, Arndt PG. Tec kinases regulate actin assembly and cytokine expression in LPS-stimulated human neutrophils via JNK activation. Cell Immunol. 2009;258(1):90–97. doi: 10.1016/j.cellimm.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mangla A, Khare A, Vineeth V, et al. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104(4):1191–1197. doi: 10.1182/blood-2004-01-0207. [DOI] [PubMed] [Google Scholar]
- 22.Jefferies CA, O’Neill LA. Bruton’s tyrosine kinase (Btk)-the critical tyrosine kinase in LPS signalling? Immunol Lett. 2004;92(1–2):15–22. doi: 10.1016/j.imlet.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang BY, Huang MM, Francesco M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011;13(4):R115. doi: 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutcheson J, Vanarsa K, Bashmakov A, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. 2012;14(6):R243. doi: 10.1186/ar4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soler-Rodriguez AM, Zhang H, Lichenstein HS, et al. Neutrophil activation by bacterial lipoprotein versus lipopolysaccharide: differential requirements for serum and CD14. J Immunol. 2000;164(5):2674–2683. doi: 10.4049/jimmunol.164.5.2674. [DOI] [PubMed] [Google Scholar]
- 27.Amersi F, Farmer DG, Shaw GD, et al. P-selectin glycoprotein ligand-1 (rPSGL-Ig)-mediated blockade of CD62 selectin molecules protects rat steatotic liver grafts from ischemia/reperfusion injury. Am J Transplant. 2002;2(7):600–608. doi: 10.1034/j.1600-6143.2002.20704.x. [DOI] [PubMed] [Google Scholar]
- 28.Tan Z, Jiang R, Wang X, et al. RORgammat+IL-17+ neutrophils play a critical role in hepatic ischemia-reperfusion injury. J Mol Cell Biol. 2013;5(2):143–146. doi: 10.1093/jmcb/mjs065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lachance G, Levasseur S, Naccache PH. Chemotactic factor-induced recruitment and activation of Tec family kinases in human neutrophils. Implication of phosphatidynositol 3-kinases. J Biol Chem. 2002;277(24):21537–21541. doi: 10.1074/jbc.M201903200. [DOI] [PubMed] [Google Scholar]
- 30.Zhou P, Ma B, Xu S, et al. Knockdown of Burton’s tyrosine kinase confers potent protection against sepsis-induced acute lung injury. Cell Biochem Biophys. 2014;70(2):1265–1275. doi: 10.1007/s12013-014-0050-1. [DOI] [PubMed] [Google Scholar]
- 31.Gilbert C, Levasseur S, Desaulniers P, et al. Chemotactic factor-induced recruitment and activation of Tec family kinases in human neutrophils. II. Effects of LFM-A13, a specific Btk inhibitor. J Immunol. 2003;170(10):5235–5243. doi: 10.4049/jimmunol.170.10.5235. [DOI] [PubMed] [Google Scholar]
- 32.Krupa A, Fol M, Rahman M, et al. Silencing Bruton’s tyrosine kinase in alveolar neutrophils protects mice from LPS/immune complex-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307(6):L435–448. doi: 10.1152/ajplung.00234.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wanner GA, Ertel W, Muller P, et al. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock. 1996;5(1):34–40. doi: 10.1097/00024382-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 34.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392(6676):565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 35.Mukhopadhyay S, Mohanty M, Mangla A, et al. Macrophage effector functions controlled by Bruton’s tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J Immunol. 2002;168(6):2914–2921. doi: 10.4049/jimmunol.168.6.2914. [DOI] [PubMed] [Google Scholar]
- 36.Horwood NJ, Mahon T, McDaid JP, et al. Bruton’s tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor alpha production. J Exp Med. 2003;197(12):1603–1611. doi: 10.1084/jem.20021845. [DOI] [PMC free article] [PubMed] [Google Scholar]