

Abstract
The interleukin-6 cytokines, acting via gp130 receptor pathways, play a pivotal role in the reduction of cardiac injury induced by mechanical stress or ischemia and in promoting subsequent adaptive remodeling of the heart. We have now identified the small proline-rich repeat proteins (SPRR) 1A and 2A as downstream targets of gp130 signaling that are strongly induced in cardiomyocytes responding to biomechanical/ischemic stress. Upregulation of SPRR1A and 2A was markedly reduced in the gp130 cardiomyocyte-restricted knockout mice. In cardiomyocytes, MEK1/2 inhibitors prevented SPRR1A upregulation by gp130 cytokines. Furthermore, binding of NF-IL6 (C/EBPβ) and c-Jun to the SPRR1A promoter was observed after CT-1 stimulation. Histological analysis revealed that SPRR1A induction after mechanical stress of pressure overload was restricted to myocytes surrounding piecemeal necrotic lesions. A similar expression pattern was found in postinfarcted rat hearts. Both in vitro and in vivo ectopic overexpression of SPRR1A protected cardiomyocytes against ischemic injury. Thus, this study identifies SPRR1A as a novel stress-inducible downstream mediator of gp130 cytokines in cardiomyocytes and documents its cardioprotective effect against ischemic stress.
Keywords: gp130, IL-6 cytokines, myocardium, stress, SPRR1A
Introduction
The interleukin (IL)-6 family of cytokines, including IL-6, IL-11, cardiotrophin-1 (CT-1), ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), and oncostatin M, play an important role in regulating survival of terminally differentiated cell types (Taga and Kishimoto, 1997). The receptors for IL-6 cytokines are composed of multi-subunit complexes that share a common signaling subunit, gp130 (Hibi et al, 1990). The activated receptor transduces the signal through janus kinases (JAKs). In turn, JAKs activate various signaling pathways including mitogen-activated protein kinases (MAPKs), signal transducers and activators of transcription 3 (STAT3), and phosphatidylinositol 3-kinase (PI-3K) pathways (Yamauchi-Takihara and Kishimoto, 2000).
In earlier studies, we identified CT-1 (Pennica et al, 1995) as a cardiac restrictive IL-6 family cytokine that promotes cardiomyocyte hypertrophy and protects myocardial cells from apoptosis (Wollert et al, 1996; Chien, 1999; Yasukawa et al, 2001, 2003). Recent clinical studies have documented that patients with congestive heart failure have elevated plasma levels of CT-1 that correspond with disease severity (Talwar et al, 2000; Ng et al, 2002). CT-1 also exhibits a significant protective effect on the isolated human myocardium against ischemic injury (Ghosh et al, 2000). In mice, ventricular-restricted ablation of the gp130 gene results in the rapid onset of dilated cardiomyopathy following acute pressure overload, with a massive induction of apoptotic death of cardiomyocytes (Hirota et al, 1999). Moreover, in experimental animal models, inhibition of gp130 signaling aggravates enterovirus-induced cardiac injury (Yasukawa et al, 2003). Taken together, a large body of evidence supports a critical role of gp130 cytokines in myocyte survival during biomechanical stress and injury, and suggests the existence of specific downstream stress-inducible effectors within cardiac muscle cells that directly mediate these effects. Although several candidate target molecules, such as manganese superoxide dismutase (Wong et al, 1988; Negoro et al, 2001) and inhibitors of apoptosis (iap, bcl-xL) (Fujio et al, 1997; Stehlik et al, 1998), have been proposed to have a cytoprotective function, induction of these molecules does not account for the entire effect of gp130 cytokines on cardio-protection. The identification of critical downstream stress-inducible factors is essential for designing new therapeutic strategies for cardiac injury in heart failure, ischemic heart disease, or myocarditis.
Accordingly, the present study identified the small proline-rich repeat protein (SPRR) gene family members SPRR1A and SPRR2A as stress-inducible proteins regulated by the gp130 signaling pathway in the heart. SPRR proteins were originally identified as markers for the terminal squamous cell differentiation where they are precursors of the cornified envelope (Baden et al, 1987). However, SPRR proteins are also expressed in various nonsquamous tissues, and their biological function is not restricted to structural proteins of the cornified envelope (Tesfaigzi and Carlson, 1999). In the current study, a cytoprotective role for SPRR1A was observed at both the level of isolated cardiomyocytes and in the intact heart exposed to ischemic injury, thereby identifying SPRR1A as a novel cytoprotective factor downstream of the gp130 pathway in the heart.
Results
Identification of SPRR1A and SPRR2A/B as gp130 signaling-inducible genes
In order to identify downstream target genes of the gp130 signaling pathway, which are not regulated by other known hypertrophic/survival signals, a serial analysis of gene expression (SAGE) was performed using RNA from primary culture of neonatal C57Bl/6 mouse cardiac myocytes stimulated for 4 h with LIF, neuregulin-1 (NRG-1), aldosterone (Aldo), angiotensin II (AngII), or control condition (untreated). These four agonists were chosen based on previous observations that each stimulus activates a different set of signaling pathways. This SAGE analysis identified 34 genes that were distinctively up- or downregulated by LIF stimulation, but not by AngII, NRG-1, and Aldo (Supplementary Table I). In order to explore the subset of cardiac genes that are selectively regulated by gp130 pathways in vivo in the stressed heart, a DNA chip analysis (Affymetrix MGU74Av2) was conducted using RNA isolated from left ventricles of cardiac-restricted gp130 knockout (gp130CKO) mice backcrossed in C57Bl/6 mouse strain and subjected to trans-aortic constriction (TAC) for 4 days. Biomechanical stress-regulated genes were first identified by comparing gene expression from TAC and sham-operated wild-type mice. Among these biomechanical stress-regulated genes, 38 genes were differentially regulated in TAC-treated gp130CKO (Supplementary Table II). SPRR1A was the only gene also specifically upregulated after LIF treatment in cardiomyocytes (SAGE analysis, see Supplementary Table I). Northern blot analysis confirmed that SPRR1A mRNA was induced by LIF stimulation (3.7±0.1-fold, n=4, P<0.01) and not by AngII, endothelin-1 (ET-1), or NRG-1 in cardiomyocytes (Figure 1A). Interestingly, three members of the SPRR gene family (SPRR1A, SPRR2A, and SPRR2B) showed markedly less induction in the gp130CKO hearts after banding, with a difference of 13.5-fold (SPRR2A), 5.3-fold (Sprr2B), and 3.9-fold (SPRR1A), respectively (Supplementary Table II). Subsequent Northern blot analysis confirmed these results (Figure 1B). SPRR2A and 2B are close homologs of similar size (88% identity at the nucleotide level) and cannot be differentiated by Northern blot or gene chip analysis. Other members of SPRR family (SPRR1B, SPRR2D, 2E, 2F, 2H, 2I, 2J, 2K, and 3) were not expressed in any of our data set. In primary culture of cardiomyocytes, SPRR1A induction was detectable as early as 1 h after stimulation and peaked 8 h after stimulation (Figure 1C). On the other hand, SPRR2A/B, whose expression level was weak, could only be detected 4 h after stimulation (Figure 1C). The weak expression level of SPRR2A/B could explain why our initial SAGE analysis did not detect this gene and prompted us to concentrate our analysis on SPRR1A. SPRR1A protein was slightly detectable before LIF stimulation, upregulated by LIF treatment in 2 h, and further stimulated for expression over 24 h (Figure 1D). Thus, we identified SPRR1A and 2A/B as early downstream targets of the gp130 pathway.
Figure 1.
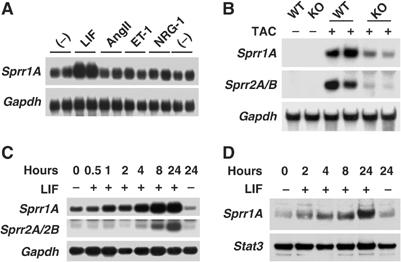
SPRR1A and 2A/2B are downstream targets of gp130 pathway. (A) Northern blot validation of SAGE analysis. Northern blots were prepared with mRNAs extracted from primary culture of mouse cardiomyocytes treated with LIF (1 nM), AngII (100 nM), ET-1 (10 nM), or NRG-1 (2 nM) for 4 h. (B) Northern blot validation of gene chip analysis in gp130CKO mice. Northern blots were prepared with mRNAs extracted from wild-type (WT) or gp130CKO (KO) mouse left ventricles subjected to TAC for 4 days or sham operated. (C, D) Time-course analysis of SPRR1A and 2A/2B mRNA (C) and protein (D) expression after LIF stimulation in neonatal mouse cardiomyocytes. Northern blots were hybridized with SPRR1A, SPRR2A/2B, and GAPDH probes. Western blots were hybridized with anti-mouse SPRR1A and anti-mouse STAT3 antibodies. STAT3 was used as a loading control. Representative blots from three independent experiments are shown.
Gp130 cytokines induce SPRR1A expression through a MAPK-dependent signaling pathway and trigger C/EBPβ and AP-1 binding to the SPRR1A promoter
The signaling pathway responsible for SPRR1A gene activation by gp130 cytokines in cardiomyocytes was investigated. Dominant-negative STAT3 completely abrogated STAT3 phosphorylation and wild-type STAT3 significantly increased STAT3 phosphorylation (Figure 2A). However, they did not affect SPRR1A upregulation by LIF (Figure 2B). Similarly, the PI-3K pathway inhibitor wortmannin and calcineurin inhibitor cyclosporin A (CsA) did not affect SPRR1A (Figure 2C and D). On the other hand, MEK1 inhibitor PD098059 and MEK1/2 inhibitor U0126 inhibited, respectively, partially and completely SPRR1A induction (Figure 2C and D). Western blot analysis confirmed the inhibitory action of U0126 and wortmannin, and showed only a partial inhibition of ERK1/2 by PD098059 (data not shown). The SPRR1A promoter was then scanned for binding sites for the transcription factor C/EBPβ (NF-IL6), the main nuclear target of gp130 signaling through the MAPK cascade (Akira, 1997). Using a phylogenetic footprinting strategy that searches for promoter sequences conserved between human and mouse, a C/EBPβ binding site was found in the proximal promoter of SPRR1A downstream of the transcription start site (Figure 2E). Chromatin immunoprecipitation (ChIP) assays confirmed a direct binding of C/EBPβ to the endogenous SPRR1A promoter after CT-1 stimulation (Figure 2F). Binding of c-Jun was also observed, according to previous reports (Figure 2F; Sark et al, 1998). Thus, the signaling pathway leading to SPRR1A activation by gp130 cytokines involved MEK1/2 and the activation of the transcription factors AP-1 and C/EBPβ. Other proinflammatory cytokines that share this pathway (interferon γ (IFNγ), and to a less extent interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα)) were also able to stimulate SPRR1A mRNA expression (Figure 2G).
Figure 2.
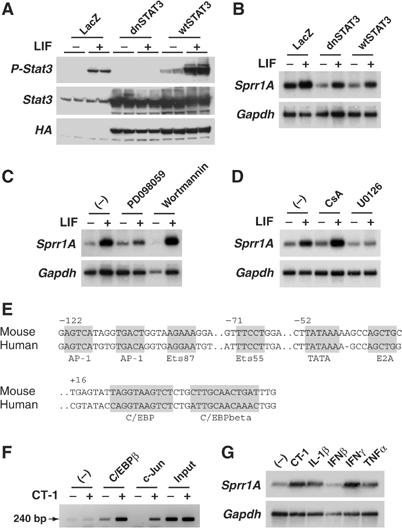
Characterization of signaling pathways responsible for LIF-induced Sprr1A gene expression in mouse neonatal cardiomyocytes. (A, B) Myocytes were infected with Ad-LacZ (LacZ), Ad-Stat3 wild type (wtSTAT3), or Ad-Stat3 dominant-negative mutant (dnSTAT3), serum-depleted for 24 h, and then stimulated with LIF (1 nM). (A) Total cell lysate was extracted 15 min after stimulation. Western blot was hybridized with anti-phospho-Stat3 (P-Stat3), anti-Stat3, and anti-HA antibodies (wtSTAT3 and dnSTAT3 contain an HA tag) to confirm STAT3 inhibition. (B) Northern blot was prepared using RNA extracted 6 h after stimulation. (C, D) Myocytes were serum-depleted for 24 h, and treated with the indicated inhibitor 1 h prior and during LIF stimulation (1 nM, 6 h). (C) Northern blot analysis from myocytes treated with PD098059 (50 μM, MEK1 inhibitor) or wortmannin (500 nM, PI-3K inhibitor). (D) Northern blot analysis from myocytes treated with CsA (1 μM, calcineurin inhibitor) or U0126 (10 μM, MEK1 and 2 inhibitor). (E) Conserved segments in SPRR1A human and mouse proximal promoter regions. Gray box indicates transcription factors binding sites and TATA box. Positions of the first nucleotide of each conserved segment are indicated. (F) ChIP assays were performed with crosslinked chromatin from myocytes stimulated or not with CT-1 (1 nM) for 4 h. Immunoprecipitations were performed with C/EBPβ antibody, c-Jun antibody, or with no antibody (−). Precipitation of Sprr1A promoter fragment was monitored by PCR (240 bp product). Crosslinked, sonicated extract prior immunoprecipitation (input) was used as positive control. (G) Northern blot analysis from myocytes untreated or treated with CT-1 (1 nM), IL-1β (5 ng/ml), IFNβ (500 U/ml), IFNγ (100 ng/ml), or TNFα (25 ng/ml) for 6 h. Representative blots from at least two independent experiments are shown. Northern blots were hybridized with SPRR1A and GAPDH probes.
SPRR1A is massively induced in myocytes adjacent to myocardial injuries
In vivo SPRR1A and 2A/2B regulation was investigated in different animal models of cardiac disease. In untreated C57Bl/6 mouse hearts, SPRR1A and 2A/2B mRNAs were not detectable (Table III). SPRR1A mRNA was markedly induced (35.4-fold) and only detectable 4 days after TAC surgery. SPRR2A/B showed a weaker and only moderate induction (2.3-fold) 4 days after TAC (Table III). Whereas SPRR1A mRNA was only present 4 days after TAC, SPRR1A protein was continuously observed from 4 days to 2 weeks after banding (Figure 3A), suggesting a tight regulation at the transcriptional level and a long half-life of SPRR1A protein in the myocardium. SPRR1A was also massively induced (16.5-fold) in the noninfarcted area of the heart 2 week after myocardial infarction and moderately induced in dilated cardiomyopathy caused by ablation of the splicing factor SC35 (Table III). On the other hand, SPRR1A and 2A/2B were not induced in physiological hypertrophy, cardiac hypertrophy related to the PI-3K pathway, and in a congenital cardiomyopathy associated with Nkx2.5 mutant mice (Table III). Using an anti-mouse SPRR1A antibody, we examined SPRR1A protein localization in the mouse myocardium 7 days after TAC. Interestingly, SPRR1A immunostaining revealed that expression was restricted to disseminated foci throughout the ventricles (Figure 3B). The specificity of this staining was confirmed by negative staining after coincubation of antibody and absorbing antigen SPRR1A protein (data not shown). Histological analysis with Masson's Trichrome staining and in situ hybridization on contiguous sections using the antisense SPRR1A mRNA revealed that SPRR1A expression was restricted to cardiomyocytes surrounding piecemeal necrotic lesions identified by mononuclear cell infiltration and fibrosis (Figure 3C). In the infarcted rat myocardium, SPRR1A protein was identified in the border zone myocardium adjacent to the infarcted area 1 week after infarction (Figure 3D). This localization was consistent with that of a gp130 activation pattern labeled by immunostaining of phosphorylated STAT3 in the rat myocardium after acute myocardial infarction (Negoro et al, 2000). Thus, these data suggest that SPRR1A is not expressed in the intact normal heart, but is massively induced in cardiomyocytes responding to biomechanical/ischemic stress.
Table 1.
Sprr1A and 2A gene expression in mouse models of cardiomyopathies
| Mouse models | Sprr1A (fold change) | Sprr2A (fold change) | Gene chip | Reference |
|---|---|---|---|---|
| Pressure overload (1 h) | NC | NC | MgU74Av2 | This paper |
| Pressure overload (5 h) | NC | NC | MgU74Av2 | This paper |
| Pressure overload (20 h) | NC | NC | MgU74Av2 | This paper |
| Pressure overload (4 days) | 35.4 | 2.3 | MgU74Av2 | This paper |
| Pressure overload (14 days) | NC | NC | MgU74Av2 | This paper |
| gp130 heart-restricted knockout versus wild type 4 days after TAC | −3.5 | −7.1 | MgU74Av2 | This paper |
| Dilated cardiomyopathy in heart-restricted knockout of SC35 | 2.2 | NC | MgU74Av2 | Ding et al (2004) |
| Myocardial infarction (1 week) | 16.5 | NC | MgU74Av2 | CardioGenomics |
| Exercised induced hypertrophy | ND | NC | MgU74Av2 | CardioGenomics |
| Overexpression of dominant-negative PI-3K | ND | NC | MgU74Av2 | CardioGenomics |
| Overexpression of constitutively active PI-3K | ND | NC | MgU74Av2 | CardioGenomics |
| Deletion of the Nk2 specific domain of the Nkx2.5 | ND | NC | Mu11k | CardioGenomics |
| Congenital heart disease in Csx/Nkx2.5 mutant embryos | ND | NC | Mu11k | CardioGenomics |
| Significant fold changes are listed (P<0.05). NC: fold change not statistically significant; ND: expression not detected. CardioGenomics: NHLBI Program for Genomic Applications, HMS Genomics of Cardiovascular Development, Adaptation, and Remodeling, Harvard Medical School. URL: http://www.cardiogenomics.org. |
Figure 3.
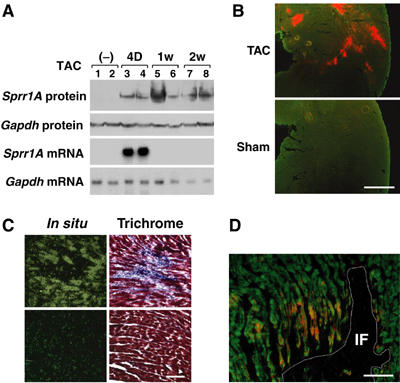
SPRR1A induction in heart after biomechanical stress. (A) Time course of expression of SPRR1A mRNA and protein in mouse ventricle after banding. Northern blot and Western blots were prepared from the left ventricle of wild-type mice subjected to TAC for the indicated time. Same animals were used for Northern and Western blots. Northern blots were hybridized with SPRR1A and GAPDH probes. Western blots were hybridized with anti-mouse SPRR1A and anti-mouse GAPDH antibodies. Representative blots from two independent experiments are shown. (B) SPRR1A expression in adult mouse left ventricle 7 days after TAC. Frozen sections of TAC and sham mouse hearts were costained with anti-SPRR1A antibody (red) and phalloidin (green). Scale bar: 500 μm. (C) Histology of ventricular area with (upper panels) and without (lower panels) SPRR1A expression. Left panels: Sections of mouse hearts 7 days after TAC were hybridized with SPRR1A antisense RNA probe. Consecutive sections were stained with Trichrome (Masson) to reveal the fibrosis and the infiltrated cells (right panels). Scale bar: 100 μm. (D) Induction of SPRR1A expression in the viable zone adjacent to the myocardial damage area. Frozen sections of rat ventricle 1 week after myocardial injury were costained with anti-SPRR1A antibody (red) and phalloidin (green). The white line delineates the infarct area (IF), which was identified by hematoxylin and eosin staining in contiguous sections. Scale bar: 100 μm.
SPRR1A protects cardiomyocytes against ischemic stress
The observation that SPRR1A was expressed in the border zone of myocardial injury prompted an investigation as to whether SPRR1A was able to protect directly cardiomyocytes against ischemic stress. Accordingly, we forced the expression of SPRR1A in cultured rat neonatal cardiomyocytes using recombinant adenovirus vectors (Adv-SPRR1A) (Figure 4A). Subsequently, we tested the effects of SPRR1A overexpression in 2-deoxyglucose (2-DG)-treated cardiomyocytes, an in vitro model of ischemia-induced cardiomyocyte cell death (Bialik et al, 1999). After 24 h of continuous 2-DG treatment, nontreated control cardiomyocytes rounded up and detached from the culture dish (Figure 4B and C). On the other hand, myocytes expressing SPRR1A maintained a polymorphic appearance, similar to cardiac myocytes treated with CT-1 (Figure 4B and C). In contrast to GFP-expressing cells, SPRR1A-expressing cells did not present any accumulation of nuclear actin as observed normally in ATP-depleted cells (Pendleton et al, 2003; Figure 4D). Moreover, in SPRR1A-expressing myocytes, myofibrils were not disrupted although displaying some signs of disorganization (Figure 4D). Cytoplasmic staining showed an accumulation of SPRR1A protein along the myofibrils (Figure 4E). The protective effect of SPRR1A against 2-DG-induced cell death was confirmed by staining simultaneously living cells with calcein AM and the nuclei of dead cells with ethidium homodimer (Figure 4F). A reduced number of apoptotic cells, measured by TUNEL staining, were also observed in Adv-SPRR1A myocytes (Figure 4G). On the other hand, SPRR1A overexpression did not significantly improve myocyte survival after reactive oxygen species treatment or serum deprivation (data not shown). Thus, in vitro, overexpression of SPRR1A protected cardiomyocytes against ischemic stress.
Figure 4.
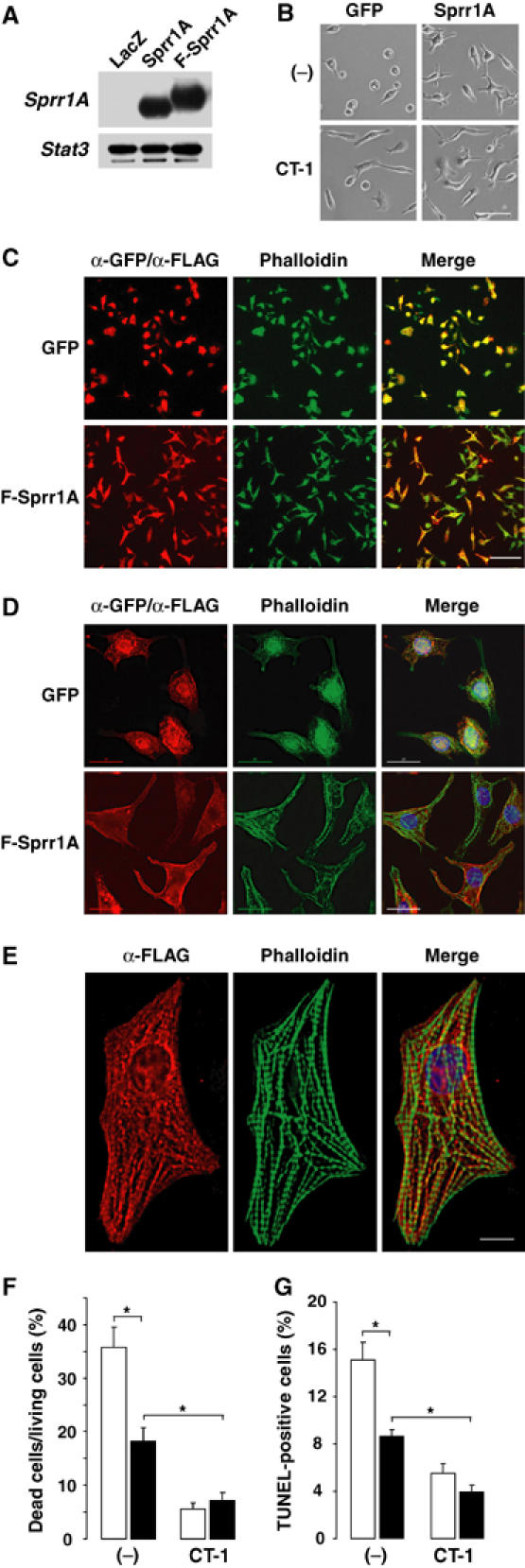
Overexpression of SPRR1A in 2-DG-treated neonatal rat cardiomyocytes. (A) Western blot with an anti-SPRR1A antibody confirmed adenovirus-mediated SPRR1A expression in neonatal myocytes infected with Ad-LacZ (LacZ), Ad-SPRR1A (Sprr1A), or Ad-FLAG-SPRR1A (F-Sprr1A). (B–F) Myocytes were transfected with Ad-SPRR1A (Sprr1A), Ad-SPRR1A (Sprr1A), or Ad-FLAG-SPRR1A (F-Sprr1A) and kept 24 h in the presence of 2-DG with or without CT-1 (1 nM). (B) Myocyte morphology under light microscopy. Scale bar: 50 μm. (C–E) Immunofluorescence costaining for ectopic expression of F-Sprr1A (α-FLAG antibody) or GFP (α-GFP antibody) and endogenous F-actin (phalloidin). (C) Low magnification (scale bar: 100 μm) under regular fluorescence after 2-DG treatment. (D) High magnification (deconvolution; scale bar: 20 μm) after 2-DG treatment. (E) Evidence of accumulation of SPRR1A protein along myofibrils (normal culture condition). SPRR1A (red) stained with anti-FLAG antibody and F-actin (green) stained with phalloidin (deconvolution; scale bar: 8 μm). (F) Number of dead cells/living cells ratio. (G) TUNEL staining. TUNEL and DAPI stainings are available as Supplementary Figure 4H. (F, G) Ad-GFP=open bar; Ad-SPRR1A=closed bar. The results are shown as mean±s.e., *P<0.01 (n=15 frames with ∼100 cells each). Experiments were repeated three times.
In vivo SPRR1A gene transfer suppresses ischemia-induced global cardiac injury
We further investigated the cell protective effects of ectopic SPRR1A expression on ischemia-induced cardiac injury using a novel strategy of adenovirus vector-mediated high-efficiency in vivo transcoronary gene transfer in the rat myocardium developed by our group (Iwanaga et al, 2004). An adenovirus vector carrying a LacZ marker gene (Adv-LacZ) was used in control animals. β-gal activity staining and immunofluorescence staining with anti-SPRR1A or anti-β-galactosidase antibodies confirmed that we could achieve approximately 50% in vivo gene transfer efficiency in the myocardium using our protocol (Figure 5A and B), consistent with our previous results (Iwanaga et al, 2004). At 4–5 days after gene transfer, the heart was excised and exposed to an ex vivo 30 min ischemia/3 h reperfusion (I/R) procedure using Langendorff perfusion (Trost et al, 1998) to assess the cell protective effect of SPRR1A against global cardiac ischemic stress. Triphenyl tetrazolium (TTC) staining revealed that SPRR1A was able to protect the isolated heart from irreversible ischemic injury after I/R (Figure 5C and D) and creatine kinase (CK) release from damaged cardiomyocytes was reduced in the SPRR1A-transfected group (Figure 5E). No significant change in heart weight/body weight and left ventricle weight/body weight ratios was observed in SPRR1A-infected hearts (data not shown). Compared to the Adv-LacZ-treated hearts, the hearts treated with Adv-SPRR1A showed a remarkable reduction in the degree of cell myofibril degradation after I/R (Figure 5F), further suggesting a protective effect of SPRR1A against cardiac cytopathic damages. Taken together, these in vitro and ex vivo data indicate that SPRR1A is capable of protecting cardiomyocytes against ischemia or I/R injuries.
Figure 5.
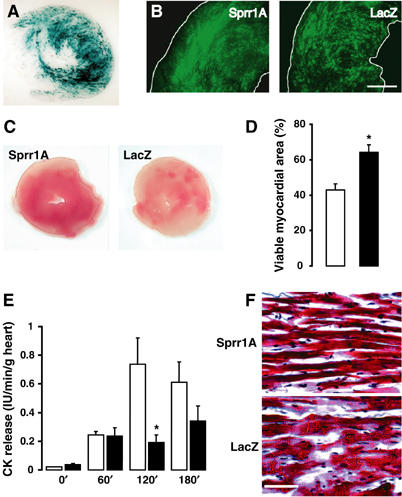
Overexpression of SPRR1A in rat hearts subjected to ex vivo ischemia–reperfusion. Rat hearts were infected with Ad-SPRR1A or Ad-LacZ using the trans-coronary procedure. At 1 week after surgery, hearts were subjected to 30 min of ischemia, followed by 180 min of reperfusion. (A) Representative image of β-gal activity in Ad-LacZ-infected rat left ventricle after ischemia–reperfusion. (B) Representative immunostaining with anti-SPRR1A (left) or anti-LacZ (right) antibody after ischemia–reperfusion. Scale bar: 1 mm. (C) Representative sections of Ad-Sprr1A and Ad-LacZ hearts stained with TTC after I/R, revealing various degrees of myocardial ischemia. Tissue that stained positive appears red and represents viable myocardium. (D) Quantification represents the percentage of viable myocardium from the total area of TTC-stained section (open bar=Ad-LacZ; closed bar=Ad-SPRR1A; n=7 in each group). (E) CK release (IU/min/g heart) at 0, 60, 120, and 180 min of reperfusion (open bar=Ad-LacZ, n=4; closed bar=Ad-SPRR1A, n=5). (F) Double-blinded histological analysis revealed a decrease of cell damage (myolysis) in Ad-SPRR1A-infected hearts after I/R. Arrows indicate myolysis in Ad-LacZ-infected hearts. Sections were stained with Trichrome (Masson). Rats where infected with Ad-LacZ (open bar) or Ad-SPRR1A (closed bar). Scale bar: 100 μm. The results are shown as means±s.e., *P<0.05.
Discussion
A growing body of evidence suggests that cytokines play a critical role in the initiation, integration, and maintenance of the myocardial response to genetic and environmental stress that leads to the loss of viable heart muscle cells and the associated onset of heart failure (Mann, 2003). In this regard, gp130 receptor signaling, which promotes hypertrophy and cell survival and ultimately prevents the onset of dilated cardiomyopathy (Wollert et al, 1996; Hirota et al, 1999), is a major pathway involved in cardiomyocyte protection. A recent study in an animal model of acute myocardial infarction revealed that gp130 signaling is activated in the surviving border area of the myocardium adjacent to the infarcted area (Negoro et al, 2000). Cardiomyocytes in this area are exposed to a high level of stress that includes hypoxia and mechanical stretch, but ultimately maintain viability (Cheng et al, 1996; Olivetti et al, 1996; Bialik et al, 1997). The present study now identifies SPRR1A as a downstream mediator of the gp130 signaling that becomes strongly induced in the surrounding border zone of myocardial injury during the in vivo biomechanical stress of pressure overload and after myocardial infarction (Figure 6). We further show that overexpression of SPRR1A in cardiomyocytes prevents ischemia-induced cardiac injury in vitro and in vivo.
Figure 6.
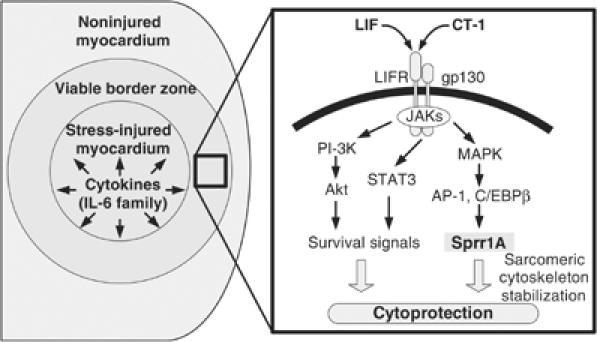
Working model for SPRR1A activation in the myocardium. In response to myocardial injuries, IL-6 cytokines are released and act on the surrounding cells (left panel). The binding of IL-6 cytokines (CT-1, LIF) to gp130/LIFR receptor complex activates STAT3, PI-3K, and MAPK signaling pathways (right panel). A MAPK signaling pathway activates the transcription factors C/EBPβ and AP-1. In turn, they bind to SPRR1A promoter and induce SPRR1A gene transcription. SPRR1A confers a protective effect to the cardiomyocytes, possibly by stabilizing the actin cytoskeleton.
The SPRR gene family members, including SPRR1A and SPRR2A, are localized in close proximity on human chromosome 1q22–q23 and mouse chromosome 3 (93.1–93.3 Mb), a chromosomal region referred to as the epidermal differentiation complex (Mischke et al, 1996). SPRR proteins have originally been identified as marker for terminal squamous cell differentiation, where they are precursors of the cornified envelope (Kartasova and van de Putte, 1988). However, later analysis in nonsquamous tissues indicated that the biological function of SPRR proteins is not restricted to squamous cell differentiation (Tesfaigzi and Carlson, 1999). For instance, Sprr1A is strongly expressed at the growth cones of axotomized neurons and is involved in axonal outgrowth (Bonilla et al, 2002). Moreover, recent microarray analysis in models of gastric injury showed a significant induction of SPRR2A in mucosal tissue infected by Helicobacter pylori (Mueller et al, 2003). Therefore, it is likely that SPRR genes are induced in response to stress injury and contribute to cell protection, tissue remodeling, or tissue regeneration.
In keratinocytes, SPRR genes are upregulated by inducers of epithelial cell differentiation (Tesfaigzi and Carlson, 1999). Induction of SPRR genes by the protein kinase C (PKC) activator phorbol-13-myristate-12-acetate (PMA) has been extensively studied (An et al, 1993; Deng et al, 2000). Using RT–PCR, a recent analysis showed that the SPRR1B gene is dominantly induced by PMA in this cell type (Vuong et al, 2002). In contrast, the responses of SPRR1A and 2A to PMA are much weaker. We did not observe an induction of the strong PMA-responsive form SPRR1B in the stressed heart, and in cultured cardiomyocytes. PKC-activating natural ligands, like AngII and ET-1, also fail to regulate SPRR1A gene expression in cardiomyocytes. Thus, the PKC pathway may not be a major pathway to regulate SPRR1A expression in the heart. On the other hand, we have shown that MEK1 inhibitors PD098059 and U0126 blunt significantly the SPRR1A response after LIF stimulation. These findings are consistent with previous studies in human conducting airway epithelial cells where PD098059 blocked Sprr1 gene induction (Deng et al, 2000). AP-1 and Ets transcription factors have been extensively studied as regulators of SPRR1A expression (Sark et al, 1998). In cardiomyocytes after CT-1 stimulation, we observed binding of the AP-1 component c-Jun as well as C/EBPβ, a downstream target of gp130 signaling, to the promoter region of SPRR1A gene.
The functional roles of SPRR proteins are known in squamous tissues, where SPRR proteins are substrates of transglutaminase (TGase) I-catalyzed crosslinking reactions in the formation of the crosslinked envelope (Tesfaigzi and Carlson, 1999). We did not detect TGase I expression either in the myocardium (with or without aortic banding) or in primary cultures of cardiac myocytes (data not shown). However, several studies have linked the ubiquitously expressed form of TGase, TGase II, to heart failure and cardiomyopathies (Iwai et al, 1995; Small et al, 1999). Assessment of dexamethasone-induced apoptosis in thymocytes from TGase II knockout mice and overexpression of TGase II in L929 cells suggests that TGase II may be involved in stabilizing apoptotic cells (Piredda et al, 1997; Nanda et al, 2001). Therefore, it is tempting to speculate that SPRR proteins may serve as a crosslinking substrate for TGase II in its role of stabilizing myocytes entering apoptotic pathways. Contrary to SPRR1A and 2A/B, TGase II expression was not changed in the TAC-treated myocardium (data not shown). Further studies are needed to clarify a potential relationship of SPRRs and the TGase gene family in cardiomyocytes.
Another potential role for SPRR1A lies in its association with the actin cytoskeleton. Recently, SPRR1A was suggested to play a critical role in the dynamic regulation of actin cytoskeleton assembly (Bonilla et al, 2002). Our data have shown that ectopic expression of SPRR1A prevents the disruption of myofibrils and the accumulation of nuclear actin following ischemic stress. Moreover, we observed a localization of SPRR1A protein along myofilaments. It is becoming increasingly clear that cytoskeletal destabilization plays a significant role in the development of cardiomyocyte dysfunction (Chien, 2000; Towbin and Bowles, 2002). In Duchenne and Becker myopathies and other forms of muscular dystrophies with genetic defects in muscle cytoskeletal genes, mechanical stress is required to disrupt the plasma membrane and destabilize the myofilament structure (Durbeej and Campbell, 2002). We recently demonstrated that defects in the cardiac cytoskeleton, including the Z disc and titin/titin-associated protein structure, result in stress sensor malfunction in cardiomyocytes, which may result in a failure in adaptation to environmental stress and the onset of heart failure (Knoll et al, 2002). Thus, the cytoprotective action of SPRR1A found in this study might relate to the mechanical stabilization of myofilament/Z disc structure in cardiomyocytes, thereby preventing cells from irreversible damage.
IL-6 family cytokines are pleiotropic cytokines involved in many pathophysiological events. In the heart, IL-6 family cytokines are consistently and rapidly expressed by cardiomyocytes and other nonmyocyte cell types in response to many forms of cardiac diseases, suggesting that these molecules constitute part of an intrinsic cardiac stress response system (Mann, 2003). The current study provides the first evidence that the SPRR1A and 2A/B proteins are downstream target of gp130 cytokine receptor signaling, and that SPRR1A confers cardiomyocyte protection in response to environmental stress (Figure 6). The concept of SPRR1A and 2A as downstream effectors of the stress response mediated by IL-6 family cytokines may likely be extended to other organs or other cell types. Indeed, in the nervous system, CNTF, another IL-6 cytokine, prevents the degeneration of motor neurons after axotomy (Sendtner et al, 1990) and peripheral axonal damage is known to activate massive transcription of SPRR1A (Bonilla et al, 2002). Therefore, the induction of SPRR1A expression by IL-6 cytokines could be a central mechanism of an ‘innate' defense system in response to injury. Thus, induction of SPPR genes may serve a novel cell protective strategy against various pathological injuries in many cell types and in many organs.
Materials and methods
Serial analysis of gene expression
Primary culture of cardiac myocytes from C57Bl/6 mice (The Jackson Laboratory, Bar Harbor, ME) were prepared as described (Wollert et al, 1996) and serum-starved for 24 h and then treated with either LIF (1 nM), NRG-1 (2 nM), Aldo (1 μM), or AngII (100 nM) or were untreated (control condition) for 4 h. MAPK (ERK1/2, p38, and JNK) phosphorylation was checked on Western blot in order to confirm that each agonist was activating selective signaling pathways. SAGE libraries were prepared with 20 μg of poly (A) mRNA as described (Robert-Nicoud et al, 2001). A total of 250 350 SAGE tags were sequenced (63 445 tags for the control library, 62 676 for the LIF library, 61 611 for the NRG-1 library, 60 466 for the Aldo library, and 62 618 for the AngII library). A total of 2927 different tags, representing 2506 different genes, were found five or more times among the four libraries. This number represents the actual number of genes assessed for a significant up- or downregulation. The tags, the numbers of which were significantly different with a two-fold change in all four comparisons (i.e., LIF versus control, LIF versus NRG-1, LIF versus AngII, and LIF versus Aldo) by Monte Carlo simulation analysis, were considered as tags corresponding to the transcripts specifically regulated by LIF. NCBI mapping tables were used for SAGE tag identification. SAGE data are available at http://www-imm.ucsd.edu/research/sage/.
Animal models and in vivo surgical studies
All of the animal experimental protocols received formal Institutional Review Board approval. Transverse thoracic aortic constriction (TAC) was performed as described previously (Yasukawa et al, 2001) on 8-week-old adult mice (C57Bl/6xSJL, The Jackson Laboratory, Bar Harbor, ME) or gp130 cardiomyocyte-restricted conditional knockout (gp130CKO) mice (Hirota et al, 1999). Gp130CKO mice were generated by crossbreeding floxed gp130 mice with mice that harbor a knock-in of the Cre recombinase at MLC2v genomic locus (Hirota et al, 1999). Then, gp130CKO mice were backcrossed in C57Bl/6 mouse line for five times. gp130CKO mice were selected with the genetic identification of Gp130lox/loxMLC2vCre/+. Gp130lox/loxMLC2v+/+ mice were used for the control group. Myocardial infarction was introduced in male Sprague–Dawley rats (250–300 g) by ligating the left coronary artery as described elsewhere (Oh et al, 1993). At 7 days after coronary ligation, the animals were killed. Left ventricles from surgically treated mice and rats were weighed and either quickly frozen in liquid nitrogen for total RNA and protein extraction or fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) overnight. Fixed hearts were then washed three times in PBS, placed in 30% sucrose/PBS for 1 h, and frozen in OCT.
Microarray gene expression experiments
The experiments are described in accordance with the minimal information about a microarray experiment (MIAME) in Supplementary data. For microarray analysis, we chose mice presenting equivalent level of pressure overload (surgically created trans-aortic gradient, 45–60 mmHg), similar hypertrophic responses (left ventricle weight/body weight ratio, 0.44–0.50%), and no early systemic signs of heart failure and fatality (Supplementary Tables IV and V). Gene chip analysis was performed in duplicate on TAC and sham-operated mice using Affymetrix MgU74Av2 chip. Experimental data are available at http:www.ebi.ac.uk/arrayexpress (ArrayExpress). The accession numbers are E-MEXP-104 (pressure overload-induced cardiomyopathy in G130 knockout mice) and E-MEXP-105 (pressure overload-induced cardiomyopathy).
Immunoblot and immunofluorescence staining analyses
Western blots using total cell extracts were prepared as described previously (Yasukawa et al, 2001). Anti-STAT3 and anti-phospho-STAT3 antibodies were purchased from New England Biolabs Inc. Rabbit polyclonal anti-mouse SPRR1A antibody, corresponding preimmune serum, and purified SPRR1A protein were kindly provided by SM Strittmatter (Bonilla et al, 2002). Anti-α-actinin sarcomeric, anti-FLAG (Sigma-Aldrich), Alexa Fluor 488 phalloidin, Alexa Fluor 488 goat anti-mouse IgG, and Alexa Fluor 594 donkey anti-rabbit IgG (Molecular Probes) were used for immunofluorescence staining analysis.
Promoter analysis and ChIP assay
Human and mouse promoter sequences spanning from nucleotide −500 to +300 were aligned in Consite (Sandelin et al, 2004) and four conserved fragments were extracted with a cutoff of 68% identity. In order to identify the putative transcription factors binding sites, the conserved promoter regions were run in MatInspector (Quandt et al, 1995). ChIP was performed according to the protocol of the ChIP assay kit (Upstate #17-295) on 4 h CT-1 (1 nM) stimulated and unstimulated mouse myocytes. Rabbit polyclonal antibodies against C/EBPβ sc-150x and c-Jun (sc-1694) (Santa Cruz Biotechnology) were used for immunoprecipitation. The PCR primers used were 5′-AGTCATAGGTGACTGGTAAG-3′ (up) and 5′-CCACATCAACTCTGTAATAG-3′ (down). The PCR conditions were as follows: 94°C for 180 s; 35 cycles at 94°C for 30 s, 56°C for 60 s, and 72°C for 60 s; final elongation at 72°C for 5 min.
In situ RNA hybridization
Mouse SPRR1A full-length cDNA was cloned into pCR-4-TOPO using RACE-PCR (GeneRacer kit, Invitrogen, Carlsbad, CA). 35S-UTP-labeled antisense RNA probes were generated using (T7) RNA polymerase. In situ hybridization was performed on mouse heart sections 7 days after TAC and on isolated mouse neonate heart cells. These specimens were incubated with 10 000 cpm/μl probes at 60°C overnight followed by washing. Development was performed after 10-day incubation at 4°C with LM-1 emulsion (Amersham Biosciences).
Recombinant adenovirus vectors and in vitro survival assay
Adenovirus vectors containing the cDNA coding for LacZ, GFP, or mouse SPRR1A were prepared as described previously (Yasukawa et al, 2001). Recombinant adenoviruses were used to infect primary culture of neonatal rat cardiomyocytes at a multiplicity of infection of 25–50 plaque-forming units per cell for 8 h as described (Yasukawa et al, 2001). For 2-DG experiments, cultured neonatal cardiac myocytes were infected with Adv-SPRR1A or Adv-GFP and placed in glucose-free and serum-free media in the presence of 2-DG (3 mM) for an additional 24 h. TUNEL assay (Promega, #G3250) and LIVE/DEAD Viability/Cytotoxicity kit (Molecular Probes) was used to check cell survival. Experiments were performed in triplicate and repeated three times.
In vivo adenovirus vector-mediated gene transfer followed by ex vivo ischemia/reperfusion
In vivo gene transfer was performed in male Sprague–Dawley rat (250–300 g) hearts using Adv-SPRR1A or Adv-LacZ. Methods were developed through modifications of our hamster gene transfer protocol (Hoshijima et al, 2002; Ikeda et al, 2002) and details are described elsewhere (Iwanaga et al, 2004). At 1 week after the gene transfer procedure, hearts were isolated and subjected to 30 min of ischemia followed by 3 h of reperfusion using a Langendorff perfusion system as described previously (Trost et al, 1998). Finally, the ventricles were isolated and subjected to immunohistological analysis as described above. Fresh heart sections were stained in 1% TTC solution to measure the infracted area. The analyst was blind to the experimental protocol.
Supplementary Material
Supplementary Table I
Supplementary Table II
Supplementary Table IV
Supplementary Table V
Supplementary Figure 4H
Supplementary material
Acknowledgments
We acknowledge Y Gu for her excellence in animal surgery and J Chrast for her outstanding technical assistance in virus vector production. We thank Dr Bonilla and Professor Strittmatter for providing us with anti-SPRR1A antibody. S Pradervand was supported by the Swiss Foundation for Grants in Biology and Medicine, the Novartis Foundation, and the Roche Research Foundation.
References
- Akira S (1997) IL-6-regulated transcription factors. Int J Biochem Cell Biol 29: 1401–1418 [DOI] [PubMed] [Google Scholar]
- An G, Tesfaigzi J, Chuu YJ, Wu R (1993) Isolation and characterization of the human spr1 gene and its regulation of expression by phorbol ester and cyclic AMP. J Biol Chem 268: 10977–10982 [PubMed] [Google Scholar]
- Baden HP, Kubilus J, Phillips SB, Kvedar JC, Tahan SR (1987) A new class of soluble basic protein precursors of the cornified envelope of mammalian epidermis. Biochim Biophys Acta 925: 63–73 [DOI] [PubMed] [Google Scholar]
- Bialik S, Cryns VL, Drincic A, Miyata S, Wollowick AL, Srinivasan A, Kitsis RN (1999) The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes. Circ Res 85: 403–414 [DOI] [PubMed] [Google Scholar]
- Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN (1997) Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest 100: 1363–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, Strittmatter SM (2002) Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci 22: 1303–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Kajstura J, Nitahara JA, Li B, Reiss K, Liu Y, Clark WA, Krajewski S, Reed JC, Olivetti G, Anversa P (1996) Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res 226: 316–327 [DOI] [PubMed] [Google Scholar]
- Chien KR (1999) Stress pathways and heart failure. Cell 98: 555–558 [DOI] [PubMed] [Google Scholar]
- Chien KR (2000) Genomic circuits and the integrative biology of cardiac diseases. Nature 407: 227–232 [DOI] [PubMed] [Google Scholar]
- Deng J, Chen Y, Wu R (2000) Induction of cell cornification and enhanced squamous-cell marker SPRR1 gene expression by phorbol ester are regulated by different signaling pathways in human conducting airway epithelial cells. Am J Respir Cell Mol Biol 22: 597–603 [DOI] [PubMed] [Google Scholar]
- Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, Yeakley JM, Cheng H, Xiao RP, Ross J, Chen J, Fu XD (2004) Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J 23: 885–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Campbell KP (2002) Muscular dystrophies involving the dystrophin–glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev 12: 349–361 [DOI] [PubMed] [Google Scholar]
- Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T (1997) Signals through gp130 upregulate bcl-x gene expression via STAT1-binding cis-element in cardiac myocytes. J Clin Invest 99: 2898–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Ng LL, Talwar S, Squire IB, Galinanes M (2000) Cardiotrophin-1 protects the human myocardium from ischemic injury. Comparison with the first and second window of protection by ischemic preconditioning. Cardiovasc Res 48: 440–447 [DOI] [PubMed] [Google Scholar]
- Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T (1990) Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 63: 1149–1157 [DOI] [PubMed] [Google Scholar]
- Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J Jr, Muller W, Chien KR (1999) Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97: 189–198 [DOI] [PubMed] [Google Scholar]
- Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y, Iwatate M, Li M, Wang L, Wilson JM, Wang Y, Ross J Jr, Chien KR (2002) Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med 8: 864–871 [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Gu Y, Iwanaga Y, Hoshijima M, Oh SS, Giordano FJ, Chen J, Nigro V, Peterson KL, Chien KR, Ross J Jr (2002) Restoration of deficient membrane proteins in the cardiomyopathic hamster by in vivo cardiac gene transfer. Circulation 105: 502–508 [DOI] [PubMed] [Google Scholar]
- Iwai N, Shimoike H, Kinoshita M (1995) Genes up-regulated in hypertrophied ventricle. Biochem Biophys Res Commun 209: 527–534 [DOI] [PubMed] [Google Scholar]
- Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, Date MO, Chrast J, Matsuzaki M, Peterson KL, Chien KR, Ross J Jr (2004) Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest 113: 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartasova T, van de Putte P (1988) Isolation, characterization, and UV-stimulated expression of two families of genes encoding polypeptides of related structure in human epidermal keratinocytes. Mol Cell Biol 8: 2195–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111: 943–955 [DOI] [PubMed] [Google Scholar]
- Mann DL (2003) Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol 65: 81–101 [DOI] [PubMed] [Google Scholar]
- Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A (1996) Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (‘epidermal differentiation complex') on human chromosome 1q21. J Invest Dermatol 106: 989–992 [DOI] [PubMed] [Google Scholar]
- Mueller A, O'Rourke J, Grimm J, Guillemin K, Dixon MF, Lee A, Falkow S (2003) Distinct gene expression profiles characterize the histopathological stages of disease in Helicobacter-induced mucosa-associated lymphoid tissue lymphoma. Proc Natl Acad Sci USA 100: 1292–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM (2001) Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276: 20673–20678 [DOI] [PubMed] [Google Scholar]
- Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y, Hirota H, Kishimoto T, Yamauchi-Takihara K (2001) Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 104: 979–981 [DOI] [PubMed] [Google Scholar]
- Negoro S, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T, Yamauchi-Takihara K (2000) Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res 47: 797–805 [DOI] [PubMed] [Google Scholar]
- Ng LL, O'Brien RJ, Demme B, Jennings S (2002) Non-competitive immunochemiluminometric assay for cardiotrophin-1 detects elevated plasma levels in human heart failure. Clin Sci (London) 102: 411–416 [PubMed] [Google Scholar]
- Oh BH, Ono S, Rockman HA, Ross J Jr (1993) Myocardial hypertrophy in the ischemic zone induced by exercise in rats after coronary reperfusion. Circulation 87: 598–607 [DOI] [PubMed] [Google Scholar]
- Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P (1996) Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 28: 2005–2016 [DOI] [PubMed] [Google Scholar]
- Pendleton A, Pope B, Weeds A, Koffer A (2003) Latrunculin B or ATP depletion induces cofilin-dependent translocation of actin into nuclei of mast cells. J Biol Chem 278: 14394–14400 [DOI] [PubMed] [Google Scholar]
- Pennica D, King KL, Shaw KJ, Luis E, Rullamas J, Luoh SM, Darbonne WC, Knutzon DS, Yen R, Chien KR (1995) Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci USA 92: 1142–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piredda L, Amendola A, Colizzi V, Davies PJ, Farrace MG, Fraziano M, Gentile V, Uray I, Piacentini M, Fesus L (1997) Lack of ‘tissue' transglutaminase protein cross-linking leads to leakage of macromolecules from dying cells: relationship to development of autoimmunity in MRLlpr/lpr mice. Cell Death Differ 4: 463–476 [DOI] [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T (1995) MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res 23: 4878–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Nicoud M, Flahaut M, Elalouf JM, Nicod M, Salinas M, Bens M, Doucet A, Wincker P, Artiguenave F, Horisberger JD, Vandewalle A, Rossier BC, Firsov D (2001) Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proc Natl Acad Sci USA 98: 2712–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Wasserman WW, Lenhard B (2004) ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res 32: W249–W252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sark MW, Fischer DF, de Meijer E, van de Putte P, Backendorf C (1998) AP-1 and ets transcription factors regulate the expression of the human SPRR1A keratinocyte terminal differentiation marker. J Biol Chem 273: 24683–24692 [DOI] [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H (1990) Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345: 440–441 [DOI] [PubMed] [Google Scholar]
- Small K, Feng JF, Lorenz J, Donnelly ET, Yu A, Im MJ, Dorn GW II, Liggett SB (1999) Cardiac specific overexpression of transglutaminase II (G(h)) results in a unique hypertrophy phenotype independent of phospholipase C activation. J Biol Chem 274: 21291–21296 [DOI] [PubMed] [Google Scholar]
- Stehlik C, de Martin R, Binder BR, Lipp J, Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T (1998) Cytokine induced expression of porcine inhibitor of apoptosis protein (iap) family member is regulated by NF-kappa B. Biochem Biophys Res Commun 243: 827–832 [DOI] [PubMed] [Google Scholar]
- Taga T, Kishimoto T (1997) Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 15: 797–819 [DOI] [PubMed] [Google Scholar]
- Talwar S, Squire IB, Downie PF, O'Brien RJ, Davies JE, Ng LL (2000) Elevated circulating cardiotrophin-1 in heart failure: relationship with parameters of left ventricular systolic dysfunction. Clin Sci (Lond) 99: 83–88 [PubMed] [Google Scholar]
- Tesfaigzi J, Carlson DM (1999) Expression, regulation, and function of the SPR family of proteins. A review. Cell Biochem Biophys 30: 243–265 [DOI] [PubMed] [Google Scholar]
- Towbin JA, Bowles NE (2002) The failing heart. Nature 415: 227–233 [DOI] [PubMed] [Google Scholar]
- Trost SU, Omens JH, Karlon WJ, Meyer M, Mestril R, Covell JW, Dillmann WH (1998) Protection against myocardial dysfunction after a brief ischemic period in transgenic mice expressing inducible heat shock protein 70. J Clin Invest 101: 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong H, Patterson T, Adiseshaiah P, Shapiro P, Kalvakolanu DV, Reddy SP (2002) JNK1 and AP-1 regulate PMA-inducible squamous differentiation marker expression in Clara-like H441 cells. Am J Physiol Lung Cell Mol Physiol 282: L215–225 [DOI] [PubMed] [Google Scholar]
- Wollert KC, Taga T, Saito M, Narazaki M, Kishimoto T, Glembotski CC, Vernallis AB, Heath JK, Pennica D, Wood WI, Chien KR (1996) Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J Biol Chem 271: 9535–9545 [DOI] [PubMed] [Google Scholar]
- Wong GH, Goeddel DV, Nakano M, Knowlton AA, Yokoyama T, Lesslauer W, Mann DL, Low-Friedrich I, Weisensee D, Mitrou P, Schoeppe W, Stehlik C, de Martin R, Binder BR, Lipp J, Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T (1988) Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science 242: 941–944 [DOI] [PubMed] [Google Scholar]
- Yamauchi-Takihara K, Kishimoto T (2000) A novel role for STAT3 in cardiac remodeling. Trends Cardiovasc Med 10: 298–303 [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Hoshijima M, Gu Y, Nakamura T, Pradervand S, Hanada T, Hanakawa Y, Yoshimura A, Ross J Jr, Chien KR (2001) Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest 108: 1459–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H, Yajima T, Duplain H, Iwatate M, Kido M, Hoshijima M, Weitzman MD, Nakamura T, Woodard S, Xiong D, Yoshimura A, Chien KR, Knowlton KU (2003) The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury. J Clin Invest 111: 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table I
Supplementary Table II
Supplementary Table IV
Supplementary Table V
Supplementary Figure 4H
Supplementary material