

Abstract
Systemic lupus erythematosus (SLE) is a chronic, life threatening autoimmune disorder, leading to multiple organ pathologies and kidney destruction. Analyses of numerous murine models of spontaneous SLE have revealed a critical role for endosomal Toll-like receptors (TLRs) in the production of autoantibodies and development of other clinical disease manifestations. Nevertheless, the corresponding TLR9-deficient autoimmune-prone strains consistently develop more severe disease pathology. Injection of BALB/c mice with 2,6,10,14-tetramethylpentadecane (TMPD), commonly known as pristane, also results in the development of SLE-like disease. We now show that Tlr9−/− BALB/c mice injected intraperitoneally with TMPD develop more severe autoimmunity than their TLR-sufficient cohorts. Early indications include an increased accumulation of TLR7-expressing Ly6Chi inflammatory monocytes at the site of injection, upregulation of interferon regulated gene expression in the peritoneal cavity, and an increased production of myeloid lineage precursors (CMP and GMP) in the bone marrow. TMPD-injected Tlr9−/− BALB/c mice develop higher autoantibody titers against RNA, neutrophil cytoplasmic antigens (ANCA), and myeloperoxidase (MPO) than TMPD-injected WT BALB/c mice. The TMP-injected Tlr9−/− mice, and not the WT mice, also develop a marked increase in glomerular IgG deposition and infiltrating granulocytes, much more severe glomerulonephritis, and a reduced lifespan. Together the data point to a major role for TLR7 in the response to self-antigens in this model of experimental autoimmunity. Therefore the BALB/c pristane model recapitulates other TLR7-driven spontaneous models of SLE and is negatively regulated by TLR9.
Introduction
SLE is a systemic autoimmune disorder, promoted by a combination of genetic susceptibilities and environmental factors. Numerous spontaneous models of SLE that have been used to explore the genetic basis for this disease and to further analyze SLE pathogenesis. Systemic autoimmunity can also be induced in non-autoimmune-prone mice with a single injection of the pro-inflammatory hydrocarbon oil 2, 6, 10, 14 tetramethylpentadecane (TMPD), also known as pristane, and these mice exhibit many features of human lupus (1). Immunostimulatory hydrocarbon oils such as squalene have been used as vaccine adjuvants and in some instances these vaccines have been associated with the induction of autoimmunity in both animals (2) and humans (3–6). Squalene by itself can also induce autoantibody production (4). Therefore the TMPD model has relevance to human disease.
As in other models of murine SLE, disease onset and progression in TMPD-injected mice is highly dependent on endosomal TLRs. In B6 mice, the i.p. injection of TMPD has been found to induce the rapid and persistent influx of both Ly6Chi inflammatory monocytes and Ly6Cmed Ly6G+ granulocytes into the peritoneal cavity, and the extravasation of Ly6Chi inflammatory monocytes detected at 14 days post-injection is dependent on both type I IFNs and TLR7 (7). Moreover, in contrast to TMPD-injected WT B6 mice, TMPD-injected Tlr7−/− B6 mice failed to make autoantibodies reactive with Sm/RNPs, argonaute (ago2) and other RNA-binding proteins, even though antibodies against dsDNA, as detected in a Crithidia Lucilia kinetoplast immunofluorescent assay, were still produced. In addition, much less IgG was deposited in the kidneys of these Tlr7−/− B6 mice, and as a result they developed less severe nephritis, and exhibited markedly improved survival rates (7, 8). Overall, TLR7-deficiency clearly protects B6 mice from TMPD-induced inflammation and autoimmunity.
The impact of TLR9-deficiency on TMPD-injected mice is less clearcut. As predicted from in vitro studies (9, 10), TLR9-deficient autoimmune-prone mice do not make anti-dsDNA or anti-nucleosome antibodies, as detected by homogeneous nuclear and mitotic plate staining of HEp-2 cells. Shlomchik and colleagues demonstrated that loss of TLR9 expression in MRL/lpr mice correlated with loss of binding to chromatin and nucleosomes, but not dsDNA (11). However, quite unexpectedly, in essentially all spontaneous models of SLE (including MRL/lpr, B6/lpr, Ali5 B6, Nba2.Yaa, B6.Nba2, and WASp-deficient B6 mice), Tlr9−/− mice produce significantly increased titers of autoantibodies directed against RNA-associated autoantigens and invariably develop more severe renal disease (11–17). By contrast, TMPD Tlr9−/− B6 mice have been reported to develop less severe peritoneal inflammation than the TLR-sufficient control group (7), and also less severe disease pathology (18). However, TMPD-injected BALB/c mice more closely mimic the renal complications and other clinical manifestations of human disease than TMPD-injected B6 mice (1). Therefore we decided to reexamine the role of TLR9 in the TMPD-treated BALB/c model of lupus. We show here that TMPD-treated Tlr9−/− BALB/c exhibit a more rapid progression to lupus nephritis and decreased survival compared to TLR-sufficient TMPD-treated control groups. Disease severity is preceded by an early increase in the number of Ly6Chigh inflammatory monocytes, a stronger interferon signature, and increased TMPD driven myelopoiesis. Therefore TMPD-injected Tlr9−/− BALB/c mice do in fact develop exacerbated autoimmune disease and provide a useful model for evaluating the negative regulatory role of TLR9 in murine SLE.
Material and Methods
Mice
Wild-type BALB/c mice were purchased from Jackson Lab. Tlr7−/− and Tlr9−/− mice, kindly provided by Dr. S. Akira, were backcrossed 10 generations to the BALB/c background. All mice were bred and maintained at the Department of Animal Medicine of the University of Massachusetts Medical School in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care, and all protocols were approved by the Institutional Animal Care and Use Committee.
TMPD injection
For long term experiments (5 to 6 months) 8–12 week-old female mice received a single i.p. injection of 0.5 ml of 2,6,10,14-Tetramethylpentadecane (TMPD, Sigma) (19). For short term experiments (4 and 14 days) age-matched female or male mice, as indicated in the figure legends, were injected at 6–8 weeks of age.
Flow cytometry
Single cell suspensions obtained from the peritoneal cavity, spleen, kidney and bone marrow were stained with the following antibodies: anti-CD11b (M1/70), anti-CD86 (GL1) and anti-Ly-6G (1A8) from BD Biosciences, anti-CD138 (281–2) and anti-GL7 (eBioscience), anti-Ly-6C (ER-MP20, Serotec). Hematopoietic subsets (HSPCs, CMPs, GMPs) were stained as described in (20). For TLR protein expression, cells were fixed with 2% PFA, permeabilized with 0.1% saponin and then stained with the biotinylated monoclonal anti-mTLR7 A94B10. A fluorescently labeled Streptavidin tetramer (SouthernBiotech) was added as detecting reagent. Flow cytometric analysis was carried out using a BD LSRII with Diva software (BD) and analysis was conducted with FlowJo Software (Tree Star).
Gene expression
RNA from total peritoneal lavage cells 4 days post-TMPD injection was extracted using the RNeasy Minikit (Qiagen) and cDNA was made by reverse transcription using the iScript cDNA Synthesis Kit (Biorad). qRT-PCR was performed using SYBR Green PCR Master Mix (Biorad) as described previously (19). IP10, ISG15, CCL5, Mx1, IRF7, and IFI204 expression are presented relative to beta-actin expression. Primer sequences are as follows: IP10, 5′-gctgccgtcattttctgc, 3′-tctcactggcccgtcatc; ISG15, 5′-agtcgacccagtctctgactct, 3′-ccccagcatcttcaccttta; CCL5, 5′-tgcagaggactctgagacagc, 3′-gagtggtgtccgagccata; Mx1, 5′-gagcaagtcttcttcaaggatc, 3′-gggaggtgagctcctcagt; IRF7, 5′-agcgtgagggtgtgtcct, 3′-tcttcgtagagactgttggtgct; IFI204, 5′-gccagccctaagatctgtgat, 3′-tctttcggttcactgttttcttg; and beta-actin, 5′-ctaaggccaaccgtgaaaag, 3′-accagaggcatacagggaca.
Generation of BM-macrophages and dendritic cells
M-CSF and GM-CSF-derived bone marrow macrophages (BMDMs) and dendritic cells (BMDCs) were induced by culturing with 40ng/ml M-CSF (R&D Systems) or 20 ng/ml GM-CSF (Immunotools), respectively, in 10% FCS, 1% Ciprofloxin containing RPMI1640 medium for 7 days. Differentiated BMDMs and BMDCs were seeded on 96-well cell-culture plates (5×104 cells/well), and stimulated with the doses indicated in Figure 4 of LPS (from Escherichia coli 0111:B4), Pam3CSK4 (Sigma-Aldrich), R848, RNA40 or CpG ODN1826 (InvivoGen) at 37°C in a 5% CO2 atmosphere for 24 hours before collecting supernatants. IL-6 concentrations in the supernatants were determined by ELISA (eBioscience) according to the manufacturer’s instructions.
Figure 4. Differential outcome of TLR9-deficiency on TLR7-driven responses of BM-derived macrophages and dendritic cells.

(A) M-CSF derived BMDMs and (B) GM-CSF derived BMDCs, generated from BALB/c WT, Tlr9−/− or Tlr7−/− respectively, were stimulated with titrations of ligands for TLR2 (Pam3CSK4), TLR4 (LPS), TLR7 (RNA40, R848), and TLR9 (CpG ODN1826) and after 24h the supernatants were collected and analyzed for IL-6 concentration by ELISA. Data are representative of three independent experiments performed in triplicates (mean +SEM).
ELISPOT assay
IgG2a specific Ab-forming cells (AFCs) of cells isolated from spleens of mice 5 months post pristane injection were measured by ELISPOT assay as previously described (21).
Antibody and Autoantibody Titers
Total serum IgG1, IgG2a, IgG2b, IgG3, IgM and kappa light chain were measured by ELISA as described previously (22). ANA were detected by immunofluorescence on HEp-2 slides (Antibodies Inc.) (19). ANCA were detected by immunofluorescence on ethanol fixed granulocytes (AESKUSLIDES) with serum diluted at 1:50 and using FITC-conjugated rat anti-mouse kappa (Southern Biotech) in vectashield antifade mounting medium (Vector laboratories). Antibodies against Myeloperoxidase (MPO) were detected by ELISA by using recombinant human MPO antigen (Ala49-Ser745; R&D Systems) coated wells, and a goat anti-mouse IgG HRP secondary antibody (Sigma A8924) with TMB substrate (Dako) as detecting reagent. RNA was detected by ELISA using poly-L-Lysine coated wells (Sigma P4832) coated with yeast RNA (Sigma R6750), as described previously, and reported as μg equivalent of the anti-RNA autoantibody BWR4 (23).
Renal pathology and cell infiltration
Albuminuria was measured by ELISA as described (19). Sections of paraffin embedded kidneys were stained with H&E and scored in a blinded fashion to determine a glomerular and interstitial inflammation score as described (19). For immunofluorescence, kidneys were fixed in 4% formalin solution O/N and frozen in Tissue Tek Embedding Medium (Sakura O.C.T. Compound) after dehydration in 30% sucrose solution. Total immune complex deposition was detected with AlexaFluor 488 F(ab’)2 fragment goat anti-mouse IgG (H+L) antibody (LifeTechnlogies), IgG2a or IgG2b glomerular immune complexes were detected with FITC-conjugated goat (Fab’)2 anti-mouse IgG2a, or goat anti-mouse IgG2b (γ2b chain specific) antibody (SouthernBiotech). Infiltrating neutrophils were detected by using a monoclonal rat anti-mouse Ly6G AlexaFluor647 conjugated antibody (Biolegend). Nuclei were counterstained with DAPI (Invitrogen). Images were captured on a Nikon E600 inverted light microscope or on a Leica TCS SP8 microscope (Leica Microsystems, Buffalo Grove, IL, USA) and processed in Adobe Photoshop. Using the NIS-Elements imaging software BR3.10 (Nikon), 15–20 randomly picked glomeruli per mouse were circled and analyzed for the mean fluorescence intensity (MFI) emitted in the green channel. Additionally, 20–30 randomly picked glomeruli per mouse were circled and the percentage of Ly6G+ glomeruli of the total number of glomeruli counted was determined.
For flow cytometry on renal single cell suspensions, kidneys were cut into small pieces, transferred to Gentle MACS tubes (Miltenyi, Bergisch-Gladbach, Germany) and digested in a Collagenase type-I (10mg/ml), DNase (200U/ml), HBSS mixture for 30 minutes at room temperature. Renal tissue fragments were then processed using the Gentle MACS program ‘D’ for 30 seconds, incubated for further 15 minutes at room temperature within the digestion buffer, processed a second time using the Gentle MACS program ‘D’ for 30 seconds. Cells were filtered through a 70μm filter, washed twice with cold HBSS, and red blood cells were lysed. Single cell suspensions were counted and stained for flow cytometric analysis.
Statistical analysis
All data were analyzed by non-parametric Mann-Whitney U test or Kruskal-Wallis test, as appropriate, using GraphPad Prism Software (GraphPad Software, San Diego, CA). Experiments are reported as mean +/− SEM. Differences are designated as one asterisk if p <0.05, as two asterisk if p<0.01 and three asterisk if p<0.001. Multiple comparisons were analyzed by one-way ANOVA, followed by Bonferroni multi-comparison test or Kruskal-Wallis test.
Results
Rapid progression of lupus nephritis and decreased survival in TMPD-treated Tlr9−/− BALB/c mice
In spontaneous models of murine SLE, Tlr9−/− mice consistently develop exacerbated renal disease. To directly compare TMPD-injected BALB/c mice to these other models, WT and Tlr9−/− were injected with either PBS or TMPD and urine samples were collected at monthly intervals and analyzed for proteinuria. By 116–137 days post TMPD-injection, over 30% of TMPD-injected Tlr9−/− mice, and none of the TMPD-injected WT BALB/c mice, showed elevated levels of albuminuria. The frequency of proteinuric mice as well as urine albumin concentrations increased with time and by 160–180 days post TMPD injection, over 70% of the surviving Tlr9−/− mice had urine albumin levels that were at least a 100-fold over physiologic urine protein levels. A much lower frequency of the TMPD-injected WT and mice developed proteinuria and the urine albumin levels in these mice were less than 10-fold over baseline (Figure 1A and Supplemental Figure 1A). None of the PBS injected WT or Tlr9−/− mice developed proteinuria over this time course. At 5 months post-TMPD injection, 5 mice from each group were euthanized and kidney sections were compared to PBS-injected mice for histological evidence of renal disease. The Tlr9−/− BALB/c kidneys showed more severe glomerulonephritis associated with crescent formation and sclerosis (Figure 1B and C). The TMPD-injected WT BALB/c kidneys exhibited less pathology. Consistent with severe nephritis, the TMPD-injected Tlr9−/− mice showed a markedly reduced survival rate compared to the TMPD-injected WT BALB/c mice (Figure 1D). Importantly, even our oldest (> 6 mon) untreated WT and Tlr9−/− mice failed to exhibit any sign of renal disease, reduced survival rates, or immune activation (Figure 1B–D and Supplemental Figure 1). Therefore TLR9 deficiency on the BALB/c background alone does not predispose to systemic autoimmunity. Rather, TLR9 expression exerts a protective function in the context of TMPD-induced autoimmunity in BALB/c mice.
Figure 1. Accelerated development of renal disease and shortened life span in TLR9 deficient TMPD-treated mice.
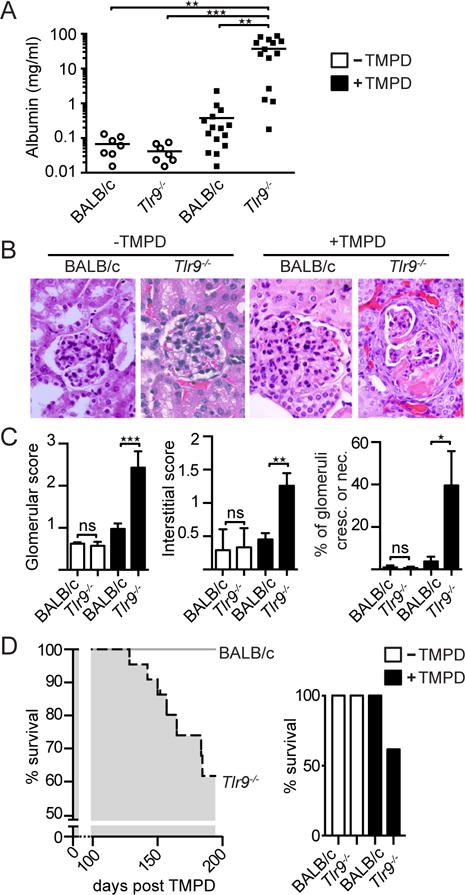
(A) Urine albumin (mg/ml) in samples obtained from untreated or TMPD-treated BALB/c WT or Tlr9−/− mice 160–180 days post TMPD was determined by ELISA. (B) H&E stain of representative glomeruli in kidney sections obtained from untreated or TMPD-treated BALB/c WT or Tlr9−/− mice 155 days post treatment. (C) Glomerular and interstitial scores for renal pathology, as well as the percent of glomeruli per mouse with crescentic or necrotic pathologies, were determined at 155 days after TMPD injection. (D) Survival curve of TMPD-treated BALB/c WT (n=23; no deaths) versus Tlr9−/− mice (n=22; 7 deaths). Statistics were done using the non-parametric Kruskal-Wallis test.
Increased numbers of TLR7-expressing Ly6Chigh inflammatory monocytes in Tlr9−/− TMPD treated mice
Prior studies suggested that TLR9-deficient B6 mice developed an attenuated acute response to TMPD (7, 18). Considering the apparent negative regulatory role of TLR9 on both nephritis and survival in TMPD-injected BALB/c mice, we decided to re-examine the initial inflammatory response elicited by TMPD in BALB/c mice. BALB/c WT, BALB/c Tlr9−/−, and BALB/c Tlr7−/− mice were injected i.p. with TMPD and cells collected from the peritoneal cavity were examined 4 days later. Compared to PBS injected WT BALB/c mice, all 3 TMPD-injected strains showed a strong increase in the total number of peritoneal exudate cells (PEC), as well as CD11b+ myeloid lineage cells. Although not statistically significant, the Tlr9−/− mice tended to have more, not fewer total and CD11b+ cells (Figure 2A, B). Untreated WT and Tlr9−/− mice had comparably low numbers of total and CD11b+ peritoneal washout cells.
Figure 2. Myeloid lineage abnormalities in TLR9 deficient TMPD-treated mice.
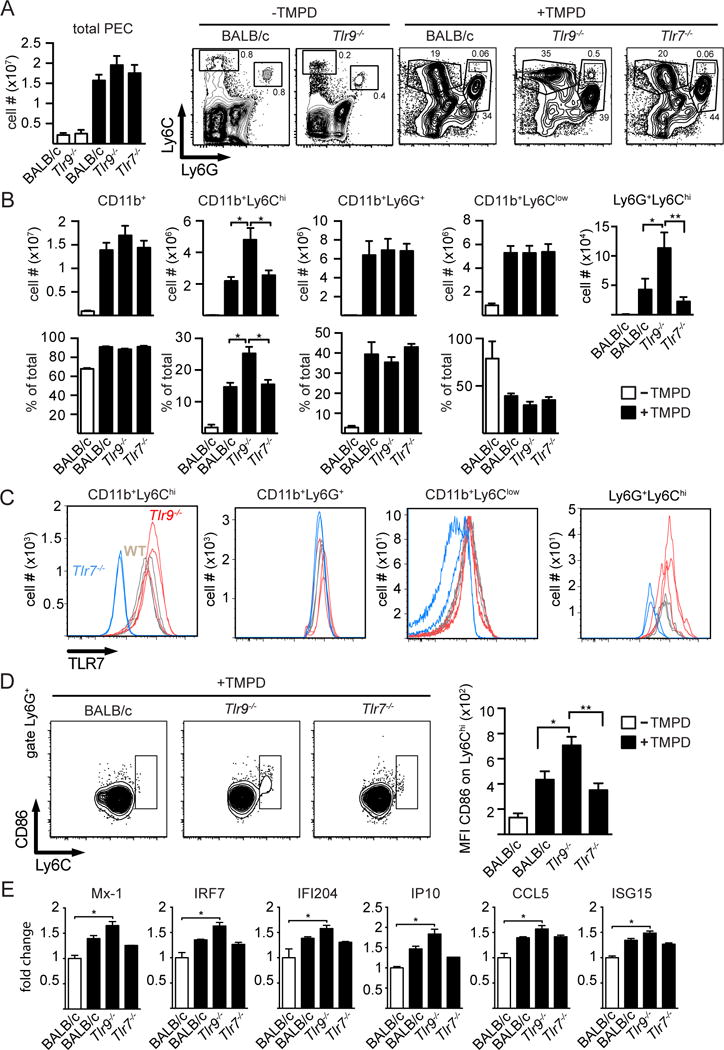
(A) Total number of cells collected from the peritoneum of untreated BALB/c WT mice (n=8, all female mice) and Tlr9−/− mice (n=5, all females) (open bars) or TMPD-treated BALB/c WT (n=7, 5 females, 2males), Tlr9−/− (n=6 females, 2 males) or Tlr7−/− mice (n=7, all females) (filled bars) (mean+SEM) 4 days after TMPD injection, and representative Ly6C/Ly6G FACS analysis of CD11b+ cells obtained from the indicated TMPD-injected mice; (B) Total number of CD11b+, CD11b+ Ly6Chi (inflammatory monocytes), CD11b+ Ly6G+ (granulocytes), CD11b+ Ly6Clow (macrophages) cells recovered from the peritoneum of untreated BALB/c mice (n=2) (open bars) or TMPD-treated BALB/c (n=7), Tlr9−/− (n=8) or Tlr7−/− (n=7) mice (filled bars) (mean+SEM). (C) Peritoneal exudate cell subsets from TMPD-treated BALB/c WT (n=3, all females), Tlr9−/− (n=3, all females) or Tlr7−/− mice (n=3, all females) were stained for total intracellular TLR7 levels using the biotinylated monoclonal anti-mTLR7 antibody A9410 and analyzed by flow cytometry. (D) Ly6G+ gated granulocytes stained for CD86 and Ly6C and graphs are depicting the CD86 MFI of the Ly6Chigh granulocyte subpopulation. (E) Mx1, IRF7, IFI 204, IP10, CCL5 and ISG15 mRNA expression of total PECs from day 4 TMPD-injected mice of the indicated genotypes (+/− SEM, BALB/c WT TMPD n=3 females, Tlr9−/− TMPD n=4 females, Tlr7−/− TMPD n=3 females). Statistics were done using a One-way ANOVA with Bonferroni multiple comparison test.
The CD11b+ cells were further analyzed for the expression of Ly6C and Ly6G. Prior studies showed that CD11b+, Ly6Chi, Ly6G− cells represent a population of inflammatory monocytes, while the CD11b+, Ly6G+ cells represent inflammatory granulocytes (19, 24). There was a significant increase in the relative and total number of Ly6Chigh inflammatory monocytes in TMPD-injected Tlr9−/− mice, when compared to either the TMPD-injected WT or Tlr7−/− mice (Figure 2A and B). Reeves and colleagues have previously shown that the number of inflammatory monocytes is decreased in B6 TMPD-injected Tlr7−/− mice compared to WT mice at day 14 post injection, and the same is true for BALB/c Tlr7−/− mice (data not shown). To further examine the role of TLR7 in this model, we stained the day 4 peritoneal cell subsets for TLR7 (Figure 2C). Compared to the Tlr7−/− negative control, both WT and Tlr9−/− mice inflammatory Ly6Chi monocytes expressed high levels of TLR7, with the Tlr9−/− cells tending toward slightly higher levels.
While the total number of Ly6G+ granulocytes was unaltered between the three different groups 4 days post TMPD-injection (Figure 2A, B), we detected a distinct Ly6Chi CD86hi subpopulation within the Ly6Ghi granulocyte gate in the Tlr9−/− TMPD-treated mice. This population was less apparent in the cells collected from TMPD-treated WT BALB/c and Tlr7−/− mice (Figure 2A, B). Although the Ly6Cint granulocytes expressed barely detectable levels of TLR7, the Ly6G+ Ly6Chi cells expressed significantly more TLR7, with the Tlr9−/− cells again expressing more than the WT cells (Figure 2C). They also expressed higher levels of CD86 than the Ly6Cint granulocytes (Figure 2D).
Altogether, these data indicate that TLR9 deficiency results in an early increase in the number of inflammatory monocytes accumulating in the peritoneal cavity, and the appearance of a distinct subpopulation of activated Ly6G+ cells, both of which express high levels of TLR7. The accumulation of inflammatory Ly6Chi monocytes in the peritoneal cavity at day 14 post TMPD has been shown to be highly dependent on type I IFN (7). Therefore the early increase in this population in the TMPD-injected Tlr9−/− mice suggested a stronger type I response in the absence of TLR9. We next tested the day 4 expression levels of six IFN-stimulated genes (ISGs), including Mx1, IRF7, Ifi204, IP10, CCL5 and ISG15. ISG expression levels were higher in the Tlr9−/− mice than either the TLR-sufficient or Tlr7−/− mice (Figure 2E), consistent with the notion of increased IFN activity.
TLR9 deficiency exacerbates TMPD driven myelopoiesis
TLR7 transgenic mice exhibit a peripheral expansion of inflammatory monocytes and neutrophils that is dependent on increased myelopoiesis in the bone marrow (BM) secondary to TLR7 signaling and type I IFN production (25). To determine whether TLR9 deficiency affects the production of myeloid progenitor subsets in the bone marrow following TMPD injection, BM cells were collected 14 days post TMPD injection and the lineageneg cells were evaluated for the frequency of myeloid precursor subsets (MP). Importantly, the Tlr9−/− mice consistently had an increased frequency of MP, with a significant increase in the percentage of the two myeloid progenitor subsets, common myeloid precursors (CMPs) and granulocyte myeloid precursors (GMPs), in the BM of the Tlr9−/− compared to WT BALB/c mice 4 days after pristane injection (Figure 3A, B). By contrast, the frequency of myeloid progenitor subsets in untreated WT and Tlr9−/− mice was indistinguishable. Together these results are consistent with an increase in type I IFN production in TMPD-injected Tlr9−/− mice but not untreated Tlr9−/− mice.
Figure 3. TLR9-deficiency exacerbates TMPD driven myelopoiesis.
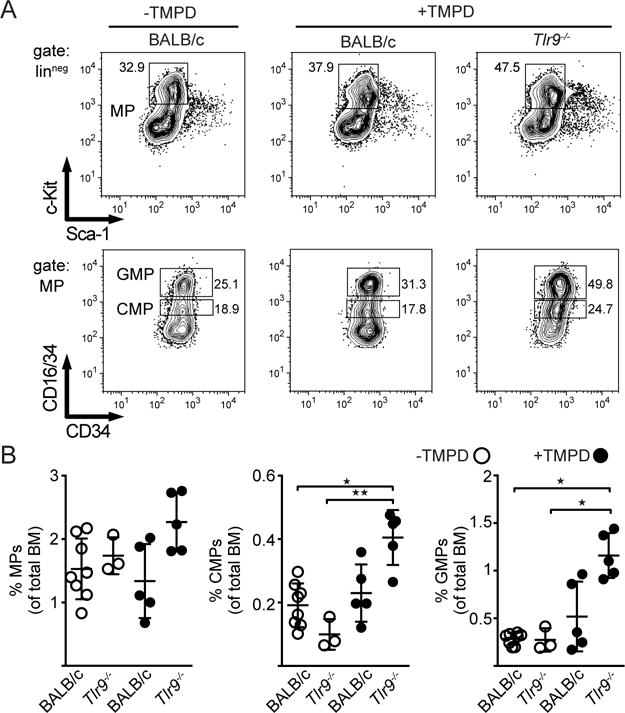
(A) Phenotype of lineageneg -bone marrow myeloid progenitors (MP) from day 14 TMPD-injected mice. The upper row panels depict live/singlet/linneg cells stained for c-Kit and Sca-1. The lower row panels are gated on MP and stained for CD16/34 versus CD34 to differentiate between granulocyte-myeloid progenitors (GMPs) and common myeloid progenitors (CMPs). (B) Percentage of total MPs, GMPs and CMPs of untreated BALB/c WT (n=5 females) and Tlr9−/− mice (n=3 females) (open circles), or day 14 TMPD-treated BALB/c WT (n=5 females) and Tlr9−/− mice (n=5 females) (filled circles) (mean+SEM). Statistics were done using the non-parametric Kruskal-Wallis test.
TLR9-deficient BM-derived macrophages, but not bone marrow derived dendritic cells, produce more IL-6 in response to TLR ligands
To further explore the effect of TLR9-deficiency on TLR7-driven responses, we stimulated M-CSF-generated BM-derived macrophages (Mø) and GM-CSF-generated BM-derived dendritic cells (DC) with increasing concentrations of the small molecule TLR7 ligand R848 or with increasing concentrations of the stimulatory RNA fragment RNA 40. The extent of activation was then quantified by measuring the amount of IL-6 produced over the next 24 hrs. Tlr9−/− macrophages produced more IL-6 than WT macrophages, and the response was entirely TLR7-dependent as no IL-6 was produced by Tlr7−/− macrophages (Figure 4A). Responses to TLR2 and TLR4 ligands were comparable. These data suggest that TLR9-deficiency results in higher TLR7 activity. However, over the entire titration curve, Tlr9−/− DCs did not respond better to the TLR7 ligands than WT DCs (Figure 4B). Together, the data show that the capacity of TLR9 to negatively regulate TLR7 responses is highly cell type dependent.
TLR9-deficient TMPD-injected mice produce autoantibodies specific for RNA-associated autoantigens
Similar to other SLE-prone strains, TMPD-injected mice routinely develop hypergammaglobulinema and autoantibody production. Tlr9−/− autoimmune prone mice routinely show a particular increase in IgG2 complement fixing antibodies (15). To determine whether TLR9-deficiency affected antibody production in TMPD-injected mice, the frequency of antibody producing cells in the spleen at 5 month post pristane injection was determined by ELISpot. We found that the spleens from the Tlr9−/− mice had a 3-fold greater frequency of IgG2a-producing cells than spleens from the TLR-sufficient BALB/c mice (Figure 5A), even though we could not detect a statistically significant difference in circulating IgG2a titers (Supplemental Figure 1D).
Figure 5. TMPD-injected Tlr9−/− mice produce autoantibodies specific for RNA-associated autoantigens.
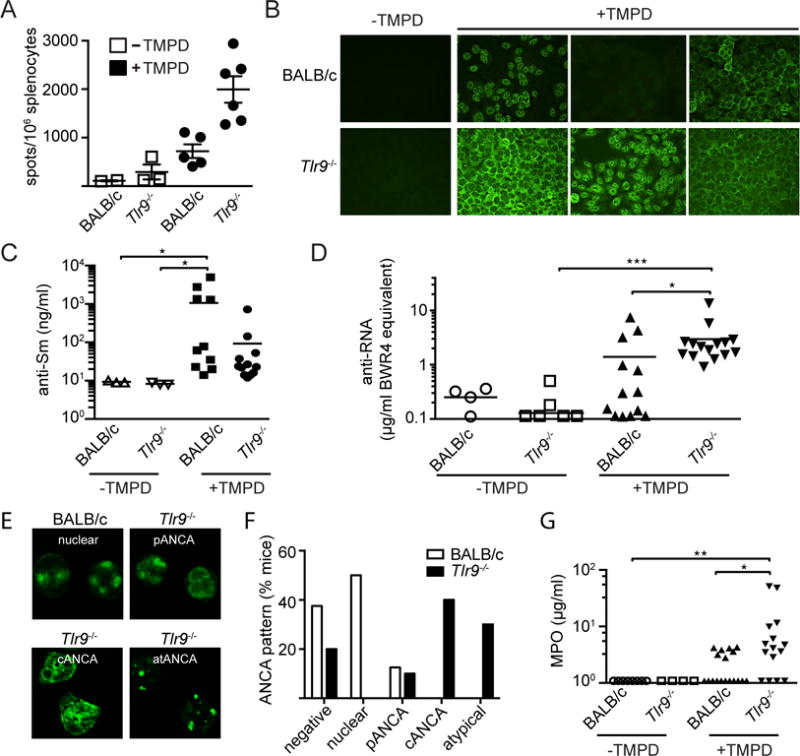
(A) Spleens were harvested from non-treated versus TMPD-treated WT or Tlr9−/− mice 5 month post injection and the number of IgG2a+ AFCs was measured by ELISPOT as described (21). (B) Representative images of HEp2 ANA staining patterns from untreated and TMPD-treated BALB/c WT and Tlr9−/− mice (BALB/c WT n=11; Tlr9−/− n=14; BALB/c WT TMPD n=37; Tlr9−/− TMPD n=37). (C) Serum anti-Sm titers from untreated and TMPD-treated BALB/c WT and Tlr9−/− mice 5 month post TMPD treatment. (D) Anti-RNA autoantibody levels in sera from untreated and TMPD-treated BALB/c WT and Tlr9−/− mice 5 month post TMPD treatment were determined by ELISA. (E) Representative images of ANCA staining patterns of sera obtained from TMPD-treated BALB/c WT and Tlr9−/− mice; and (F) quantification based on ANCA quality as indicated. (G) MPO autoantibody titers from untreated and TMPD-treated BALB/c WT and Tlr9−/− mice 5 month post TMPD treatment (BALB/c WT n=8; Tlr9−/− n=4; BALB/c WT TMPD n=18; Tlr9−/− TMPD n=16). Throughout this figure each shape corresponds to an experimental female mouse. Statistics were done using the non-parametric Kruskal-Wallis test.
TMPD-injected WT BALB/c mice routinely produce autoantibodies that show a predominantly speckled nuclear HEp-2 staining pattern, often associated with autoantibodies reactive with SmRNP (Figure 5B). A smaller percentage of the TMPD-injected Tlr9−/− mice gave a speckled nuclear pattern and this tendency correlated with a loss of nuclear SmRNP reactivity, as determined by an SmRNP ELISA (Figure 5C). Instead they shifted to a more prominent cytoplasmic staining pattern, characteristic of anti-RNA autoantibodies, and developed higher RNA autoantibody titers, as detected by an RNA ELISA (Figure 5D). Both speckled nuclear and cytoplasmic staining patterns are thought to be TLR7-dependent (15). Neither the WT or Tlr9−/− control groups produced autoantibodies as detected by ANA or RNA ELISA. Together the data indicate that loss of TLR9 results in a shift in autoantibody specificity as well as an increase in autoantibody production.
Increased frequency of neutrophil specific autoantibodies and glomerular granulocytic infiltrates associated with accelerated renal disease in the Tlr9−/− mice
The increased frequency of CMPs and GMPs in the BM of the TMPD-injected Tlr9−/− mice indicated an increased turnover of granulocytes and the potential for an increase in the release of granulocyte-derived cell debris that could trigger autoantibody production. Anti-neutrophil cytoplasmic antibodies (ANCAs) are frequently observed in vasculitis patients (26) but have also been described in animal models (27, 28) and in SLE patients with severe renal disease (29–31). Sera from TMPD-injected Tlr9−/− BALB/c and TLR-sufficient BALB/c mice were therefore assayed for ANCA reactivity by immunofluorescence staining of fixed neutrophils. Approximately half the BALB/c sera stained neutrophils, and those sera that were positive showed a predominantly nuclear staining pattern. However, a much higher frequency of the TMPD-injected Tlr9−/− sera showed strong ANCA reactivity with cytoplasmic antigens (c-ANCA), perinuclear (p-ANCA) and/or atypical ANCA staining patterns (Figure 5E, F). Since a major antigenic specificity of ANCAs is myeloperoxidase (MPO) (32) we further tested the sera in a MPO ELISA. Here again a higher frequency of the Tlr9−/− sera were positive (69%) compared to a lower level MPO reactivity of most TLR-sufficient BALB/c TMPD-treated mice (34%) (Figure 5G,).
Due to the association between ANCA antibodies and nephritis scores, we decided to compare kidneys obtained from TMPD-injected Tlr9−/− and WT mice at 5 months post treatment for evidence of autoantibody deposition and granulocyte infiltration. Even though we could not detect a significant increase in the circulating levels of total IgG or IgG2a in the Tlr9−/− mice compared to WT mice (Supplemental Fig 1D), we found a dramatic increase in the percentage of glomeruli with IgG, IgG2a and IgG2b deposits as determined by immunofluorescent staining (Figure 6A). The average mean fluorescent intensity signal per glomerulus was also increased in the TMPD-treated Tlr9−/− mice (Figure 6A).
Figure 6. The exacerbated kidney pathology in Tlr9−/− TMPD treated mice correlates with increased immune complex deposition and glomerular granulocyte accumulation.
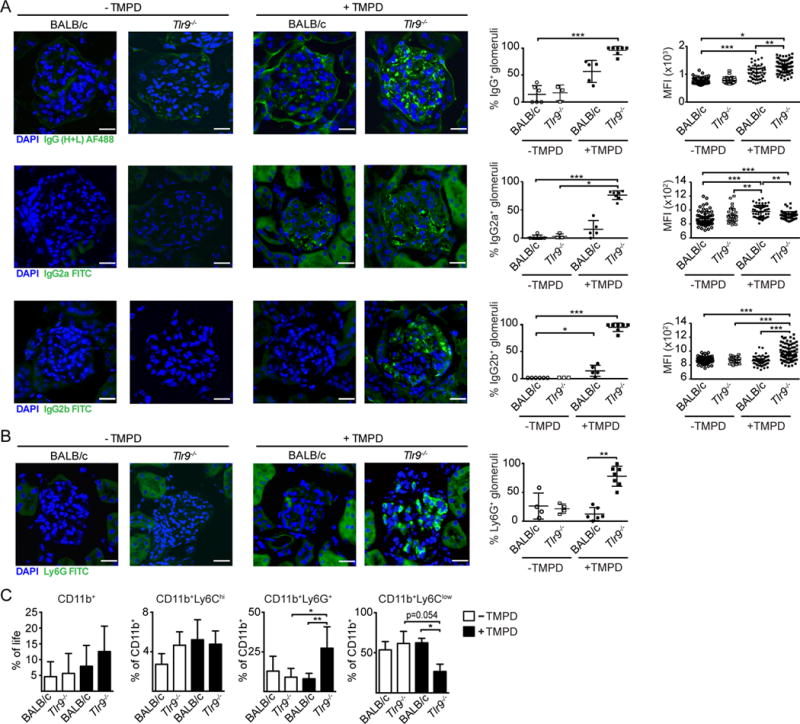
(A) Representative confocal images of kidneys from untreated BALB/c WT and Tlr9−/− mice and BALB/c WT and Tlr9−/− mice 5 month post TMPD-treatment stained with DAPI (blue), anti-total IgG (H+L), anti-IgG2a or anti-IgG2b (all green). The graphs depict the percentage of stained glomeruli and the average mean fluorescence intensity per glomerulus of the positive glomeruli (each dot represents the mean out of 10–30 randomly picked glomeruli per individual mouse). (B) Representative confocal images of kidneys from WT and Tlr9−/− mice 5 month post TMPD-treatment stained with DAPI (blue) and anti-Ly6G (green). (C) Percentage of CD11b+ cells within the live gate isolated from the kidney and percentages of Ly6Chigh monocytes, Ly6G+ granulocytes and Ly6Clow cells within this CD11b+ gate from untreated BALB/c mice (n=5) and untreated Tlr9−/− mice (n=5) (open bars) or TMPD-treated BALB/c (n=8), or Tlr9−/− (n=6) mice (filled bars) (mean+SEM). Statistics were done using a One-way ANOVA with Tukey’s multiple comparison test.
We further examined renal cellular composition 5 month post TMPD-treatment between the different groups. Kidney sections were stained with Ly6G and infiltrating granulocytes were detected by immunofluorescence. Substantial numbers of granulocytes were only detected in the TMPD-injected Tlr9−/− mice (Figure 6B; Supplemental Figure 2). Kidneys of untreated and TMPD-treated mice were also digested with a collagenase/DNase mixture to obtain single cell suspensions, which were stained for lymphoid (B cells, T cells) and myeloid (monocytes/macrophages, neutrophils) cells and analyzed by flow cytometry. We did not detect a difference in either the number of lymphoid cells, or the formation of ectopic lymphoid tissue within kidneys of the TMPD-treated groups (data not shown). Numbers of migrating inflammatory monocytes (Ly6Chigh) and renal macrophages were comparable in the WT and Tlr9−/− mice. However, there was a strong increase in the percentage of infiltrating neutrophils, as detected by Ly6G, only in the TMPD-treated Tlr9−/− mice and not the TMPD-treated WT mice (Figure 6C). Neither the untreated WT or untreated Tlr9−/− mice showed any evidence of IgG deposition or granulocyte infiltration. Together, these data indicate that TLR9-deficiency in TMPD induced autoimmunity leads to severe kidney damage associated with the deposition of IgG immune complexes and granulocyte recruitment.
Discussion
Inducible models of SLE serve an important role in SLE research as they provide a system for exploring the early events in the onset of disease without the need for extensive backcrossing to strains with additional risk alleles. TMPD-induced systemic autoimmunity has often been used in this context. However, the effect of TMPD is strain dependent and most of the murine studies involving TMPD have utilized B6 mice. TMPD-treated B6 mice often develop hemorrhagic lesions in the lung, and exhibit little evidence of glomerulonephritis or other forms of renal disease (33) (34). Moreover, in contrast to spontaneous models of SLE, TMPD-induced autoimmunity was found to be attenuated in Tlr9−/− B6 mice (18). TMPD-treated BALB/c mice do not develop lung lesions and do eventually develop mild nephritis. We now show that TMPD-treated Tlr9−/− BALB/c mice develop more severe renal disease than TLR-sufficient mice, along with other clinical manifestations of disease, and exhibit a reduced lifespan. TMPD-injected BALB/c mice therefore resemble genetically programmed models of SLE (11, 12, 14–17), and can be used to explore the basis for the negative regulatory role of TLR9 in murine SLE.
Since the time of disease induction is clearly delineated by the day of TMPD injection, this model allows us to monitor the very early events leading to systemic autoimmunity. Importantly, within 4 days of TMPD injection, flow cytometry revealed clear differences between the WT and Tlr9−/− mice with regard in the quality and number of inflammatory cells extravasating into the peritoneal cavity. The total number of inflammatory monocytes that accumulated in the peritoneal cavity was increased two-fold in the Tlr9−/− mice. Reeves and colleagues have reported that these inflammatory monocytes are an early source of type I IFN (24), at least at the RNA level and we have reported that they produce many additional proinflammatory cytokines, including IL-6 (19). In the context of TMPD autoimmunity IL-6 is clearly important since SLE like disease is ameliorated in IL-6 deficient mice (35). We now show that inflammatory Ly6Chi monocytes express very high levels of TLR7 protein, and that Tlr9−/− BM-derived macrophages produce more IL-6 in response to TLR7 ligands than WT BMDCs, despite the fact that the WT BMDCs also express very high levels of TLR7. These data are consistent with the notion that TLR9 and TLR7 compete for binding to the chaperone protein Unc93B1, and in the absence of TLR9, TLR7 can respond more effectively (36, 37). However we also found that Tlr7−/− BMDMs produced more IL-6 in response to a small molecule TLR9 ligand than WT BMDCs, leaving the distinction between TLR7 and TLR9 uncertain. Furthermore, in contrast to BMDMs, Tlr9−/− and WT BM-derived DCs produced comparable levels of IL-6. Therefore the capacity of TLR9 to negatively regulate TLR7 responses is highly cell type dependent.
Several studies have pointed to a key role for B cells in the exacerbated disease exhibited by TLR9 deficient MRL/lpr and WASp−/− mice (13, 23), pointing to distinct functional outcomes of TLR9 vs TLR7 activated B cells. Nevertheless, WT and Tlr9−/− B cells express similar levels of TLR7, and R848 induces comparable levels of proliferation in WT and Tlr9−/− B cells (21). Purified WT and Tlr9−/− B cells have also been reported to make comparable levels of IL-6 in response to a TLR7 ligand (38). Clearly function does not correlate with protein levels and additional studies involving a more extensive analysis of TLR-expressing primary cell types will be required to fully understand the molecular basis for the apparent negative regulatory role of TLR9 in SLE.
The TMPD-injected Tlr9−/− mice also showed indications of a stronger IFN signature, as reflected by increased expression of IFN-inducible genes, including Mx1 and IRF7. Both MyD88-deficient and IFNaR-deficient TMPD-injected mice, fail to produce either Mx1 or IRF-7, indicating that these genes are induced through a TLR-dependent pathway and dependent on a type I IFN (7). TMPD-injected Tlr9−/− mice, but not uninjected Tlr9−/− mice also had an increased frequency of CMPs and GMPs in the bone marrow than their WT counterparts. This apparent IFN signature is consistent with the dramatically elevated IFNα serum titers previously reported for Tlr9−/− MRL/lpr mice (39). TMPD has been shown to induce cell death both in vitro and in vivo and the cell debris generated by TMPD exposure could contribute to an inflammatory reaction (19, 40). B cells and dendritic cells appear to be particularly sensitive to pristane (40). In response to this inflammatory stimulus, it is then possible that TLR7 more effectively activates IRF5, or other transcription factors that lead to the production of IFN and/or IFN-inducible genes, than TLR9. We propose that the elevated type I titers lead to increased myelopoiesis/granulopoiesis, a more rapid turnover of neutrophil lineage cells, the increased production of anti-neutrophil antibodies, and a much more extensive granulocyte glomerular infiltrate, which all together promote glomerulonephritis, renal sclerosis and crescent formation.
We also consistently found an increased frequency of Ly6Chi Ly6G+ CD86high cells in the Tlr9−/− mice. Ly6C expression in neutrophils is associated with stringent cell migration and activation (ROS production, release of pro-inflammatory cytokines) by a potential interaction with the Src family kinase Fgr (41) (42) (43). In addition, the more activated phenotype of the Ly6Chigh granulocyte subpopulation might be linked to the acquisition of an APC-like phenotype and subsequent T cell activation, as has previously been described in a mouse model of chronic colitis (44). The upregulation of CD86 on these Tlr9−/− Ly6G+ cells is significant, as activated neutrophils have been shown to upregulate MHC class II, prime T cells and activate B cells (45), and even exert B cell helper function (46). This population exhibits greater side scatter than the inflammatory monocytes or granulocytes for reasons that remain to be determined. Further characterization of this cell subset will be a focus of future studies.
In contrast to previous studies with Tlr9−/− B6 mice (38), we did not detect any evidence of spontaneous disease in Tlr9−/− BALB/c mice, as indicated by the absence of splenomegaly, autoantibody production, increased frequency of myeloid progenitors or histological indications of renal disease. These differences may reflect shifts in the microbiome between colonies. Alternatively, it is possible that the propensity of Tlr9−/− B6 mice to develop modest SLE-like clinical manifestations reflects the increased signal strength of the TLR9 allele expressed by B6 mice (47).
Type I IFNs have been definitively implicated in granulopoiesis, and the increase in myelopoietic progenitors in the bone marrow in the absence of TLR9 is well in line with the recently observed “emergency myelopoiesis” resulting from chronic TLR7 stimulation (25). Since neutrophils have a short lifespan, the increase in neutrophil numbers must lead to an increase in neutrophil death and a potential source of neutrophil-associated autoantigens. In fact, we found increased titers of ANCA autoantibodies including anti-MPO. These ANCAs not only recognize antigens that are contained within neutrophils but also within lysosomes of monocytes (48). It is also possible that glomerular neutrophils in the TMPD-treated TLR9 KO mice promote renal injury through the production of neutrophil extracellular traps (NETs) (49, 50) respiratory burst and degranulation (reviewed by (32). Nucleic acid-associated immune complexes have been shown to promote NET formation, especially in neutrophils exposed to high levels of IFN (51). NETs may also serve as a source of autoantigen in an inflammatory setting. Importantly, auto-antibodies against MPO are associated with a so called pauci-immune form of vasculitis (REF ZHAO). Strikingly, these anti-MPO autoantibodies are highly pathogenic and can cause glomerular necrosis and crescent formation even in the absence of T- and B-cells (28).
Exactly why there is a strong difference in IgG deposition between WT and TLR9−/− mice in the kidneys is not clear. One possibility is that the shift in the autoantibody repertoire to RNA-associated antibodies in TLR9−/− mice favors the formation of circulating immune complexes, however we could not detect any difference in circulating IC levels using a standard C1q binding assay (data not shown). Alternatively, in the absence of TLR9, autoantibodies may acquire physical properties that enhance binding to FcgR+ cells present in the kidney. Alternatively, renal disease may directly reflect the capacity of ANCA or other autoantibodies to directly bind neutrophils or other damaged cells in the kidney, thereby promoting an immune complex feed-forward process of renal inflammation. Neutrophil migration to kidneys has been used to define active lupus nephritis, and the presence of neutrophil-specific proteins in the urine of SLE patients serves as a surrogate marker for disease activity (52). This study now identifies a useful model, TMPD-injected BALB/c mice, for rigorously examining the connections between TLR9-deficiency, type I IFN production, the increased production of neutrophils, anti-neutrophil antibodies, and the development and progression of renal disease. It may also help decipher the pathogenesis of hydrocarbon oil induced autoimmunity in predisposed individuals and contribute to the development of safer vaccines.
Supplementary Material
Acknowledgments
This work was supported by NIH grants AR066808 (to A.M.-R.), DK090558 (to R.B.), HL093262 (to E.L.), Lupus Research Institute (to A.M.-R.), German Research Foundation grants SFB TR 57 (to E.L.) and DFG BO 4325/1-1 (to L.B.).
Abbreviations used
- ANA
anti-nuclear antibody
- ANCA
anti-neutrophil cytoplasmic antibody
- Ago 2
argonaute 2
- CMP
common myeloid progenitors
- GMP
granulocyte monocyte progenitors
- HSPC
hematopoietic stem progenitor cell
- Ifi204
interferon activated gene 204
- ISGs
IFN stimulated genes
- ISG15
IFN-stimulated gene 15
- MP
multipotent progenitors
- MPO
myeloperoxidase
- Mx1
MX dynamin-like GTPase 1, PEC, peritoneal exudate cells
- SLE
systemic lupus erythematosus
- TLR
Toll-like receptor
- TMPD
2,6,10,14-Tetramethylpentadecane
- Viperin
virus inhibitory protein, endoplasmic reticulum-associated, interferon-inducible
References
- 1.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30:455–464. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koppang EO, Bjerkas I, Haugarvoll E, Chan EK, Szabo NJ, Ono N, Akikusa B, Jirillo E, Poppe TT, Sveier H, Torud B, Satoh M. Vaccination-induced systemic autoimmunity in farmed Atlantic salmon. J Immunol. 2008;181:4807–4814. doi: 10.4049/jimmunol.181.7.4807. [DOI] [PubMed] [Google Scholar]
- 3.Dahlgren J, Takhar H, Anderson-Mahoney P, Kotlerman J, Tarr J, Warshaw R. Cluster of systemic lupus erythematosus (SLE) associated with an oil field waste site: a cross sectional study. Environmental health: a global access science source. 2007;6:8. doi: 10.1186/1476-069X-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuroda Y, Nacionales DC, Akaogi J, Reeves WH, Satoh M. Autoimmunity induced by adjuvant hydrocarbon oil components of vaccine. Biomedicine & pharmacotherapy = Biomédecine & pharmacothérapie. 2004;58:325–337. doi: 10.1016/j.biopha.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 5.van Assen S, Bijl M. Immunization of patients with autoimmune inflammatory rheumatic diseases (the EULAR recommendations) Lupus. 2012;21:162–167. doi: 10.1177/0961203311429555. [DOI] [PubMed] [Google Scholar]
- 6.Shoenfeld Y, Agmon-Levin N. ‘ASIA’ - autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun. 2011;36:4–8. doi: 10.1016/j.jaut.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, Kellner ES, Nacionales D, Barker T, Kelly-Scumpia K, van Rooijen N, Kumar H, Kawai T, Satoh M, Akira S, Reeves WH. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savarese E, Steinberg C, Pawar RD, Reindl W, Akira S, Anders HJ, Krug A. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107–1115. doi: 10.1002/art.23407. [DOI] [PubMed] [Google Scholar]
- 9.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. The Journal of experimental medicine. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 11.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, Hudkins KL, Alpers CE, Liggitt D, Rawlings DJ. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. 2014;192:4525–4532. doi: 10.4049/jimmunol.1400098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lartigue A, Courville P, Auquit I, Francois A, Arnoult C, Tron F, Gilbert D, Musette P. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. 2006;177:1349–1354. doi: 10.4049/jimmunol.177.2.1349. [DOI] [PubMed] [Google Scholar]
- 15.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, Reith W, Mohan C, Kotzin BL, Izui S. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34:339–348. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Yu P, Wellmann U, Kunder S, Quintanilla-Martinez L, Jennen L, Dear N, Amann K, Bauer S, Winkler TH, Wagner H. Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int Immunol. 2006;18:1211–1219. doi: 10.1093/intimm/dxl067. [DOI] [PubMed] [Google Scholar]
- 18.Summers SA, Hoi A, Steinmetz OM, O’Sullivan KM, Ooi JD, Odobasic D, Akira S, Kitching AR, Holdsworth SR. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. Journal of Autoimmunity. 2010;35:291–298. doi: 10.1016/j.jaut.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Bossaller L, Rathinam VAK, Bonegio R, Chiang PI, Busto P, Wespiser AR, Caffrey DR, Li QZ, Mohan C, Fitzgerald KA, Latz E, Marshak-Rothstein A. Overexpression of Membrane-Bound Fas Ligand (CD95L) Exacerbates Autoimmune Disease and Renal Pathology in Pristane-Induced Lupus. Journal of Immunology. 2013;191:2104–2114. doi: 10.4049/jimmunol.1300341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, Tall AR. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nundel K, Green NM, Shaffer AL, Moody KL, Busto P, Eilat D, Miyake K, Oropallo MA, Cancro MP, Marshak-Rothstein A. Cell-intrinsic expression of TLR9 in autoreactive B cells constrains BCR/TLR7-dependent responses. J Immunol. 2015;194:2504–2512. doi: 10.4049/jimmunol.1402425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nundel K, Busto P, Debatis M, Marshak-Rothstein A. The role of Bruton’s tyrosine kinase in the development and BCR/TLR-dependent activation of AM14 rheumatoid factor B cells. J Leukoc Biol. 2013;94:865–875. doi: 10.1189/jlb.0313126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nickerson KM, Cullen JL, Kashgarian M, Shlomchik MJ. Exacerbated Autoimmunity in the Absence of TLR9 in MRL.Faslpr Mice Depends on Ifnar1. Journal of immunology (Baltimore, Md: 1950) 2013;190:3889–3894. doi: 10.4049/jimmunol.1203525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, van Rooijen N, Moldawer L, Satoh M, Reeves WH. A novel type I IFN-producing cell subset in murine lupus. J Immunol. 2008;180:5101–5108. doi: 10.4049/jimmunol.180.7.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buechler MB, Teal TH, Elkon KB, Hamerman JA. Cutting edge: Type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J Immunol. 2013;190:886–891. doi: 10.4049/jimmunol.1202739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kettritz R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clinical And Experimental Immunology. 2012;169:220–228. doi: 10.1111/j.1365-2249.2012.04615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heeringa P, Brouwer E, Klok PA, Huitema MG, van den Born J, Weening JJ, Kallenberg CG. Autoantibodies to myeloperoxidase aggravate mild anti-glomerular-basement-membrane-mediated glomerular injury in the rat. Am J Pathol. 1996;149:1695–1706. [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. The Journal of clinical investigation. 2002;110:955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nasr SH, D’Agati VD, Park HR, Sterman PL, Goyzueta JD, Dressler RM, Hazlett SM, Pursell RN, Caputo C, Markowitz GS. Necrotizing and crescentic lupus nephritis with antineutrophil cytoplasmic antibody seropositivity. Clin J Am Soc Nephrol. 2008;3:682–690. doi: 10.2215/CJN.04391007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nassberger L, Sjoholm AG, Jonsson H, Sturfelt G, Akesson A. Autoantibodies against neutrophil cytoplasm components in systemic lupus erythematosus and in hydralazine-induced lupus. Clin Exp Immunol. 1990;81:380–383. doi: 10.1111/j.1365-2249.1990.tb05342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao MH, Liu N, Zhang YK, Wang HY. Antineutrophil cytoplasmic autoantibodies (ANCA) and their target antigens in Chinese patients with lupus nephritis. Nephrol Dial Transplant. 1998;13:2821–2824. doi: 10.1093/ndt/13.11.2821. [DOI] [PubMed] [Google Scholar]
- 32.Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10:463–473. doi: 10.1038/nrrheum.2014.103. [DOI] [PubMed] [Google Scholar]
- 33.Chowdhary VR, Grande JP, Luthra HS, David CS. Characterization of haemorrhagic pulmonary capillaritis: another manifestation of Pristane-induced lupus. Rheumatology (Oxford) 2007;46:1405–1410. doi: 10.1093/rheumatology/kem117. [DOI] [PubMed] [Google Scholar]
- 34.Barker TT, Lee PY, Kelly-Scumpia KM, Weinstein JS, Nacionales DC, Kumagai Y, Akira S, Croker BP, Sobel ES, Reeves WH, Satoh M. Pathogenic role of B cells in the development of diffuse alveolar hemorrhage induced by pristane. Lab Invest. 2011;91:1540–1550. doi: 10.1038/labinvest.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–990. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, Matsumoto M, Akira S, Yoshida N, Miyake K. Unc93B1 Restricts Systemic Lethal Inflammation by Orchestrating Toll-like Receptor 7 and 9 Trafficking. Immunity. 2011;35:69–81. doi: 10.1016/j.immuni.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife. 2013;2:e00291. doi: 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, Alexopoulou L. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U S A. 2014;111:1497–1502. doi: 10.1073/pnas.1314121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nickerson KM, Christensen SR, Cullen JL, Meng W, Luning Prak ET, Shlomchik MJ. TLR9 Promotes Tolerance by Restricting Survival of Anergic Anti-DNA B Cells, Yet Is Also Required for Their Activation. Journal of immunology (Baltimore, Md: 1950) 2013;190:1447–1456. doi: 10.4049/jimmunol.1202115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvani N, Caricchio R, Tucci M, Sobel ES, Silvestris F, Tartaglia P, Richards HB. Induction of apoptosis by the hydrocarbon oil pristane: implications for pristane-induced lupus. Journal of immunology (Baltimore, Md: 1950) 2005;175:4777–4782. doi: 10.4049/jimmunol.175.7.4777. [DOI] [PubMed] [Google Scholar]
- 41.Lee PY, Wang JX, Parisini E, Dascher CC, Nigrovic PA. Ly6 family proteins in neutrophil biology. J Leukoc Biol. 2013;94:585–594. doi: 10.1189/jlb.0113014. [DOI] [PubMed] [Google Scholar]
- 42.Fumagalli L, Zhang H, Baruzzi A, Lowell CA, Berton G. The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J Immunol. 2007;178:3874–3885. doi: 10.4049/jimmunol.178.6.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowell CA, Berton G. Resistance to endotoxic shock and reduced neutrophil migration in mice deficient for the Src-family kinases Hck and Fgr. Proc Natl Acad Sci U S A. 1998;95:7580–7584. doi: 10.1073/pnas.95.13.7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostanin DV, Kurmaeva E, Furr K, Bao R, Hoffman J, Berney S, Grisham MB. Acquisition of antigen-presenting functions by neutrophils isolated from mice with chronic colitis. J Immunol. 2012;188:1491–1502. doi: 10.4049/jimmunol.1102296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oehler L, Majdic O, Pickl WF, Stockl J, Riedl E, Drach J, Rappersberger K, Geissler K, Knapp W. Neutrophil granulocyte-committed cells can be driven to acquire dendritic cell characteristics. J Exp Med. 1998;187:1019–1028. doi: 10.1084/jem.187.7.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, Comerma L, Chorny A, Shan M, Xu W, Magri G, Knowles DM, Tam W, Chiu A, Bussel JB, Serrano S, Lorente JA, Bellosillo B, Lloreta J, Juanpere N, Alameda F, Baro T, de Heredia CD, Toran N, Catala A, Torrebadell M, Fortuny C, Cusi V, Carreras C, Diaz GA, Blander JM, Farber CM, Silvestri G, Cunningham-Rundles C, Calvillo M, Dufour C, Notarangelo LD, Lougaris V, Plebani A, Casanova JL, Ganal SC, Diefenbach A, Arostegui JI, Juan M, Yague J, Mahlaoui N, Donadieu J, Chen K, Cerutti A. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2012;13:170–180. doi: 10.1038/ni.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mills RE, Lam VC, Tan A, Cresalia N, Oksenberg N, Zikherman J, Anderson M, Weiss A, Hermiston ML. Unbiased modifier screen reveals that signal strength determines the regulatory role murine TLR9 plays in autoantibody production. J Immunol. 2015;194:3675–3686. doi: 10.4049/jimmunol.1500026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao L, David MZ, Hyjek E, Chang A, Meehan SM. M2 macrophage infiltrates in the early stages of ANCA-associated pauci-immune necrotizing GN. Clin J Am Soc Nephrol. 2015;10:54–62. doi: 10.2215/CJN.03230314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, Gröne H-J, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nature medicine. 2009;15:623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Science Translational Medicine. 2011;3:73ra20–73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunner HI, Mueller M, Rutherford C, Passo MH, Witte D, Grom A, Mishra J, Devarajan P. Urinary neutrophil gelatinase-associated lipocalin as a biomarker of nephritis in childhood-onset systemic lupus erythematosus. Arthritis Rheum. 2006;54:2577–2584. doi: 10.1002/art.22008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.