

Summary
Conjugated oligomers of 3,4-ethylenedioxythiophene (EDOT) are attractive materials for tissue engineering applications and as model systems for studying the properties of the widely used polymer poly(3,4-ethylenedioxythiophene). We report here the facile synthesis of a series of keto-acid end-capped oligo-EDOT derivatives (n = 2–7) through a combination of a glyoxylation end-capping strategy and iterative direct arylation chain extension. Importantly, these structures not only represent the longest oligo-EDOTs reported but are also bench stable, in contrast to previous reports on such oligomers. The constructs reported here can undergo subsequent derivatization for integration into higher-order architectures, such as those required for tissue engineering applications. The synthesis of hetero-bifunctional constructs, as well as those containing mixed-monomer units, is also reported, allowing further complexity to be installed in a controlled manner. Finally, we describe the optical and electrochemical properties of these oligomers and demonstrate the importance of the keto-acid in determining their characteristics.
Keywords: thiophene, oligomer, conjugated polymer, C-H activation, end capping, synthesis
UN Sustainable Development Goals: SDG3: Good health and well-being, SDG7: Affordable and clean energy
Graphical Abstract
Highlights
-
•
A keto-acid end-capping strategy has been used to create stable oligo-EDOTs
-
•
Oligomers can be synthesized in a facile manner via iterative direct arylation
-
•
Hetero-bifunctional and mixed-monomer constructs can be controllably synthesized
-
•
The keto-acid end group determines oligomer optical and electrochemical properties
The Bigger Picture
The production of materials that can aid the repair, regrowth, or replacement of damaged tissue is a key challenge in tissue engineering. In this context, conjugated polymers have been proposed as attractive materials for the engineering of electroactive tissues such as the heart. Although there has been much progress in the field, the use of conjugated polymers is still hindered by their high heterogeneity, stiffness, poor solubility, and lack of chemical functionality. Therefore, there is a pressing need to produce new routes to create constructs such as those reported here, which offer homogeneity, stability, ease of synthesis, and most importantly, flexibility of design. This versatility allows the incorporation of conjugated structures into the higher-order biomaterial architectures required for tissue engineering, as well as tunable solubility and material properties. It is anticipated that this report will open the door to an exciting new chapter in the use of EDOT in biology.
The synthesis and characterization of a series of keto-acid end-capped conjugated oligomers (n = 2–7) based around the monomer EDOT is reported. The use of direct arylation chain extension allows the synthesis of stable structures, which represent the longest reported EDOT oligomers to date with tunable properties based around the versatile end-capping group and monomer composition. These constructs can undergo subsequent derivatization, allowing them to be integrated into functional materials, such as those required for tissue engineering applications.
Introduction
Conjugated polymers (CPs) are promising materials for tissue engineering applications.1, 2, 3, 4 However, further developments are required in order to allow their full potential to be realized in the biomedical field. Although initial investigations have shown CPs to be able to modulate cellular growth,5 migration,6 and differentiation,7, 8 as well as protein adhesion and conformation,9 difficulties remain as a consequence of their poor material characteristics, difficult processing, and lack of biodegradability.1, 2, 10 Further, the production of constructs bearing reactive functionalities for integration into more complex scaffold architectures remains challenging.2
In order to address these issues, there is increasing interest in the use of oligomers rather than polymeric systems. Although oligomers are often more synthetically complex,11 they offer the benefits of a defined molecular structure, improved solubility, tunability, and additional chemical functionality.2, 12 Oligomers can also act as mono-disperse model systems for studying the electronic and optical properties of the parent polymer, for which such investigations can be hindered.13
Poly(3,4-ethylenedioxythiophene) (PEDOT) is a particularly attractive material for tissue engineering because of its electrical and chemical stability and high conductivity when doped with polymeric ionomers such as polystyrene sulfonate.14, 15 Although the synthesis of thiophene-based oligomers has been widely reported,11, 16, 17, 18, 19 those of EDOT (1; Scheme 1) have generated comparatively little interest, largely as a consequence of the poor oxidative stability and low solubility of the oligomers.20, 21 Mesityl,22 phenyl,21 n-hexyl,23 and trimethylsilyl24 capping groups have all been reported. However, longer oligomers were found to be unstable in solution, very poorly soluble, and difficult to purify, limiting their utility. Indeed, there remains only a single report on the synthesis of a pentameric species, but no synthetic details were reported24 (Figure 1A). Furthermore, the end caps utilized offer no opportunities for further chemical derivatization and subsequent incorporation into more complex structures.
Scheme 1.

EDOT Glyoxylation and Functionalization
(A) Treatment of EDOT with oxalyl chloride and subsequent treatment with the desired nucleophile generates functional end-capped derivatives, which can then undergo bromination (route A). Alternatively, amide or ester coupling can be undertaken from a common intermediate 10 to give the desired monomers (route B).
(B) Functionalized brominated monomers synthesized.
Figure 1.

Previous EDOT End Caps and the Concept of This Report
(A) Previous reports of the synthesis of EDOT oligomers.
(B) Keto-acid-capped oligomers presented in this work. These oligomers were synthesized by direct arylation.
Here, we report the facile synthesis and characterization of bench-stable oligo-EDOT derivatives, up to n = 7, produced via a glyoxylation keto-acid end-capping strategy and iterative C–H activation chemistry. Importantly, this allows the production of hetero-bifunctional constructs with a wide range of functional handles for further modification (Figure 1B). These motifs allow additional integration into more challenging substrates, such as those required for tissue engineering applications.
Results and Discussion
Oligomer Synthesis
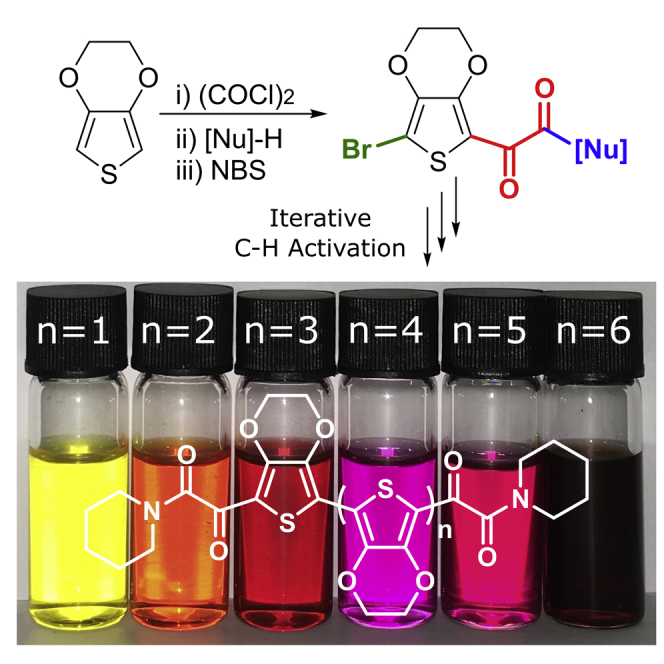
Our initial designs were inspired by reports of thiophene glyoxylation with oxalyl chloride.25 We reasoned that the intermediate glyoxylyl chloride 2 could be reacted in situ with a range of nucleophiles to generate α-functionalized EDOT derivatives (Scheme 1). Importantly, the choice of nucleophile would have little influence on aromatic stability, allowing for a range of diverse constructs to be produced. After treatment of EDOT with 1 equiv of oxalyl chloride, the intermediate chloride 2 reacted smoothly with piperidine to generate the tertiary keto-amide 3 (Scheme 1A; Figures S6 and S7) in good yield. Subsequent bromination with N-bromosuccinimide yielded the di-functional monomer 4 on a multi-gram scale (Figures S8 and S9).22
A range of functionalized monomers could be produced by this method, including secondary amines (5), hindered tertiary amines (6), esters (7, 8, and 9), and monomers bearing functional groups for further modifications (Scheme 1, route A; Figures S10–S19, S73, S74, S91, S92, S99, S100, and S129–S132). In addition, hydrolysis of brominated-EDOT methyl ester 7 and subsequent amide or ester coupling allowed the synthesis of a range of di-functional monomers from a common intermediate 10 (Scheme 1, route B; Figures S20 and S21). Thus, monomers containing orthogonal reactive groups for further conjugation, such as alkynes (11), alkenes (12), azides (13), and protected alcohols (14), thiols (15), and amines (16), could all be produced in good yields in a simple fashion (Figures S22–S37, S125–S128, and S133–S150).26
Next, we investigated the chain extension of brominated monomer 4 to form dimer 19. The most popular strategies for undertaking such reactions utilize Kumada,27 Negishi,28 or Stille29 couplings. However, problems such as poor functional-group tolerance, monomer instability, and high reagent toxicity result in significant limitations, particularly for use in biological applications.22, 30, 31 As such, we chose to investigate the use of direct arylation, which has emerged in recent years as a powerful tool for constructing conjugated systems.32, 33 Pleasingly, 4 was found to be partially converted to 19 in the presence of 1.5 equiv of EDOT 1 in N,N-dimethylformamide (DMF) at 130°C for 1 hr (Scheme 2A; Figures S38 and S39). Importantly, the reaction was catalyzed by a readily available combination of Pd(OAc)2, pivalic acid, and potassium carbonate, thus negating the need for expensive or air-sensitive catalysts and ligands or the use of specialist techniques.34
Scheme 2.

Synthesis of Piperidine End-Capped Homo-bifunctional EDOT Oligomers
(A) Chain extension of mono-functional (n = 1–3) piperidine-capped oligomers.
(B) Convergent coupling to generate bifunctional piperidine end-capped constructs (n = 2–7).
Investigating the reaction further, we found yields to be increased significantly through the use of 4 equiv of EDOT, the excess of which could be readily re-isolated through column chromatography. At lower loadings, a significant amount of the symmetrical di-capped trimer 20 was produced as a result of further reaction of 19 with 4 (Figures S40 and S41). Although small amounts of this side product were still produced at higher EDOT loadings, yields were significantly lowered, and separation was readily achieved. Further iterations of bromination and direct arylation allowed the production of brominated dimer 21 and trimer 22 on a gram scale, both of which were found to be bench stable (Figures S42–S45). Bromination to form brominated trimer 23 was also possible, although its low solubility and stability prevented characterization and required its immediate use once prepared, as discussed later.
With these mono-capped building blocks in hand, we investigated the synthesis of di-capped oligomers (Scheme 2B). Heating a mixture of brominated and non-brominated monomers 4 and 3 (1.1 equiv) under the same conditions required for chain extension cleanly produced di-capped dimer 24 (Figures S46 and S47). Similarly, trimer 20 was produced from 4 and dimer 19. Alternatively, 20 could be produced from the reaction of 2 equiv of either monomer 3 or brominated monomer 4 with 2,5-dibromo-EDOT 25 or EDOT 1, respectively, in an optimized version of the previously discussed chain-extension side reaction.
By suitable choice of starting materials, di-capped oligomers (n = 2–5; 24, 20, 26, and 27) were all readily produced and easily isolated by column chromatography (Figures S48–S50). Extending the scope further to the use of brominated trimer 23, used immediately without purification, allowed the synthesis of hexamer 28, whereas coupling of trimer 22 with 2,5-dibromo-EDOT 25 allowed the synthesis of heptamer 29, the first time the synthesis of EDOT oligomers of such lengths has been reported (Figures S51 and S52). Oligomers up to n = 6 were found to be bench and air stable and therefore could be easily handled, purified, and analyzed; no change in structure was observed by UV-Vis or 1H-NMR spectroscopy after 2 months of storage at room temperature. Heptamer 29 was produced with reduced purity (∼80% as judged by 1H NMR) but retained stability. Although oligomers of n = 2–5 were also found to be stable in solution, after long periods in chlorinated solvents (>2 weeks), a broadened UV-Vis absorption indicated that hexamer 28 and heptamer 29 had undergone partial degradation.
Oligomer solubility was found to decrease with increasing chain length, and aggregation in solution became significant at longer lengths. However, it remained high enough to allow manipulation in solution and the use of typical synthetic techniques such as phase extraction and column chromatography. Oligomers of n = 2–5 were soluble at concentrations of >20 mM in dichloromethane (DCM), and hexamer 28 was soluble at concentrations of >5 mM, whereas heptamer 29 could be solubilized at concentrations up to 0.5 mM. It is important to note that solubility is strongly influenced by the choice of end group and can be readily improved by the introduction of a flexible solubilizing linker to the functional group of interest, as discussed later. Finally, we analyzed oligomers 20 and 26 by inductively coupled plasma mass spectrometry (ICP-MS) to determine the levels of residual palladium present. As for other heavy metals, palladium contamination in pharmaceuticals and biomedical devices is tightly regulated because of the potential for toxic side effects. Palladium contamination was found to be at a low level of 7.4 ± 0.5 ppm for trimer 20 and 1.2 ± 0.5 ppm for tetramer 26. Although it is difficult to make a direct comparison between a substrate intended for applications in tissue engineering and an active pharmaceutical ingredient (API), it is useful to note that these low levels of contamination are below the 10 ppm limit set by the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use and the US Pharmacopeia for acceptable levels of palladium in APIs.35 Furthermore, because no extensive effort was taken to remove palladium from the samples, it is likely that these levels could be reduced further. For example, the use of palladium chelators during purification or the use of heterogeneous catalysts would be expected to lead to a significant reduction in contamination in any structures intended for biological applications.36, 37, 38
Although the ability to create symmetrical oligo-EDOTs with non-functional end groups is a useful tool for modeling the properties of PEDOT, the true utility of the method described above for the synthesis of di-piperidine-capped oligomers is in the synthesis of hetero-bifunctional constructs, which can be selectively derivatized at both ends, allowing their integration into more complex architectures. To demonstrate this, we first synthesized a series of unsymmetrical oligomers capped with a piperidine motif at one terminus and diisopropylamine at the other (see Scheme S1). Coupling differently terminated oligomers as described above produced oligomers of n = 2–5 (30–33) in a limited number of steps (Figures S53–S59 and S93–S98).
During these experiments, a number of observations were made. Firstly, although a temperature of 130°C was required for the chain extension and oligomer synthesis with brominated piperidine-based species, for diisopropyl-functionalized oligomers, 90°C was found to be sufficient to give complete conversion within 1 hr of reaction, leading to cleaner reaction products. Indeed, for all other end-capping groups investigated during this work, 90°C was high enough to facilitate reaction.39 Secondly, although couplings generally proceeded cleanly, the amount of side products produced increased with increasing oligomer length. The major side product was found to stem from the instability of the brominated species, resulting in partial dehalogenation and subsequent homo-coupling and, to a lesser extent, homo-coupling of the non-brominated reaction partner. Such side reactions have been studied extensively40 and are also known to occur during Stille and Suzuki polymerizations.41 Although outside the scope of this work (which focuses on the use of unoptimized, simple, and cheap catalyst systems), it is likely that such products could be minimized through judicious choice of both metal and ligand.42
To create functional oligomers primed for further reaction and derivatization, we considered that a number of common reactive handles would not be amenable to the chain extension and bromination procedures described above.43 It would therefore be advantageous to be able to install functionality at a late stage after oligomer synthesis. Thus, we investigated the use of orthogonal ester-protecting groups to provide latent functionality. Initial attempts to react methyl ester 7 with an excess of EDOT 1 led not only to chain extension but also to a significant amount (∼40%) of ester cleavage (see Scheme S2A). However, switching to iso-propyl ester 8 lead to a clean conversion to dimer 34 at 90°C, followed by subsequent bromination and extension to yield trimer 35 (reaction at 130°C as described for piperidine oligomers led to complete ester cleavage; see Scheme S2B; Figures S60–S63, S101, and S102). Similarly, the orthogonally protected tert-butyl ester 9 could undergo iterative chain extension and bromination to yield brominated dimer 36 (Figures S64, S65, and S111–S114).
With these substrates in hand, we were able to synthesize di-capped, orthogonally protected oligomers 37–40 with n = 2–5 in a short number of steps and in good yields (Scheme 3A; see Scheme S2C for full details and Figures S66–S71). Although the synthesis of tetramer 39 and pentamer 40 was confirmed by mass spectrometry, the propensity of the constructs to aggregate in solution prevented analysis by 13C NMR. As an alternative, constructs possessing a solubility-enhancing triethylene glycol chain could also be produced as discussed above (41; Scheme 3B; Figures S72 and S103–S110). Here, the significant difference in end-group polarity greatly aided purification, offering a potential means of enhancing purity during particularly difficult separations. This representative example demonstrates an important advantage of the synthesis reported in this work; because the choice of end group is an important determinant in the material properties of the synthesized constructs, simply choosing an appropriate end cap can alter factors such as the solubility of the material to reflect the desired application.
Scheme 3.

Synthesis of Orthogonal-Ester-Functionalized Hetero-bifunctional Oligomers
(A) Synthesis of unsymmetrical, orthogonally protected oligo-EDOT diesters 37–40 with iso-propyl and tert-butyl end groups.
(B) Triethylene glycol ester-capped tetra-EDOT 41 with improved solubility.
Amide coupling after sequential ester deprotection, first in the presence of trifluoroacetic acid to remove the tert-butyl group and then in the presence of sodium hydroxide to cleave the iso-propyl ester, allowed the subsequent synthesis of unsymmetrical constructs bearing reactive functionality for further modification (see Scheme S3; Figures S115 and S116). As a result of the mild amide- or ester-forming conditions required, this method is applicable to the late-stage hetero-functionalization of the oligomers reported with a wide range of reactive or functional groups, such as those shown in Scheme 1. The potential applications of this methodology are diverse. The ability to create hetero-bifunctional oligomers of a tunable length and bearing handles for further modification allows the modular synthesis of more complex structures. For example, the integration of such constructs into biologically active scaffolds2 or the production of amphiphilic, self-assembling morphologies44, 45 offers exciting possibilities in the fields of both the material and biomedical sciences.
Finally, we wished to investigate the application of our methodology to the synthesis of mixed oligomers composed of different monomer units, which could possess interesting properties. In particular, we considered the rigidity of EDOT oligomers, which are known to lead to highly planar structures with enhanced π conjugation.23 We reasoned that disrupting planarity in a controlled fashion could tune the properties of the resultant material. Structurally related dialkoxythiophene monomers such as 3,4-dimethoxythiophene (DMT, 43) and 3,4-propylenedioxythiophene (ProDOT, 44) were found to be suitable substrates for our glyoxylation and chain-extension procedures. We therefore introduced a single DMT moiety in an EDOT-pentameric structure to create three structural isomers: 45, 46, and 47 (Scheme 4; see Scheme S4 for full details and Figures S75–S83 and S117–S124). The simple manner in which such compounds can be created allows the rapid construction of a library of dialkoxythiophene-based constructs for investigating the effects of structure, substituents, and isomerization on the chemical and electrical properties of CPs.
Scheme 4.

Synthesis of Dimethoxythiophene-Containing Isomers
Pentameric EDOT oligomers containing a single DMT unit were synthesized for the generation of the structural isomers 45, 46, and 47.
Oligomer Characterization
Solutions of the di-piperidine-capped oligomers described above (24, 20, and 26–29) in DCM were analyzed by UV-Vis and fluorescence spectroscopy. Within the range investigated, the optical properties of the materials were found to be independent of concentration, indicating that aggregation was not occurring. As expected, a gradual red shift in the onset of absorbance was observed with increasing chain length (Figure 2), although a blue shift in absorbance maxima for heptamer 29 was observed, most likely because of the presence of impurities in the sample. Furthermore, the spectra possessed well-defined vibronic structures, a widely reported feature of EDOT oligomers not shared by unsubstituted thiophene structures.21, 23, 46 When compared with the parent C–H capped oligomers biEDOT 51 and terEDOT 52, mono-piperidine-capped dimer 19 and trimer 22 displayed a large red shift in absorbance (see Figure S1). This effect was even more pronounced for the di-capped oligomers 24 and 20. A red shift in absorbance of >100 nm indicated that conjugation of the thiophene core with the keto-acid end group, to create an acceptor-donor-acceptor triad, played a major role in influencing the properties of the synthesized oligomers, leading to a significant narrowing of the optical gap (Eopt).47, 48
Figure 2.

Oligomer Optical Characterization
(A) Normalized UV-Vis (solid line) and fluorescence (dashed line) spectra of di-piperidine-capped oligomers 24, 20, and 26–29.
(B) Correlation of inverse chain length and Eopt for oligomers 24, 20, and 26–29 (adjusted R2 = 0.9828).
(C) Summary of Eopt for a series of di-functionalized oligomers.
When compared with those of previously reported EDOT end-capped oligomers, the absorption spectra were strongly red shifted in relation to the corresponding mesityl, phenyl, hexyl, and trimethylsilyl structures highlighted in Figure 1.21, 22, 23, 24 The remarkably low-energy Eopt of the structures reported here is considered to be a consequence of the lowering in energy of the lowest unoccupied molecular orbital (LUMO) as a result of the electron-withdrawing nature of the keto-acid moiety, as discussed later. Oligomer capping with primary amines to yield secondary amides was found to result in a further lowering of Eopt (Figure 2C, entry 8; Figures S2 and S151–S158). This effect was enhanced through capping with more electron-poor ester groups, resulting in an Eopt as low as 1.88 eV for the iso-propyl ester di-capped pentamer 56 (Figure 2C, entry 10; Figures S84–S86 and S159–S161).
The constrained six-membered ring of EDOT is known to result in favorable attractive intramolecular S–O interactions between repeating units.23, 49 This effect is reduced upon the introduction of the more structurally flexible methoxy units of DMT. Therefore, as predicted, the introduction of a single DMT residue into an EDOT pentamer led to an increase in Eopt as a result of disruption of the highly planar EDOT-repeating structure. This effect was found to be position dependent such that the length of the longest continuous EDOT chain determined the degree of disruption. When compared with the pentaEDOT oligomer 27, DMT-containing isomer 45 (four continuous residues) exhibited a ΔEopt = +0.013 eV, whereas isomer 47 (two continuous residues) possessed an increased ΔEopt = +0.057 eV (Figure 2C, entries 11–13). This widening of the optical gap was further enhanced in an oligomer consisting of end-capped penta-DMT 57 (ΔEopt = +0.122 eV) or the analogous penta-ProDOT oligomer 58 (ΔEopt = +0.44 eV) (Figure 2C, entries 14 and 15; Figures S87, S88, and S164–S173). These results support our hypothesis that the oligomer properties can be tuned through the suitable choice and positioning of alternative monomer units.
Next, we investigated the solution electrochemical properties of selected oligomers by cyclic voltammetry. Di-piperidine-capped oligomers 24, 20, and 26–28 (n = 2–6) were all investigated. However, because of the low solubility of EDOT-heptamer 29 and its reduced purity, weak signal intensity was observed during measurements, and therefore this structure was not further investigated. Cyclic voltammograms (CVs) demonstrated a decrease in the first oxidation potential with increasing chain length, supporting the results obtained by UV-Vis spectroscopy (Figure 3A). Linear correlations were found between the first and second oxidation potentials and the inverse chain length (Figure 3B; see Table S1). The oxidation of oligomers 24, 20, and 26 (n = 2–4) was electrochemically quasi-reversible, whereas pentamer 27 and hexamer 28 displayed improved electrochemical reversibility (Figure 3). Furthermore, CVs of penta-DMT 57 and penta-ProDOT 58 allowed comparison with penta-EDOT 27 (see Figure S3). As was seen for the optical gap, the first oxidation potential was found to follow the trend EDOT < ProDOT < DMT. These results further support the higher effective conjugation of EDOT oligomers and a degree of planarity disruption induced by the high torsional strain of DMT-based structures.50 The ease with which the oxidation potentials can be tuned, through both alteration of oligomer length and monomer composition, offers intriguing possibilities for applications not only in tissue engineering but also in creating sensitive and selective organic bioelectronics.51, 52
Figure 3.

Cyclic Voltammetry Characterization
(A) Cyclic voltammograms of piperidine-capped oligomers 24, 20, and 26–28. CVs were recorded at a scan rate of 100 mV s−1 with oligomer concentrations of 1 mM in DCM containing 0.1 M Bu4NPF6.
(B) Correlation of inverse chain length and first and second oxidation potentials for oligomers 24, 20, and 26–28 (adjusted R2 = 0.9680 and 0.9725, respectively).
Finally, we undertook computational density functional theory (DFT) calculations to further probe the influence of the keto-acid end groups on oligomer properties.53 The trends observed in the calculated HOMO-LUMO gaps during these studies reproduced the structural and length dependencies observed during experimental measurements. Initial calculations on carboxy-terminated EDOT pentamer 59 validated our hypothesis that the keto-acid end group played an important role in extending π conjugation (Figure 4). This was particularly true for the LUMO—the electron-withdrawing nature of the end group led to a large orbital localization across the ketone group. Partial distribution of the LUMO across the terminal carboxyl indicated that the choice of an ester or amide linkage might influence the electrical properties of oligomeric constructs. Thus, compared with an analogous amide substrate, the presence of a more electron-deficient ester group would be expected to lower the LUMO level, leading to a decreased HOMO-LUMO gap (see Figure S4). This supports our experimental observation of a lower Eopt for iso-propyl ester di-capped oligomers than for amide-capped structures.
Figure 4.
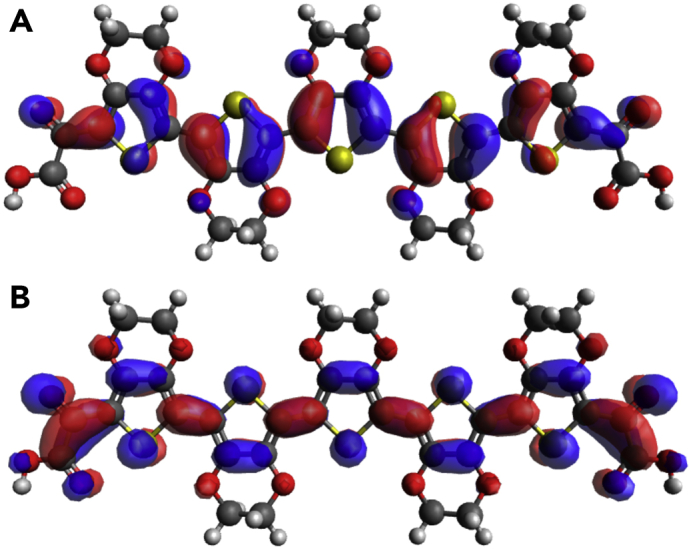
DFT Orbital Projections
(A) HOMO orbital distribution of carboxy-capped pentamer 59.
(B) LUMO orbital distribution.
DFT also provided rationale for the increase in Eopt observed for tertiary-amide-capped structures. To accommodate the steric bulk of both the piperidine and diisopropylamine substituents, the dicarbonyl groups were found to be significantly disrupted from the antiperiplanar orientation observed for other substituents. This led to dihedral angles of as little as 131° for diisopropyl-capped dimer 60 and 142° for piperidine-capped dimer 24 (see Figures S5, S89, S90, S162, and S163). As a result, conjugation was partially disrupted, leading to an increase in the HOMO-LUMO gap, supporting the observed increase in Eopt. Replacement of EDOT with DMT or ProDOT offered two different mechanisms by which disruption of the expected planar configuration could potentially occur. In the case of DMT, the high torsional strain of consecutive units was found to lead to a slight twisting of the backbone for longer oligomer structures, therefore decreasing effective conjugation. In contrast, calculations predicted a slight deflection of the alkoxy substituents in the ProDOT structure (174° and 180° dihedral angle in EDOT and DMT, respectively) to accommodate an expanded seven-membered ring. The resultant cumulative decrease in electron donation from these substituents might explain the slight increase in Eopt observed for the ProDOT derivatives described above.
Conclusions
We have developed a glyoxylation end-capping strategy that allows the rapid installation of keto-amides and keto-esters at the end of oligomeric-EDOT chains. The resultant materials retain solubility and are bench stable, in contrast to previous reports of oligo-EDOT derivatives. These developments allow us to report the synthesis of hexa- and heptameric EDOT constructs for the first time. Furthermore, the use of iterative chain extension allows the construction of hetero-bifunctional constructs bearing orthogonally reactive handles for further modification. Characterization of the structures produced demonstrated the important role played by the keto-acid end group in determining oligomer properties. The remarkably low optical gap observed for the oligomeric structures was attributed to the important role played by the extended conjugated system, particularly in lowering the LUMO energy, as demonstrated by DFT calculations. Notably, through suitable choice of oligomer length, end group, and monomer composition, the optical, electronic, and physical properties of a construct can be readily tuned both across a wide range and with fine control. This ability to undertake a flexible and modular approach to structural design creates intriguing opportunities in the synthesis of novel materials. Work to explore the full possibilities of this powerful methodology is currently ongoing in our group for the integration of tunable conjugated materials into tissue engineering scaffolds.
Experimental Procedures
General Method for EDOT Glyoxylation
Oxalyl chloride (850 μL, 10 mmol) was added drop-wise to a solution of EDOT (1.05 mL, 10 mmol) in dioxane (30 mL). The mixture was heated to 100°C for 1 hr and then allowed to cool to room temperature. The requisite amine (15 mmol) and base (50 mmol) were then added, and the mixture was stirred for 3 hr. After this time, the mixture was diluted with DCM (150 mL) and washed with water (100 mL), and the organics were dried with MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography, and pure fractions were concentrated in vacuo.
General Method for Monomer Bromination
EDOT derivative (5 mmol) was dissolved in a mixture of tetrahydrofuran (THF, 5 mL) and acetic acid (3 mL). If solubility was poor, a further 25 mL of THF was added. The mixture was placed in the dark, and N-bromosuccinimide (6 mmol) was added. After being stirred for 2 hr, the mixture was either precipitated in water (150 mL), causing precipitation of the product, which could be collected by filtration, or diluted with DCM (150 mL) and washed with saturated NaHCO3 (3 × 100 mL), dried with MgSO4, filtered, and concentrated in vacuo. Column chromatography was then undertaken if required, although the products were usually sufficiently pure for further use.
General Method for Chain Extension
Brominated monomer (1 mmol), pivalic acid (0.5 mmol), palladium(II) acetate (0.05 mmol), and potassium carbonate (10 mmol) were charged under nitrogen. Dry DMF (2 mL) and EDOT (4 mmol) were then added, and the mixture was heated to 90°C for 2 hr. After cooling to room temperature, the mixture was diluted with DCM (50 mL) and washed with water (2 × 50 mL) and brine (50 mL). The organics were dried with MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography, and pure fractions were concentrated in vacuo.
General Method for Oligomer Synthesis
Brominated oligomer (1 mmol), hydrogen-capped oligomer (1.2 mmol), pivalic acid (0.5 mmol), palladium(II) acetate (0.05 mmol), and potassium carbonate (10 mmol) were charged under nitrogen. Dry DMF (2 mL) was added, and the mixture was heated to 90°C for 2 hr. After cooling to room temperature, the mixture was diluted with DCM (50 mL) and washed with water (2 × 50 mL) and brine (50 mL). The organics were dried with MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography, and pure fractions were concentrated in vacuo.
Author Contributions
C.D.S. performed all experiments and wrote the manuscript. M.A.B. performed CV measurements. A.A. performed UV-Vis measurements. C.D.S., M.A.B., A.A., and C.B.N. analyzed and interpreted data. C.D.S., D.M., and M.M.S. developed the ideas. All authors commented on the manuscript. M.M.S. supervised the project.
Acknowledgments
Prof. John McArthur of the ICP Cross-Faculty Elemental Analysis Facility and Dr. Kersti Karu of the Mass Spectrometry Facility at University College London are thanked for running ICP-MS analyses. Prof. M. Heeney and Drs. A.G. Guex and C.S. Wood are thanked for critical evaluation of the manuscript. M.M.S. and C.D.S. acknowledge the British Heart Foundation Cardiovascular Regenerative Medicine Center (RM/13/1/30157). D.M. was supported by Marie Skłodowska-Curie Actions under the Seventh Framework Programme (FP7) through the Intra-European Marie Curie Fellowship “MultiFun CP” under grant agreement no. 328897. M.A.B. was supported by the Freemasons Foundation of New Zealand through the Royal Society of New Zealand-Rutherford Foundation. M.M.S. acknowledges support from the ERC FP7 Consolidator Grant “Naturale CG” under grant agreement no. 616417 and a Wellcome Trust Senior Investigator Award (098411/Z/12/Z) for funding. The research data supporting this publication are available online at https://doi.org/10.5281/zenodo.163505.
Published: January 12, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, 173 figures, 2 tables, and 4 schemes and can be found with this article online at http://dx.doi.org/10.1016/j.chempr.2016.12.003.
Supplemental Information
References and Notes
- 1.Guimard N.K., Gomez N., Schmidt C.E. Conducting polymers in biomedical engineering. Prog. Polym. Sci. 2007;32:876–921. [Google Scholar]
- 2.Guo B., Glavas L., Albertsson A.-C. Biodegradable and electrically conducting polymers for biomedical applications. Prog. Polym. Sci. 2013;38:1263–1286. [Google Scholar]
- 3.Balint R., Cassidy N.J., Cartmell S.H. Conductive polymers: towards a smart biomaterial for tissue engineering. Acta Biomater. 2014;10:2341–2353. doi: 10.1016/j.actbio.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Pashuck E.T., Stevens M.M. Designing regenerative biomaterial therapies for the clinic. Sci. Transl. Med. 2012;4:1–12. doi: 10.1126/scitranslmed.3002717. [DOI] [PubMed] [Google Scholar]
- 5.Wong J.Y., Langer R., Ingber D.E. Electrically conducting polymers can noninvasively control the shape and growth of mammalian cells. Proc. Natl. Acad. Sci. USA. 1994;91:3201–3204. doi: 10.1073/pnas.91.8.3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gumus A., Califano J.P., Wan A.M.D., Huynh J., Reinhart-King C.A., Malliaras G.G. Control of cell migration using a conducting polymer device. Soft Matter. 2010;6:5138–5142. [Google Scholar]
- 7.Gilmore K.J., Kita M., Han Y., Gelmi A., Higgins M.J., Moulton S.E., Clark G.M., Kapsa R., Wallace G.G. Skeletal muscle cell proliferation and differentiation on polypyrrole substrates doped with extracellular matrix components. Biomaterials. 2009;30:5292–5304. doi: 10.1016/j.biomaterials.2009.06.059. [DOI] [PubMed] [Google Scholar]
- 8.Srivastava N., Venugopalan V., Divya M.S., Rasheed V.A., James J., Narayan K.S. Neuronal differentiation of embryonic stem cell derived neuronal progenitors can be regulated by stretchable conducting polymers. Tissue Eng. Part A. 2013;19:1984–1993. doi: 10.1089/ten.tea.2012.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan A.M.D., Schur R.M., Ober C.K., Fischbach C., Gourdon D., Malliaras G.G. Electrical control of protein conformation. Adv. Mater. 2012;24:2501–2505. doi: 10.1002/adma.201200436. [DOI] [PubMed] [Google Scholar]
- 10.Green R.A., Hassarati R.T., Goding J.A., Baek S., Lovell N.H., Martens P.J., Poole-Warren L.A. Conductive hydrogels: mechanically robust hybrids for use as biomaterials. Macromol. Biosci. 2012;12:494–501. doi: 10.1002/mabi.201100490. [DOI] [PubMed] [Google Scholar]
- 11.Koch F.P.V., Smith P., Heeney M. “Fibonacci’s route” to regioregular oligo(3-hexylthiophene)s. J. Am. Chem. Soc. 2013;135:13695–13698. doi: 10.1021/ja4057932. [DOI] [PubMed] [Google Scholar]
- 12.Lin Y., Zhan X. Oligomer molecules for efficient organic photovoltaics. Acc. Chem. Res. 2016;49:175–183. doi: 10.1021/acs.accounts.5b00363. [DOI] [PubMed] [Google Scholar]
- 13.Müllen K., Wegner G. Wiley-VCH; 1998. Electronic Materials: The Oligomer Approach. [Google Scholar]
- 14.Yamato H., Ohwa M., Wernet W. Stability of polypyrrole and poly(3,4-ethylenedioxythiophene) for biosensor application. J. Electroanal. Chem. 1995;397:163–170. [Google Scholar]
- 15.Strakosas X., Sessolo M., Hama A., Rivnay J., Stavrinidou E., Malliaras G.G., Owens R.M. A facile biofunctionalisation route for solution processable conducting polymer devices. J. Mater. Chem. B. 2014;2:2537–2545. doi: 10.1039/c3tb21491e. [DOI] [PubMed] [Google Scholar]
- 16.Izumi T., Kobashi S., Takimiya K., Aso Y., Otsubo T. Synthesis and spectroscopic properties of a series of b-blocked long oligothiophenes up to the 96-mer: revaluation of effective conjugation length. J. Am. Chem. Soc. 2003;125:5286–5287. doi: 10.1021/ja034333i. [DOI] [PubMed] [Google Scholar]
- 17.Kreyes A., Amirkhani M., Lieberwirth I., Mauer R., Laquai F., Landfester K., Ziener U. The longest β-unsubstituted oligothiophenes and their self-assembly in solution. Chem. Mater. 2010;22:6453–6458. [Google Scholar]
- 18.Zhang L., Colella N.S., Liu F., Trahan S., Baral J.K., Winter H.H., Mannsfeld S.C., Briseno A.L. Synthesis, electronic structure, molecular packing/morphology evolution, and carrier mobilities of pure oligo-/poly(alkylthiophenes) J. Am. Chem. Soc. 2013;135:844–854. doi: 10.1021/ja3104796. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L., Colella N.S., Cherniawski B.P., Mannsfeld S.C.B., Briseno A.L. Oligothiophene semiconductors: synthesis, characterization, and applications for organic devices. ACS Appl. Mater. Inter. 2014;6:5327–5343. doi: 10.1021/am4060468. [DOI] [PubMed] [Google Scholar]
- 20.Sotzing G.A., Reynolds J.R., Steel P.J. Poly(3,4-ethylenedioxythiphene) (PEDOT) prepared via electrochemical polymerization of EDOT, 2,2’-Bis(3,4-ethylenedioxythiophene) (BiEDOT), and their TMS derivatives. Adv. Mater. 1997;9:795–798. [Google Scholar]
- 21.Apperloo J.J., Groenendaal L.B., Verheyen H., Jayakannan M., Janssen R.A.J., Dkhissi A., Beljonne D., Lazzaroni R., Brédas J.L. Optical and redox properties of a series of 3,4-ethylenedioxythiophene oligomers. Chemistry. 2002;8:2384–2396. doi: 10.1002/1521-3765(20020517)8:10<2384::AID-CHEM2384>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 22.Hicks R.G., Nodwell M.B. Synthesis and electronic structure investigations of alpha,omega-bis(arylthio)oligothiophenes: toward understanding wire-linker interactions in molecular-scale electronic materials. J. Am. Chem. Soc. 2000;122:6746–6753. [Google Scholar]
- 23.Turbiez M., Frère P., Roncali J. Stable and soluble oligo(3,4-ethylenedioxythiophene)s end-capped with alkyl chains. J. Org. Chem. 2003;68:5357–5360. doi: 10.1021/jo0345493. [DOI] [PubMed] [Google Scholar]
- 24.Wasserberg D., Meskers S.C.J., Janssen R.A.J., Mena-Osteritz E., Bäuerle P. High-resolution electronic spectra of ethylenedioxythiophene oligomers. J. Am. Chem. Soc. 2006;128:17007–17017. doi: 10.1021/ja066920k. [DOI] [PubMed] [Google Scholar]
- 25.Merkul E., Dohe J., Gers C., Rominger F., Müller T.J.J. Three-component synthesis of ynediones by a glyoxylation/Stephens-Castro coupling sequence. Angew. Chem. Int. Ed. Engl. 2011;50:2966–2969. doi: 10.1002/anie.201007194. [DOI] [PubMed] [Google Scholar]
- 26.Spicer C.D., Davis B.G. Selective chemical protein modification. Nat. Commun. 2014;V5:P4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- 27.Stefan M.C., Bhatt M.P., Sista P., Magurudeniya H.D. Grignard metathesis (GRIM) polymerization for the synthesis of conjugated block copolymers containing regioregular poly(3-hexylthiophene) Polym. Chem. 2012;3:1693–1701. [Google Scholar]
- 28.Xu S., Kim E.H., Wei A., Negishi E. Pd- and Ni-catalyzed cross-coupling reactions in the synthesis of organic electronic materials. Sci. Technol. Adv. Mater. 2014;15:44201. doi: 10.1088/1468-6996/15/4/044201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carsten B., He F., Son H.J., Xu T., Yu L. Stille polycondensation for synthesis of functional materials. Chem. Rev. 2011;111:1493–1528. doi: 10.1021/cr100320w. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H., Liu C.-Y., Luo S.-C., Zhu B., Wang T.-H., Hsu H.-F., Yu H.-H. Facile syntheses of dioxythiophene-based conjugated polymers by direct C–H arylation. Macromolecules. 2012;45:7783–7790. [Google Scholar]
- 31.Vechorkin O., Proust V., Hu X. Functional group tolerant Kumada-Corriu-Tamao coupling of nonactivated alkyl halides with aryl and heteroaryl nucleophiles: catalysis by a nickel pincer complex permits the coupling of functionalized Grignard reagents. J. Am. Chem. Soc. 2009;131:9756–9766. doi: 10.1021/ja9027378. [DOI] [PubMed] [Google Scholar]
- 32.Schipper D.J., Fagnou K. Direct arylation as a synthetic tool for the synthesis of thiophene-based organic electronic materials. Chem. Mater. 2011;23:1594–1600. [Google Scholar]
- 33.Segawa Y., Maekawa T., Itami K. Synthesis of extended π-systems through C-H activation. Angew. Chem. Int. Ed. Engl. 2015;54:66–81. doi: 10.1002/anie.201403729. [DOI] [PubMed] [Google Scholar]
- 34.Lafrance M., Fagnou K. Palladium-catalyzed benzene arylation: incorporation of catalytic pivalic acid as a proton shuttle and a key element in catalyst design. J. Am. Chem. Soc. 2006;128:16496–16497. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]
- 35.United States Phamacopeia Convention . United States Pharmacopeia; 2015. <232> Elemental Impurities - Limits. [Google Scholar]
- 36.Torborg C., Beller M. Recent applications of palladium-catalyzed coupling reactions in the pharmaceutical, agrochemical, and fine chemical industries. Adv. Synth. Catal. 2009;351:3027–3043. [Google Scholar]
- 37.Recho J., Black R.J.G., North C., Ward J.E., Wilkes R.D. Statistical DoE approach to the removal of palladium from active pharmaceutical ingredients (APIs) by functionalized silica adsorbents. Org. Process. Res. Dev. 2014;18:626–635. [Google Scholar]
- 38.Cano R., Schmidt A.F., McGlacken G.P. Direct arylation and heterogeneous catalysis; ever the twain shall meet. Chem. Sci. 2015;6:5338–5346. doi: 10.1039/c5sc01534k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bura T., Blaskovits J.T., Leclerc M. Direct (hetero)arylation polymerization: trends and perspectives. J. Am. Chem. Soc. 2016;138:10056–10071. doi: 10.1021/jacs.6b06237. [DOI] [PubMed] [Google Scholar]
- 40.Lombeck F., Komber H., Gorelsky S.I., Sommer M. Identifying homocouplings as critical side reactions in direct arylation polycondensation. ACS Macro Lett. 2014;3:819–823. doi: 10.1021/mz5004147. [DOI] [PubMed] [Google Scholar]
- 41.Hong W., Chen S., Sun B., Arnould M.A., Meng Y., Li Y. Is a polymer semiconductor having a “perfect” regular structure desirable for organic thin film transistors? Chem. Sci. 2015;6:3225–3235. doi: 10.1039/c5sc00843c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan Y., Hartwig J.F. Assessment of the intermediacy of arylpalladium carboxylate complexes in the direct arylation of benzene: evidence for C-H bond cleavage by “ligandless” species. J. Am. Chem. Soc. 2011;133:3308–3311. doi: 10.1021/ja1113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson Seechurn C.C.C., Kitching M.O., Colacot T.J., Snieckus V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. Engl. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 44.Jatsch A., Schillinger E.-K., Schmid S., Bäuerle P. Biomolecule assisted self-assembly of π-conjugated oligomers. J. Mater. Chem. 2010;20:3563–3578. [Google Scholar]
- 45.Bell O.A., Wu G., Haataja J.S., Brömmel F., Fey N., Seddon A.M., Harniman R.L., Richardson R.M., Ikkala O., Zhang X., Faul C.F. Self-assembly of a functional oligo(aniline)-based amphiphile into helical conductive nanowires. J. Am. Chem. Soc. 2015;137:14288–14294. doi: 10.1021/jacs.5b06892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medina B.M., Wasserberg D., Meskers S.C.J., Mena-Osteritz E., Bäuerle P., Gierschner J. EDOT-type materials: planar but not rigid. J. Phys. Chem. A. 2008;112:13282–13286. doi: 10.1021/jp809632z. [DOI] [PubMed] [Google Scholar]
- 47.Bredas J.-L. Mind the gap! Mater. Horiz. 2014;1:17–19. [Google Scholar]
- 48.Kularatne R.S., Magurudeniya H.D., Sista P., Biewer M.C., Stefan M.C. Donor-acceptor semiconducting polymers for organic solar cells. J. Polym. Sci. Part A. Polym. Chem. 2013;51:743–768. [Google Scholar]
- 49.Nielsen C.B., Angerhofer A., Abboud K.A., Reynolds J.R. Discrete photopatternable pi-conjugated oligomers for electrochromic devices. J. Am. Chem. Soc. 2008;130:9734–9746. doi: 10.1021/ja7112273. [DOI] [PubMed] [Google Scholar]
- 50.Lomas J.S., Adenier A., Gao K., Maurel F., Vaissermann J. Hydrogen bonding and steric effects on rotamerization in 3,4-alkylenedioxy-, 3-alkoxy- and 3,4-dialkoxy-2-thienyldi(tert-butyl)methanols: an NMR, IR and X-ray crystallographic study. J. Chem. Soc. Perkin Trans. 2002;2:216–224. [Google Scholar]
- 51.Rivnay J., Owens R.M., Malliaras G.G. The rise of organic bioelectronics. Chem. Mater. 2014;26:679–685. [Google Scholar]
- 52.Simon D.T., Gabrielsson E.O., Tybrandt K., Berggren M. Organic bioelectronics: bridging the signaling gap between biology and technology. Chem. Rev. 2016;116:13009–13041. doi: 10.1021/acs.chemrev.6b00146. [DOI] [PubMed] [Google Scholar]
- 53.Sun H., Autschbach J. Electronic energy gaps for π-conjugated oligomers and polymers calculated with density functional theory. J. Chem. Theor. Comput. 2014;10:1035–1047. doi: 10.1021/ct4009975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.