

Abstract
Mutations in the UBQLN2 gene, which encodes a member of the ubiquitin-like protein family (ubiquilin-2), have been identified in patients with dominant X-linked amyotrophic lateral sclerosis (ALS) and ALS with frontotemporal dementia (FTD). We analyzed mutations in the UBQLN2 gene in a Chinese cohort of 515 patients with sporadic ALS (sALS). A novel missense mutation (p.M392V) was detected in one sALS patient. The p.M392V mutation substitutes a highly conserved residue, has not been reported in the population databases, and previously, at the same residue, a missense mutation p.M392I was detected in two Turkey ALS patients and was considered to be pathogenic, so the M392V is a variant of uncertain significance (VOUS) for ALS. We also found a deletion mutation (p.P500_G502del), which seems to be benign. In conclusion, our data suggest that mutations in the UBQLN2 gene are rare in Chinese sALS patients.
Introduction
It has been known that UBQLN2 gene is associated with amyotrophic lateral sclerosis (ALS) and ALS with frontotemporal dementia (FTD) [1]. Studies of ALS cohorts from different ethnic groups have been conducted to identify the pathogenicity of UBQLN2 mutations in ALS patients, and it is now recognized that UBQLN2 mutations are not a common cause of ALS [2–7].
To date, Korea and Taiwan have conducted genetic surveys in ALS patients, and their results suggested that mutations in UBQLN2 gene are rare [8, 9]. As for mainland China, a huge ethnic group of East Asian, few data are available. Thus, we assessed the site and frequency of UBQLN2 gene mutations in a cohort of mainland Chinese patients with sALS.
Methods
A total of 515 patients from mainland China were recruited in the study from January 2013 to December 2014. They were diagnosed with definite, probable or lab-supported probable ALS as per El Escorial criteria, and appeared to have sALS without clinical manifestation of FTD. Patients’ clinical data and blood samples were collected during their first visit to the outpatient department, and we have access to the information for identifying a special individual participant during the study. All patients had previously been screened for mutations in known ALS genes including SOD1, FUS, TARDBP, OPTN and DCTN1. All participants provided written informed consent in this study, which was approved by the Peking University Third Hospital Ethics Committee. Age- and sex-matched individuals without neurological disease were used as controls (n = 203).
The entire coding region of UBQLN2 including the 5’ and 3’-UTR was amplified using five overlapping amplicons (S1 File) and further sequenced. Polymerase chain reactions (PCR) were performed using I-5TM 2X High-Fidelity Master Mix (CMLAB) as per manufacturer’s guidelines. PCR products were sequenced at Tsingke biological technology corporation. All sequence variations were confirmed by independent PCR reactions using primers from both forward and reverse directions. The PXX domain and the STI1 domain which contains the discovered missense mutation c.1174A>G were further sequenced in another 286 controls.
Discovered mutations were checked for presence in the public SNP databases dbSNP, the 1000 Genomes Project, Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC).
The potential impact of missense mutations on the structure and function of the encoded proteins was evaluated using two bioinformatic prediction programs: PolyPhen-2 and SIFT. To assess whether the altered amino acids were conserved, orthologous peptide sequences were obtained for the human UBQLN2 gene from five different species and aligned using the CLUSTALW2 server.
Results
We found a missense mutation (c.1174A>G; p.M392V) of UBQLN2 in one sALS patient (2014–042) and a deletion mutation (c.1500-1508delCATAGGCCC; p.P500_G502del) in a patient with sALS (2013–368) (Fig 1). None of these two variants were found in the 489 (203+286) healthy controls, and the two residues were both in conserved domains. As for clinical information, patient 2014–042 was a 62-year-old man who presented with a 32-month history of progressive upper limb weakness. The patient was diagnosed as ALS-FAS and had an amyotrophic lateral sclerosis function rating scale-revised (ALSFRS-R) score of 46 at diagnosis. In a two years follow-up visit, the patient now has an ALSFRS-R score of 35 and shows no sign of FTLD symptoms. Patient 2013–368 was a 63-year-old man with a 20-month history of slowly progressive limb weakness and had an ALSFRS-R score of 47 at diagnosis. At the time of report, the patient has an ALSFRS-R score of 44 and showed no sign of FTLD symptoms. In addition, we also detected a silent variant in one patient and one control: c.51T>A (p.P17P) (2/718; rs371163085), which seems to be a benign polymorphism.
Fig 1. UBQLN2 mutations in Chinese sALS patients.
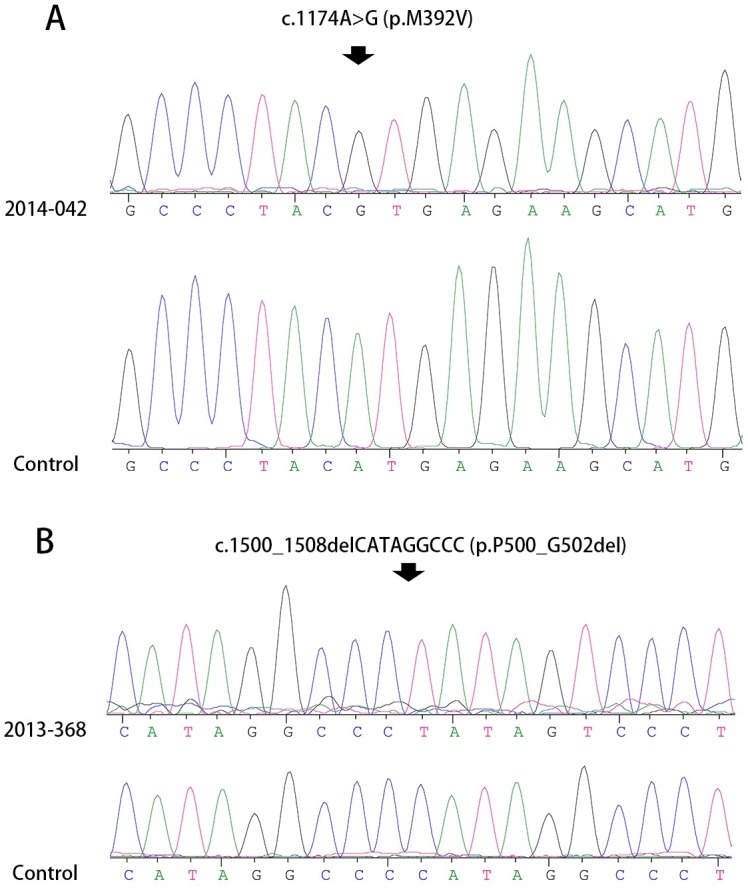
(A) Patient 2014–042 had a c.1174A>G (p.M392V) missense mutation. (B) Patient 2013–368 carried a deletion mutation (c.1500_1508delCATAGGCCC, p.P500_G502del).
Discussion
The missense variant we detected, p.M392V, is a novel variant of uncertain significance (VOUS), as it was predicted to be possibly damaging (by Polyphen-2) or tolerated (by SIFT). The patient’s mother died at the age of 60 while his father was still alive at the time of report, and both of them did not show signs of weakness or atrophy. It is a pity that we did not get the DNA samples of the parent’s parents to investigate the transmission of the mutation. Interestingly, in the same position, a mutation p.M392I was detected in Turkey ALS patients, and it was thought to be deleterious [7]. However, the onset age of the patients carrying the p.M392I mutation were fairly young (14 and 16 years old) and one of them showed a special subtype of ALS: Madras-type MND. By contrast, our patient showed a typical ALS-FAS phenotype. This may be due to that different amino acid change could lead to a different phenotype. None of these two mutations were found in public SNP resources and in controls. So, according to the ACMG Standards and guidelines for the interpretation of sequence variants [10], the p.M392V variant is a VOUS for ALS. The mutation is located at a quite conservative region (Fig 2), so further analyses are needed to clarify the pathogenicity of the p.M392V variant of UBQLN2, for example, to test the parental samples for the variant or functional studies of the p.M392V protein.
Fig 2. The location and genetic conservation of the mutations in UBQLN2 gene.
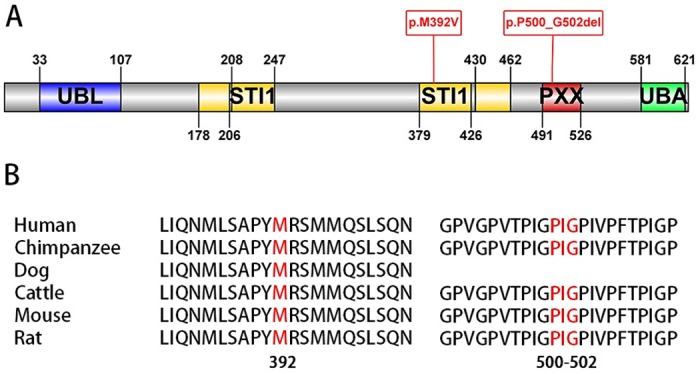
(A) Ubiquilin-2 protein indicating structural and functional domains: UBL (ubiquitin-like domain), STI1 (heat-shock-chaperonin binding motif), PXX (proline-rich region) and UBA (ubiquitin-associated domain). Our detected mutations are displayed in red. (B) The conservation of ubiquilin-2 protein in different species. The mutated residues are displayed in red.
The deletion mutation, c.1500_1508.delCATAGGCCC, was firstly reported by Millecamps in a French fALS female proband. The proband’s mutation was at the heterozygote state, and her father, also an ALS patient, did not carry this mutation. Rather, they detected the mutation at the heterozygous state in a female control subject, so the mutation did not seem to be deleterious [3]. Besides, just before the mutation side, the p.P497_G499del mutation, which causes same amino acids change, was mapped in the ExAC database (global 10/55335, East Asian 2/4156). This further confirmed that the mutation seems to be benign for ALS.
In conclusion, the present study demonstrates the low frequency of UBQLN2 variants in mainland Chinese sALS patients. Previously, it has been estimated that UBQLN2 gene accounts for less than one percent of sALS in populations of European ancestry [11]. As for the Asian region, Kim et al. reported a missense variant (c.942T>A, p.D314E) existing both in 2 unrelated sALS patients and 5 healthy control individuals of Korean population, indicating that this variant might be a rare polymorphism [8]; and no Taiwanese patient was found to have any mutation of UBQLN2 gene in Soong’s research [9]. Thus, our conclusion is in accordance with the results of globe and the Asian region, suggesting that causative mutations are rare in the UBQLN2 gene in sALS patients. We did not find an explicit genotype-phenotype association in sALS patients with UBQLN2 mutations, but we assume that different mutation locus and different amino acid change of UBQLN2 gene can lead to diverse clinical manifestations. Further functional study is need to elucidate the pathogenicity of the p.M392V missense variant of UBQLN2 gene.
Supporting Information
The amplified five overlapping PCR fragments cover the entire coding sequence (1,872bp), 125bp of the 5’-UTR and 293bp of the 3’-UTR.
(DOC)
Acknowledgments
We thank the patients and their families for participating in this project. Special thanks to Dr. Yi-Chung Lee from National Yang-Ming University for his advice to this project.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Natural Sciences Foundation of China [81030019] (URL: http://www.nsfc.gov.cn/). No individuals employed or contracted by the funders (other than the named authors) played any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–5. 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daoud H, Suhail H, Szuto A, Camu W, Salachas F, Meininger V, et al. UBQLN2 mutations are rare in French and French-Canadian amyotrophic lateral sclerosis. Neurobiology of aging. 2012;33(9):2230.e1–e5. [DOI] [PubMed] [Google Scholar]
- 3.Millecamps S, Corcia P, Cazeneuve C, Boillee S, Seilhean D, Danel-Brunaud V, et al. Mutations in UBQLN2 are rare in French amyotrophic lateral sclerosis. Neurobiology of aging. 2012;33(4):839.e1–3. [DOI] [PubMed] [Google Scholar]
- 4.van Doormaal PT, van Rheenen W, van Blitterswijk M, Schellevis RD, Schelhaas HJ, de Visser M, et al. UBQLN2 in familial amyotrophic lateral sclerosis in The Netherlands. Neurobiology of aging. 2012;33(9):2233.e7–e8. [DOI] [PubMed] [Google Scholar]
- 5.Gellera C, Tiloca C, Del Bo R, Corrado L, Pensato V, Agostini J, et al. Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. Journal of neurology, neurosurgery, and psychiatry. 2013;84(2):183–7. 10.1136/jnnp-2012-303433 [DOI] [PubMed] [Google Scholar]
- 6.McLaughlin RL, Kenna KP, Vajda A, Byrne S, Bradley DG, Hardiman O. UBQLN2 mutations are not a frequent cause of amyotrophic lateral sclerosis in Ireland. Neurobiology of aging. 2014;35(1):267.e9–11. [DOI] [PubMed] [Google Scholar]
- 7.Ozoguz A, Uyan O, Birdal G, Iskender C, Kartal E, Lahut S, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiology of aging. 2015;36(4):1764.e9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HJ, Kwon MJ, Choi WJ, Oh KW, Oh SI, Ki CS, et al. Mutations in UBQLN2 and SIGMAR1 genes are rare in Korean patients with amyotrophic lateral sclerosis. Neurobiology of aging. 2014;35(8):1957.e7–8. [DOI] [PubMed] [Google Scholar]
- 9.Soong BW, Lin KP, Guo YC, Lin CC, Tsai PC, Liao YC, et al. Extensive molecular genetic survey of Taiwanese patients with amyotrophic lateral sclerosis. Neurobiology of aging. 2014;35(10):2423.e1–6. [DOI] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics. 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience. 2014;17(1):17–23. 10.1038/nn.3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The amplified five overlapping PCR fragments cover the entire coding sequence (1,872bp), 125bp of the 5’-UTR and 293bp of the 3’-UTR.
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.