

Abstract
The high-affinity biosynthetic pathway for converting acetate to acetyl-coenzyme A (acetyl-CoA) is catalyzed by the central metabolic enzyme acetyl-coenzyme A synthetase (Acs), which is finely regulated both at the transcriptional level via cyclic AMP (cAMP)-driven trans-activation and at the post-translational level via acetylation inhibition. In this study, we discovered that cAMP directly binds to Salmonella enterica Acs (SeAcs) and inhibits its activity in a substrate-competitive manner. In addition, cAMP binding increases SeAcs acetylation by simultaneously promoting Pat-dependent acetylation and inhibiting CobB-dependent deacetylation, resulting in enhanced SeAcs inhibition. A crystal structure study and site-directed mutagenesis analyses confirmed that cAMP binds to the ATP/AMP pocket of SeAcs, and restrains SeAcs in an open conformation. The cAMP contact residues are well conserved from prokaryotes to eukaryotes, suggesting a general regulatory mechanism of cAMP on Acs.
Keywords: acetyl-CoA synthetase, acetylation, crystal structure, cyclic AMP (cAMP), post-translational modification (PTM)
Introduction
Acetyl-coenzyme A (acetyl-CoA) is a central metabolic intermediate that modulates the balance of anabolism and catabolism by functioning as the fuel for energy generation or as the precursor of lipogenesis for energy storage in many organisms (1). Acetyl-CoA is also deemed a second messenger for physiological regulation by supplying an acetyl group for protein acetylation (1, 2), an important mechanism of post-translational modification (PTM). Acetyl-coenzyme A synthetase (Acs)5 (EC 6.2.1.1) belongs to the acyl- or aryl-CoA synthetase family and catalyzes the biosynthesis of acetyl-CoA from acetate, coenzyme A (CoA), and ATP, and is well conserved from bacteria to mammals. The Acs of Escherichia coli scavenges environmental acetate with high affinity (≤10 mm in general) and converts it to acetyl-CoA during glucose limitation (3). In humans, Acs is implicated in disposing of alcohol in normal hepatocytes (1, 4) and maintaining cell growth under metabolic stress conditions in certain tumors (5, 6). Acs is also reported to be involved in chromatin regulation (7) and bacterial chemotaxis (8, 9) by supplying acetyl-CoA as the acetylation donor.
Acs is strictly regulated at both transcriptional and post-translational levels in enteric bacteria. Under carbon limitation, cAMP-CRP (cAMP receptor protein) binds upstream of the acs proximal promoter (acsP2) and initiates transcription of acs (10). However, this cAMP-CRP-dependent activation may be inhibited by the nucleoid proteins IHF and FIS by competitive binding to the CRP-binding sites or other unknown steric hindrances (11). The activity of Acs is regulated by acetylation catalyzed by the protein acetyltransferase Pat and deacetylation catalyzed by the NAD+-dependent deacetylase CobB. Pat transfers an acetyl group from acetyl-CoA onto the Salmonella enterica Acs (SeAcs) Lys609 and blocks the first half of the catalytic reaction, acetyl-AMP formation (12), whereas CobB restores activity by specifically removing this acetyl group (13).
cAMP is a master second messenger that is involved in rapid response to environmental nutritional changes in bacteria (14, 15). The acetate scavenging by Acs is an alternative pathway for acetyl-CoA production under a low-glucose condition, and cAMP regulates Acs in multiple ways. Besides the direct transcription activation of the acs gene by cAMP-CRP as described above (10), cAMP indirectly modulates Acs activity by activating transcription of the pat gene in the presence of non-phosphotransferase system carbon sources in enteric bacteria (16), or by activating Pat activity via binding to its allosteric site in mycobacteria (17, 18).
In this study, we discovered that cAMP inhibits the activity of SeAcs by directly binding to the enzyme and competing against one of its substrates, ATP. The binding of cAMP increases SeAcs Lys609 acetylation, which further inhibits SeAcs activity. The acetylation is apparently caused by concomitant promotion of Pat-dependent acetylation and inhibition of CobB-dependent deacetylation. We further determined a crystal structure of SeAcs complexed with cAMP and CoA, and the crystal structure not only confirms that cAMP binds to the conserved ATP/AMP binding pocket of SeAcs, but also suggests cAMP, in addition to CoA, restrains SeAcs in an open conformation. This structure provides possible scenarios for the mechanism that promotes SeAcs Lys609 acetylation. In addition, we found cAMP also inhibited S. enterica propionyl-CoA synthetase (SePrpE) and long-chain acyl-CoA synthetase (SeFadD), which are from the same protein family of acyl- or aryl-CoA synthetases. As the residues of cAMP-binding site are generally conserved in this family, we infer that cAMP possibly inhibits other acyl- or aryl-CoA synthetases in S. enterica.
Results
cAMP Binds to SeAcs Directly
We measured the thermodynamic parameters of the proposed interaction between cAMP and non-acetylated SeAcs isolated from a pat-null S. enterica strain using the isothermal titration calorimetry (ITC) method to test whether cAMP binds directly to Acs. The result shows that cAMP directly interacted with SeAcs, with one cAMP-binding site for each SeAcs (blue in Fig. 1A). The dissociation constant (Kd) was estimated to be 164 ± 11 μm, which is very close to the physiological concentration of cAMP in live bacterial cells under starvation conditions (19, 20), implicating a physiological relevance. We next measured the interaction between cAMP and acetylated SeAcs isolated from a cobB-null S. enterica strain, and the Kd was estimated to be 297 ± 15 μm (red in Fig. 1A).
FIGURE 1.

cAMP directly binds to SeAcs and inhibits its activity by competing with substrate ATP. A, representative real-time ITC heat change (upper and middle panels) and the integration (lower panel) of each titration of cAMP to SeAcs or acetylated SeAcs (ac-SeAcs). B, representative real-time ITC heat change (upper and middle panels) and the integration (lower panel) of each titration of ATP to SeAcs or ac-SeAcs. C, cAMP inhibits SeAcs at a low concentration of ATP (1 mm CoA, 20 μm ATP, and 20 mm acetate), but not at either a low concentration of acetate (1 mm CoA, 1 mm ATP, and 20 μm acetate), or a low concentration of CoA (20 μm CoA, 1 mm ATP, and 20 mm acetate). The SeAcs activity in the absence of cAMP was treated as 100%. D, the representative Lineweaver-Burk plot shows cAMP inhibits SeAcs by competing with the substrate ATP. The inset panel is a Dixon plot of apparent Km versus cAMP concentration. The Ki of cAMP is estimated from the linear regression. The experiments were repeated in triplicate, and the data are presented as mean ± S.D., ***, p < 0.001.
Given that the 0.8-fold increase in Kd of cAMP binding to acetylated SeAcs compared with the non-acetylated enzyme, we investigated if acetylation of SeAcs also affects the binding of its natural substrate, ATP. The results indicate a similar 2.4-fold increase in Kd of ATP binding to the acetylated SeAcs (1.09 ± 0.079 mm; red in Fig. 1B) compared with the non-acetylated SeAcs (322 ± 19 μm; blue in Fig. 1B). Therefore, both cAMP and ATP preferably bind to the non-acetylated SeAcs.
We next measured the acetylation level of Lys609, the primary site on SeAcs under acetylation regulation. There is a clear difference in the Lys609 acetylation level of SeAcs isolated from pat-null or cobB-null S. enterica cells (Fig. 2, A and B). As Lys609 participates in the catalytic reaction of SeAcs, it is not surprising that acetylation at Lys609 affects the binding of ATP to SeAcs. The results also suggest that cAMP might bind to a location similar to that of ATP in SeAcs.
FIGURE 2.

cAMP promotes acetylation of SeAcs Lys609in vitro. A, the antibody used is specific to acetylated SeAcs Lys609. The same amounts of SeAcs isolated from cobB-null or pat-null S. enterica strains were separated by SDS-PAGE, transferred to the nitrocellulose membrane, then blotted with the antibody specific to the acetylated SeAcs Lys609 in the presence or absence of unmodified peptide (KTRSGKIMRRI), or acetylated peptide (KTRSG(Kac)IMRRI), and detected using an ImageQuant LAS 4000 (GE Healthcare). Coomassie Brilliant Blue staining was used for loading controls. B, semiquantification of the acetylation level of SeAcs Lys609 purified from CobB-null or pat-null S. enterica cells. C, cAMP activates SePat-dependent acetylation and enhances the activity inhibition upon SeAcs. Recombinant SeAcs purified from the pat-null S. enterica strain is treated by acetylation in the presence or absence of cAMP followed by activity measurement and Western blotting detection. The SeAcs activity in the absence of cAMP, acetyl-CoA, and SePat (Lane 1) was treated as 100%. Coomassie Brilliant Blue-stained proteins on SDS-PAGE gels are shown below as a loading control. D, cAMP inhibits SeCob-dependent deacetylation and weakens the activity recovery of SeAcs. Recombinant SeAcs purified from a cobB-null S. enterica strain was treated by deacetylation in the presence or absence of cAMP followed by activity measurement and Western blotting detection. The SeAcs activity (lane 4) was treated as 100%. Each experiment was repeated in triplicate, and the data are presented as mean ± S.D., ***, p < 0.001; **, p < 0.01.
cAMP Inhibits SeAcs by Competing with the Substrate ATP
We measured SeAcs activity (isolated from pat-null S. enterica cells) in the presence or absence of cAMP to test whether binding of cAMP inhibits the activity of SeAcs, SeAcs catalyzes the formation of acetyl-CoA using CoA, acetate, and ATP as substrates. The potential inhibitory effect of cAMP on Acs was tested using a subsaturated concentration for each of the substrates. cAMP substantially inhibited SeAcs activity only in an enzymatic reaction system with a low concentration of ATP (Fig. 1C), suggesting cAMP inhibits SeAcs via an ATP-competitive mechanism.
To further confirm that cAMP inhibits SeAcs by competing against the substrate ATP, we measured the apparent Km constants of ATP under various cAMP concentrations. The Lineweaver-Burk plot for cAMP shows a characteristic signature of a competitive inhibitor (Fig. 1D), in agreement with the above observation that cAMP barely inhibits SeAcs when ATP is saturated (Fig. 1C).
The inhibition constant (Ki) for cAMP-SeAcs was estimated as 185 ± 25 μm by plotting the apparent Km (toward substrate ATP) against cAMP concentration (inset of Fig. 1D), which is similar to the ITC estimated Kd (164 ± 11 μm; blue in Fig. 1A).
cAMP Promotes Lys609 Acetylation of SeAcs
Acetylation of the catalytic lysine residue (Lys609 in SeAcs) is an efficient way to modulate Acs activity (12, 13). cAMP has been reported to enhance Acs acetylation by activating the lysine acetyltransferase Pat in mycobacteria (17, 18) via binding to the allosteric site in the cyclic nucleotide (cNMP)-binding domain of Pat. This domain is unique to mycobacteria species (17), and we speculate that the mechanism by which cAMP affects the Acs in enteric bacteria, such as SeAcs, must be different.
To determine whether direct binding of cAMP to SeAcs affects its Lys609 acetylation level, we performed in vitro enzymatic acetylation assays using purified non-acetylated SeAcs (from a pat-null S. enterica strain) and acetyl-CoA as the substrates. The acetylation assays were catalyzed by purified S. enterica protein acetyltransferase (SePat), with or without cAMP in the reaction system. The level of SeAcs acetylation was determined by Western blotting using an antibody that specifically recognizes acetylated Lys609 (Fig. 2A). As shown in Fig. 2C, SePat increases the acetylation level of SeAcs Lys609 in vitro (lane 4), and cAMP promotes Pat-dependent acetylation substantially (lane 7). The addition of cAMP to the acetylation assay system further decreases SeAcs activity, suggesting SeAcs acetylation promoted by cAMP also contributes to the inhibitory effect of cAMP on SeAcs. Because a cNMP-binding domain is absent in SePat (17), we infer that cAMP promotes SeAcs acetylation through direct binding to its ATP/AMP pocket as well.
Lysine acetyltransferase Pat and deacetylase CobB exist normally as a pair to modulate cellular protein acetylation levels in most bacteria (21). To test if cAMP also affects deacetylation of SeAcs by SeCobB, we performed in vitro enzymatic deacetylation assay using purified acetylated SeAcs from a cobB-null S. enterica strain as the substrate, NAD+ as a coenzyme, and purified S. enterica Sir2-like lysine deacetylase (SeCobB) as the catalytic enzyme. The result in Fig. 2D confirmed that SeCobB was able to remove the acetyl group on Lys609 of SeAcs (lane 4), and rescued SeAcs activity. However, decrease in the acetylation level as well as the recovery of SeAcs activity by CobB treatment was inhibited remarkably in the presence of cAMP (lane 7). Because cAMP does not bind to SeCobB (data not shown), we infer that cAMP inhibits CobB-dependent deacetylation by directly binding to SeAcs.
cAMP Binds to the ATP/AMP Binding Pocket of SeAcs
To understand the structural basis of SeAcs inhibition by cAMP, we determined the crystal structure of SeAcs complexed with cAMP, CoA, and acetate (Fig. 3, Table 1) at 1.65-Å resolution. The structure defined the binding site of cAMP on SeAcs, and the effects of cAMP and CoA binding on SeAcs conformation. The simulated annealing omit Fo − Fc electron density map shows unambiguous electron density of cAMP within the ATP/AMP binding pocket of SeAcs (Fig. 3, A and E), in agreement with our observation that cAMP inhibits SeAcs by competing with the substrate ATP (Fig. 1, C and D). The map also shows unambiguous electron density for adenosine and diphosphate moieties of CoA within the adjacent CoA binding pocket (right in Fig. 3A). However, no electron density for acetate was observed within the acetate-binding site of SeAcs, probably due to effect of steric hindrance by cAMP.
FIGURE 3.

Crystal structure of SeAcs in complex with cAMP and CoA. A, simulated-annealing omit Fo − Fc electron density map (green) contoured at 3.0 σ for cAMP (left) and CoA (right) molecules. Blue, orange, and red represent nitrogen, phosphorus, and oxygen atoms, respectively. Cyan and green represent carbon atoms of cAMP and CoA, respectively. B, overall structure of SeAcs in complex with cAMP (cyan) and CoA (green). White and red ribbons represent the N-terminal and C-terminal domains of SeAcs, respectively; red sphere, Lys609; cyan sphere, cAMP; green sphere, CoA. C, superimposition of our SeAcs-cAMP-CoA crystal structure (red and gray) with the crystal structure of SeAcs complexed with AMP, CoA, and acetate (PDB code 2P2F; cyan). D, superimposition of our SeAcs-cAMP-CoA crystal structure (red and gray) with the crystal structure of yeast Acs in complex with AMP (PDB code 1RY2; green). E, close-up view of the cAMP (cyan) binding pocket with superimposed propyl-AMP (red) from the crystal structure of SeAcs complexed with propyl-AMP, CoA, and acetate (PDB code 1PG4), and with superimposed AMP (purple) from the crystal structure of SeAcs in complex with AMP (PDB code 2P2F).
TABLE 1.
Statistics of crystal structure of SeAcs in complex with cAMP and CoA
| SeAcs-cAMP-CoA | |
|---|---|
| Data collection | |
| Space group | P 21 |
| Cell dimensions | |
| a, b, c (Å) | 60.2, 144.3, 71.7 |
| α, β, γ (°) | 90.0, 91.7, 90.0 |
| Resolution (Å) | 40.00–1.65 (1.68–1.65)a |
| Rsym or Rmerge | 0.146 (0.616)a |
| I/σI | 8.0 (2.0)a |
| Completeness (%) | 1.000 (1.000)a |
| Redundancy | 3.3 (3.3)a |
| CC1/2 in highest shell | 0.630 |
| Refinement | |
| Resolution (Å) | 40.00–1.65 |
| No. reflections | 144575 |
| Rwork/Rfree | 0.169/0.207 |
| No. atoms | |
| Protein | 9998 |
| Ligand/ion | 138 |
| Water | 1113 |
| B-factors (Å2) | |
| Protein | 17.2 |
| Ligand/ion | 29.6 |
| Water | 25.3 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.148 |
a Highest resolution shell is shown in parentheses.
Our crystal structure revealed that in the presence of cAMP and CoA, the C-terminal domain of SeAcs adopted an open conformation (red in Fig. 3B), exposing Lys609 to the solvent. Lys609 plays an important role in the first half of the reaction catalyzed by SeAcs and is subject to negative regulation by acetylation (13). We compared the “open” conformation in our crystal structure of SeAcs complex with the previously reported open conformation in a crystal structure of SeAcs in complex with CoA, acetate, and AMP (Protein Data Bank (PDB) code 2P2F) (22); and the two open conformations are essentially identical. The Lys609 residue is located at the same position, and the side chains of Lys609 are both partially disordered in the two structures (Fig. 3C).
We also compared our open conformation with a previously determined “closed” conformation of a crystal structure of yeast Acs in complex with AMP. We observed a large rotation of ∼100° in the Acs C-terminal domain, which includes Lys675 (Lys609 in SeAcs), in the closed conformation (Fig. 3D), as a result Lys675 is buried into the active center. The exposed conformation of Lys609 in our current structure would allow Lys609 to be more accessible for acetylation, consistent with the observation that the acetylation of SeAcs was remarkably increased in the presence of cAMP (Fig. 2C).
Contacts between cAMP and residues of SeAcs are shown in Fig. 4, A and B. The adenine moiety of cAMP makes the same interactions with SeAcs as the adenine moiety of AMP makes in the crystal structure of SeAcs in complex with AMP, CoA, and acetate (PDB 2P2F; Fig. 3E) (22). These interactions are also the same for the adenine moiety of 5′-propyl-AMP in the crystal structure of SeAcs in complex with 5′-propyl-AMP and CoA (PDB 1PG4; Fig. 3E) (23). These interactions include: stacking effects of the adenine base to side chain atoms of Trp413 and Ile512 from one side and main chain atoms of Val386, Glu388, and Pro389 from the other side; one H-bond through the N6 atom of the adenine with Asp411; and one water-mediated H-bond through the N3 atom of the adenine with Asp500 (Fig. 4, A and B). The ribose moiety of cAMP makes three H-bonds through its 2′ OH group with Asp500, Gln415, and Arg515, respectively, and one H-bond through its 5′ O atom with Asn521. The phosphate group of cAMP contacts SeAcs by one H-bond with Thr416, one salt-bridge bond with Arg526, and Van der Waals interactions with Thr311 and Glu417.
FIGURE 4.

Detail interactions between SeAcs and cAMP. A, stereo-presentation of detail interactions between SeAcs and cAMP. White ribbon, SeAcs backbone; white stick, SeAcs carbon atoms; cAMP is colored as in panel A. B, schematic presentation of the possible interaction between SeAcs and cAMP. Red dashed lines, H-bonds; blue arcs, Van der Waals interactions. C, sequence alignment of the cAMP-binding pocket in Acs enzymes from different species. The number at the beginning of each line indicates the residue position relative to start of each protein sequence. Residues involved in interactions with cAMP are denoted with colored dots (red, H-bond interaction; black, Van der Waals interaction). Residues conserved in different species are shaded red.
cAMP binds to the highly conserved ATP/AMP binding pocket of Acs and shares most contact residues with AMP (Figs. 3E and 4, A and B). Therefore it is not surprising that most contact residues of cAMP are conserved among bacterial species, yeast, and humans (Fig. 4C). We thereby propose that the regulatory effect of cAMP on Acs is possibly an important mechanism that has been preserved during evolution.
Trp413, Gln415, Asp500, and Arg526 in SeAcs Are Determinants for cAMP Binding and Promotion of SeAcs Acetylation
Although the SeAcs crystal structure shows 14 total residues interact with cAMP either by H-bonds or Van der Waals forces (Fig. 4), it is still unknown which site(s) is critical. To further define the determinants of cAMP binding, we performed ITC experiments analyzing the binding affinity of cAMP to SeAcs derivatives bearing alanine substitution of residues that are involved in side chain interactions. As shown in Table 2, the derivatives of SeAcs W413A, Q415A, D500A, and R526A lost their ability to bind cAMP; whereas D411A, R515A, and N521A displayed decreased affinity; and T416A displayed a similar affinity to wild-type SeAcs.
TABLE 2.
Equilibrium dissociation constants (Kd) of cAMP to SeAcs derivatives
| SeAcs | Kd |
|---|---|
| μm | |
| WT | 164 ± 11 |
| W413A | NDa |
| Q415A | ND |
| D500A | ND |
| R526A | ND |
| T416A | 200 ± 50 |
| D411A | 1500 ± 300 |
| R515A | 1400 ± 300 |
| N521A | 590 ± 50 |
a ND, not detectable.
To verify that cAMP promotes SeAcs Lys609 acetylation through direct binding of cAMP to the ATP/AMP pocket in SeAcs, we performed similar in vitro Pat-dependent acetylation and CobB-dependent deacetylation assays using SeAcs derivatives W413A, Q415A, and D500A, which lost their cAMP binding ability. The W413A, Q415A, and D500A derivatives of SeAcs isolated from pat-null S. enterica cells were acetylated in vitro by SePat to a similar level to wild-type SeAcs (Fig. 5A, lanes 2 versus 3, 5 versus 6, 8 versus 9, and 11 versus 12). The W413A, Q415A, and D500A SeAcs derivatives isolated from cobB-null S. enterica cells were deacetylated by SeCobB in vitro to a similar level of undetectable acetylation to wild-type SeAcs (Fig. 5B, lanes 2 versus 3, 5 versus 6, 8 versus 9, and 11 versus 12). However, in contrast to wild-type SeAcs (Fig. 5, A and B, lane 1 versus 2), cAMP failed to affect either Pat-catalyzed acetylation (Fig. 5A, lanes 4 versus 5, 7 versus 8, and 10 versus 11) or CobB-catalyzed deacetylation (Fig. 5B, lanes 4 versus 5, 7 versus 8, and 10 versus 11) of the Lys609 on these SeAcs derivatives. As W413A, Q415A, and D500A derivatives of SeAcs were defective in cAMP binding (Table 2), the above results provide further support that the modulation of SeAcs acetylation by cAMP is dependent on direct cAMP binding.
FIGURE 5.
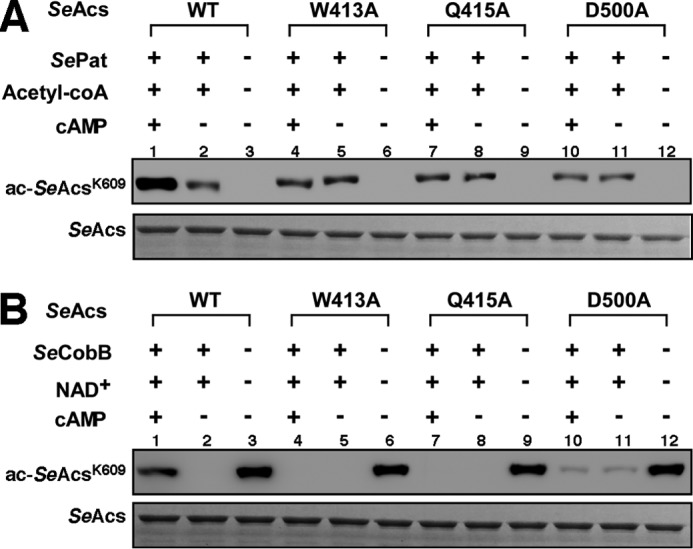
cAMP has no effect on acetylation promotion of mutant SeAcs. A, cAMP activates SePat-dependent acetylation of Lys609 on wild-type SeAcs but has no effect on mutant SeAcs in vitro. B, cAMP inhibits SeCobB-dependent deacetylation of Lys609 on wild-type SeAcs but has no effect on mutant SeAcs in vitro. Top band, acetylation detection by Western blotting using anti-SeAcs Lys609ac antibody; bottom band, Coomassie Brilliant Blue-stained proteins on SDS-PAGE gels are shown as a loading control. Each experiment was independently repeated three times.
cAMP Generally Inhibits Other Acyl- or Aryl-CoA Synthetases
Acs belongs to a large acyl- or aryl-CoA synthetase protein family (EC 6.2.1-), which all contain a two-domain architecture and harbors a conserved ATP/AMP binding pocket (24). To explore whether cAMP might inhibit other members of the acyl- or aryl-CoA synthetase family in S. enterica, we tested the effect of cAMP against two other acyl-CoA synthetases from S. enterica, SePrpE and SeFadD. The potential inhibitory activities were measured in the presence of subsaturation ATP concentration. The activities of SePrpE and SeFadD were inhibited by cAMP by 80 and 70%, respectively (Fig. 6A). These results support our hypothesis that cAMP possibly regulates other members of the acyl- or aryl-CoA synthetase family in S. enterica.
FIGURE 6.
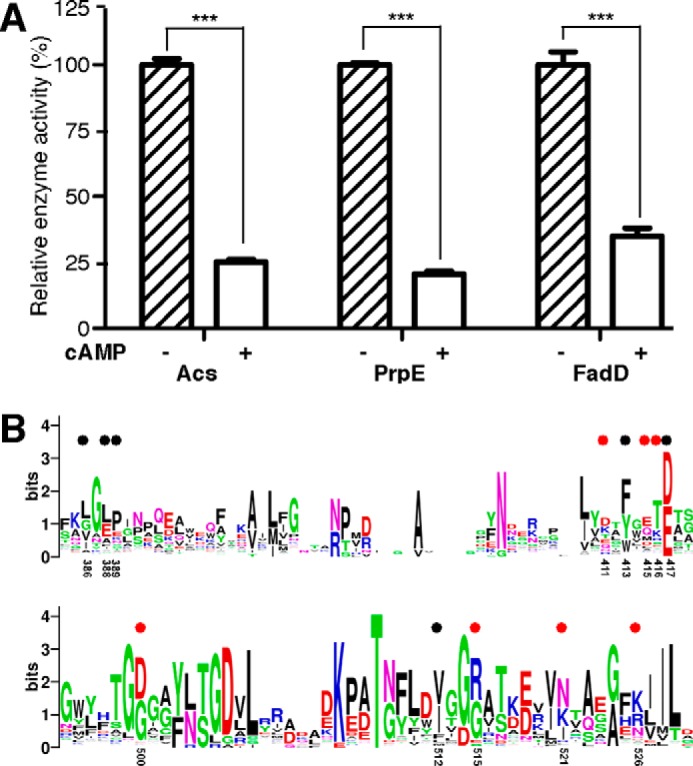
cAMP inhibits the activity of other AMP-forming enzymes harboring ATP/AMP binding pockets. A, cAMP inhibits enzymatic activity of SePrpE and SeFadD at a low concentration of ATP (1 mm CoA, 20 μm ATP, and 20 mm acetate), the activities of Acs, PrpE, and FadD in the absence of cAMP were treated as 100%, respectively. Each experiment was independently repeated in triplicate, and the data are presented as mean ± S.D., ***, p < 0.001. B, sequence alignment of 1190 acyl- or aryl-CoA synthetases retrieved from the UniPortKB database (search keyword: EC 6.2.1.- CoA) suggests that the cAMP-binding pocket of Acs is generally conserved in other members of the family. Residues involved in interactions with cAMP are denoted by colored dots as described in the legend to Fig. 4. Residues are numbered as in S. enterica.
We aligned the protein sequences of all available acyl- or aryl-CoA synthetases (1190 records) from the UniPortKB database, and found that 13 of the 14 cAMP contact residues are generally conserved (Fig. 6B). We speculate that the regulation by cAMP might be a more widespread occurrence in this protein family in organisms apart from S. enterica.
Discussion
This study establishes that cAMP modulates Acs activity through a novel mechanism. We show that cAMP occupies the ATP/AMP binding pocket of SeAcs, inhibiting SeAcs activity by competitive binding against its natural substrate ATP. We found that cAMP cooperates with Pat/CobB to regulate SeAcs acetylation and activity. Finally, we observed a similar inhibitory effect of cAMP against SePrpE and SeFadD, both harboring the ATP/AMP-binding domain and forming AMP during catalysis, and thus, this novel regulatory mechanism is proposed to be extendable to acyl- or aryl-CoA synthetase family enzymes, subject to future confirmation.
cAMP adjusts cellular anabolism and catabolism in some bacteria to allow adaptation to an environment with a non-preference carbon source. cAMP activates/represses the transcription of about 200 genes by directly binding to and allosterically activating CRP (25). One of them is the gene that encodes Acs, which converts acetate to acetyl-CoA to fuel the life when glucose is absent and the environmental concentration of acetate is low. This reaction is costly and is deemed to be regulated by cAMP (10). Acs consumes one ATP for activating one acetate, releasing one AMP and one PPi. Two more ATPs in total are consumed by adenylate kinase to catalyze the recycling of AMP to ADP (the ATP precursor), and by pyrophosphate phosphohydrolase to catalyzes PPi hydrolysis (26). Uncontrolled overexpression of Acs has been reported to cause a low-energy physiology state and arrest growth of S. enterica cells (26). Therefore, either the acs gene expression level or Acs activity has to be tightly regulated.
Previous studies have revealed activity-based negative regulation of cAMP upon Acs through PTM. cAMP coordinates expression of yfiQ genes in E. coli, which encodes the lysine acetyltransferase Pat (16). Pat completely abolishes the activity of Acs by adding an acetyl group to the catalytic lysine residue in Acs active center (13). cAMP also allosterically activates the activity of the protein acetyltransferase Pat in Mycobacterium tuberculosis and Mycobacterium smegmatis by targeting the Pat cNMP-binding domain, hence inhibiting the activity of Acs (17, 18). The new cAMP regulation mechanism identified in this study provides an alternative means for directly sensing the cellular ATP/cAMP levels, and also a rapid way to rescue energy deficiency by inhibiting Acs activity. We found binding of cAMP could influence the acetylation potential of Acs by promoting Pat-dependent acetylation and inhibiting CobB-dependent deacetylation concomitantly, thus enhancing the PTM inhibition of Acs activities.
SeAcs enzymatic activity is subject to post-translational regulation by acetylation of the catalytic Lys609 residue (13). Lys609 is located at the C-terminal domain of SeAcs, which adopts open and closed conformations by rotating ∼100° relative to the N-terminal domain (22, 23, 27). The C-terminal domain of SeAcs is proposed to fold into a closed conformation during the first half-reaction (converting ATP and acetate into acetyl-AMP), in which Lys609 inserts into the active center and makes critical contacts with the substrates (27). The C-terminal domain adopts an open conformation in the second half-reaction (converting acetyl-AMP and CoA into acetyl-CoA and AMP), in which Lys609 moves out from the active center and is exposed to solvent (27).
The two conformations have been captured in the crystal structures of SeAcs complexed with CoA, acetate, and AMP (open conformation; PDB code 2P2F) (22), and the yeast Acs in complex with AMP (closed conformation; PDB code 1RY2) (27), respectively. It is proposed that binding CoA triggers the conformation change (22). In the current crystal structure of SeAcs in complex with cAMP and CoA, the C-terminal domain of SeAcs is held to an open conformation, which exposes Lys609 to the protein surface. A recently reported crystal structure of SeAcs in complex with SePat (28) revealed that SeAcs adopts an open conformation during Lys609 acetylation exactly as that of our crystal structure, which may account for enhanced acetylation by SePat via cAMP binding. However, it is puzzling why cAMP also inhibits the SeCobB catalyzed deacetylation of SeAcs. Because no crystal structures of either bona fide acetylated SeAcs or SeAcs complexed with SeCobB are available, it is unclear how Lys609 acetylation affects SeAcs conformation and what conformation of SeAcs would adopt during deacetylation. We speculate that SeCobB might favor a slightly different conformation of SeAcs from the open one observed in our structure, and the presence of cAMP hinders the conformation flexibility.
This study also broadens our knowledge of the complexity of cAMP signaling. This second messenger was initially discovered as a regulator for carbohydrate metabolism in bacteria (29, 30), and then increasingly recognized as an important factor involved in other biological processes such as cell division, cell motility, and microbial virulence (31–33). cAMP has been implicated into multiple events in humans, ranging from metabolism (34) to memory formation (35) and innate immunity (36). The major outcome of cAMP signaling is regulating gene expression, either by directly modulating transcription through cAMP-CRP in bacteria, or by indirectly controlling gene expression through protein kinase A as an intermediate in eukaryotes (37). Besides gene expression, cAMP also controls gating of some ion channels (38) and activates guanine exchanging factors (39). For all the target proteins discussed above, cAMP binds to a conserved cNMP-binding domain (40) shared by all of them, characterized by a β barrel flanked by α helixes folded from ∼120 continuous residues. The cAMP binding pocket of Acs discovered in our crystal structure exhibits a typical Rossmann-fold (Fig. 3B, six β sheets and three α helixes form the cAMP binding pocket), which is radically different from the fold of the conserved cNMP-binding domain. Moreover, this new cAMP binding pocket is well conserved based on our sequence alignments of Acs from bacteria, yeast, and humans (Fig. 4C). Therefore, we propose that the regulation mechanism of cAMP on Acs is evolutionally conserved from bacteria to mammals.
The newly identified cAMP binding pocket is also generally conserved in other acyl- or aryl-CoA synthetases (Fig. 6B), as implicated by the sequence alignment of 1190 acyl- or aryl-CoA synthetase family members across multiple species. We tested cAMP inhibitions of two other family members from S. enterica experimentally, and the inhibition effects upon SePrpE and SeFadD were similar to that for SeAcs. We infer that the regulatory mechanism of cAMP on Acs might be extendable to other family members of acyl- or aryl-CoA synthetases in S. enterica by competitively binding the substrate pockets of these enzymes. We speculate that the regulatory mechanism by cAMP on this protein family might also exist in other organisms.
The regulation of Acs by cAMP binding is yet to be characterized in vivo. However, the Kd of cAMP measured in this study is comparable with the physiological concentration found in bacterial cells under glucose depletion conditions (19, 20), which implicates the physiological relevance of cAMP. It is intriguing that Acs is regulated by cAMP in such a complex network involving several different layers of transcriptional activation, PTM, and direct inhibition. The concentration of cAMP is ever changing in cells corresponding to its growth phase and the growth environment (Table 3). In particular, cAMP is low when glucose is abundant in the culture medium, the concentration increases as glucose is consumed, and it reaches a maximum when glucose is depleted. It was reported that cAMP activates the transcription of Acs at a micromolar level but represses this transcription when cAMP is present a millimolar level (41, 42). We therefore speculate that one dominant regulatory means is preferred at a certain level of cAMP concentration for Acs activities in vivo. Once the concentration of cAMP reaches to more than 100 μm, a hostile environment may be represented and cells need to curtail most energy costs including inhibiting acetate scavenging by directly binding cAMP to the Acs protein.
TABLE 3.
Different intracellular cAMP concentrations correspond to the change of bacterial growth phase and culture environment
| E. coli strains | Supplements in LB medium | Growth condition | cAMP | References |
|---|---|---|---|---|
| μm | ||||
| ATCC 8739 | 1% Glucose | Pre-logarithmic phase | 0.5 | 19 |
| ATCC 8739 | 1% Glucose | Incubation for 60 min | 170 | 19 |
| W3110 | 0.04% Glucose and 0.2% lactose | Depletion of glucose | 15 | 48 |
| MC4100 | 0.02% Glucose | Depletion of glucose | 500 | 49 |
| MC4100 | 0.02% Glucose | Depletion of glucose | 125 | 20 |
| CHE9 | 0.2% Glucose-6-P | Mid-logarithmic phase | 0.38 | 50 |
| CHE9 | 0.2% Glucose | Mid-logarithmic phase | 0.39 | 50 |
| CHE9 | 0.2% Glycerol | Mid-logarithmic phase | 1.3 | 50 |
| CHE9 | 0.2% Glycerol to 0.2% glucose | Incubation for 20 min | 0.1 | 50 |
Our data present a new means of Acs regulation allowing direct sensing and rapid response by bacterial to cellular energy level. Our crystal structure also uncovered a new cAMP binding pocket, adding another layer to the complexity of cAMP signaling.
Experimental Procedures
Materials
Bacteria strains and plasmids used in this study are described and listed in supplemental Tables S1 and S2. Antibody of anti-acetylated SeAcs Lys609 was kindly gifted from Prof. Chen Yang. Acetate, acetyl-CoA, CoA, NAD+, cAMP, ATP, myokinase, pyruvate kinase, lactate dehydrogenase, and other chemicals were purchased as high purity from Sigma-Aldrich. Modified sequencing grade trypsin was purchased from Promega (Madison, WI).
Construction of cobB-null and pat-null Strains
The completely knock-out mutants of ΔcobB and Δpat were constructed in S. enterica G2466 by adapting a method described by Datsenko and Wanner (43). Genetic information of strains and primer sequences are listed in supplemental Tables S1 and S3. Obtained deletion mutations were verified by PCR.
Protein (SeAcs, SePat, SeCobB, SePrpE, and SeFadD) Expression and Purification
The acs (STM4275), pat (STM2651), cobB (STM1221), prpE (STM0371), and fadD (STM1818) genes were PCR amplified from S. enterica G2466 genome and cloned into the pET22b-tac or pET28b-tac vector (supplemental Table S2). Recombinants were then introduced into S. enterica G2466 wild-type or the indicated mutant cells, and successful transformants were selected by plating the cells on LB plates containing 100 μg/ml of ampicillin. Single colonies were first grown in 5 ml LB overnight at 37 °C, and then inoculated to 250 ml LB. Protein expressions were induced with isopropyl β-d-thiogalactoside to a final concentration of 0.5 mm overnight at 18 °C. Cells were harvested and resuspended in lysis buffer containing 50 mm Tris (pH 7.5), 300 mm KCl, 10% (v/v) glycerol, and 1 mm PMSF, and disrupted using an EmulsiFlex-C5 cell disruptor (AVESTIN, Inc., Ottawa, Canada). Cellular debris was removed by centrifuging at 13,000 × g for 30 min at 4 °C. The recombinant proteins were purified with affinity chromatography using a nickel-nitrilotriacetic acid Superflow column (Qiagen) and dialyzed into a buffer containing 50 mm Tris (pH 7.5), 50 mm NaCl, 1 mm DTT, and 10% (v/v) glycerol. After purification, the target proteins with sufficient purity (above 95%) were quantified and kept frozen at −80 °C. The variants of SeAcs were constructed via PCR site-directed mutagenesis (Agilent, Inc.) and then expressed in S. enterica G2466 and purified in the same way as that of wild-type SeAcs. Specially, SeAcs used in Pat-dependent acetylation assay and enzymatic inhibition assay was produced from the pat-null S. enterica G2466 strain, whereas SeAcs used in CobB-dependent deacetylation assay was produced from cobB-null S. enterica G2466 strain.
SeAcs for Crystallization
A single colony of BL21(DE3) transformed with plasmid Ph 3 (pET22b-tac-SeAcs) was used to inoculate 20-ml LB broth containing 100 μg/ml of ampicillin, and cultures were incubated overnight at 37 °C with shaking. LB broth (1 liter) was inoculated with a 1:100 ratio, incubated at 37 °C with shaking until A600 = 0.8. Protein expression was induced by addition of isopropyl β-d-thiogalactoside to 1 mm, and cultures were incubated for 3 h at 37 °C. Cells were harvested by centrifugation, re-suspended in lysis buffer (20 mm Tris, pH 7.7, 500 mm NaCl, 5% glycerol, and 5 mm 2-mercaptoethanol), and lysed using an Avestin EmulsiFlex-C5 cell disrupter (Avestin, Inc.). The supernatant after centrifugation was loaded onto a column packed with 1 ml of nickel-nitrilotriacetic acid-agarose (Qiagen, Inc.). The column was sequentially washed with 20 ml of lysis buffer containing 0, 5, and 10 mm imidazole, and eluted with 20 ml of lysis buffer containing 40 mm imidazole. The eluted fraction was dialyzed 10 h against 1 liter of 10 mm EPPS, pH 8.0, 0.2 mm TECP, and 1 mm EDTA for three times. Fractions were pooled, concentrated to ∼15 mg/ml using 10-kDa MWCO Amicon Ultra-15 centrifugal ultrafilters (Millipore, Inc.), and stored at −80 °C.
In Vitro SeAcs Enzymatic Assay
SeAcs enzymatic activity was measured by a spectrophotometric coupled assay at 37 °C as described in Ref. 22. During the assay, SeAcs first converts acetate, CoA, and ATP to acetyl-CoA and AMP. Then, myokinase converts AMP to ADP. Pyruvate kinase converts ADP and phosphoenolpyruvate to pyruvate and ATP. Finally, lactate dehydrogenase reduces pyruvate and oxidizes NADH to NAD+. The change of NADH level is detected spectrometrically at 340 nm. The enzymatic reaction mixtures (90 μl) contain 50 mm HEPES (pH 7.5), 5 mm MgCl2, 1 mm TCEP, 0.02 μm SeAcs, 5 units of myokinase, 1 unit of pyruvate kinase, 1.5 units of lactate dehydrogenase, 3 mm phosphoenolpyruvate, and 0.1 mm NADH. Reaction mixtures were incubated at 37 °C for 2 min, and then the assay was initiated by adding 10 μl of substrate mixtures (final concentration: 1 mm CoA, 1 mm ATP, and 20 mm acetate).
To determine the ratio of cAMP inhibition, the potential inhibitory effect of cAMP (1 mm) on SeAcs was tested using subsaturated concentrations for one substrate (i.e. 20 μm for CoA, 20 μm for ATP, or 20 μm for acetate), and saturated concentrations for other substrates (i.e. 1 mm for CoA, 1 mm for ATP, or 20 mm for acetate). To determine the dissociate constant Ki of cAMP toward SeAcs, different concentrations of cAMP (0, 0.15, 0.3, 0.45, 0.6 mm) was added to the reaction mixtures. For each cAMP concentration, the apparent Km value of SeAcs toward the substrate of ATP was measured by adding 10 μl of substrate mixture (final concentration: 20 mm acetate, 1 mm CoA, and varying ATP: 20, 40, 80, 120, 200, 300, 400, 600, 1000, 1500, and 2000 μm). The reactions were continually monitored by measuring absorbance at A340 every 2 s for 10 min using a microplate reader (Thermo Multiskan FC, Thermo-Fisher Inc.). The initial velocities were analyzed using Prism 5 (GraphPad Software Inc.), and the apparent Km values in the presence of inhibitors were estimated by non-linear regression using the Michaelis-Menten equation. The mode of cAMP inhibition was determined by a Lineweaver-Burk plot and the substrate binding affinity Ki was estimated (intercept on x axis) in a Dixon plot with an apparent Km toward ATP against the concentration of cAMP as described in Ref. 44. Each point was measured in triplicate.
In Vitro SeAcs Acetylation and Deacetylation Assay
Pat-dependent SeAcs acetylation assays were carried out as follows: reaction mixtures (100 μl) containing 50 mm Tris (pH 7.5), 200 μm TCEP, 200 μm acetyl-CoA, 0.5 μm SePat, 1 μm SeAcs, and in the presence or absence of 1 mm cAMP were incubated at 37 °C for 30 min. The acetylation level of SeAcs Lys609 was measured by Western blotting using a monoclonal antibody specific to the acetylated SeAcs Lys609. Meanwhile, the activity of treated SeAcs was detected by adding 10 μl of acetylation reaction mixtures (containing 2 pmol of SeAcs) to the enzymatic reaction mixtures (90 μl) containing 50 mm HEPES (pH 7.5), 5 mm MgCl2, 1 mm TCEP, 5 units of myokinase, 1 unit if pyruvate kinase, 1.5 units of lactate dehydrogenase, 3 mm phosphoenolpyruvate, 0.1 mm NADH, 1 mm CoA, 1 mm ATP, and 20 mm acetate. cAMP (1 mm) was additionally supplied in the enzymatic reaction mixture if it was present in the acetylation reaction mixtures.
CobB-dependent SeAcs deacetylation assays were carried out as follows: reaction mixtures (100 μl) containing 50 mm Tris (pH 7.5), 200 μm TCEP, 1 mm NAD+, 0.3 μm SeCobB, and 1 μm SeAcs and in the presence or absence of 1 mm cAMP, were incubated at 37 °C for 60 min. The acetylation level of SeAcs Lys609 was measured by Western blotting. Meanwhile, the activity of treated SeAcs was detected by adding 10 μl of deacetylation reaction mixtures (containing 2 pmol of SeAcs) to the enzymatic reaction mixtures as described above.
Western Blotting
Standard Western blotting procedures were followed for protein acetylation analysis. Proteins were separated by SDS-PAGE, transferred onto NC membrane (Millipore), subjected to immunoblotting with the homemade antibody specific to the acetylated SeAcs Lys609, and detected by ImageQuant LAS 4000 (GE Healthcare). Coomassie Blue staining (SDS-PAGE) was used for loading controls.
In Vitro SePrpE, SeFadD Enzymatic Assay
The SePrpE and SeFadD enzymatic activities were measured by a similar spectrophotometric-coupled assay as described above. Reaction mixtures (90 μl) contain 50 mm HEPES (pH 7.5), 5 mm MgCl2, 1 mm TCEP, 10 nm SePrpE or 800 nm SeFadD, 5 units of myokinase, 1 unit of pyruvate kinase, 1.5 units of lactate dehydrogenase, 3 mm phosphoenolpyruvate, 0.1 mm NADH, and cAMP (1 mm for determining ratio of inhibition). Reaction mixtures (90 μl) were incubated at 37 °C for 2 min, and initiated by adding 10 μl of substrate mixture (final concentration: 1 mm CoA, 20 mm sodium propionate, and 20 μm ATP for SePrpE; 1 mm CoA, 0.8 mm oleic acid, and 80 μm ATP for SeFadD). The reactions were continually monitored by measuring absorbance at A340 every 2 s for 10 min using a microplate reader (Thermo Multiskan FC, ThermoFisher Inc.).
Isothermal Titration Calorimetry
ITC experiments were carried out using a Microcal iTC200 microcalorimeter (GE Healthcare). SeAcs and SeAcs derivatives were dialyzed against a buffer containing 20 mm HEPES (pH 7.5), 100 mm NaCl, and 1 mm TCEP and concentrated to 100 μm by ultrafiltration. cAMP or ATP was dissolved in the same buffer to the concentration of 4 mm. During the experiments, 200 μl of 100 μm SeAcs or SeAcs derivatives was titrated with 2 μl of 4 mm cAMP for 20 steps. The resulting titration curves were fitted with MicroCal ORIGIN software. The dissociation constant Kd was estimated by non-linear regression with ORIGIN 7.0 software (OriginLab Inc.).
SeAcs Crystallization
SeAcs (180 μm) was incubated with cAMP (2 mm), coenzyme A (1 mm), and acetate (1 mm), and incubated 1 h at room temperature. Crystal growth conditions of the complex were screened with commercial solutions (Emerald Biosystems, Inc. and Hampton Research, Inc.) using a sitting-drop vapor-diffusion technique (drop: 0.2 μl of protein complex plus 0.2 μl of reservoir solution; reservoir: 60 μl of commercial screening solution; 22 °C). Small shell-like crystals appeared within 3 days during screening, and were able to reach a dimension of 0.2 × 0.4 × 0.1 mm when growing in drops of larger size (2 μl; 1 μl of protein complex plus 1 μl of reservoir) using hanging-drop vapor diffusion with reservoir (0.2 m KH2PO4, pH 4.8, 20% PEG3350). Crystals were harvested by transferring into reservoir solution containing 12.5% (v/v) (2R,3R)-(−)-2,3-butanediol (Sigma), and then flash-cooled in liquid nitrogen.
Structure Determination
Diffraction data were collected from cryo-cooled crystals at SSRF beamline BL17U1 (51). Data were processed using HKL2000 (45). The structure was solved by molecular replacement with Molrep (46) using the crystal structure of SeAcs (PDB code 1PG4) (23) as the search model, and refined with Phenix (47) and Coot (47). cAMP and coenzyme A were built into the model during the late stage of refinement. The final SeAcs-cAMP-CoA model, has been refined to Rwork and Rfree of 0.169 and 0.207, respectively.
Author Contributions
X. H. conceived the study, performed most in vitro biochemical experiments and data analysis; L. S. purified and crystallized SeAcs-cAMP-CoA, and determined the crystal structure; P. L., Q. W., X. C., J. W., and M. W. assisted in data analysis; W. Z., Y. Z., and G. Z. planned experiments, and wrote the manuscript. All the authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the staff of beamline BL17U1 at the Shanghai Synchrotron Radiation Facility, Shanghai, People's Republic of China, for assistance during data collection. We thank Dr. Andrew M. Gulick for pAC10 construction.
This work was supported by Natural Science Foundation for Youth Grant 31400039, Postdoctoral Science Foundation of China Grant 2012M510787, and the Chinese Thousand Talents Program grant. The authors declare no conflict of interest with the contents of this article.

This article contains supplemental Tables S1–S3.
The atomic coordinates and structure factors (code 5JRH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Acs
- acetyl-coenzyme A synthetase
- PTM
- post-translational modification
- CRP
- cAMP receptor protein
- ITC
- isothermal titration calorimetry
- cNMP
- cyclic nucleotide
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- PDB
- Protein Data Bank
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Pietrocola F., Galluzzi L., Bravo-San Pedro J. M., Madeo F., and Kroemer G. (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 [DOI] [PubMed] [Google Scholar]
- 2. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., and Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Starai V. J., and Escalante-Semerena J. C. (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell Mol. Life Sci. 61, 2020–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cederbaum A. I. (2012) Alcohol metabolism. Clin. Liver Dis. 16, 667–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Comerford S. A., Huang Z., Du X., Wang Y., Cai L., Witkiewicz A. K., Walters H., Tantawy M. N., Fu A., Manning H. C., Horton J. D., Hammer R. E., McKnight S. L., and Tu B. P. (2014) Acetate dependence of tumors. Cell 159, 1591–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schug Z. T., Peck B., Jones D. T., Zhang Q., Grosskurth S., Alam I. S., Goodwin L. M., Smethurst E., Mason S., Blyth K., McGarry L., James D., Shanks E., Kalna G., Saunders R. E., et al. (2015) Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takahashi H., McCaffery J. M., Irizarry R. A., and Boeke J. D. (2006) Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 23, 207–217 [DOI] [PubMed] [Google Scholar]
- 8. Barak R., Prasad K., Shainskaya A., Wolfe A. J., and Eisenbach M. (2004) Acetylation of the chemotaxis response regulator CheY by acetyl-CoA synthetase purified from Escherichia coli. J. Mol. Biol. 342, 383–401 [DOI] [PubMed] [Google Scholar]
- 9. Yan J., Barak R., Liarzi O., Shainskaya A., and Eisenbach M. (2008) In vivo acetylation of CheY, a response regulator in chemotaxis of Escherichia coli. J. Mol. Biol. 376, 1260–1271 [DOI] [PubMed] [Google Scholar]
- 10. Beatty C. M., Browning D. F., Busby S. J., and Wolfe A. J. (2003) Cyclic AMP receptor protein-dependent activation of the Escherichia coli acsP2 promoter by a synergistic class III mechanism. J. Bacteriol. 185, 5148–5157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Browning D. F., Beatty C. M., Sanstad E. A., Gunn K. E., Busby S. J., and Wolfe A. J. (2004) Modulation of CRP-dependent transcription at the Escherichia coli acsP2 promoter by nucleoprotein complexes: anti-activation by the nucleoid proteins FIS and IHF. Mol. Microbiol. 51, 241–254 [DOI] [PubMed] [Google Scholar]
- 12. Starai V. J., and Escalante-Semerena J. C. (2004) Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J. Mol. Biol. 340, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 13. Starai V. J., Celic I., Cole R. N., Boeke J. D., and Escalante-Semerena J. C. (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 14. McKnight G. S. (1991) Cyclic AMP second messenger systems. Curr. Opin. Cell Biol. 3, 213–217 [DOI] [PubMed] [Google Scholar]
- 15. Shenoy A. R., and Visweswariah S. S. (2006) New messages from old messengers: cAMP and mycobacteria. Trends Microbiol. 14, 543–550 [DOI] [PubMed] [Google Scholar]
- 16. Castaño-Cerezo S., Bernal V., Blanco-Catalá J., Iborra J. L., and Cánovas M. (2011) cAMP-CRP co-ordinates the expression of the protein acetylation pathway with central metabolism in Escherichia coli. Mol. Microbiol. 82, 1110–1128 [DOI] [PubMed] [Google Scholar]
- 17. Nambi S., Basu N., and Visweswariah S. S. (2010) cAMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 285, 24313–24323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee H. J., Lang P. T., Fortune S. M., Sassetti C. M., and Alber T. (2012) Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat. Struct. Mol. Biol. 19, 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makman R. S., and Sutherland E. W. (1965) Adenosine 3′,5′-phosphate in Escherichia coli. J. Biol. Chem. 240, 1309–1314 [PubMed] [Google Scholar]
- 20. Notley-McRobb L., Death A., and Ferenci T. (1997) The relationship between external glucose concentration and cAMP levels inside Escherichia coli: implications for models of phosphotransferase-mediated regulation of adenylate cyclase. Microbiology 143, 1909–1918 [DOI] [PubMed] [Google Scholar]
- 21. Hu L. I., Lima B. P., and Wolfe A. J. (2010) Bacterial protein acetylation: the dawning of a new age. Mol. Microbiol. 77, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reger A. S., Carney J. M., and Gulick A. M. (2007) Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46, 6536–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gulick A. M., Starai V. J., Horswill A. R., Homick K. M., and Escalante-Semerena J. C. (2003) The 1.75-Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 [DOI] [PubMed] [Google Scholar]
- 24. Schmelz S., and Naismith J. H. (2009) Adenylate-forming enzymes. Curr. Opin. Struct. Biol. 19, 666–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng D., Constantinidou C., Hobman J. L., and Minchin S. D. (2004) Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 32, 5874–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan C. H., Garrity J., Crosby H. A., and Escalante-Semerena J. C. (2011) In Salmonella enterica, the sirtuin-dependent protein acylation/deacylation system (SDPADS) maintains energy homeostasis during growth on low concentrations of acetate. Mol. Microbiol. 80, 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jogl G., and Tong L. (2004) Crystal structure of yeast acetyl-coenzyme A synthetase in complex with AMP. Biochemistry 43, 1425–1431 [DOI] [PubMed] [Google Scholar]
- 28. Tucker A. C., Taylor K. C., Rank K. C., Rayment I., and Escalante-Semerena J. C. (2014) Insights into the specificity of lysine acetyltransferases. J. Biol. Chem. 289, 36249–36262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Görke B., and Stülke J. (2008) Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624 [DOI] [PubMed] [Google Scholar]
- 30. Sutherland E. W., and Robison G. A. (1969) The role of cyclic AMP in the control of carbohydrate metabolism. Diabetes 18, 797–819 [DOI] [PubMed] [Google Scholar]
- 31. D'Ari R., Jaffé A., Bouloc P., and Robin A. (1988) Cyclic AMP and cell division in Escherichia coli. J. Bacteriol. 170, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villarreal J. M., Hernández-Lucas I., Gil F., Calderón I. L., Calva E., and Saavedra C. P. (2011) cAMP receptor protein (CRP) positively regulates the yihU-yshA operon in Salmonella enterica serovar Typhi. Microbiology 157, 636–647 [DOI] [PubMed] [Google Scholar]
- 33. Botsford J. L., and Harman J. G. (1992) Cyclic AMP in prokaryotes. Microbiol. Rev. 56, 100–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McDonough K. A., and Rodriguez A. (2011) The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 10, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ortega-Martínez S. (2015) A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front. Mol. Neurosci. 8, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Serezani C. H., Ballinger M. N., Aronoff D. M., and Peters-Golden M. (2008) Cyclic AMP: master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 39, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takio K., Smith S. B., Krebs E. G., Walsh K. A., and Titani K. (1982) Primary structure of the regulatory subunit of type II cAMP-dependent protein kinase from bovine cardiac muscle. Proc. Natl. Acad. Sci. U.S.A. 79, 2544–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaupp U. B., and Seifert R. (2002) Cyclic nucleotide-gated ion channels. Physiol. Rev. 82, 769–824 [DOI] [PubMed] [Google Scholar]
- 39. de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., and Bos J. L. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 [DOI] [PubMed] [Google Scholar]
- 40. Rehmann H., Wittinghofer A., and Bos J. L. (2007) Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 8, 63–73 [DOI] [PubMed] [Google Scholar]
- 41. Fic E., Bonarek P., Gorecki A., Kedracka-Krok S., Mikolajczak J., Polit A., Tworzydlo M., Dziedzicka-Wasylewska M., and Wasylewski Z. (2009) cAMP receptor protein from Escherichia coli as a model of signal transduction in proteins: a review. J. Mol. Microbiol. Biotechnol. 17, 1–11 [DOI] [PubMed] [Google Scholar]
- 42. Małecki J., Polit A., and Wasylewski Z. (2000) Kinetic studies of cAMP-induced allosteric changes in cyclic AMP receptor protein from Escherichia coli. J. Biol. Chem. 275, 8480–8486 [DOI] [PubMed] [Google Scholar]
- 43. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dixon M. (1953) The determination of enzyme inhibitor constants. Biochem. J. 55, 170–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 46. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Inada T., Kimata K., and Aiba H. (1996) Mechanism responsible for glucose-lactose diauxie in Escherichia coli: challenge to the cAMP model. Genes Cells 1, 293–301 [DOI] [PubMed] [Google Scholar]
- 49. Death A., and Ferenci T. (1994) Between feast and famine: endogenous inducer synthesis in the adaptation of Escherichia coli to growth with limiting carbohydrates. J. Bacteriol. 176, 5101–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Epstein W., Rothman-Denes L. B., and Hesse J. (1975) Adenosine 3′-5′-cyclic monophosphate as mediator of catabolite repression in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 72, 2300–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Q., Yu F., Huang S., Sun B., Zhang K., Liu K., Wang Z., Xu C., Wang S., Yang L., Pan Q., Li L., Zhou H., Cui Y., Xu Q., et al. (2015) The macromolecular crystallography beamline of SSRF. Nucl. Sci. Tech. 26, 010102 [Google Scholar]