

Abstract
Although an enormous and still growing number of biologically diverse methyltransferases have been reported and identified, a comprehensive understanding of the enzymatic methyl transfer mechanism is still lacking. Glycine N-methyltransferase (GNMT), a member of the family that acts on small metabolites as the substrate, catalyzes methyl transfer from S-adenosyl-l-methionine (AdoMet) to glycine to form S-adenosyl-l-homocysteine and sarcosine. We report primary carbon (12C/14C) and secondary (1H3/3H3) kinetic isotope effects at the transferred methyl group, together with 1H3/3H3 binding isotope effects for wild-type GNMT and a series of Tyr21 mutants. The data implicate a compaction effect in the methyl transfer step that is conferred by the protein structure. Furthermore, a remarkable similarity of properties is observed between GNMT and catechol O-methyltransferase, despite significant differences between these enzymes with regard to their active site structures and catalyzed reactions. We attribute these results to a catalytically relevant reduction in the methyl donor–acceptor distance that is dependent on a tyrosine side chain positioned behind the methyl-bearing sulfur of AdoMet.
Introduction
Enzymes are proteins that catalyze the large majority of chemical reactions that occur in the cell, at rates accelerated far beyond what can be achieved by analogous molecular catalysts.1 The precise molecular mechanism of these rate enhancements is still under debate, with proposals put forth over the past 60 years2−13 that include transition-state stabilization,3 reactant destabilization,3,4 electrostatic effects,6 protein motions,7,9,11,13 and protein-facilitated quantum mechanical tunneling.8,12 The contribution of protein motions to create a compact active site that modulates the donor–acceptor distance is plausible for various enzymatic systems.8,9 Although evidence for mediation of the donor–acceptor distance in the methyl transfer catalyzed by catechol O-methyltransferase (COMT) has been reported recently,14,15 the generality of this finding to other enzymatic methyl transfer reactions remains to be demonstrated.
Methyl transfer reactions that utilize S-adenosyl-l-methionine (AdoMet) play a vital role in diverse and essential physiological processes, including the reversible methylation of histones during transcription and the processing of small metabolites.16,17 The enzyme glycine N-methyltransferase (GNMT), compromising up to 3% of total cytosolic protein, catalyzes a methylation of glycine to sarcosine (N-methylglycine) and has been linked to toxicology and cancer.18 GNMT is increasingly viewed as a central player in cellular regulation via its impact on the ratio of cellular AdoMet to S-adenosyl-l-homocysteine (AdoHcy).19 GNMT could also provide an excellent model system in which to investigate the mechanism of AdoMet-based methyl transfer reactions to nitrogen-based nucleophilic substrates, as occurs among the widely distributed RNA, DNA, and histone methyltransferases.
The kinetic mechanism of GNMT has been characterized as an ordered addition of AdoMet followed by glycine, Scheme 1.20−22 This enzyme is distinctive as a dimer of dimers, in contrast to the more common monomeric form of class I AdoMet-dependent enzymes that include COMT.21 Within the apo form of GNMT, the active site of each monomer is capped by a N-terminal U-loop from an adjacent subunit, representing a closed conformation. Upon addition of AdoMet, the N-terminal domain becomes highly disordered, swinging into the solvent region, and providing an entrance to the active center for the substrate (Scheme S1).21 The subsequent chemical reaction is expected to occur via an SN2-type transfer of the methyl group of AdoMet to the amine of glycine, in strict analogy with other members of this enzyme class.21,23
Scheme 1.

The comparative enzyme, COMT, has been previously characterized in some detail, in particular with regard to the factors that determine the efficiency of the methyl transfer step.14,15,24,25 In a recent study from this laboratory, we proposed a role for ground-state compaction that is linked to the ionization of the substrate to form an ion pair between the oxyanion of the substrate and the charged sulfur of the cofactor.15 There are a number of notable differences between COMT and GNMT that extend beyond the targeted atom (O for COMT and N for GNMT) that include the aforementioned state of protein oligomerization (monomer for COMT and homotetramer for GNMT), the extent of exposure of the active site (the active site of GNMT lies deeper than that of COMT), the importance of loop movement (significant movement in GNMT), and a requirement for a metal ion (Mg2+ for COMT, none for GNMT). Despite these extensive differences, both enzymes belong to the AdoMet methyltransferase class I family, sharing a structural core composed of a Rossmann fold with its characteristic center seven-stranded β-sheet and two side layers of helices to form an αβα sandwich.26,27
Although considerable experimental and computational effort was previously invested in the GNMT system, a detailed mechanism for catalysis of the methyl group transfer remains to be established.21,23,28−39 In a quantum mechanics/molecular mechanics (QM/MM) and molecular dynamics (MD) simulation of GNMT, the possible role of active site compaction, as postulated for COMT, was examined, leading to a computed internuclear distance along the reaction coordinate that was incompatible with such a conclusion.38 In the present work, both the wild type (WT) and site-specific mutants at an active site tyrosine of GNMT have been characterized with regard to catalytic efficiency, primary and secondary kinetic isotope effects (KIEs), and binary and abortive ternary binding isotope effects (BIEs), with the goal of interrogating the geometry of the reaction coordinate. The resulting data demonstrate an impact of Y21 on catalytic rate constants and kinetic isotope effects that is highly analogous to effects seen previously with COMT. The aggregate data indicate a remarkable convergence of the behavior between COMT and GNMT that is critically dependent on the size and charge of a tyrosine side chain positioned behind the reactive sulfur of the cofactor.
Results
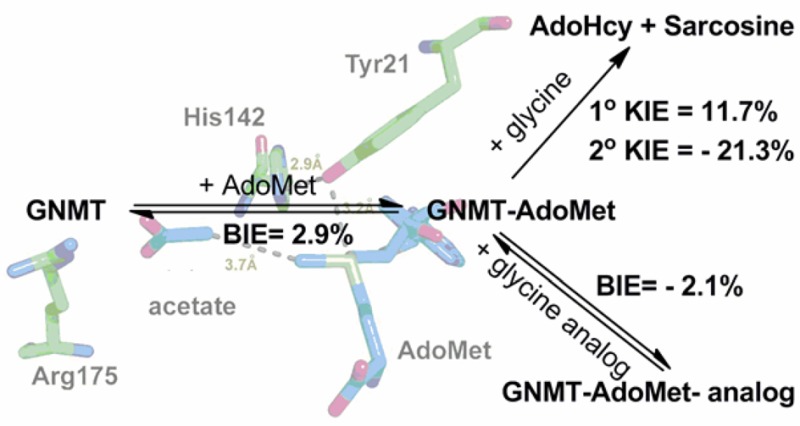
The crystal structure of GNMT with AdoMet and an inhibitor (acetate) indicates that the sulfur atom of AdoMet is in close proximity to a tyrosine residue at position 21 (Figure 1).21 By analogy to the earlier studies of COMT, a series of mutants at Y21 were generated as a probe of their impact on the catalytic rate acceleration. The catalytic turnover number (kcat) under saturation of both the cofactor and substrate, as measured radiometrically using [methyl-3H]AdoMet (Table 1), shows a 7-fold reduction with Y21F, followed by a 260-fold reduction with all of the smaller side chains at position 21. Second-order rate constants were collected for both AdoMet and glycine at saturating concentrations of the alternate substrate. An ordered binding of the cofactor, followed by glycine, has been demonstrated for WT GNMT,20−22,30 and it was possible that some randomness in binding order would be introduced via mutations at Y21; however, the measurement of kcat/Km(glycine) under conditions of saturating AdoMet ensures that the same physical processes are being studied, which involve the binding of glycine to a preformed E–AdoMet complex and its subsequent transformation to sarcosine. The impact of Phe is similar when measuring kcat/Km of both the cofactor and substrate (around a 10-fold decrease), whereas the average impact of the smaller residues is much greater: 1.6 × 103 for kcat/Km(AdoMet) and 6.6 × 104 for kcat/Km(Gly). Regardless of the side chain substitution at Tyr21, the primary 12C/14C KIE is identical within experimental error of a value of 1.13 ± 0.007 (Table 2). This large value is near the experimental maximum KIEs reported for typical SN2 reactions, indicating a fully rate-determining methyl transfer step under the condition of kcat/Km(glycine). The secondary CH3/CT3 KIE at the transferred methyl group is 0.787 ± 0.005 for WT GNMT, highly similar to the measured CH3/CT3 KIE of 0.791 ± 0.012 for WT COMT.14 The most straightforward interpretation of such an inverse secondary KIE is an increase in out-of-plane bending vibrations that arises from a tight positioning between the incoming nucleophile and leaving group.24,25,40,41 Mutation of Tyr21 to Phe and other mutants leads to a progressive shift of this secondary KIE toward unity, displaying 0.809 for Y21F and an average value of 0.861 for Gly, Ala, Val, Ser, and Thr at position 21, approaching the value of unity observed in the solution reaction.24,25,40 Once again, in a manner analogous to that of COMT, an approximately linear relationship is seen between the magnitude of the secondary KIE and the catalytic efficiency of glycine reacting with the enzyme AdoMet complex, kcat/Km(glycine), Figure 2. A similar trend is observed when the secondary KIE is correlated with kcat/Km(AdoMet) (Figure S1). As a frame of reference for the magnitude and trends in the secondary CH3/CT3 KIE, the magnitude of the CH3/CT3 BIEs for binary and ternary complex formation was also determined (Table 2). In contrast to the inverse values for the KIEs, the BIEs are all normal, with the exception of the WT ternary complex, where a value within experimental error of unity is observed. Overall, no compelling trends could be discerned either between binary and ternary complexes or among the series of mutants generated at position 21.
Figure 1.
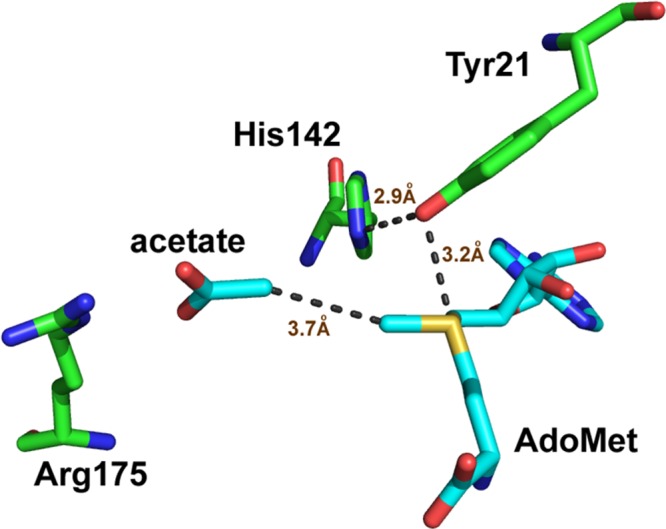
Active site of rat GNMT (PDB 1NBH).
Table 1. Summary of Kinetic Parameters for the Methylation of Glycine by the Recombinant Rat GNMT and Its Mutants.
| AdoMet |
glycine |
||||
|---|---|---|---|---|---|
| GNMT | kcat (min–1) | Km (μM) | kcat/Km (M–1 s–1) | Km (mM) | kcat/Km (M–1 s–1) |
| WT | 174.5 ± 23.2 | 108 ± 18 | 26935 ± 5738 | 1.1 ± 0.2 | 2616 ± 537 |
| Y21F | 25.2 ± 1.3 | 174 ± 43 | 2409 ± 609 | 1.7 ± 0.8 | 248 ± 117 |
| Y21G | 0.93 ± 0.30 | 696 ± 37 | 22.3 ± 7.2 | 33.6 ± 5.3 | 0.46 ± 0.17 |
| Y21A | 0.59 ± 0.18 | 539 ± 96 | 18.3 ± 6.4 | 26.6 ± 4.4 | 0.37 ± 0.13 |
| Y21V | 0.58 ± 0.07 | 582 ± 31 | 16.7 ± 2.2 | 38.8 ± 7.5 | 0.25 ± 0.06 |
| Y21S | 0.65 ± 0.18 | 803 ± 70 | 13.6 ± 3.9 | 23.4 ± 2.3 | 0.46 ± 0.14 |
| Y21T | 0.81 ± 0.31 | 1136 ± 86 | 11.9 ± 4.7 | 22.9 ± 3.7 | 0.59 ± 0.25 |
Table 2. Summary of Isotope Effects for Recombinant Rat GNMT and Its Mutants.
| GNMT | primary KIE, k12C/k14C | secondary KIE, kCH3/kCT3 | binary BIEa (GNMT–AdoMet) | ternary BIEb (GNMT–AdoMet–acetate) |
|---|---|---|---|---|
| WT | 1.117 ± 0.008 | 0.787 ± 0.005 | 1.029 ± 0.013 | 1.008 ± 0.011 |
| Y21F | 1.119 ± 0.013 | 0.809 ± 0.006 | 1.041 ± 0.018 | 1.042 ± 0.003 |
| Y21G | 1.146 ± 0.023 | 0.867 ± 0.013 | 1.021 ± 0.044 | 1.027 ± 0.009 |
| Y21A | 1.132 ± 0.007 | 0.832 ± 0.013 | 1.031 ± 0.012 | 1.025 ± 0.020 |
| Y21V | 1.117 ± 0.010 | 0.865 ± 0.017 | 1.034 ± 0.013 | 1.033 ± 0.017 |
| Y21S | 1.132 ± 0.015 | 0.859 ± 0.020 | 1.028 ± 0.023 | 1.041 ± 0.021 |
| Y21T | 1.127 ± 0.019 | 0.881 ± 0.015 | 1.027 ± 0.016 | 1.030 ± 0.015 |
Binding isotope effect (CH3/CT3) for the GNMT–AdoMet binary complex.
Binding isotope effect (CH3/CT3) for the GNMT–AdoMet–acetate ternary complex.
Figure 2.
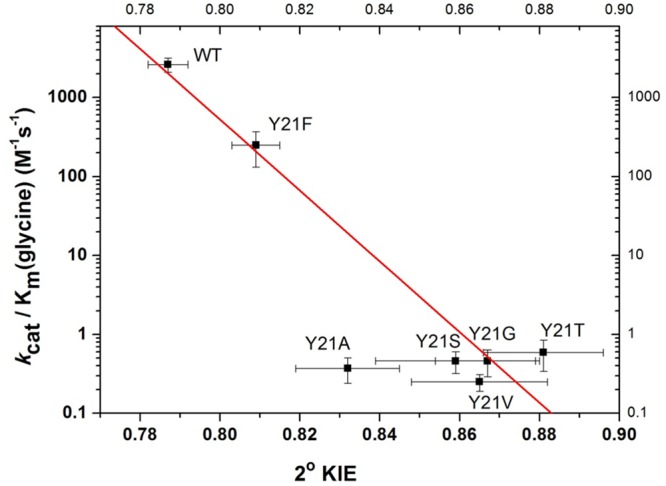
Relationship between the kcat/Km for glycine and secondary KIE for methylation of glycine by the recombinant rat GNMT and Y21 mutants. r2 = 0.93 except for the Y21A mutants. The observation that Y21A is an outliner may indicate a change in active site hydration for this variant (cf. ref (49)).
Discussion
The enzymes COMT and GNMT belong to the class I family of AdoMet-dependent methyltransferases,26,27 in which the cofactor binds prior to the substrate within a classical Rossmann fold. Recent computational studies of COMT indicate an unusual set of properties that include a short (ca. 2.7 Å) methyl donor–acceptor distance that becomes elongated following a series of mutations at a single tyrosine that resides behind the sulfur of bound AdoMet.15 Concomitant with elongation of the donor–acceptor bond, the catalytic efficiency declines by 3 orders of magnitude in an unexpected linear fashion. With the goal of better understanding this behavior, we undertook a detailed examination of the related GNMT that differs from COMT in a number of mechanistically significant ways that include the transfer of a methyl group from AdoMet to a nitrogen vs oxyanion acceptor and the absence of the active site divalent metal ion that anchors and activates one of the ring oxygens of the substrate for COMT. These changes at the active site would appear to be of paramount importance in determining the mechanism and transition state for methyl transfer. In the case of COMT, both oxygens on the ring of the catechol substrate undergo chelation to Mg2+, which is further coordinated to two aspartate side chain and one asparagine side chain (Asp 141, Asp 169, and Asn170) and a water molecule.42 On the basis of extensive X-ray characterizations42−47 and a recent computational study,15 one of the catechol oxygens is concluded to undergo ionization to form a substrate-derived oxyanion prior to entering into reaction with the methyl group of AdoMet. According to this scenario, there will be a net negative charge on the attacking nucleophile in COMT. By contrast, the substrate for GNMT is glycine (pKa1 = 2.3 and pKa2 = 9.6), which exists in a zwitterionic form at pH 7 with the amino group protonated and the carboxylate ionized. Although the charge status of the transferred methyl group at different stages of reaction can be reasonably inferred, the situation for the reactive nitrogen of the glycine substrate is less clear. Given the lack of reactivity of a protonated amino group on glycine, it will by necessity undergo deprotonation to form a suitable nucleophilic center for reaction with AdoMet; this may occur either prior to or after binding, to generate a neutral amine within the enzyme–substrate complex. The remaining ionized carboxylate of glycine may be expected to be accommodated by hydrogen bonding with possible interactions to active site residues Tyr33, Asn138, Arg175, and Tyr220 (Figure S2). Although an active site general base has not been unambiguously identified in GNMT, a hydrogen-bonding network between the amino group of glycine and Tyr242, Tyr220, and the substrate carboxylate can be inferred by inserting glycine into the position occupied by acetate within the solved ternary complex structure (Figure S3). This network would be expected to assist in further deprotonation of a bound, initially neutral amine form of the substrate, as it undergoes nucleophilic addition to the methyl group of the AdoMet. The resulting charge on the attacking nitrogen nucleophile, while unknown currently, is expected to range from neutral (for a deprotonation that is a fully concerted reaction with nucleophilic attack at the methyl carbon of AdoMet) to slightly positive (in the event that deprotonation lags behind the formation of the new nitrogen–carbon bond).
While the above list of differences between GNMT and COMT might have predicted very different experimental outcomes for GNMT and COMT, the present analysis of kinetic parameters and their isotope effects for GNMT (Table 1) indicates quite similar properties. As shown, GNMT undergoes a large decrease in all of its kinetic parameters as the tyrosine side chain that resides behind the sulfur of the cofactor is altered, with the parameter that undergoes the largest impact being kcat/Km for glycine (reduced >104-fold relative to that of the WT). The magnitude and trends in the secondary tritium kinetic isotope effect on kcat/Km(glycine), Table 2, are also very similar to those of COMT, being inverse in value for the WT enzyme and rising toward unity in the family of mutants studied. Once again in analogy with COMT, an approximately straight line can be drawn between kcat/Km(glycine) and the magnitude of the secondary KIE (Figure 2): with the exception of Y21A, all of the smaller aliphatic side chains are within experimental error of the line drawn through the WT and Y21F. The overall decrease in the rate constant is, in fact, 10-fold greater than for COMT, reflecting the larger initial rates for WT GNMT (10-fold for kcat and 3-fold for kcat/Km(glycine)). Importantly, the magnitude of the methyl carbon isotope effect is elevated to values consistent with a fully rate-limiting methyl transfer reaction for all variants. Finally, a comparison of binding isotope effects (BIEs) to KIEs indicates opposing trends, with the BIEs either within experimental error or slightly greater than unity. Thus, with regard to every property interrogated, GNMT parallels the earlier reported effects with COMT.
The fact that the measured BIEs are normal and opposite in direction to the experimental KIEs with GNMT may be due to the need to employ a substrate analogue (acetate) with weak enough binding to allow the labeled AdoMet to undergo rapid equilibration between its bound and free states (cf. ref (33)). However, there are subtle differences between the WT and variants at Tyr21 in the case of GNMT, Table 2. All of the mutants yield the same magnitude of BIE, and furthermore, this remains unchanged when the properties of binary and ternary complexes are compared. This feature may be different for the WT, where measurements indicate a slightly reduced BIE for the binary complex that undergoes further reduction toward unity in the ternary complex. Given the small magnitude of the measurements in relation to the limits of experimental error (ca. 1%), it is difficult to reach a firm conclusion, yet the ternary complex data with the WT may suggest a trend in which the enzyme is capable of achieving an active site that is closer to the catalytically relevant configuration(s) despite the use of an analogue lacking the nucleophilic amino group.
In attempting to rationalize the rate and KIE data for GNMT in the context of COMT, it is important to question whether factors other than a degree of transition-state compaction that is dependent on the side chain of position 21 could produce the experimental trends. We thus turned to density functional theory (DFT) (Table 3) to compute the magnitude of equilibrium isotope effects (EIEs) for transfer of a methyl group from sulfur to nitrogen (eq 1, where R1 is CO2H or CO2– and R2 is either absent or represents a proton, leading to a positive charge on nitrogen; see the structures in Table 3), finding values that will vary depending on the ionization state of the substrate’s carboxylate and, most importantly, the charge assigned to its nitrogen atom. For an ionized carboxylate, the EIEs are predicted to be 0.762–0.781 in the event of formation of a fully positive charge on the nuclephilic nitrogen or 1.090–1.104 when proton transfer is complete at the time of N-methyl formation. Clearly, the degree of positive charge on the nucleophile nitrogen atom at the transition state will have an impact on the expected magnitude of the KIE, especially under the condition of a late transition state in which the methyl group is almost fully transferred.
| 1 |
Table 3. Density Functional Theory (DFT) Simulated Equilibrium Isotope Effects in Different Media (DC = Dielectric Constant) for the Methyl Group Transfer from AdoMet to Glycine.
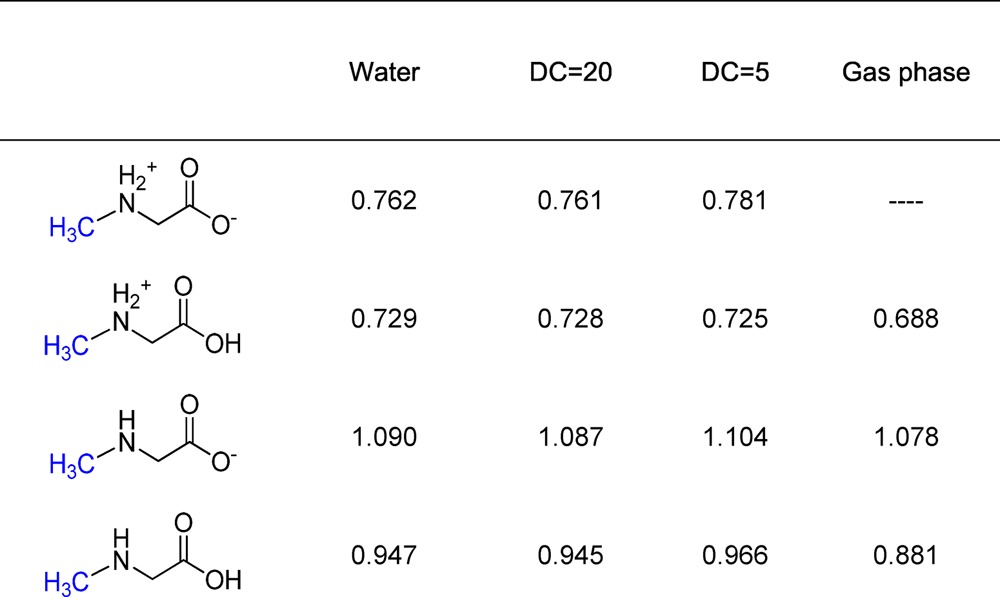
In the latter context, we note that GNMT is a better catalyst than COMT (which uses a metal ion to deprotonate its substrate), making it likely that GNMT participates in base catalysis to accelerate its reaction. This makes the EIE of 0.761 (DC (dielectric constant) = 20) an unlikely outcome for GNMT, with a concerted reaction predicting an EIE of 1.087 a far more likely boundary condition. A measurement of the solvent isotope effect on the individual kinetic parameters as a function of pH may be informative in this regard, and will form the basis of future detailed investigations. A second indication of the nature of the transition state in GNMT is the observation that the magnitudes of the carbon isotope effects (12C/14C KIE) are all quite large and within experimental error of each other. This rules out a very late transition state for WT GNMT and, furthermore, argues that the observed trends in the secondary tritium isotope effects are not arising from extreme differences in transition-state structure.
One key similarity between GNMT and COMT is the presence of a tyrosine side chain that resides behind the sulfur atom of bound AdoMet (Figure 1 and Figure S4). As shown for COMT, the β-methylene of Tyr68 lies behind the sulfur of AdoMet, with its ring hydroxyl pointing toward a remote carboxylate (Figure S4), whereas in GNMT the ring hydroxyl of Tyr21 actually points toward the sulfur atom of the cofactor; these different binding modes, when coupled to the similar sensitivity of kinetic parameters to mutation at Tyr, strongly suggest a dominant role for steric bulk in directing the reactivity of the methyl functional group of AdoMet. This is supported by the data for GNMT, Table 1, where replacement of Tyr by Phe leads to a relatively small impact on all of the kinetic parameters relative to the impact of substituting Tyr21 with a large series of smaller aliphatic side chains of varying hydrophobicity and H-bonding properties. While it is tempting to try to interpret differences in behavior among these aliphatic side chain replacements, with the exception of Y21A, the secondary KIE values are all within experimental error of one another. The origin of the deviation of Y21A from the remaining small side chain variants is difficult to assess in the absence of structural data, though it has previously been proposed that replacement of Tyr by smaller side chains including Ala may lead to water binding and the resulting restoration of a portion of native enzyme behavior (cf. refs (48) and (49)). We propose that the pattern of rates and KIEs vs mutagenesis in both GNMT (at Tyr21) and COMT (at Tyr68) is a consequence of a role of this side chain in constraining the position of the sulfur of the reactive cofactor in relation to the attacking substrate.
As already discussed in the case of COMT, the WT structure appears able to achieve an internuclear distance between the carbon of the transferred methyl group and the nucleophile of the substrate that is ca. 0.3–0.4 Å shorter than predicted from the carbon to oxygen van der Waals distance. Computational studies of ground-state structures, using the graphics processing unit (GPU)-extended methodology, have indicated that a very large portion of the enzyme is necessary to reproduce this ground-state compaction, and that active site snugness/compaction is a likely cooperative event involving a large number of protein side chains. In a recent computational study by Williams and co-workers,50 the role of this compaction along the axis of reaction (referred to as axial interactions) has been compared to the possible impact of hydrogen-bonding interaction between the transferred CH3 and hydrogen-bonding functional groups in an orthogonal plane (referred to as equatorial interactions). One important result of the computation of equatorial binding interactions is the observation of an increase in the size of the secondary KIE (toward less inverse values) as the strength of the equatorial interaction increases. While these computations are capable of showing a decreased barrier height and accompanying increase in rate for the WT enzyme, the expected trend in the secondary KIE with the less active, mutated forms of the enzyme is predicted to be opposite that observed here for GNMT and in the earlier studies of COMT. More specifically, the simulation results by Williams and co-workers50 show that a decrease in catalytic efficiency via a decrease in equatorial compaction will lead to more inverse KIEs, rather than the experimentally observed trends of more normal KIEs (Figure 2). This difference between results from experiment and those from simulation might be due to the lack of involvement of a solvation environment in the model, or an inherent gap in properties between the computed and enzymatic reactions, with the latter having features such as catalysis-linked protein motions that are not yet fully understood. Interestingly, a careful examination of the trends in ref (50) reveals that it is compaction along the reaction axis, rather than the relatively small impact of equatorial CH···O interactions, that may be expected to dominate the comparative behavior of the WT enzyme in relation to its mutants within the proximal Tyr side chain.
Conclusions
Previous X-ray crystal structures and NMR data reveal a possible role for ground-state hydrogen bonds of the methyl group of AdoMet that could be the source of BIEs greater than unity in COMT and GNMT.15,51,52 The crystal structure of GNMT with AdoMet shows that the methyl group in AdoMet is close enough to interact with the residues Tyr21, Gly137, and Tyr194, within respective distances of 3.8, 3.7, and 3.7 Å from the methyl carbon to either the hydroxyl group of the tyrosines or the backbone oxygen of glycine. These putative CH···O hydrogen bonds appear to strengthen when the substrate analogue acetate is present, with the ternary crystal structure indicating carbon to oxygen distances at 3.3, 3.6, and 3.5 Å, respectively (Figure 3). To the degree that such CH···O interactions would decrease the vibrational frequencies of the methyl group of AdoMet,51,52 BIEs will be elevated from unity (Table 2). However, with the exception of the ternary complex of the WT, the remainder of the measured BIEs are all within experimental error of each other. It may be of interest that the ternary BIE for the WT has become less inverse, with a possible implication of increased compaction within this singular ground-state complex.
Figure 3.
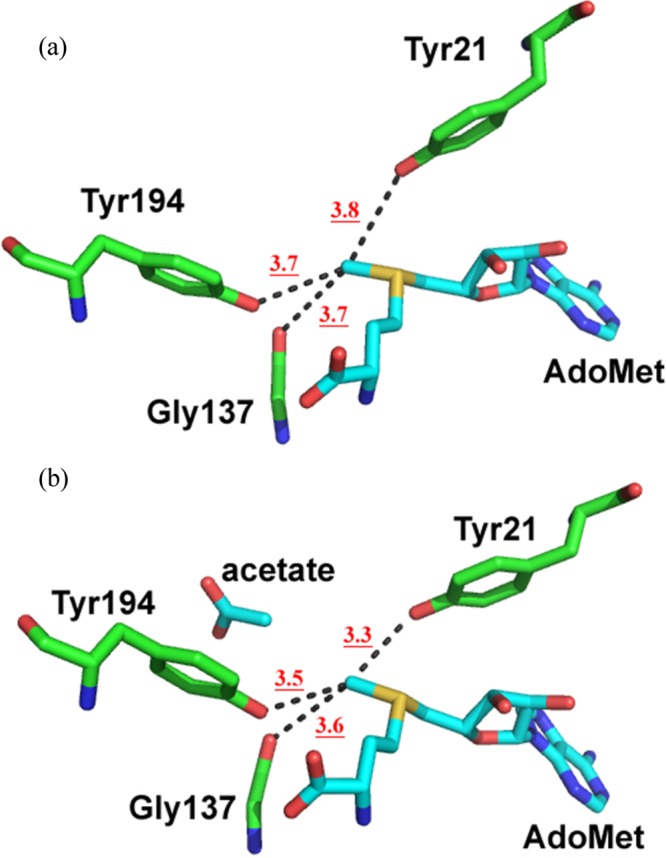
CH···O hydrogen bond (dashed lines) in the (a) binary GNMT–AdoMet complex (PDB 1NBI) and (b) ternary GNMT–AdoMet–acetate complex (PDB 1NBH).
The outcome is quite different for the inverse KIEs (Scheme 2), which show robust trends for GNMT that are highly similar to those for COMT. The most important aspect of these KIE measurements is their dependence on the presence of a tyrosine side chain immediately proximal to the sulfur of the cofactor, with decreases in the second-order rate constant for reaction of the enzyme–AdoMet complex with free glycine paralleling increases in the secondary KIEs toward unity. As discussed above, while the magnitude of the inverse secondary KIE for the WT is within the range of the computed equilibrium isotope effect for transfer of a methyl group from sulfur to nitrogen, this will only be true in the case of a very late transition state and in the absence of any base catalysis. Importantly, the consistently large magnitudes of the primary KIEs together with the trends in the secondary KIEs upon mutation of the proximal tyrosine indicate a different origin. We attribute the experimental effects in both GNMT and the previously studied COMT to differences in protein structure that are dependent on the proximal tyrosine and lead to an elongation of the attacking nucleophile to the transferred methyl along the axis of reaction. For COMT, it was possible to attribute a large part of the “Tyr effect” to an activated ground state, via a very close approach of the ionized oxyanion of the substrate to the cationic sulfur of the cofactor,15 a feature that is highly unlikely for GNMT. Nonetheless, the final impact of the proximal tyrosine on the kinetic properties of GNMT reproduces those seen in COMT. In light of the unique oligomeric structure for GNMT, which functions as a tetramer in contrast to the monomeric forms of the other methyltransfeases within this class, a movement of its N-terminal loop that includes and is dependent on Tyr 21 may be the critical element that ensures the compactness/tightness of the active site in the WT; while loop closure will also need to occur with the Tyr21 mutants for the reaction to proceed, the mutated variants have lost their ability to achieve a constrained donor–acceptor distance that both ensures high turnover rates and generates magnitudes for the KIE that lie significantly below those seen in solution reactions.
Scheme 2.

We propose that the new data for GNMT provide strong support for the introduced model of COMT reactivity, in which small decreases in active site compactness/tightness, either in the ground state or in the transition state, are capable of producing large increases in catalytic efficiency; furthermore, this behavior appears dependent on the size and positioning of a proximal amino acid side chain located directly behind the sulfur bearing the methyl group of AdoMet.15 It will be exciting to see the extent to which future experimental efforts, which include X-ray crystallographic studies of mutant forms of COMT and GNMT, together with biophysical probes of changes in functionally relevant protein motions as a function of mutation, can provide both structural and dynamical insights into the experimentally observed kinetic behaviors reported herein and earlier.14,15
Experimental Section
Materials
S-Adenosyl-l-[methyl-3H]methionine (specific activity 80 Ci/mmol), S-[carboxyl-14C]adenosyl-l-methionine (specific activity 55 mCi/mmol), S-adenosyl-l-[methyl-14C]methionine (specific activity 55 mCi/mmol), and [2-3H]glycine (specific activity 60 Ci/mmol) were from American Radiolabeled Chemicals Inc. [1-14C]Glycine (specific activity 50 mCi/mmol) was from ViTrax Inc. The radioactive AdoMet was purified using a C18 HPLC column in three steps as described in our previous paper.53 The cocktail for liquid scintillation counting was ECOLITE(+) LSC from MP Biomedicals LLC.
Expression and Purification of Rat GNMT
The plasmid sample of rat GNMT in the pET-17b vector was a generous gift from Prof. Conrad Wagner (Vanderbilt University). All mutagenesis of rat GNMT were performed using a commercial kit (QuickChange site-directed mutagenesis kit, Stratagene), and the primers containing an NNK sequence (N = A, T, G, or C, and K = T or G) used are (GGGATCCCCGACCAGNNKGCGGATGGGGAGGCC and GGCCTCCCCATCCGCMNNCTGGTCGGGGATCCC). This is a saturated mutagenesis procedure selecting more than 60 colonies. The randomized plasmids were isolated by miniprep and sequenced by T7 forward and reverse primers to verify the presence of the expected mutation and the absence of unwanted mutations.
Recombinant rat GNMTs were transformed and expressed in Escherichia coli BL21 (DE3) cells (Stratagene). Transformed BL21 (DE3) cells were grown in LB medium containing ampicillin (100 μg/mL). When the absorbance at 600 nm was around 0.6, the cells were induced with 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) and grown at 20 °C overnight with shaking at 200 rpm. The cells were harvested via centrifugation at 8000g for 15 min at 4 °C. The cell pellets were stored at −80 °C for future purification.
The purification procedure used here was revised according to previous reports.31,34 The cell pellets were resuspended in at least 5-fold Bugbuster buffer (Novagen) in 20 mM Tris–HCl (pH 8.0) with 5 mM β-mercaptoethanol, and the cell suspension was incubated on a shaking platform for 20–30 min at room temperature. The suspended solution was centrifuged at 20000g for 20 × 2 min at 4 °C. The clear supernatant was filtered with a 0.2 μM filter (Millipore) and then applied to a 5 mL × 2 HiTrap DEAE fast flow column (GE Healthcare) which was pre-equilibrated with 20 mM Tris–HCl (pH 8.0) and 5 mM β-mercaptoethanol. The GNMT was not retained on the column, and the pass-through fraction was collected. Then ammonium sulfate (19.0 g in every 100 mL of buffer) was added to achieve 35% saturation and the resulting mixture incubated on ice for 30 min. The precipitated proteins were removed by centrifugation at 20000g for 15 min, and a second portion of ammonium sulfate (15 g in every 100 mL of buffer) was added to the supernatant to achieve 50% saturation. After 30 min on ice, the mixture was centrifuged at 20000g for 15 min. The obtained pellet containing GNMT was dissolved in a minimum volume of size exclusion column buffer [20 mM Tris–HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl, 5 mM β-mercaptoethanol] and loaded into the Sephacryl S-200 HiPrep 26/60 (GE Healthcare), which was equilibrated with at least three column volume size exclusion column buffer in advance. The GNMT was eluted as the second peak, and the fractions were collected and concentrated into storage buffer (100 mM phosphate, pH 7.4, 1 mM EDTA, 0.02% NaN3, 20% glycerol, 5 mM DTT). The protein sample was aliquoted and stored at −80 °C until further use. Protein concentrations were calculated using the Bradford assay. UV–vis spectra were recorded on a Cary 50 Bio spectrophotometer.
Steady-State Kinetic Measurements
The activity of GNMT was measured radiometrically using [methyl-3H]AdoMet. The reaction mixture contained recombinant GNMT protein, S-adenosyl-l-[methyl-3H]methionine, and glycine in phosphate buffer (40 mM, pH 7.4) and 5 mM DTT at 37 °C. The concentration of [methyl-3H]AdoMet was varied in the range of 25–2600 μM while the glycine concentration was kept fixed at 20 mM for the WT and Y21F mutant and 200 mM for all other mutants. Similarly, the concentration of glycine was varied in the range of 0.2–200 mM while the [methyl-3H]AdoMet concentration was kept fixed at 2600 μM. After termination of the reaction with 4 M HCl (final concentration 10%), the reaction mixture was spun prior to HPLC analysis. The samples were manually loaded into the HPLC system based on an amino column (Phenomenex Luna 5 μm NH2, 100 Å) and then eluted by phosphate/acetonitrile solvent (28% 5 mM phosphate, pH 7.2, 72% MeCN) with a flow rate of 1 mL/min. The retention time was 11.7 min for sarcosine, 16.4 min for glycine, and 29.1 min for AdoMet. The portion of the unreacted AdoMet and product sarcosine were collected directly into 20 mL scintillation vials followed by addition of eluted buffer to achieve a final volume of 5 mL of eluted buffer. A 15 g portion of the scintillation cocktail was added, and the samples were determined by scintillation counting (Packard Tri-Carb 2700TR) for 5 min. The reaction without addition of any enzyme was performed as a blank. The concentrations of product were obtained on the basis of the product sarcosine standard curve. Data obtained were plotted by regression analysis using Origin 8.0 to give the corresponding Vmax and Km.
Competitive Primary and Secondary Kinetic Isotope Effect (KIE) Measurements
The KIEs for GNMT were determined as previously described using the competitive isotopes method.14 Briefly, KIEs were measured by mixing 50 μM AdoMet which was radioactively labeled at the methyl group (methyl-14C for the primary KIE; methyl-3H for the secondary KIE) and 100 μM glycine ([2-3H]glycine for the primary KIE; [1-14C]glycine for the secondary KIE, which will act as a tracer). The counts per minute (CPM) ratio of 3H to 14C was around 10:1 (320000 CPM:32000 CPM) for the primary KIE and 5:1 (600000 CPM:120000 CPM) for the secondary KIE. The recombinant GNMT protein was added to the initial reaction at 37 °C in 5 mM DTT and 50 mM phosphate buffer (pH 6.8). At different time points, 50 μL aliquots were removed and quenched with 5 μL of 4 M HCl. Concentrated WT GNMT was added to the reaction mixture to achieve 100% conversion (t∞) of AdoMet, and more than three t0 and t∞ values were obtained. Independent experiments were carried out at least three times for each mutant to obtain the KIE. The quenched aliquots were stored at −20 °C prior to separation by HPLC. The separation of the samples was the same as stated in the section “Steady-State Kinetic Measurements” above. The KIE was calculated according to the following equation:54−56
| 2 |
Competitive Equilibrium Binding Isotope Effect (BIE) Measurements
The binding isotope effects (BIEs) were measured using the ultrafiltration method described by the Schramm group.57−60 Generally, the solution (320 μL) for the measurement consisted of 50 μM recombinant GNMT protein and a 30 μM concentration of the mixture of prepurified S-adenosyl-l-[methyl-3H]methionine and S-[carboxyl-14C]adenosyl-l-methionine in 50 mM phosphate buffer with 5 mM 1,4-dithiothreitol (pH 6.8), and the solution was incubated for 20 min at room temperature [for the BIE with acetate, sodium acetate was added at this step]. Three 100 μL aliquots were removed and added to the upper wells of the ultrafiltration apparatus, and around 22 psi of nitrogen gas (N2) was applied for 45 min, at which point around half of the solution had passed through the dialysis membrane into the lower well. Samples (25 μL) from the top and bottom wells were taken by using a Hamilton syringe into a 20 mL scintillation vial with 500 μL of distilled water. A 5 g portion of the scintillation cocktail was added to each sample, and the samples were analyzed by a liquid scintillation counter (Packard Tri-Carb 2700TR) for at least 10 cycles (10 min per cycle). The counting channels for 3H and 14C were 0–12 and 35–156 eV, respectively. The BIEs were calculated from the following equation,57,58 where 14Ctop and 3Htop are the counts for the 14C and 3H in the top wells, respectively, and 14Cbottom and 3Hbottom are the counts for the 14C and 3H in the bottom wells, respectively:
| 3 |
Calculation of the Equilibrium Isotope Effect
Equilibrium isotope effect calculations were carried out on the reactant AdoMet and the resulting product sarcosine by obtaining the zero-point energy of the standard and tritium-substituted species using vibrational analysis implemented in Gaussian 09.61 These calculations were carried out at B3LYP/6-31+g** with a polarizable continuum model (IEFPCM) for the solvent. Subsequent force constant calculations were performed, frequencies were computed for isotopologues of interest, and the resulting frequencies were used to calculate the EIE.
Acknowledgments
We thank Prof. Conrad Wagner (Vanderbilt University) for the plasmid sample of GNMT and Prof. Vern L. Schramm (Albert Einstein College of Medicine of Yeshiva University) for sharing the ultrafiltration apparatus. We are grateful for funding of this laboratory by the National Institutes of Health (NIH) to J.P.K. (Grants GM025765 and GM039296).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b03462.
Figures and schemes showing the conversion between open and closed conformations for dimer structures of rat GNMT, relationship between the kcat/Km for AdoMet and the secondary KIE for methylation of glycine by the recombinant rat GNMT and Y21 mutants, interactions between a bound carboxylate and active site side chains in GNMT, modeled interaction of glycine in the active site of GNMT, and active site of COMT (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Radzicka A.; Wolfenden R. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Cannon W. R.; Singleton S. F.; Benkovic S. J. Nat. Struct. Biol. 1996, 3, 821–833. 10.1038/nsb1096-821. [DOI] [PubMed] [Google Scholar]
- Bruice T. C.; Lightstone F. C. Acc. Chem. Res. 1999, 32, 127–136. 10.1021/ar960131y. [DOI] [Google Scholar]
- Bruice T. C.; Benkovic S. J. Biochemistry 2000, 39, 6267–6274. 10.1021/bi0003689. [DOI] [PubMed] [Google Scholar]
- Garcia-Viloca M.; Gao J.; Karplus M.; Truhlar D. G. Science 2004, 303, 186–195. 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H. B.; Olsson M. H. M. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Nature 2007, 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Klinman J. P. Chem. Phys. Lett. 2009, 471, 179–193. 10.1016/j.cplett.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes G. G.; Benkovic S. J.; Hammes-Schiffer S. Biochemistry 2011, 50, 10422–10430. 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V. L. Annu. Rev. Biochem. 2011, 80, 703–732. 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynamics in Enzyme Catalysis; Klinman J. P., Hammes-Schiffer S., Eds.; Springer-Verlag: Berlin, Heidelberg, 2013. [Google Scholar]
- Klinman J. P.; Kohen A. Annu. Rev. Biochem. 2013, 82, 471–496. 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protein Conformational Dynamics; Han K.-L., Zhang X., Yang M.-j., Eds.; Springer International Publishing: New York, Dordrecht, London, 2014. [Google Scholar]
- Zhang J.; Klinman J. P. J. Am. Chem. Soc. 2011, 133, 17134–17137. 10.1021/ja207467d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Kulik H. J.; Martinez T. J.; Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 7954–7959. 10.1073/pnas.1506792112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni G. L. Annu. Rev. Biochem. 1975, 44, 435–451. 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- Chiang P. K.; Gordon R. K.; Tal J.; Zeng G. C.; Doctor B. P.; Pardhasaradhi K.; McCann P. P. FASEB J. 1996, 10, 471–480. [PubMed] [Google Scholar]
- Yen C.-H.; Lin Y.-T.; Chen H.-L.; Chen S.-Y.; Chen Y.-M. A. Toxicol. Appl. Pharmacol. 2013, 266, 67–75. 10.1016/j.taap.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Luka Z.; Mudd S. H.; Wagner C. J. Biol. Chem. 2009, 284, 22507–22511. 10.1074/jbc.R109.019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi K.; Fujioka M. Biochemistry 1987, 26, 8496–8502. 10.1021/bi00399a069. [DOI] [PubMed] [Google Scholar]
- Takata Y.; Huang Y. F.; Komoto J.; Yamada T.; Konishi K.; Ogawa H.; Gomi T.; Fujioka M.; Takusagawa F. Biochemistry 2003, 42, 8394–8402. 10.1021/bi034245a. [DOI] [PubMed] [Google Scholar]
- Luka Z. Vitam. Horm. 2008, 79, 325–345. 10.1016/S0083-6729(08)00411-1. [DOI] [PubMed] [Google Scholar]
- Velichkova P.; Himo F. J. Phys. Chem. B 2005, 109, 8216–8219. 10.1021/jp0443254. [DOI] [PubMed] [Google Scholar]
- Gray C. H.; Coward J. K.; Schowen K. B.; Schowen R. L. J. Am. Chem. Soc. 1979, 101, 4351–4358. 10.1021/ja00509a051. [DOI] [Google Scholar]
- Hegazi M. F.; Borchardt R. T.; Schowen R. L. J. Am. Chem. Soc. 1979, 101, 4359–4365. 10.1021/ja00509a052. [DOI] [Google Scholar]
- Cheng X. D.; Roberts R. J. Nucleic Acids Res. 2001, 29, 3784–3795. 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J. L.; McMillan F. M. Curr. Opin. Struct. Biol. 2002, 12, 783–793. 10.1016/S0959-440X(02)00391-3. [DOI] [PubMed] [Google Scholar]
- Konishi K.; Fujioka M. J. Biol. Chem. 1988, 263, 13381–13385. [PubMed] [Google Scholar]
- Ogawa H.; Gomi T.; Fujioka M. Comparative Biochemistry and Physiology B-Biochemistry & Molecular Biology 1993, 106, 601–611. 10.1016/0305-0491(93)90137-T. [DOI] [PubMed] [Google Scholar]
- Fu Z. J.; Hu Y. B.; Konishi K.; Takata Y.; Ogawa H.; Gomi T.; Fujioka M.; Takusagawa F. Biochemistry 1996, 35, 11985–11993. 10.1021/bi961068n. [DOI] [PubMed] [Google Scholar]
- Ogawa H.; Gomi T.; Takata Y.; Date T.; Fujioka M. Biochem. J. 1997, 327, 407–412. 10.1042/bj3270407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayek R.; Newcomer M. E.; Wagner C. Protein Sci. 1998, 7, 1326–1331. 10.1002/pro.5560070608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Komoto J.; Konishi K.; Takata Y.; Ogawa H.; Gomi T.; Fujioka M.; Takusagawa F. J. Mol. Biol. 2000, 298, 149–162. 10.1006/jmbi.2000.3637. [DOI] [PubMed] [Google Scholar]
- Luka Z.; Wagner C. Protein Expression Purif. 2003, 28, 280–286. 10.1016/S1046-5928(02)00710-6. [DOI] [PubMed] [Google Scholar]
- Pakhomova S.; Luka Z.; Grohmann S.; Wagner C.; Newcomer M. E. Proteins: Struct., Funct., Genet. 2004, 57, 331–337. 10.1002/prot.20209. [DOI] [PubMed] [Google Scholar]
- Luka Z.; Pakhomova S.; Luka Y.; Newcomer M. E.; Wagner C. Protein Sci. 2007, 16, 1957–1964. 10.1110/ps.072921507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luka Z.; Pakhomova S.; Loukachevitch L. V.; Egli M.; Newcomer M. E.; Wagner C. J. Biol. Chem. 2007, 282, 4069–4075. 10.1074/jbc.M610384200. [DOI] [PubMed] [Google Scholar]
- Soriano A.; Castillo R.; Christov C.; Andres J.; Moliner V.; Tunon I. Biochemistry 2006, 45, 14917–14925. 10.1021/bi061319k. [DOI] [PubMed] [Google Scholar]
- Panuszko A.; Smiechowski M.; Stangret J. J. Chem. Phys. 2011, 134, 115104. 10.1063/1.3567202. [DOI] [PubMed] [Google Scholar]
- Rodgers J.; Femec D. A.; Schowen R. L. J. Am. Chem. Soc. 1982, 104, 3263–3268. 10.1021/ja00376a003. [DOI] [Google Scholar]
- Westaway K. C. Adv. Phys. Org. Chem. 2006, 41, 217–273. 10.1016/S0065-3160(06)41004-2. [DOI] [Google Scholar]
- Rutherford K.; Le Trong I.; Stenkamp R. E.; Parson W. W. J. Mol. Biol. 2008, 380, 120–130. 10.1016/j.jmb.2008.04.040. [DOI] [PubMed] [Google Scholar]
- Vidgren J.; Svensson L. A.; Liljas A. Nature 1994, 368, 354–358. 10.1038/368354a0. [DOI] [PubMed] [Google Scholar]
- Bonifacio M. J.; Archer M.; Rodrigues M. L.; Matias P. M.; Learmonth D. A.; Carrondo M. A.; Soares-da-Silva P. Mol. Pharmacol. 2002, 62, 795–805. 10.1124/mol.62.4.795. [DOI] [PubMed] [Google Scholar]
- Palma P. N.; Rodrigues M. L.; Archer M.; Bonifacio M. J.; Loureiro A. I.; Learmonth D. A.; Carrondo M. A.; Soares-da-Silva P. Mol. Pharmacol. 2006, 70, 143–153. 10.1124/mol.106.023119. [DOI] [PubMed] [Google Scholar]
- Tsuji E.; Okazaki K.; Takeda K. Biochem. Biophys. Res. Commun. 2009, 378, 494–497. 10.1016/j.bbrc.2008.11.085. [DOI] [PubMed] [Google Scholar]
- Ellermann M.; Lerner C.; Burgy G.; Ehler A.; Bissantz C.; Jakob-Roetne R.; Paulini R.; Allemann O.; Tissot H.; Gruenstein D.; Stihle M.; Diederich F.; Rudolph M. G. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 253–260. 10.1107/S0907444912001138. [DOI] [PubMed] [Google Scholar]
- DuBois J. L.; Klinman J. P. Biochemistry 2006, 45, 3178–3188. 10.1021/bi052025m. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Sigala P. A.; Fenn T. D.; Herschlag D. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1960–1965. 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson P. B.; Williams I. H. Angew. Chem., Int. Ed. 2016, 55, 3192–3195. 10.1002/anie.201511708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S.; Yesselman J. D.; Al-Hashimi H. M.; Trievel R. C. J. Biol. Chem. 2011, 286, 18658–18663. 10.1074/jbc.M111.232876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S.; Dirk L. M. A.; Yesselman J. D.; Nimtz J. S.; Adhikari U.; Mehl R. A.; Scheiner S.; Houtz R. L.; Al-Hashimi H. M.; Trievel R. C. J. Am. Chem. Soc. 2013, 135, 15536–15548. 10.1021/ja407140k. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Klinman J. P. Anal. Biochem. 2015, 476, 81–83. 10.1016/j.ab.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant K. L.; Klinman J. P. Biochemistry 1989, 28, 6597–6605. 10.1021/bi00442a010. [DOI] [PubMed] [Google Scholar]
- Bandaria J. N.; Cheatum C. M.; Kohen A. J. Am. Chem. Soc. 2009, 131, 10151–10155. 10.1021/ja902120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B. Y.; Maley F.; Kohen A. Biochemistry 2007, 46, 14188–14197. 10.1021/bi701363s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murkin A. S.; Tyler P. C.; Schramm V. L. J. Am. Chem. Soc. 2008, 130, 2166–2167. 10.1021/ja7104398. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Schramm V. L. Biochemistry 2011, 50, 4813–4818. 10.1021/bi200638x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murkin A. S.; Birck M. R.; Rinaldo-Matthis A.; Shi W. X.; Taylor E. A.; Almo S. C.; Schramm V. L. Biochemistry 2007, 46, 5038–5049. 10.1021/bi700147b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B. E.; Schramm V. L. J. Am. Chem. Soc. 2003, 125, 4785–4798. 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.