

Abstract
Structural characterization of transmembrane proteins in isotropic bicelles has become an increasingly popular application of solution NMR spectroscopy, as the fast-tumbling bicelles are membrane-like yet can often yield spectral quality comparable to those of detergent micelles. While larger bicelles are closer to the true lipid bilayer, it remains unclear how large the bicelles need to be to allow accurate assessment of protein transmembrane partition in lipid bilayer. Here, we address the above question from the perspective of protein residing in the bicelles, through systematic measurement of protein chemical shift and transmembrane partition at different lipid:detergent ratios (q), ranging from 0.3 to 0.7, using the transmembrane domain of human Fas receptor as model system. We found that the lipid environment of the bicelles, as reflected by the protein chemical shift, begins to be perturbed when the q is reduced to below 0.6. We also implemented a solvent paramagnetic relaxation enhancement (PRE) approach for bicelles to show that the protein transmembrane partition in bicelles with q = 0.5 and 0.7 are very similar, but at q = 0.3 the solvent PRE profile is significantly different. Our data indicate that q values between 0.5 and 0.6 are good compromise between high resolution NMR and closeness to the membrane environment, and allow accurate characterization of protein position in lipid bilayer.
Keywords: membrane proteins, bicelle size, transmembrane partition, solvent PRE, NMR spectroscopy
Graphical Abstract
A robust method is described to assess the optimal bicelle size(s) to accurately determine the position of oligomeric transmembrane protein domains in lipid bilayer. This method combines solvent paramagnetic agent titration and paramagnetic relaxation data fitting with sigmoid function. Our results show that bicelles with lipid/detergent ratio (q) between 0.5 and 0.6 ensure an almost-ideal lipid bilayer environment, while at the same time providing favorable relaxation properties that are compatible with high resolution NMR spectroscopy.
Introduction
Transmembrane domains (TMDs) of many cell surface receptors are not merely inert single-helix anchors; their oligomerization often play essential roles in receptor assembly in the membrane and receptor signaling across the membrane. The modes of association between the transmembrane (TM) helices directly link the conformational state of the extracellular receptor domain to that of the intracellular signaling domains, thus modulating the transmission of signals across the membrane[1]. The membrane-associated regions of TM receptors have historically been very difficult to crystalize, except for a few very recent breakthroughs[2], and they are also too small for the state-of-the-art cryo-electron microscopy technology. For these reasons, solution NMR has been frequently used for deriving high resolution structural information for these small and hydrophobic systems[3]. The combination of bicelles and solution NMR is particularly attractive because bicelles have the potential to offer an essentially lipid bilayer environment while concurrently being compatible with high resolution NMR spectroscopy that enables de novo structure determination of TM proteins.
Bicelles are composed of long-chain phospholipids that form the lipid bilayer disc and detergents that enclose the exposed hydrophobic side of the disc, such that the aggregate is soluble and fast tumbling in water[4]. The lipids are commonly long-chain phospholipids such as dimyristoylphosphatidylcholine (DMPC). As for detergents, dihexanoylphosphatidyl-choline (DHPC), 3-(cholamido-propyl) dimethylammonio-2-hydroxy-1-propane-sulfonate (CHAPSO)[5], and 6-cyclohexyl-1-hexyl-phosphocholine (Cyclofos-6)[6] have been shown to be compatible with bicelle formation, among which DHPC is by far the most used in structural studies of TM proteins. In an ideal bicelle (lipid and detergent well segregated), the size of the bicelle can be estimated based purely on geometric arguments[7], i.e., the radius of the bilayer region of the bicelle (R) can be calculated directly from the lipid:detergent molar ratio (q)[8].
While it is generally agreed upon that larger q values yield more ideal bicelles, it remains vague how large the q needs to be to provide a planar bilayer environment for the proteins residing within. For solution NMR studies, smaller bicelles are obviously preferred, and essentially all successful bicelle applications in solving atomic resolution structures of TMDs have been achieved using bicelles with q = 0.5[1c, 3f, 9]. In those earlier studies, however, bicelles were used mainly as effective solubilization system of membrane proteins, and the potential of bicelles in providing information about protein partition in lipid bilayer, which is an important aspect of TMD structure, has not been fully exploited. In this study, we use the homo-trimeric TMD of the human Fas receptor as a model system to identify the optimal bicelle size that compromises high resolution NMR and the ability to allow accurate assessment of protein TM partition in lipid bilayer.
Results and Discussion
Fas is an apoptosis-inducing death receptor in the tumor necrosis factor receptor superfamily[10], and its TMD structure was previously determined by solution NMR in DMPC/DHPC bicelles[1c]. The structure shows that the TM helices pack around a proline-containing sequence motif to form homotrimer with C3 symmetry. We first examined the effect of bicelle q on the Fas TMD NMR spectrum using the ‘q-titration’ method[11] because chemical shift is exquisitely sensitive to the surrounding surfactant environment, especially when the bicelle changes from lipid-disk-like (lipids and detergents segregated) to mixed-micelle-like (lipids and detergents mixed). We began with the Fas TMD reconstituted in DMPC/DHPC bicelles with q = 0.7 and monitored the spectral changes as we gradually reduced the q to 0.3 by titrating the sample with DHPC. The 2D 1H-15N TROSY-HSQC spectrum of the Fas TMD essentially did not change when reducing the q from 0.7 to 0.6, but further reducing the q to 0.5 and below caused substantial changes in chemical shifts (Fig. 1 and S1). Residues in the N- and C-termini (residues 173–175 and 193–195) exhibited the largest changes.
Figure 1.

Chemical shift changes of Fas TMD induced by changing bicelle q. The central region of the 2D 1H-15N TROSY-HSQC spectrum is shown for q = 0.7 (black), 0.6 (purple), 0.5 (green), 0.4 (red) and 0.3 (orange). On the left of each 2D spectrum, the methyl signals of DMPC and DHPC from the 1D 1H spectrum are provided to indicate bicelle q. The bottom right plot shows chemical shift changes at various q values relative to q = 0.7 for residues 179, 187, 189 and 194.
This result indicates that the lipid environments experienced by the TMD in bicelles with q = 0.7 and 0.6 are essentially the same; it also implies that the DMPC and DHPC molecules are well segregated, as increase in DHPC could not affect the protein that is exclusively associated with the lipid bilayer region of the bicelles. Conversely, the steady resonance shifts observed from q = 0.5 to q = 0.3 suggest that in this q range the TMD chemical environment is more affected due to increasing mixing of DHPC into the DMPC bilayer region.
As independent validation of increased bicelle size at larger q, the effective TMD rotational correlation time (τc) was measured using the TRACT method[12] (Fig. S2). We found that the apparent protein τc at 30°C, as probed by the N-terminal residue, is 13, 20, and 32 ns for q values of 0.3, 0.5, and 0.7, respectively. These results explain why smaller bicelles are generally preferred for high-resolution NMR studies of TMDs. However, only ideal bicelles with discoidal profile at higher q afford the opportunity to characterize the TMD transmembrane partition.
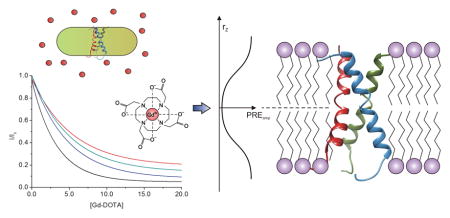
We thus examined the partition of the Fas TMD in bicelles with q values of 0.7, 0.5, and 0.3, in order to provide a guideline for the selection of the optimal q value for this type of application. Accurate determination of protein partition in lipid bilayer remains a challenging task in structural biology. In solution NMR studies, measuring nuclear Overhauser enhancement (NOE) between protein and lipid acyl chains and headgroup has been the most common method to address this problem[13]. This approach, however, can only provide a rather qualitative description of lipid/detergent distribution around the protein due to dynamic nature of the lipid and detergent molecules. Alternatively, paramagnetic agents such as Gd-DTPA-BMA) Gd-DOTA, O2, and Mn2+ ion have been successfully used to probe membrane immersion depth of protein domains[14]. The majority of these studies, however, have been designed for detergent micelles and thus could not be employed for the purpose of our study. We thus implemented a solvent paramagnetic relaxation enhancement (PRE) approach for bicelle applications to overcome the technical hurdle.
In this approach (schematically illustrated in Fig. 2a), the TMD is reconstituted in bicelles and titration of paramagnetic agents outside the bicelles is performed to generate solvent paramagnetic field of varying strength. The resulting residue-specific signal reduction profiles due to PRE are then analyzed collectively to determine the protein region that resides at the center of the bilayer. This method has a number of requirements for it to be of practical use: (i) the paramagnetic agent must be water-soluble and bicelle-inaccessible, so that the PRE it generates decreases with distance from the bicelle surface to the core, and by not having membrane partition the paramagnetic molecule avoids potential non-specific interactions with the protein that could mislead PRE interpretation. Paramagnetic agents such as Gd- DOTA) and Gd-DTPA-BMA satisfy these requirements; (ii) the role of the paramagnetic agent is to probe protein immersion depth along the bicelle normal (or longitudinal axis). Hence, the bicelles need to be sufficiently large to exclude lateral contribution from the surrounding paramagnetic field. This is critical because for very small bicelles, e.g., lipid/detergent ratio (q) < 0.3, we would encounter the mixed micelle situation in which it is essentially impossible to deconvolute the longitudinal and lateral components of the solvent PRE.
Figure 2.

The paramagnetic agent titration strategy for probing membrane immersion depth. a) Schematic illustration of titrating bicelle-reconstituted TMD with Gd- DOTA. R and r are the radii of the planar region and the rim, respectively. b) PREs induced by Gd-DOTA titration measured in 2D 1H-15N TROSY-HSQC spectra of Fas TMD reconstituted in q = 0.5 bicelles. The PRE (or I/I0, as defined in the text) vs. Gd-DOTA concentration is shown for residues 186 (buried) and 198 (exposed).
A critical aspect of the experimental design was performing paramagnetic agent titration rather than a single addition, as a wide range of paramagnetic field strengths (from very weak to strong) is required to probe both solvent-exposed and buried protein regions. We titrated bicelle-reconstituted Fas TMD with Gd-DOTA at Gd-DOTA:TMD ratio from 1 to 40. At each of the titration points, a 2D 1H-15N TROSY-HSQC spectrum was recorded to measure residue-specific PRE, defined here as I/I0, where I and I0 are the intensities of a peak in the presence and absence of the paramagnetic agent, respectively. The NMR titration generated a PRE vs. Gd-DOTA concentration profile (Fig. 2b) for each of the assigned residues of the TMD, which could be described by the following exponential decay (Eq. 1)[14b]:
| Eq. 1 |
where I and I0 are as defined above, [Gd-DOTA] is the concentration of the paramagnetic agent, τ is the decay constant, and PREamp is the amplitude of PRE which can be experienced by each of the reporting nuclear spins in the protein. In this study, we chose backbone amide protons as the dominant PRE reporters. Fitting the I/I0 vs. [Gd-DOTA] data points to Eq. 1 yielded residue-specific PREamp (Fig. 2b and Table S2). The complete PREamp vs. (residue number) plot is shown in Fig. 3a.
Figure 3.

Residue-specific PRE and position-specific PRE. a) PREamp vs. residue number for Fas TMD reconstituted in q = 0.5 bicelles. b) The NMR structure of the Fas TMD homotrimer with the symmetry axis aligned with the bicelle normal, showing the amide protons (red spheres) for which PREamp has been determined. The dashed lines point to the projected positions (rz) of the amide protons onto the C3 axis. c) PREamp vs. rZ plot. The rZ reference point is arbitrarily chosen, e.g., rZ = 0 for the N-terminus.
In previous studies of solvent PRE, it was shown for water-soluble protein that, if the protein structure is known, the PREamp for each of the 1H in protein can be predicted by integrating the PRE between the 1H and an infinitesmal volume outside surrounding the protein structure (with the r−6 distance dependence) over a defined cube in which the protein structure resides[15]. This PRE calculation, however, would be extremely difficult to implement for a bicelle-reconstituted protein due to the added unknowns about the protein position relative to the bicelle as well as the potentially dynamic morphology of the fast tumbling bicelles.
For these reasons, we preferred a simple mathematical fitting approach to analyze the PREamp data with the practical goal of determining the distance between each TMD residue and the center of the lipid bilayer of the bicelle. To convert PREamp vs. (residue number) (Fig. 3a) to PREamp vs. (bilayer immersion depth), we calculated, for each residue i, the distance along the bilayer normal (rz) from the amide proton to an arbitrary reference point based on the protein structure. Since the Fas TMD is a homotrimer, its symmetry axis is parallel to the bilayer normal, which allowed convenient calculation of rz (Fig. 3b and S3). The PREamp vs. rz plot shows a sigmoidal profile (Fig. 3c). We thus fitted the PREamp vs. rz data with the symmetric sigmoid equation, which has been used previously to approximate the O2 concentration gradient across a lipid bilayer[14d, 16]. Here, the equation has been implemented to describe PREamp as a function of |rz| (Eq. 2):
| Eq. 2 |
where and are the limits within which PREamp can vary for a particular protein system, is the inflection point (the distance from the bilayer center at which PREamp is halfway between and ), and SLOPE is a parameter which reports the steepness of the curve at the inflection point. The Eq. 2 empirically describes three PRE regimes: the PRE-saturated regime near the bicelle surface and the PRE-insensitive regime in the lipid core, connected by the PRE-sensitive regime that shows the r−6 dependence to the first-order approximation. Moreover, by the symmetry assumption of the fast-tumbling bicelles, the bilayer center should lie at the middle of the bottom flat region of Eq. 2. This means that if the bilayer center is correctly assigned to the protein, the PRE data should fit very well to this symmetric function.
Based on the above rationale, we systematically slid the Fas TMD along the bicelle normal relative to the bilayer center to achieve best fit to Eq. 2, as illustrated in Fig. 4a and S4. At each TMD position the adjusted coefficient of determination (R2adj) was obtained, as shown in Fig. 4b in the case of q = 0.5. The TMD position that yielded the highest R2 adj was assigned as the protein position in the bilayer region of the bicelles (Fig. 4c, S5, and Table S3).
Figure 4.

Assignment of the bilayer center to the Fas TMD via data fitting. a) Schematic illustration of the symmetric sigmoidal profile used to describe PREamp along the bilayer normal (rz). The protein is translated along the bilayer normal to generate PREamp vs rz data that best fit the sigmoidal function. b) The adjusted coefficient of determination (R2 adj) from data fitting versus deviation from the true bilayer center. The data is from q = 0.5 bicelles. The plot shows that R2 adj is a reliable indicator of the protein position with an error around ±0.5 Å. c) The optimized fit of the PREamp vs. rz data to Eq. 2 from (b). The gray-striped box represents the thickness of the DMPC bilayer (considering both hydrocarbon and headgroup regions)[17].
The above solvent PRE measurement and analysis were performed for bicelles with q values of 0.3, 0.5, and 0.7 (residue-specific PREamp shown in Fig. S6). For q = 0.7, data fitting show that the Ile184 amide proton lies almost exactly at the middle of the bilayer (Fig. 5a). The bilayer center derived from the q = 0.5 bicelle is only slightly shifted by 0.9 Å compared to the results from q = 0.7 bicelle. For q = 0.3, however, the PREamp vs. rz profile is drastically different from that of q = 0.5 or 0.7 bicelle, and the bilayer center from data fitting is shifted by 2.5 Å compared to that of the q = 0.7 bicelle.
Figure 5.

Fas TMD transmembrane partition analysis at different bicelle q. a) Comparison of PREamp vs rz data fitting from the q = 0.7 (black), 0.5 (red) and 0.3 (blue) bicelles. In the zoomed region, the rz distance of Ile184 from the bilayer center (rz=0) is shown. b) Position of the Fas TMD in lipid bilayer. The solid and dashed lines represent the center and the boundaries of DMPC bilayer, respectively. The Ile184 1HN is shown as red sphere.
The different results obtained for the tested q values can be explained by the following arguments. The analysis of solvent PRE using the sigmoidal function is based on the premise that the bicelle has a sufficiently large planar bilayer region to exclude most, if not all, of the lateral solvent PRE, such that the longitudinal PRE is the dominant factor in probing TMD immersion depth. In the case of bicelle with q = 0.7, the theoretical bilayer radius R is ~30.4 Å (or R + r ≈ 50 Å)[8], which should in principle eliminate lateral paramagnetic effect. The position of the human Fas TMD in the bilayer region of the bicelle, as derived from the q = 0.7 bicelle, should thus provide an accurate picture of the Fas TMD in the context of the lipid bilayer. The TM helices are completely immersed in the lipid bilayer (Fig. 5b). The critical Pro185 sidechain important for trimer assembly is almost exactly at the center of the bilayer.
The result that bicelles with q = 0.7 and 0.5 yielded very similar sigmoidal profiles and positions with respect to the bilayer center indicates that both bicelles have defined discoidal shapes. On the contrary, the very different sigmoidal profile of the bicelle with q = 0.3 suggests that the shape of this bicelle deviates significantly from that of an ideal bicelle and is more mixed-micelle-like. Notably, the PRE-saturated regime region at q = 0.3 reported in Fig. 5a shows significant upward shift relative to those at q = 0.5 and 0.7. This can be explained by larger lateral paramagnetic effect on the N- and C-terminal ends of the TMD near the bicelle surface. As for the comparison between q = 0.5 and q = 0.7, the PRE-saturated regimes are the same, although the PRE-insensitive regime at q = 0.5 shows larger PREamp than that at q = 0.7. This observation can be explained by the fact that, for the protein terminal regions near the bicelle surface, the longitudinal component of PRE dominates over the lateral component, and thus no significant changes are observed; conversely, in the bilayer core, where the longitudinal component of PRE is weak, the small increase in the lateral PRE at q = 0.5 is no longer negligible, resulting in the upward shift of the PRE-insensitive regime.
Conclusions
In conclusion, we have shown, from the perspective of a bicelle-reconstituted TMD, that DMPC/DHPC bicelles with q ≥ 0.6 provide the same phospholipid environment for the protein as indicated by NMR chemical shifts, and that this environment is very close to the planar lipid bilayer as shown by our solvent PRE analysis. Although protein chemical shifts at q = 0.5 show small differences relative to those at q = 0.6 and 0.7, the overall TM partition of the protein is essentially the same, suggesting that bicelles with q = 0.5 are still very close to ideal bicelles. This conclusion obtained from the perspective of bicelle-reconstituted protein is consistent with earlier biophysical studies of empty bicelles[8], which showed that DMPC/DHPC bicelle with q = 0.5 adopts a discoidal shape. In consideration of the significantly better Fas TMD τc at q = 0.5 (20 ns at 30 °C) compared to that at q = 0.7 (32 ns), we propose that q values between 0.5 and 0.6 should be a good compromise between high resolution liquid-state NMR and closeness to the lipid bilayer environment. We thus recommend using bicelles of this q values to obtain TM partition of membrane-embedded protein domains. Finally, the above conclusion is based on results derived for DMPC/DHPC bicelles at 30 °C, which represents one of the most common conditions for solution NMR studies of TMDs. Since melting temperatures of frequently used bicelle lipids such as DMPC, POPC, and POPG are well below 30 °C[17–18], the temperature effect on our analysis should be negligible if it is above the lipid phase transition temperature. The general analysis method reported here should also apply to bicelle detergents different from DHPC, though the optimized q may differ slightly[19] and thus requires experimental validation.
Experimental Section
Protein expression, purification and reconstitution
The (15N, 2H)-labeled human Fas TMD was expressed and purified as previously described[1c]. The lyophilized protein (1–2 mg) was dissolved in hexafluoro-isopropanal (HFIP) with approximately 9 mg DMPC, followed by drying of the solution under a nitrogen stream to achieve a thin film. The thin film was then dissolved in 3 ml of an 8 M urea solution containing approximately 27 mg DHPC, followed by dialysis against 20 mM sodium phosphate buffer at pH 6.8 to remove the denaturant. After dialysis, DHPC was added to adjust the ratio of DMPC:DHPC (q) to the desired value. The solution with reconstituted Fas TMD was concentrated using centricon to prepare the final NMR sample containing 500 μM Fas TMD, 60 mM DMPC, 200, 120 or 85 mM DHPC (for q = 0.3, 0.5 or 0.7, respectively) and 20 mM sodium phosphate (pH 6.8). 10% (v/v) D2O was added for the NMR lock. The final DMPC/DHPC ratio of the samples was determined by integrating the resolved methyl peaks of DMPC and DHPC in the 1D 1H NMR spectra.
DHPC titration
The Fas TMD was reconstituted in q = 0.7 bicelles and DHPC was progressively added to reduce the bicelle size. The detergent was taken from a concentrated stock solution (660 mM DHPC) made in the same buffer of the protein sample and it was added in small aliquots (few μL per step) to minimize possible dilution effects. To monitor the progress of the titration by NMR, a 2D 1H-15N TROSY-HSQC spectrum was recorded at 600 MHz (Table S1) at each of the following q values: 0.7, 0.6, 0.5, 0.4 and 0.3. The chemical shift assignments of the human Fas TMD was taken from the Biological Magnetic Resonance Bank (BMRB)[20], entry 25930[1c].
τc determination
The Fas TMD was reconstituted in q = 0.7, 0.5 and 0.3 bicelles. For each q, a series of 1D 1H-15N TRACT spectra was recorded at 600 MHz (Table S1) with the following relaxation time: 4, 12, 20, 28, 36, 44, 52, 64, 76 and 100 ms. For each time point, the TROSY and anti-TROSY signals were acquired separately.
Gd-DOTA titrations
The Fas TMD was reconstituted in q = 0.7, 0.5 and 0.3 bicelles and titrated against known concentration of Gd-DOTA. The titrant was taken from a concentrated stock solution (600 mM Gd-DOTA) in the same buffer as that of the protein sample, and it was added in small aliquots (few μL per step) to minimize possible dilution effects. To monitor the progress of the titration by NMR, a 2D 1H-15N TROSY-HSQC spectrum was recorded at 600 MHz (Table S1) at each of the following titrant concentrations: 0.5, 1.0, 1.5, 2.0, 4.0, 6.0, 8.0, 10.0, 15.0 and 20.0 mM.
NMR data acquisition, processing, and analysis
The NMR experiments were performed at 14.1 T on a Bruker Avance III HD spectrometer operating at 600.13 MHz 1H, 150.90 MHz 13C, and 60.81 MHz 15N frequencies and on a Bruker Avance I spectrometer operating at 600.47 MHz 1H, 150.99 MHz 13C, and 60.85 MHz 15N frequencies, both equipped with a cryogenically cooled probe head. All the measurements were performed at 303.0 K. The most relevant acquisition parameters of the experiments are reported in Table S1. The NMR data sets were processed using nmrPipe[21] and the resulting NMR spectra were analyzed with Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). Topspin was used to measure integrals in 1D spectra. Chemical shift variations were quantified using the formula Peak in tensitiesfo rmweurlea measured at peak local maximum using quadratic interpolation to identify peak center. Origin (OriginLab, Northampton, MA) was used to fit the experimental data.
Derivation of Equation 1
The general form of the exponential decay reported in Eq. 1 is:
By definition, PREamp describes the variation of the I/I0 ratios when the concentration of the paramagnetic agent is zero and approaches infinity:
Since peak intensities in the NMR spectra were normalized so that , then . Rearranging this last expression for and substituting it into the general form of the exponential decay shown above yields Eq. 1 in the main text.
Supplementary Material
Footnotes
This work was supported by NIH grants HL103526 and GM116898 to J.J.C.
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, Groves JT, Kuriyan J. Cell. 2013;152:543–556. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mi LZ, Lu C, Li Z, Nishida N, Walz T, Springer TA. Nat Struct Mol Biol. 2011;18:984–989. doi: 10.1038/nsmb.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fu Q, Fu TM, Cruz AC, Sengupta P, Thomas SK, Wang S, Siegel RM, Wu H, Chou JJ. Mol Cell. 2016;61:602–613. doi: 10.1016/j.molcel.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kim C, Schmidt T, Cho EG, Ye F, Ulmer TS, Ginsberg MH. Nature. 2012;481:209–213. doi: 10.1038/nature10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Trenker R, Call ME, Call MJ. J Am Chem Soc. 2015;137:15676–15679. doi: 10.1021/jacs.5b11354. [DOI] [PubMed] [Google Scholar]; b) Knoblich K, Park S, Lutfi M, van 't Hag L, Conn CE, Seabrook SA, Newman J, Czabotar PE, Im W, Call ME, Call MJ. Cell reports. 2015;11:1184–1192. doi: 10.1016/j.celrep.2015.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Call ME, Chou JJ. Structure. 2010;18:1559–1569. doi: 10.1016/j.str.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) MacKenzie KR, Prestegard JH, Engelman DM. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]; c) Han X, Bushweller JH, Cafiso DS, Tamm LK. Nature structural biology. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]; d) Lorieau JL, Louis JM, Bax A. Proc Natl Acad Sci U S A. 2010;107:11341–11346. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Call ME, Wucherpfennig KW, Chou JJ. Nature immunology. 2010;11:1023–1029. doi: 10.1038/ni.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Lau TL, Kim C, Ginsberg MH, Ulmer TS. EMBO J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Sanders CR, 2nd, Schwonek JP. Biochemistry. 1992;31:8898–8905. doi: 10.1021/bi00152a029. [DOI] [PubMed] [Google Scholar]; b) Sanders CR, Hare BJ, Howard KP, Prestegard JH. Progress In Nuclear Magnetic Resonance Spectroscopy. 1994;26:421–444. [Google Scholar]
- 5.Sanders CR, Prosser RS. Structure. 1998;6:1227–1234. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
- 6.Lu Z, Van Horn WD, Chen J, Mathew S, Zent R, Sanders CR. Molecular pharmaceutics. 2012;9:752–761. doi: 10.1021/mp2004687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vold RR, Prosser RS. JOURNAL OF MAGNETIC RESONANCE SERIES B. 1996;113:267–271. doi: 10.1006/jmrb.1996.0108. [DOI] [PubMed] [Google Scholar]
- 8.Glover KJ, Whiles JA, Wu G, Yu N, Deems R, Struppe JO, Stark RE, Komives EA, Vold RR. Biophysical journal. 2001;81:2163–2171. doi: 10.1016/s0006-3495(01)75864-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG, Arseniev AS. J Biol Chem. 2008;283:6950–6956. doi: 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]; b) Dev J, Park D, Fu Q, Chen J, Ha HJ, Ghantous F, Herrmann T, Chang W, Liu Z, Frey G, Seaman MS, Chen B, Chou JJ. Science. 2016;353:172–175. doi: 10.1126/science.aaf7066. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. Nature. 2012;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H, Hymowitz SG. In: Handbook of cell signaling. 2. Bradshaw RA, Dennis EA, editors. Vol. 1. Academic Press; Oxford: 2009. pp. 265–275. [Google Scholar]
- 11.Son WS, Park SH, Nothnagel HJ, Lu GJ, Wang Y, Zhang H, Cook GA, Howell SC, Opella SJ. Journal of magnetic resonance. 2012;214:111–118. doi: 10.1016/j.jmr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee D, Hilty C, Wider G, Wuthrich K. Journal of magnetic resonance. 2006;178:72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 13.a) Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fernandez C, Hilty C, Wider G, Wuthrich K. Proc Natl Acad Sci U S A. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Respondek M, Madl T, Gobl C, Golser R, Zangger K. J Am Chem Soc. 2007;129:5228–5234. doi: 10.1021/ja069004f. [DOI] [PubMed] [Google Scholar]; b) Hilty C, Wider G, Fernandez C, Wuthrich K. Chembiochem : a European journal of chemical biology. 2004;5:467–473. doi: 10.1002/cbic.200300815. [DOI] [PubMed] [Google Scholar]; c) Teriete P, Franzin CM, Choi J, Marassi FM. Biochemistry. 2007;46:6774–6783. doi: 10.1021/bi700391b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Al-Abdul-Wahid MS, Verardi R, Veglia G, Prosser RS. Journal of biomolecular NMR. 2011;51:173–183. doi: 10.1007/s10858-011-9551-z. [DOI] [PubMed] [Google Scholar]
- 15.a) Hernandez G, Teng CL, Bryant RG, LeMaster DM. J Am Chem Soc. 2002;124:4463–4472. doi: 10.1021/ja017340k. [DOI] [PubMed] [Google Scholar]; b) Pintacuda G, Otting G. J Am Chem Soc. 2002;124:372–373. doi: 10.1021/ja016985h. [DOI] [PubMed] [Google Scholar]
- 16.Marsh D. Proc Natl Acad Sci U S A. 2001;98:7777–7782. doi: 10.1073/pnas.131023798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kucerka N, Nieh MP, Katsaras J. Biochimica et biophysica acta. 2011;1808:2761–2771. doi: 10.1016/j.bbamem.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 18.Pencer J, Nieh MP, Harroun TA, Krueger S, Adams C, Katsaras J. Biochimica et biophysica acta. 2005;1720:84–91. doi: 10.1016/j.bbamem.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Mineev KS, Nadezhdin KD, Goncharuk SA, Arseniev AS. Langmuir : the ACS journal of surfaces and colloids. 2016;32:6624–6637. doi: 10.1021/acs.langmuir.6b00867. [DOI] [PubMed] [Google Scholar]
- 20.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. Nucleic acids research. 2008;36:D402–408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Journal of biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.