

ABSTRACT
Enteropathogenic Escherichia coli (EPEC) is a gastrointestinal pathogen that utilizes a type III secretion system (T3SS) to inject an array of virulence effector proteins into host enterocytes to subvert numerous cellular processes for successful colonization and dissemination. The T3SS effector NleD is a 26-kDa zinc metalloprotease that is translocated into host enterocytes, where it directly cleaves and inactivates the mitogen-activated protein kinase signaling proteins JNK and p38. Here a library of 91 random transposon-based, in-frame, linker insertion mutants of NleD were tested for their ability to cleave JNK and p38 during transient transfection of cultured epithelial cells. Immunoblot analysis of p38 and JNK cleavage showed that 7 mutant derivatives of NleD no longer cleaved p38 but maintained the ability to cleave JNK. Site-directed mutation of specific regions surrounding the insertion sites within NleD revealed that a single amino acid, R203, was essential for cleavage of p38 but not JNK in a direct in vitro cleavage assay, in transiently transfected cells, or in EPEC-infected cells. Mass spectrometry analysis narrowed the cleavage region to within residues 187 and 213 of p38. Mutation of residue R203 within NleD to a glutamate residue abolished the cleavage of p38 and impaired the ability of NleD to inhibit AP-1-dependent gene transcription of a luciferase reporter. Furthermore, the R203 mutation abrogated the ability of NleD to dampen interleukin-6 production in EPEC-infected cells. Overall, this work provides greater insight into substrate recognition and specificity by the type III effector NleD.
KEYWORDS: NleD, metalloprotease, JNK, p38, EPEC, type III effector proteins, mitogen-activated protein kinases
INTRODUCTION
Enteropathogenic Escherichia coli (EPEC) is an extracellular gastrointestinal pathogen that causes significant diarrheal disease in infants, particularly in developing countries. EPEC adheres intimately to the mucosal surface of enterocytes and injects virulence proteins (effectors) directly into host cells via a type III secretion system (T3SS). The genes required to form a functional T3SS, along with genes encoding a subset of effector proteins, are located on a 35-kb genomic pathogenicity island (PAI) called the locus of enterocyte effacement (LEE). Other effector proteins translocated by the EPEC T3SS are located throughout the genome on distinct genomic PAIs and are termed non-LEE-encoded (Nle) effectors. Many Nle effectors from EPEC display novel enzymatic activity and mediate the subversion of host immune signaling processes, such as NF-κB and mitogen-activated protein kinase (MAPK) activation, apoptosis, and necroptosis. For example, NleE is an S-adenosyl-l-methionine-dependent transferase that specifically modifies the NF-κB adaptor proteins TAB2/3, eliminating ubiquitin chain binding activity and thus inhibiting downstream NF-κB activation and interleukin-8 (IL-8) production during EPEC infection (1–3). NleB is an N-acetylglucosamine transferase that inhibits extrinsic apoptosis by adding a single GlcNAc moiety to a conserved arginine residue on the Fas-associated death domain (FADD) protein, as well as other host death domain proteins, including TRADD and RIPK1. This activity predominantly blocks death receptor signaling and subsequent inflammation and apoptosis during infection, thus promoting cell survival and pathogen persistence (4, 5).
EPEC also produces two zinc metalloprotease T3SS effectors, NleC and NleD, which were first identified to be effectors secreted by the LEE-encoded T3SS of the EPEC-like mouse pathogen Citrobacter rodentium (6). Zinc-containing metalloproteases are a group of proteolytic enzymes found across eukaryotes and prokaryotes and are important in maintaining physiological homeostasis within the organism (7). Collectively known as the “zincins,” they contain a consensus motif, HEXXH, where the histidine residues bind a zinc ion and promote nucleophilic attack on peptide bonds using a water molecule at the active site (8). Recent molecular studies have shown that NleC and NleD require the HEXXH motif for cleavage of host innate immune-signaling proteins. NleC specifically cleaves and inactivates NF-κB Rel proteins, including p65 and p50, and thus contributes to the inhibition of IL-8 production during EPEC infection (9–15). NleD cleaves and inactivates the mitogen-activated protein (MAP) kinases c-Jun amino-terminal kinase (JNK) and p38 but not extracellular signal-related kinases (ERK) and contributes modestly to the inhibition of IL-8 production and UV-induced apoptosis in EPEC infection (15).
The MAP kinases are members of discrete signaling pathways that mediate responses to multiple extracellular stimuli, including UV light, heat, osmotic shock, inflammatory cytokines (tumor necrosis factor [TNF] and interleukin-1 [IL-1]), and growth factors (colony-stimulating factor 1 [CSF1]) (16). Three distinct members of the MAPK family exist in mammals: ERK, JNK, and p38/stress-activated protein kinases (SAPK). Activation of JNK and/or p38 contributes to a number of important cellular processes, including inflammation, apoptosis, cell differentiation, and cytokine production (17).
Apart from the metalloprotease motif, little is known about other functional regions of NleD. In this study, we screened a library of transposon and site-directed mutants of NleD to identify amino acids critical for its proteolytic activity against p38 and JNK. We found that aside from amino acids within the catalytic site (H142ELLH146), R203 was essential for direct p38 cleavage and inhibition of AP-1 activation and IL-6 production by NleD but dispensable for JNK cleavage. In addition, we demonstrated direct cleavage of p38 by NleD and propose that cleavage is likely to occur between residues 187 and 213 of p38.
RESULTS
Effect of pentapeptide insertions on proteolytic activity of NleD.
To map the contribution of amino acid residues of NleD to substrate binding and proteolytic cleavage, an extensive pentapeptide scanning mutagenesis screen was performed. A library of 80 mutants was generated using a mutation generation system kit (catalog number F-701; Thermo Scientific) and screened for the insertion of 5 additional amino acids at random positions in NleD originating from EPEC O127:H6 strain E2348/69. An additional 11 pentapeptide mutants were generated by site-directed mutagenesis (SDM) to fill the gaps where there were more than 5 amino acids between mutants obtained with the transposon-mediated mutagenesis system (Fig. 1; see also Table S1 in the supplemental material). In total, 91 mutants of pGFP-NleD with pentapeptide insertions at 78 different positions were made. The resultant pGFP-NleD derivatives were transfected into HEK293T cells, and the cell lysates were analyzed by immunoblotting for cleavage of JNK and p38 (Fig. 1 and Table S1). pGFP-NleD was selected as the template vector for the mutagenesis screen, as expression can easily be visualized by immunoblotting using anti-green fluorescent protein (anti-GFP) antibodies or directly by immunofluorescence microscopy, if required.
FIG 1.

Selected NleD transposon mutants that display altered proteolytic activity toward JNK and/or p38. (A) Immunoblots of JNK and p38 cleavage in cells transiently transfected with GFP-NleD or GFP-NleD mutant derivatives. Cell lysates were probed with anti-JNK and anti-p38 antibodies. Arrows, cleavage products. Anti-GFP antibody was used to show the expression of GFP-NleD and GFP-NleD variants. Actin was used as a loading control. The immunoblots are representative of those from at least 3 independent experiments. UT, untransfected. (B) Schematic diagram mapping transposon insertion sites in NleD that alter JNK and/or p38 cleavage. Dark gray arrowheads, little or no cleavage of JNK or p38; light gray arrowheads, cleavage of JNK but little or no cleavage of p38; white arrowheads, total cleavage of both JNK and p38. aa, amino acids.
NleD from EPEC E2348/69 is 232 amino acids in length, and the HEXXH motif spans amino acids 142 to 146. Mutants harboring insertions of pentapeptides within the 59 amino acids of the N-terminal region of NleD showed proteolytic activity comparable to that of wild-type NleD (Table S1), whereas insertions at 22 different positions downstream of amino acid 59 altered the proteolytic activity of NleD to various extents (Fig. 1A and B and Table S1). Insertion of pentapeptides starting at amino acid positions 73, 196, 202, 203, and 217 in NleD abolished the cleavage of p38 and reduced the level of JNK cleavage compared to that by wild-type NleD (Fig. 1A and Table S1). Insertions starting at amino acid positions 60, 76, 100, 110, 154, 222, and 226 decreased the level of cleavage of p38 to different degrees, whereas cleavage of JNK by these mutants was comparable to that by wild-type NleD (Fig. 1A and Table S1). Insertions at amino acid positions 115 and 179 completely abolished the cleavage of p38 and JNK, as did those within or immediately preceding the H142ELLH146 catalytic motif at positions 144, 146, 147, and 150 (Fig. 1A and Table S1).
Site-directed mutagenesis of NleD and screening of mutants for loss of proteolytic activity.
To further examine potential functional regions of NleD, 16 site-directed mutants with mutations consisting of either two or three consecutive alanine substitutions were constructed using a QuikChange II site-directed mutagenesis kit. Mutation positions were selected on the basis of the pentapeptide insertion positions that yielded a significant effect on p38 or JNK cleavage. The site-directed mutants were subsequently tested for their ability to cleave p38 and JNK (Fig. 2A and B and Table S2). HEK293T cells were transfected with derivatives of pGFP-NleD, and lysates were subjected to SDS-PAGE followed by immunoblot analysis using anti-p38 or anti-JNK antibodies (Fig. 2A). NleD harboring amino acid substitutions across positions 109 to 111, 114 to 116, 146 to 148, 149 to 151, 153 to 155, 178 to 180, 202 to 204, 216 to 218, and 227 to 229 cleaved JNK as efficiently as wild-type NleD. However, substitutions across 109 to 111, 114 to 116, 146 to 148, and 216 to 218 resulted in less efficient cleavage of p38 compared to that by wild-type NleD, and substitutions across positions 149 to 151, 153 to 155, 178 to 180, and 202 to 204 rendered NleD unable to cleave p38 (Fig. 2A and B and Table S2).
FIG 2.

Screen of NleD multiple site-directed mutants for loss of proteolytic activity against JNK and/or p38. (A) Immunoblots of JNK and p38 cleavage in HEK293T cells transiently transfected with GFP-NleD or GFP-NleD multiple site-directed mutants. Cell lysates were probed with anti-JNK and anti-p38 antibodies. Anti-GFP antibody was used to show the expression of GFP-NleD and GFP-NleD variants. Arrows, cleavage products. Actin was used as a loading control. The immunoblots are representative of those from at least 3 independent experiments. UT, untransfected. (B) Schematic diagram mapping triple alanine replacement sites in NleD that alter JNK and/or p38 cleavage. Light gray arrowheads, cleavage of JNK but little or no cleavage of p38; white arrowheads, total cleavage of both JNK and p38. aa, amino acids.
Arginine 203 in NleD is required for p38 cleavage but dispensable for JNK cleavage.
Single amino acid changes were made across most of the 3 amino acid substitutions within NleD that resulted in the loss in p38 cleavage. This included the replacement of glutamate 143 within the H142EXXH146 catalytic motif by alanine (E143A) as a control for the proteolytic activity of NleD. Of these mutants, pGFP-NleD with the R203A substitution (pGFP-NleDR203A) completely lost the ability to cleave p38 but retained the ability to cleave JNK (Fig. 3 and Table S2).
FIG 3.
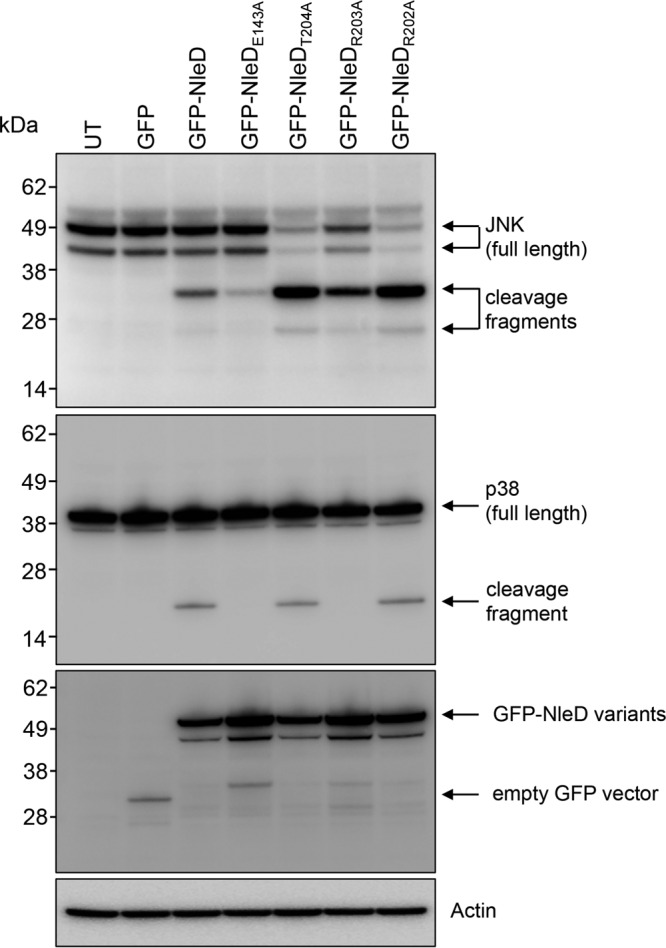
Analysis of NleD single site-directed mutants for p38 cleavage. Immunoblots of JNK and p38 cleavage in HEK293T cells transiently transfected with GFP-NleD or GFP-NleD single site-directed mutants are shown. Cell lysates were probed with anti-JNK and anti-p38 antibodies. Anti-GFP antibody was used to show the expression of GFP-NleD and GFP-NleD variants. Cleavage products are indicated by arrows. Actin was used as a loading control. The immunoblots are representative of those from at least 3 independent experiments. UT, untransfected.
Arginine 203 is conserved among metalloprotease effectors from pathogenic bacteria.
In order to determine whether R203 was conserved in homologues of NleD from other pathogenic bacteria, a BLAST analysis was performed and an alignment of proteins was performed using the ESPript (v3) program. R203 was indeed conserved across a number of diverse bacterial pathogens (Fig. 4). To ensure that the phenotype of the NleD mutant with the R203A substitution (the NleDR203A mutant) was not only due to the selection of alanine as the substitute amino acid, we replaced R203 with lysine or glutamate, each of which carries a charge similar to or opposite that of arginine, respectively. pGFP-NleDR203A, pGFP-NleDR203E, pGFP-NleDR203K, and GFP-NleDE143A were transfected into HEK293T cells and the cell lysates were analyzed by immunoblotting using anti-p38 and anti-JNK antibodies. Cleavage of p38 was abrogated upon expression of GFP-NleDR203A, GFP-NleDR203E, or GFP-NleDE143A compared to that obtained upon expression of GFP-NleD, whereas expression of GFP-NleDR203K only partially compromised p38 cleavage (Fig. 5A).
FIG 4.

Alignment of zinc metalloprotease motif and the surrounding area in NleD proteins from various Gram-negative bacteria. Sequences were identified through BLAST analysis using NleD from EPEC O127:H6 strain E2348/69 as a reference. A section of the proteins identified by BLAST analysis was aligned using the Clustal Omega program, and presentation of the alignment was performed using the ESPript (v3) program (36). *, HEXXH zinc metalloprotease site; arrow, conserved residue R203.
FIG 5.

Analysis of various NleDR203 site-directed mutants for p38 cleavage and their effect on AP-1 activation. (A) Immunoblots of JNK and p38 cleavage in HEK293T cells transiently transfected with GFP-NleD or GFP-NleD single site-directed mutants. Cell lysates were probed with anti-JNK and anti-p38 antibodies. Anti-GFP antibody was used to show the expression of GFP-NleD and GFP-NleD variants. Arrows, cleavage products. Actin was used as a loading control. The immunoblots are representative of those from at least 3 independent experiments. UT, untransfected. (B) Fold increase in AP-1-dependent luciferase activity in HEK293T cells expressing GFP or GFP-NleD variants unstimulated or stimulated with PMA for 6 h. Results are the means ± SEMs from at least three independent experiments carried out in duplicate. *, a result significantly different from the results obtained with GFP-NleD only stimulated with PMA (P < 0.05, by an unpaired, two-tailed t test).
Previous studies have suggested that NleD inhibits AP-1 activation (15); therefore, we tested NleD derivatives carrying amino acid substitutions in R203 and the adjacent amino acids, R202 and T204, for their ability to activate AP-1-dependent gene transcription of a luciferase reporter. Upon stimulation with phorbol myristate acetate (PMA) (18), AP-1 activation was evident in cells transfected with the empty vector pEGFP-C2, whereas it was not in unstimulated cells (Fig. 5B). AP-1 activation was inhibited in cells transfected with wild-type pGFP-NleD, pGFP-NleDR202A, or pGFP-NleDT204A, whereas ectopic expression of the catalytic mutant GFP-NleDE143A or GFP-NleDR203E had no significant impact on AP-1 activation upon PMA stimulation (Fig. 5B). Inhibition of AP-1 activation by GFP-NleDR203K in response to PMA was similar to that by wild-type NleD, and we noted that GFP-NleDR203A still inhibited AP-1 activation in this system, despite being unable to cleave p38 (Fig. 5A and B).
NleD directly cleaves p38.
To test the ability of NleD to cleave p38 directly, we incubated purified recombinant NleD with purified p38 and observed cleavage of the substrate by catalytically active NleD (Fig. 6A and B). Both catalytically inactive NleD (His-NleDE143A) and His-NleDR203E were unable to cleave p38 directly (Fig. 6A). In order to prove that the R203E mutant was active, we incubated purified His-NleDR203E with HT-29 cell lysate and observed cleavage of endogenous JNK but not that of p38 (Fig. S1). The in vivo-generated p38 fragments were further assessed by mass spectrometry, which both confirmed their status as p38 cleavage fragments and revealed the putative cleavage site to be between residues 187 and 213 (Fig. 6B and C, S2, and S3). Analysis of the peptide coverage of full-length p38 compared to that of the N- and C-terminal cleavage fragments revealed that peptides spanning the entire protein sequence could be identified, with peptide coverage being restricted to the N- and C-terminal regions within the N- and C-terminal fragments, respectively (Fig. S2). However, there remained 26 residues (187WYRAPEIMLNWMHYNQTVDIWSVGCI212) between the N- and C-terminal cleavage fragments that were inaccessible to analysis due to the composition of the sequence, which lacks sufficient lysine or arginine residues required for tryptic digestion and subsequent mass spectrometry analysis (Fig. S2). The peptide 213MAELLTGR220 was the most N-terminal peptide identified within the C-terminal cleavage fragment and was not present in the full-length or the N-terminal p38 cleavage fragment tryptic digest, suggesting that M213 may be the most likely cleavage site (Fig. S3). Overall, we can safely conclude only that cleavage occurs between residues 187 and 213 of p38.
FIG 6.

Direct cleavage of p38 by NleD in vitro and mass spectrometric analysis of putative cleavage site for p38. (A) Immunoblots of GST or GST-p38 incubated with His-NleD or His-NleD site-directed mutants. Arrows, p38 cleavage fragment. Reaction mixtures were probed with anti-GST antibodies, and anti-His antibodies were used to detect NleD and NleD variants. (B) A Coomassie-stained gel showing the N-terminal (N-term) and C-terminal (C-term) cleavage products of GST-p38 in the presence of His-tagged NleD after incubation for 2 h or overnight (O/N). White boxes, bands that were excised from the gel and used for further mass spectrometric analysis. (C) Identification of semitryptic peptide 213MAELLTGR220 only within the C-terminal p38 cleavage fragment supports the suggestion that cleavage of p38 occurs at or before position 213. Mox, oxidized methionine residue; a, b, and y, peptide fragment ions.
Arginine 203 in NleD is essential for p38 cleavage and inhibition of IL-6 secretion during EPEC infection.
To examine whether the NleD R203E variant had the same effect on p38 and JNK cleavage in EPEC-infected cells as in transiently transfected cells, nleD, nleDR203E, and nleDE143A were cloned into the bacterial expression vector pTrc99A that carries a C-terminal 2× hemagglutinin (HA) tag to visualize the expression of NleD by immunoblotting. The constructs were expressed in the EPEC E2348/69 ΔPP4/IE6 genetic background. This strain is missing seven effectors (nleD, nleC, nleE, nleB1, nleB2, espL, and nleG). Infection of HT-29 cells with wild-type EPEC resulted in the cleavage of p38 and JNK, whereas infection with either the T3SS mutant (ΔescN), the ΔPP4/IE6 double island mutant, or the ΔPP4/IE6 mutant complemented with nleDE143A had no effect on the cleavage of either substrate (Fig. 7A). Complementation of the ΔPP4/IE6 mutant with wild-type nleD resulted in the cleavage of both JNK and p38 in EPEC-infected HT-29 cells, whereas complementation with nleDR203E resulted only in the loss of JNK (Fig. 7A).
FIG 7.

Effect of the R203E mutation in NleD on p38 cleavage and IL-6 secretion in EPEC infection. (A) Immunoblots of JNK and p38 cleavage in HT-29 cells infected with various EPEC derivatives. Cell lysates were probed with anti-JNK and anti-p38 antibodies. Anti-HA antibody was used to show the expression of NleD-2HA and NleD-2HA variants. Arrows, cleavage products. Actin was used as a loading control. The immunoblots are representative of those from at least 3 independent experiments. (B) HT-29 cells were infected with derivatives of EPEC, as indicated, for 2 h and left unstimulated or stimulated with TNF for 8 h (gray bars). Results are the means ± SEMs from at least three independent experiments carried out in duplicate (*, P < 0.05; **, P < 0.005; P values were determined by an unpaired, two-tailed t test). UI, uninfected; UN, unstimulated.
Studies have shown that activation of p38 MAPK signaling results in the increased secretion of proinflammatory cytokines, including IL-1β, TNF, and IL-6 (19). Therefore, we examined IL-6 secretion levels in HT-29 cells infected with various EPEC derivatives. Cells infected with wild-type EPEC and stimulated with TNF produced minimal amounts of IL-6, comparable to the findings for uninfected, unstimulated cells (Fig. 6B). This was also the case for cells infected with ΔPP4/IE6 complemented with nleE, which is a potent inhibitor of inflammatory cytokine production (2) (Fig. 6B). Expression of wild-type NleD but not that of NleDE143A or NleDR203E in the ΔPP4/IE6 mutant also inhibited IL-6 production, but NleD was less effective than NleE (Fig. 7B).
DISCUSSION
Early in vivo studies showed no significant virulence phenotype for single nleC or nleD deletion mutants in C. rodentium infection of mice (20) or enterohemorrhagic E. coli (EHEC) infection of infant ruminants (21). Subsequently, both NleC and NleD were identified to be metalloprotease effectors. Whereas NleC and its cleavage of NF-κB are very well characterized (9–15, 22), to date only one study has examined the substrate specificity and enzymatic function of NleD (15). Baruch et al. showed that NleD cleaved the MAP kinases p38 and JNK and that mutation of glutamate within the metalloprotease motif (H142ELLH146) abolished the cleavage of JNK. In addition, this study demonstrated direct cleavage of JNK by NleD and identified the conserved TPY motif within the kinase activation loop of JNK to be the specific cleavage site (15). The activation of JNK/p38 influences transcription of the proinflammatory cytokines, such as IL-8 (23, 24). Previous studies showed that EPEC potently inhibits the production of proinflammatory cytokines early in infection (25, 26) and that NleE and NleC contribute significantly to this inhibition (2, 3, 12). Cleavage of JNK by NleD makes a minor contribution to this inhibition, as a modest reduction in the level of IL-8 secretion was observed in cells infected with an EPEC mutant lacking nleC, nleD, nleE, and nleB1 and complemented with wild-type nleD (15).
Here, we screened a library of insertion mutants of NleD for their ability to cleave the host substrates p38 and JNK during transient transfection of cultured epithelial cells. A total of 78 unique insertion mutants were constructed, and of these, 22 insertions affected p38 and/or JNK cleavage. Further mutagenesis in the form of multiple or single site-directed mutations was performed to assess the contribution of specific amino acids to the cleavage of host substrates. Amino acid substitutions at HNL from positions 149 to 151 [HNL(149–151)], GER from positions 153 to 155 [GER(153–155)], TVG from positions 178 to 180 [TVG(178–180)], and RRT from positions 202 to 204 [RRT(202–204)] abolished p38 cleavage, while cleavage of JNK remained unaffected. Analysis of multiple single site-directed mutants revealed that mutation of the R203 residue in NleD abolished the cleavage of p38 but not that of JNK during both transient transfection and EPEC infection, suggesting that R203 in NleD discriminates between these host substrates. An alignment of the region surrounding the zinc metalloprotease motif from a subset of effectors from various Gram-negative pathogens revealed that R203 in NleD was highly conserved. This may reflect the importance of the conserved amino acid in host substrate recognition for other metalloprotease effectors; however, there is currently no structural information available for these effectors. Therefore, the function of R203 in NleD proteolytic activity remains unknown.
NleD mediated the direct cleavage of p38, which was dependent on the catalytic motif of NleD, as mutation of E143 within the HEXXH motif abolished cleavage of the substrate. In addition, the R203E NleD mutant was also unable to directly cleave p38, further suggesting that R203 is essential for the proteolytic activity of NleD against p38. The purified R203E NleD mutant was still, however, active against JNK. A previous study identified the NleD cleavage site within the activation loop of JNK before residue Y185 of the TPY motif (15). No study has yet, however, identified the cleavage site within p38. Here, we have shown that cleavage does not occur within the TGY motif of p38 (residues 180 to 182), as we observed a clear intact peptide containing this motif within the N-terminal cleavage fragment. We did, however, narrow down the cleavage site to a region just downstream of the TGY motif, between residues 187 and 213, suggesting that the cleavage sites of p38 and JNK differ. Mass spectrometric analysis of full-length, N- and C-terminal fragments of p38 suggests that the most likely cleavage site is M213.
The loss of p38 cleavage attributed to the R203 mutation in NleD significantly affected MAPK signaling in host cells. AP-1 is a transcriptional activator located downstream of the MAP kinases p38 and JNK that mediates transcription of proinflammatory cytokines, such as IL-6, in host cells (27, 28). NleD inhibited transcription of an AP-1 luciferase reporter in response to PMA, which was dependent on the R203 residue in NleD, as radical mutation of R203 to glutamate most significantly affected NleD activity.
Since a consequence of AP-1 activation is the production of the proinflammatory cytokine IL-6 (28), we examined the ability of NleD to suppress IL-6 production. Previous studies have shown that IL-6 is required for protection against colonization, mucosal pathology, and mortality in C. rodentium infection of mice (29). A single nleD deletion mutant of EPEC was unable to cleave JNK or p38; therefore, in order to test the contribution of NleD to inflammatory cytokine production in EPEC infection, we utilized a double island deletion mutant lacking seven effectors (nleB1, nleB2, nleC, nleD, nleE, nleG, and espL) and complemented this with wild-type nleD or nleD site-directed mutants. The reason that we chose to utilize the seven-effector mutant is because NleE, NleB, and NleC all significantly contribute to the inhibition of inflammatory cytokine production (2, 12, 15). Therefore, we aimed to test the relative contribution of NleD in this genetic background. Here, wild-type EPEC inhibited IL-6 production in response to TNF stimulation, to which NleD made a significant contribution. Mutation of R203 in NleD significantly abrogated IL-6 inhibition, suggesting that p38 activation contributed to IL-6 production during EPEC infection and that NleD specifically targets p38 to dampen the IL-6 response. Interestingly, cleavage of JNK alone by NleD was insufficient to mediate the suppression of IL-6 during EPEC infection.
Overall, the potent early inhibition of inflammation in EPEC infection is a result of the translocation of multiple type III effectors that specifically target innate immune signaling pathways, including NF-κB (NleE, NleC, NleB1, NleH1, NleH2, Tir) (2–5, 12, 13, 30) and MAPK (NleC and NleD) (13, 15). Here we showed that NleD blocked IL-6 production through cleavage of the MAPK p38 during EPEC infection. Hence, NleD is yet another example of an effector that targets host immune processes and contributes to the dampening of inflammatory responses during EPEC infection.
MATERIALS AND METHODS
Bacterial strains, plasmids, cell lines, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Tables 1 and 2, respectively. All PCR primers are listed in Table 3. Bacteria were grown at 37°C in Luria-Bertani (LB) medium, Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX (Gibco), or RPMI with GlutaMAX (Gibco) where indicated, and the media were supplemented with ampicillin (100 μg/ml), kanamycin (100 μg/ml), or chloramphenicol (25 μg/ml) where necessary. HEK293T cells were grown in DMEM with GlutaMAX (Gibco) supplemented with 10% fetal calf serum (FCS; Sigma) at 37°C with 5% CO2. HT-29 cells were grown in RPMI with GlutaMAX (Gibco) supplemented with 10% FCS (Sigma) at 37°C with 5% CO2.
TABLE 1.
Bacterial strains used in this study
| Bacterial strain | Characteristics | Source or reference |
|---|---|---|
| EPEC E2348/69 | Wild-type EPEC O127:H6 | 37 |
| ΔnleD | EPEC E2348/69 ΔnleD Kmr | This study |
| ΔescN | EPEC E2348/69 ΔescN Kmr | 38 |
| ΔPP4/IE6 | EPEC E2348/69 PP4/IE6 double island deletion mutant | 2 |
| XL1-Blue | E. coli recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tetr)] | Stratagene |
| BL21 C43(DE3) | E. coli strain used for expression of proteins for affinity purification | Novagen |
TABLE 2.
Plasmids used in this study
| Plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| pEGFP-C2 | Expression vector carrying enhanced GFP, Kanr | Clontech |
| pGFP-NleD | Full-length nleD from EPEC E2348/69 in pEGFP-C2, Kanr | 2 |
| pGFP-NleDG110A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid G110 mutated to A, Kanr | This study |
| pGFP-NleDT111A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid T111 mutated to A, Kanr | This study |
| pGFP-NleDF115A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid F115 mutated to A, Kanr | This study |
| pGFP-NleDE143A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid E143 mutated to A, Kanr | This study |
| pGFP-NleDL144A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid L144 mutated to A, Kanr | This study |
| pGFP-NleDH146A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid H146 mutated to A, Kanr | This study |
| pGFP-NleDN150A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid N150 mutated to A, Kanr | This study |
| pGFP-NleDE154A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid E154 mutated to A, Kanr | This study |
| pGFP-NleDR155A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid R155 mutated to A, Kanr | This study |
| pGFP-NleDV179A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid V179 mutated to A, Kanr | This study |
| pGFP-NleDG180A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid G180 mutated to A, Kanr | This study |
| pGFP-NleDR202A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid R202 mutated to A, Kanr | This study |
| pGFP-NleDR203A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid R203 mutated to A, Kanr | This study |
| pGFP-NleDR203E | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid R203 mutated to E, Kanr | This study |
| pGFP-NleDR203K | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid R203 mutated to K, Kanr | This study |
| pGFP-NleDT204A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid T204 mutated to A, Kanr | This study |
| pGFP-NleDN217A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid N217 mutated to A, Kanr | This study |
| pGFP-NleDT218A | nleD from EPEC E2348/69 in pEGFP-C2 with amino acid T218 mutated to A, Kanr | This study |
| pGFP-NleDLDK(59–61)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the LDK(59–61) motif mutated to AAA, Kanr | This study |
| pGFP-NleDLN(72–73)AA | nleD from EPEC E2348/69 in pEGFP-C2 with the LN(72–73) motif mutated to AA, Kanr | This study |
| pGFP-NleDIES(75–77)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the IES(75–77) motif mutated to AAA, Kanr | This study |
| pGFP-NleDHR(100–101)AA | nleD from EPEC E2348/69 in pEGFP-C2 with the HR(100–101) motif mutated to AA, Kanr | This study |
| pGFP-NleDRGT(109–111)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the RGT(109–111) motif mutated to AAA, Kanr | This study |
| pGFP-NleDVF(147–148)AA | nleD from EPEC E2348/69 in pEGFP-C2 with the VF(147–148) motif mutated to AA, Kanr | This study |
| pGFP-NleDDFH(114–116)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the DFH(114–116) motif mutated to AAA, Kanr | This study |
| pGFP-NleDHVF(146–148)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the HVF(146–148) motif mutated to AAA, Kanr | This study |
| pGFP-NleDHNL(149–151)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the HNL(149–151) motif mutated to AAA, Kanr | This study |
| pGFP-NleDGER(153–155)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the GER(153–155) motif mutated to AAA, Kanr | This study |
| pGFP-NleDTVG(178–180)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the TVG(178–180) motif mutated to AAA, Kanr | This study |
| pGFP-NleDREE(195–197)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the REE(195–197) motif mutated to AAA, Kanr | This study |
| pGFP-NleDRRT(202–204)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the RRT(202–204) motif mutated to AAA, Kanr | This study |
| pGFP-NleDDNT(216–218)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the DNT(216–218) motif mutated to AAA, Kanr | This study |
| pGFP-NleDQVR(225–227)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the QVR(225–227) motif mutated to AAA, Kanr | This study |
| pGFP-NleDRLH(227–229)AAA | nleD from EPEC E2348/69 in pEGFP-C2 with the RLH(227–229) motif mutated to AAA, Kanr | This study |
| pET28a | N-terminal 6× His Tag cloning/expression vector | Novagen |
| pET-NleD | nleD from EPEC E2348/69 in pET28a, Kanr | This study |
| pET-NleDE143A | nleD from EPEC E2348/69 in pET28a with amino acid E143 mutated to A, Kanr | This study |
| pET-NleDR203E | nleD from EPEC E2348/69 in pET28a with amino acid R203 mutated to E, Kanr | This study |
| pGEX-4T-1 | N-terminal GST cloning/expression vector, Ampr | GE Healthcare |
| pGEX-p38 | Human p38 (MAPK14 transcript variant 2) in pGEX-4T-1, Ampr | This study |
| pTrc99A | Bacterial expression vector containing a lacI promoter, Ampr | Pharmacia Biotech |
| pNleD | nleD from EPEC 2348/69 in pTrc99A, Ampr | 12 |
| pNleDE143A | nleD from EPEC E2348/69 in pTrc99A with amino acid E143 mutated to A, Ampr | This study |
| pNleDR203E | nleD from EPEC E2348/69 in pTrc99A with amino acid R203 mutated to E, Ampr | This study |
| pNleD-2HA | nleD from EPEC 2348/69 in pTrc99A with a 2× C-terminal HA tag, Ampr | This study |
| pNleDE143A-2HA | nleD from EPEC E2348/69 in pTrc99A with amino acid E143 mutated to A and a 2× C-terminal HA tag, Ampr | This study |
| pNleDR203E-2HA | nleD from EPEC E2348/69 in pTrc99A with amino acid R203 mutated to E and a 2× C-terminal HA tag, Ampr | This study |
TABLE 3.
Primers used in this study
| Primer | Sequencea |
|---|---|
| Pos. 18F | GTCATCAACGTCGAGCTTGCGGCCGCAGAGCTCAATGTCAGATACAGATATCG |
| Pos. 18R | CGATATCTGTATCTGACATTGAGCTCTGCGGCCGCAAGCTCGACGTTGATGAC |
| Pos. 50F | CGTTTCCAGGGGACTGATCATGCGGCCGCAGATCATAATATATATCAGCAGATTG |
| Pos. 50R | CAATCTGCTGATATATATTATGATCTGCGGCCGCATGATCAGTCCCCTGGAAACG |
| Pos. 60F | GCAGATTGAAGCAGCACTCTGCGGCCGCACACTCGATAAGATTGGCTCTACAG |
| Pos. 60R | CTGTAGAGCCAATCTTATCGAGTGTGCGGCCGCAGAGTGCTGCTTCAATCTGC |
| Pos. 91F | CAGTGGTAATACACCTTGCGGCCGCACACCTCAACTCTTCCAGACTAGG |
| Pos. 91R | CCTAGTCTGGAAGAGTTGAGGTGTGCGGCCGCAAGGTGTATTACCACTG |
| Pos. 121F | CGATTTTCACTGTAATCTGAATTGCGGCCGCATGAATGCAGTTGAATATCCCTGTGG |
| Pos. 121R | CCACAGGGATATTCAACTGCATTCATGCGGCCGCAATTCAGATTACAGTGAAAATCG |
| Pos. 128F | CAGTTGAATATCCCTGTGGTGCGGCCGCATGTGGGGAGGGGATTAGCGTGGTG |
| Pos. 128R | CACCACGCTAATCCCCTCCCCACATGCGGCCGCACCACAGGGATATTCAACTG |
| Pos. 133F | GTGGGGAGGGGATTAGCGTTGCGGCCGCAAGCGTGGTGGACTTTCATGCGAC |
| Pos. 133R | GTCGCATGAAAGTCCACCACGCTTGCGGCCGCAACGCTAATCCCCTCCCCAC |
| Pos. 144F | CGACTATTGTTTTTCATGAGTGCGGCCGCAATGAGTTGCTCCATGTTTTCCAC |
| Pos. 144R | GTGGAAAACATGGAGCAACTCATTGCGGCCGCACTCATGAAAAACAATAGTCG |
| Pos. 167F | CCGACCAGAATCACAAAATGCGGCCGCACAAAAATACTCTCCACTTTTACTCG |
| Pos. 167R | CGAGTAAAAGTGGAGAGTATTTTTGTGCGGCCGCATTTTGTGATTCTGGTCGG |
| Pos. 191F | CTTTTTCAGAGGAGGTGCTTTCTGCGGCCGCACTTTCAGAAAATAAATTCCGCGAAGAG |
| Pos. 191R | CTCTTCGCGGAATTTATTTTCTGAAAGTGCGGCCGCAGAAAGCACCTCCTCTGAAAAAG |
| Pos. 207F | GATGCCCCGTAGAACCTCCTATGCGGCCGCATCCTACCCGCACGACTCAGCTCTTATTC |
| Pos. 207R | GAATAAGAGCTGAGTCGTGCGGGTAGGATGCGGCCGCATAGGAGGTTCTACGGGGCATC |
| LDK59-61AAAF | CAGCAGATTGAAGCAGCAGCGGCGGCGATTGGCTCTACAGAGACAGGG |
| LDK59-61AAAR | CCCTGTCTCTGTAGAGCCAATCGCCGCCGCTGCTGCTTCAATCTGCTG |
| LN72-73AAF | CAGAGACAGGGCGTGTACTCGCGGCGGCTATTGAATCAATATCC |
| LN72-73AAR | GGATATTGATTCAATAGCCGCCGCGAGTACACGCCCTGTCTCTG |
| IES75-77AAAF | CGTGTACTCCTGAATGCTGCGGCGGCGATATCCCGACTTAAATCAGAAACAGTG |
| IES75-77AAAR | CACTGTTTCTGATTTAAGTCGGGATATCGCCGCCGCAGCATTCAGGAGTACACG |
| HR100-101AAF | CCAGACTAGGAGTTATGGCAGCGGCGGATATAGATGCTGAGAACCATCG |
| HR100-101AAR | CGATGGTTCTCAGCATCTATATCCGCCGCTGCCATAACTCCTAGTCTGG |
| DFH114-116AAAF | CCATCGGGGGACTGGTTCCGCGGCGGCGTGTAATCTGAATGCAGTTGAATATCCC |
| DFH114-116AAAR | GGGATATTCAACTGCATTCAGATTACACGCCGCCGCGGAACCAGTCCCCCGATGG |
| HVF146-148AAAF | CGACTATTGTTTTTCATGAGTTGCTCGCGGCGGCGCACAATTTAAATGGGGAGCGTTTG |
| HVF146-148AAAR | CAAACGCTCCCCATTTAAATTGTGCGCCGCCGCGAGCAACTCATGAAAAACAATAGTCG |
| VF147-148AAF | GACTATTGTTTTTCATGAGTTGCTCCATGCGGCGCACAATTTAAATGGGGAGC |
| VF147-148AAR | GCTCCCCATTTAAATTGTGCGCCGCATGGAGCAACTCATGAAAAACAATAGTC |
| HNL149-151AAAF | CATGAGTTGCTCCATGTTTTCGCGGCGGCGAATGGGGAGCGTTTGAAAGTTGAGAGTTC |
| HNL149-151AAAR | GAACTCTCAACTTTCAAACGCTCCCCATTCGCCGCCGCGAAAACATGGAGCAACTCATG |
| GER153-155AAAF | CCATGTTTTCCACAATTTAAATGCGGCGGCGTTGAAAGTTGAGAGTTCCCGAC |
| GER153-155AAAR | GTCGGGAACTCTCAACTTTCAACGCCGCCGCATTTAAATTGTGGAAAACATGG |
| TVG178-180AAAF | CTCGAAGAAGCCAGGGCGGCGGCGTTGGGGGCTTTTTCAGAG |
| TVG178-180AAAR | CTCTGAAAAAGCCCCCAACGCCGCCGCCCTGGCTTCTTCGAG |
| REE195-197AAAF | GGTGCTTTCAGAAAATAAATTCGCGGCGGCGATTGGGATGCCCCGTAGAACC |
| REE195-197AAAR | GGTTCTACGGGGCATCCCAATCGCCGCCGCGAATTTATTTTCTGAAAGCACC |
| RRT202-204AAAF | CGAAGAGATTGGGATGCCCGCGGCGGCGTCCTACCCGCACGACTCAGC |
| RRT202-204AAAR | GCTGAGTCGTGCGGGTAGGACGCCGCCGCGGGCATCCCAATCTCTTCG |
| DNT216-218AAAF | GACTCAGCTCTTATTCATGATGCGGCGGCGGTGAGTCTGGGATTCCAACAGG |
| DNT216-218AAAR | CCTGTTGGAATCCCAGACTCACCGCCGCCGCATCATGAATAAGAGCTGAGTC |
| QVR225-227AAAF | CAGTGAGTCTGGGATTCCAAGCGGCGGCGCTGCATCCATTGCTTTAG |
| QVR225-227AAAR | CTAAAGCAATGGATGCAGCGCCGCCGCTTGGAATCCCAGACTCACTG |
| RLH227-229AAAF | GACAATACAGTGAGTCTGGGATTCCAACAGGTAGCGGCGGCGCCATTGCTTTAG |
| RLH227-229AAAR | CTAAAGCAATGGCGCCGCCGCTACCTGTTGGAATCCCAGACTCACTGTATTGTC |
| E143AF | GGACTTTCATGCGACTATTGTTTTTCATGCGTTGCTCCATGTTTTCCAC |
| E143AR | GTGGAAAACATGGAGCAACGCATGAAAAACAATAGTCGCATGAAAGTCC |
| G110AF | GATGCTGAGAACCATCGGGCGACTGGTTCCGATTTTCACTG |
| G110AR | CAGTGAAAATCGGAACCAGTCGCCCGATGGTTCTCAGCATC |
| T111AF | GCTGAGAACCATCGGGGGGCGGGTTCCGATTTTCACTG |
| T111AR | CAGTGAAAATCGGAACCCGCCCCCCGATGGTTCTCAGC |
| F115AF | CATCGGGGGACTGGTTCCGATGCGCACTGTAATCTGAATGCAG |
| F115AR | CTGCATTCAGATTACAGTGCGCATCGGAACCAGTCCCCCGATG |
| L144AF | GACTATTGTTTTTCATGAGGCGCTCCATGTTTTCCACAATTTAAATGG |
| L144AR | CCATTTAAATTGTGGAAAACATGGAGCGCCTCATGAAAAACAATAGTC |
| H146AF | CGACTATTGTTTTTCATGAGTTGCTCGCGGTTTTCCACAATTTAAATGG |
| H146AR | CCATTTAAATTGTGGAAAACCGCGAGCAACTCATGAAAAACAATAGTCG |
| N150AF | GTTGCTCCATGTTTTCCACGCGTTAAATGGGGAGCGTTTG |
| N150AR | CAAACGCTCCCCATTTAACGCGTGGAAAACATGGAGCAAC |
| E154AF | GTTTTCCACAATTTAAATGGGGCGCGTTTGAAAGTTGAGAG |
| E154AR | CTCTCAACTTTCAAACGCGCCCCATTTAAATTGTGGAAAAC |
| R155AF | CCACAATTTAAATGGGGAGGCGTTGAAAGTTGAGAGTTCCCGAC |
| R155AR | GTCGGGAACTCTCAACTTTCAACGCCTCCCCATTTAAATTGTGG |
| V179AF | CGAAGAAGCCAGGACTGCGGGGTTGGGGGCTTTTTC |
| V179AR | GAAAAAGCCCCCAACCCCGCAGTCCTGGCTTCTTCG |
| G180AF | CGAAGAAGCCAGGACTGTTGCGTTGGGGGCTTTTTCAGAGG |
| G180AR | CCTCTGAAAAAGCCCCCAACGCAACAGTCCTGGCTTCTTCG |
| R202AF | CGAAGAGATTGGGATGCCCGCGAGAACCTCCTACCCGCACG |
| R202AR | CGTGCGGGTAGGAGGTTCTCGCGGGCATCCCAATCTCTTCG |
| R203AF | GATTGGGATGCCCCGTGCGACCTCCTACCCGCAC |
| R203AR | GTGCGGGTAGGAGGTCGCACGGGGCATCCCAATC |
| R203EF | GATTGGGATGCCCCGTGAGACCTCCTACCCGCAC |
| R203ER | GTGCGGGTAGGAGGTCTCACGGGGCATCCCAATC |
| R203KF | GATTGGGATGCCCCGTAAGACCTCCTACCCGCAC |
| R203KR | GTGCGGGTAGGAGGTCTTACGGGGCATCCCAATC |
| T204AF | GAGATTGGGATGCCCCGTAGAGCGTCCTACCCGCACGACTCAG |
| T204AR | CTGAGTCGTGCGGGTAGGACGCTCTACGGGGCATCCCAATCTC |
| N217AF | GCTCTTATTCATGATGACGCGACAGTGAGTCTGGGATTCC |
| N217AR | GGAATCCCAGACTCACTGTCGCGTCATCATGAATAAGAGC |
| T218AF | CTCTTATTCATGATGACAATGCGGTGAGTCTGGGATTCCAACAG |
| T218AR | CTGTTGGAATCCCAGACTCACCGCATTGTCATCATGAATAAGAG |
| NleDF | AAGAATTCATGCGCCCTACGTCCCTC |
| NleDR | GGAGTCGACGCTAAAGCAATGGATGCAGTCTTAC |
| p38F | GGGGATCCATGTCTCAGGAGAGGCCCACG |
| p38R | CGCTCGAGTCAGGACTCCATCTCTTCTTGGTC |
| NleD-2HAF | AACCATGGATGCGCCCTACGTCCCTC |
| NleD-2HAR | CCGGATCCCTACGCATAATCCGGCACATCATACGGATACGCATAATCCGGCAC ATCATACGGATAAAGCAATGGATGCAGTCTTACC |
| pTrcF | AACACCCCCATCGGCG |
| pTrcR | GTAACCATTATAAGCTGC |
| pEGFP-C2F | AACACCCCCATCGGCG |
| pEGFP-C2R | GTAACCATTATAAGCTGC |
| NotI | TGCGGCCGCA |
Boldface nucleotides indicate mutated codons.
Random-insertion scanning mutagenesis and site-directed mutagenesis.
Generation of an NleD pentapeptide scanning mutagenesis library was carried out in the plasmid pGFP-NleD using a mutation generation system kit (catalog number F-701; Thermo Scientific) with an entranceposon (M1-Camr) according to the manufacturer's instructions. Three independent mutagenesis reactions were performed. E. coli strain XL1-Blue was used as the transformation host for the chemical transformation steps, and agar plates were incubated for 40 h at 37°C. The restriction enzymes EcoRI and BamHI (New England BioLabs) were used to clone nleD open reading frames containing the entranceposon into the vector backbone of pEGFP-C2 without entranceposon insertions. The 15-bp insertions in the individual nleD clones were mapped by colony PCR following the manufacturer's recommendations using primers pEGFP-C2F and pEGFP-C2R in combination with the NotI miniprimer (Table 3). The exact position of the 15-bp insertion within nleD was determined by DNA sequencing of selected transformants using primer pEGFP-C2R (Table 3).
Site-directed mutagenesis of nleD was performed in plasmid pGFP-NleD, pNleD, or pNleD-2HA as the template. The oligonucleotides used to generate the mutants are listed in Table 3. The mutant strand synthesis reaction was performed with Pfu DNA polymerase (Promega) under the following conditions: 95°C for 30 s; 18 cycles of 95°C for 30 s, 55°C for 1 min, and 68°C for 5.5 min; and a final extension at 72°C for 7 min. The amplification products were digested with DpnI (New England BioLabs) and directly transformed into chemically competent cells of E. coli strain XL1-Blue. Mutations were confirmed by DNA sequencing using either primer pair pEGFP-C2F/pEGFP-C2R or primer pair pTrcF/pTrcR (Table 3).
Construction of plasmids to express NleD and p38.
pNleD-2HA was constructed by amplifying nleD from EPEC E2348/69 genomic DNA by PCR using the primer pair NleD-2HAF/NleD-2HAR. PCR amplification consisted of an initial denaturation step at 95°C for 10 min, followed by 30 cycles of 94°C for 45 s, 50°C for 40 s, and 70°C for 1 min and then a final elongation step of 70°C for 10 min. The PCR product was digested with NcoI and BamHI and ligated into pTrc99A.
pET-NleD, pET-NleDE143A, and pET-NleDR203E were constructed by amplifying nleD, nleDE143A, and nleDR203E from pGFP-NleD, pGFP-NleDE143A, and pGFP-NleDR203E template DNA, respectively, by PCR using the primer pair NleDF/NleDR. PCR amplification consisted of an initial denaturation step at 95°C for 10 min, followed by 30 cycles of 94°C for 45 s, 50°C for 40 s, and 70°C for 1 min and then a final elongation step of 70°C for 10 min. The PCR products were digested with EcoRI and SalI and ligated into pET-28a to produce N-terminal 6×His tag fusions to NleD.
pGEX-p38 was constructed by amplifying human p38 (MAPK14 transcript variant 2) from HeLa cell cDNA using the primer pair p38F/p38R. PCR amplification consisted of an initial denaturation step at 98°C for 30 s, followed by 35 cycles of 98°C for 10 s, 63.4°C for 30 s, and 72°C for 30 s and then a final elongation step of 72°C for 5 min. The PCR product was digested with BamHI and XhoI and ligated into pGEX-4T-1 to produce an N-terminal glutathione S-transferase (GST) tag fusion to p38.
EPEC infection.
HT-29 cell monolayers were seeded into 24-well tissue culture trays (Corning) at 24 h prior to infection. EPEC derivatives were inoculated into LB broth and grown with shaking at 37°C overnight. On the day of infection, the overnight EPEC cultures were subcultured 1:75 in DMEM with GlutaMAX (Gibco) or RPMI with GlutaMAX (Gibco) and grown statically for 3 h at 37°C with 5% CO2. Where necessary, cells were induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 30 min prior to infection. Cells were washed twice with phosphate-buffered saline (PBS) and infected with EPEC cultures at an optical density at 600 nm of 0.03 for 2 h.
Transfection.
Transfections were carried out in HEK293T cells using the Fugene 6 (Promega) transfection reagent according to the manufacturer's instructions. Cells were seeded into 24-well tissue culture trays (Corning) and transfected with 0.4 μg of pEGFP-C2 derivatives 24 h later for a period of 18 h.
Dual-luciferase reporter assay.
For the AP-1 dual-luciferase assay, HEK293T cells were seeded into 24-well trays (Corning) and cotransfected with derivatives of pEGFP-C2 (0.4 μg), 0.05 μg of pRL-TK (Promega), and 0.2 μg of pAP-1-Luc (Clontech). At approximately 18 h posttransfection, cells were left untreated or stimulated with 25 ng/ml phorbol myristate acetate (PMA; Calbiochem) for 6 h. Firefly and Renilla luciferase levels were measured using a dual-luciferase reporter assay system (Promega) in a CLARIOstar microplate reader (BMG Labtech). The expression of firefly luciferase was normalized to Renilla luciferase measurements, and luciferase activity was expressed relative to that of unstimulated pEGFP-C2-transfected cells.
Immunoblot analysis.
For immunoblot analysis following EPEC infection or transfection, cells were collected and lysed in cold lysis buffer (1% Triton X-100, 50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 150 mM NaCl) with cOmplete protease inhibitor (Roche), 2 mM Na3VO4, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride and incubated on ice for 10 min to complete lysis. The samples were pelleted at 4°C, and the supernatants were added to 4×Bolt lithium dodecyl sulfate sample buffer containing 50 mM dithiothreitol (DTT) (Thermo Fisher), heated to 70°C for 10 min, and resolved on Bolt 4 to 12% bis-Tris Plus gels (Thermo Fisher) by PAGE. Proteins were transferred to nitrocellulose membranes using an iBlot2 gel transfer device (Thermo Fisher) and probed with one of the following primary antibodies: rabbit polyclonal anti-SAPK/JNK (Cell Signaling), rabbit polyclonal anti-p38 MAPK (Cell Signaling), mouse monoclonal anti-HA (Covance), mouse monoclonal anti-GFP (clones 7.1 and 13.1; Roche), and mouse monoclonal anti-β-actin (clone AC-15; Sigma). The antibodies were diluted in Tris-buffered saline (TBS) with 5% bovine serum albumin (BSA) (Sigma) and 0.1% Tween (Sigma). Proteins were detected using anti-rabbit or anti-mouse IgG secondary antibodies conjugated to horseradish peroxidase (PerkinElmer) diluted in TBS with 5% BSA and 0.1% Tween and developed with an enhanced chemiluminescence (ECL) immunoblotting reagent (Amersham). Images were visualized using an MFChemiBis imaging station (DNR Bio-Imaging Systems). At least three biological replicates were performed for all experiments.
In vitro cleavage assay.
Plasmids for the expression of 6×His-tagged proteins (pET-NleD, pET-NleDE143A, and pET-NleDR203E) or GST-tagged proteins (pGEX-4T-1 and pGEX-p38) were transformed into E. coli BL21 C43(DE3). Overnight LB cultures of BL21 containing the appropriate plasmid were used to inoculate 200 ml LB broth 1:100, in which the cultures were grown at 37°C with shaking to an optical density (A600) of 0.6. The cultures were then induced with 1 mM IPTG and grown for a further 2.5 h at 37°C (for His-NleD constructs and GST) or 18°C (for GST-p38) before being pelleted by centrifugation. Purification of proteins was performed using Novagen His · Bind and GST · Bind kits according to the manufacturer's protocols. Protein concentrations were determined using a bicinchoninic acid (BCA) kit (Thermo Scientific).
In vitro cleavage assays were performed by incubating 2 μg of purified GST or GST-p38 either alone or with 2 μg of His-NleD, His-NleDE143A, or His-NleDR203E in buffer [TBS supplemented with 1 mM Tris(2-carboxyethyl)phosphine hydrochloride] for 4 h at 37°C. Sample buffer was then added to the incubation mixtures before they were heated at 70°C for 10 min, resolved by SDS-PAGE, and subsequently transferred to nitrocellulose membranes. The membranes were probed with mouse monoclonal anti-His (clone AD1.1.10; AbD Serotec) or rabbit polyclonal anti-GST (Cell Signaling) primary antibodies and horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin secondary antibodies (PerkinElmer) and developed as described above.
For detection of the activity of purified NleD proteins within HT-29 cell lysates, HT-29 cells were grown in 10-cm dishes for 48 h before being washed with PBS and then collected and resuspended in 800 μl PBS. Cells were then lysed by passage through a 26-gauge needle 50 times and pelleted, and the supernatant was used for incubations. Forty microliters of the HT-29 cell lysate was incubated alone or with 0.5 μg of His-NleD, His-NleDE143A, or His-NleDR203E for 4 h at 37°C. Sample buffer was added to the incubation mixtures before they were heated at 70°C for 10 min and immunoblotted as described above.
Detection of IL-6 by ELISA.
For analysis of IL-6 secretion, HT-29 cell monolayers were infected for 2 h before 8 to 12 h of incubation with 50 μg/ml gentamicin with or without 20 ng/ml TNF (Calbiochem, EMD4Biosciences). The HT-29 cell supernatant was collected for subsequent analysis of IL-6 secretion. Analysis of IL-6 secretion was performed using a human IL-6 enzyme-linked immunosorbent assay (ELISA) set (Max Deluxe; BioLegend) as per the manufacturer's instruction. All experiments were performed at least three independent times with samples from three independent infections performed in duplicate.
Mass spectrometry analysis of p38 cleavage fragments.
For mass spectrometric analysis, 15 μg of GST-p38 and 1 μg of His-NleD were incubated together or individually at 37°C for 2 h or overnight. Sample buffer was then added to the incubation mixtures before they were heated at 70°C for 10 min and subjected to SDS-PAGE. Polyacrylamide gels were fixed and visualized with Coomassie G-250 according to the protocol of Kang et al. (31).
Tryptic digestion of full-length p38 and cleavage products.
Bands of interest were excised from the Coomassie-stained gels and destained in a 50:50 solution of 50 mM NH4HCO3–100% ethanol for 20 min at room temperature with shaking at 750 rpm. Destained samples were then washed with 100% ethanol, vacuum dried for 20 min, and rehydrated in 10 mM DTT in 50 mM NH4HCO3. Reduction was carried out for 60 min at 56°C with shaking. The reducing buffer was then removed, and the gel bands were washed twice in 100% ethanol for 10 min to remove the residual DTT. Reduced ethanol-washed samples were sequentially alkylated with 55 mM iodoacetamide in 50 mM NH4HCO3 in the dark for 45 min at room temperature (RT). Alkylated samples were then washed for 2 rounds with 100% ethanol and vacuum dried. Alkylated samples were then rehydrated with 12 ng/μl trypsin (Promega, Madison, WI) in 40 mM NH4HCO3 at 4°C for 1 h. Excess trypsin was removed, and gel pieces were covered in 40 mM NH4HCO3 and incubated overnight at 37°C. Peptides were concentrated and desalted using C18 stage tips (32, 33) before analysis by liquid chromatography-mass spectrometry (LC-MS).
Identification of p38 sequence coverage using reversed-phase LC-MS.
Purified peptides were resuspended in buffer A* (0.1% trifluoroacetic acid, 2% acetonitrile [MeCN]) and separated using a two-column chromatography setup composed of a PepMap100 C18 trap column (20 mm by 75 μm) and PepMap C18 analytical columns (500 mm by 75 μm) (Thermo Scientific). Samples were concentrated onto the trap column at 5 μl/min for 5 min and infused into an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific) at 300 nl/min via the analytical column using a Dionex Ultimate 3000 ultraperformance liquid chromatograph (Thermo Scientific). A 90-min gradient from 2% buffer B to 28% buffer B over 51 min and then from 28% buffer B to 40% buffer B in the next 5 min was run; it was then increased to 100% buffer B over a 2-min period, held at 100% buffer B for 2.5 min, and then dropped to 0% buffer B for another 20 min. The Lumos mass spectrometer was operated in a data-dependent mode automatically switching between the acquisition of a single Orbitrap MS scan (120,000 resolution) every 3 s and ion-trap collision-induced dissociation MS-MS, Orbitrap higher-energy collision-induced dissociation MS-MS, and Orbitrap electron-transfer/higher-energy collision dissociation for each selected precursor (maximum fill time, 100 ms; automatic gain control, 5 × 104 with a resolution of 30,000 for Orbitrap MS-MS scans).
Data analysis.
MaxQuant (v1.5.3.1) software (34) was used for identification of p38 cleavage products. A database search of the sequences against a GST-p38 protein sequence supplemented with data from an E. coli K-12 database (from UniProt, 16 March 2015) was carried out with the following search parameters: carbamidomethylation of cysteine as a fixed modification; oxidation of methionine, deamidation (NQ), and acetylation of protein N termini as variable modifications; and semitrypsin cleavage specificity. The precursor mass tolerance was set to 20 ppm for the first search and 10 ppm for the main search, and a maximum false discovery rate of 1.0% was set for protein and peptide identifications. To enhance the identification of peptides between full-length GST-p38 and cleavage products, the Match between Runs option was enabled with a precursor match window set to 2 min and an alignment window of 10 min. The resulting protein group output was processed within the Perseus (v1.4.0.6) (35) analysis environment to remove reverse matches and common protein contaminants prior to analysis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants to E.L.H. from the Australian National Health and Medical Research Council (NHMRC) (APP1100609). K.C. is supported by a Deutsche Forschungsgemeinschaft (DFG) research fellowship. J.S.P. is the recipient of an NHMRC early career research fellowship. C.G. is the recipient of an Australian Postgraduate Award (APA). T.W.F.L. is a recipient of a University of Melbourne International Research Scholarship (MIRS). N.E.S. is the recipient of an NHMRC early career research fellowship.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00620-16.
REFERENCES
- 1.Nadler C, Baruch K, Kobi S, Mills E, Haviv G, Farago M, Alkalay I, Bartfeld S, Meyer T, Ben-Neriah Y, Rosenshine I. 2010. The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog 6:e1000743. doi: 10.1371/journal.ppat.1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, Wagstaff KM, Dunstone MA, Sloan J, Whisstock JC, Kaper JB, Robins-Browne RM, Jans DA, Frankel G, Phillips AD, Coulson BS, Hartland EL. 2010. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog 6:e1000898. doi: 10.1371/journal.ppat.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L, Ding X, Cui J, Xu H, Chen J, Gong Y-N, Hu L, Zhou Y, Ge J, Lu Q, Liu L, Chen S, Shao F. 2011. Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 481:204–208. doi: 10.1038/nature10690. [DOI] [PubMed] [Google Scholar]
- 4.Li S, Zhang L, Yao Q, Li L, Dong N, Rong J, Gao W, Ding X, Sun L, Chen X, Chen S, Shao F. 2013. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501:242–246. doi: 10.1038/nature12436. [DOI] [PubMed] [Google Scholar]
- 5.Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, Lung TWF, Mansell A, Riedmaier P, Oates CVL, Zaid A, Mühlen S, Crepin VF, Marches O, Ang C-S, Williamson NA, O'Reilly LA, Bankovacki A, Nachbur U, Infusini G, Webb AI, Silke J, Strasser A, Frankel G, Hartland EL. 2013. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501:247–251. doi: 10.1038/nature12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O'Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci U S A 101:3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyoshi S-I, Shinoda S. 2000. Microbial metalloproteases and pathogenesis. Microbes Infect 2:91–98. doi: 10.1016/S1286-4579(00)00280-X. [DOI] [PubMed] [Google Scholar]
- 8.Potempa J, Pike R. 2005. Bacterial peptidases. Contrib Microbiol 12:132–180. [DOI] [PubMed] [Google Scholar]
- 9.Giogha C, Wong Fok Lung T, Mühlen S, Pearson JS, Hartland EL. 2015. Substrate recognition by the zinc metalloprotease effector NleC from enteropathogenic Escherichia coli. Cell Microbiol 17:1766–1768. doi: 10.1111/cmi.12469. [DOI] [PubMed] [Google Scholar]
- 10.Hodgson A, Wier E, Fu K, Sun X, Yu H, Zheng W, Sham H, Johnson K, Bailey S, Vallance B, Wan F. 2015. Metalloprotease NleC suppresses host NF-kappaB/inflammatory responses by cleaving p65 and interfering with the p65/RPS3 interaction. PLoS Pathog 10:e1004705. doi: 10.1371/journal.ppat.1004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mühlen S, Ruchaud-Sparagano M-H, Kenny B. 2011. Proteasome-independent degradation of canonical NFκB complex components by the NleC protein of pathogenic Escherichia coli. J Biol Chem 286:5100–5107. doi: 10.1074/jbc.M110.172254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson JS, Riedmaier P, Marches O, Frankel G, Hartland EL. 2011. A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-κB for degradation. Mol Microbiol 80:219–230. doi: 10.1111/j.1365-2958.2011.07568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sham HP, Shames SR, Croxen MA, Ma C, Chan JM, Khan MA, Wickham M, Deng W, Finlay BB, Vallance BA. 2011. Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-κB and p38 mitogen-activated protein kinase activation. Infect Immun 79:3552–3562. doi: 10.1128/IAI.05033-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. 2010. NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog 6:e1001231. doi: 10.1371/journal.ppat.1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baruch K, Gur-Arie L, Nadler C, Koby S, Yerushalmi G, Ben-Neriah Y, Yogev O, Shaulian E, Guttman C, Zarivach R, Rosenshine I. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-κB. EMBO J 30:221–231. doi: 10.1038/emboj.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zarubin T, Han J. 2005. Activation and signaling of the p38 MAP kinase pathway. Cell Res 15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 17.Morrison DK. 2012. MAP kinase pathways. Cold Spring Harbor Perspect Biol 4:a011254. doi: 10.1101/cshperspect.a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karin M. 1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 19.Guan Z, Buckman SY, Pentland AP, Templeton DJ, Morrison AR. 1998. Induction of cyclooxygenase-2 by the activated MEKK1 → SEK1/MKK4 → p38 mitogen-activated protein kinase pathway. J Biol Chem 273:12901–12908. doi: 10.1074/jbc.273.21.12901. [DOI] [PubMed] [Google Scholar]
- 20.Kelly M, Hart E, Mundy R, Marchès O, Wiles S, Badea L, Luck S, Tauschek M, Frankel G, Robins-Browne RM, Hartland EL. 2006. Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infect Immun 74:2328–2337. doi: 10.1128/IAI.74.4.2328-2337.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchès O, Wiles S, Dziva F, La Ragione RM, Schüller S, Best A, Phillips AD, Hartland EL, Woodward MJ, Stevens MP, Frankel G. 2005. Characterization of two non-locus of enterocyte effacement-encoded type III-translocated effectors, NleC and NleD, in attaching and effacing pathogens. Infect Immun 73:8411–8417. doi: 10.1128/IAI.73.12.8411-8417.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Liu Y, Sheng X, Yin P, Hu F, Liu Y, Chen C, Li Q, Yan C, Wang J. 2014. Structure and mechanism of a type III secretion protease, NleC. Acta Crystallogr D Biol Crystallogr 70:40–47. doi: 10.1107/S1399004713024619. [DOI] [PubMed] [Google Scholar]
- 23.Kang S-S, Woo SS, Im J, Yang JS, Yun C-H, Ju HR, Son CG, Moon E-Y, Han SH. 2007. Human placenta promotes IL-8 expression through activation of JNK/SAPK and transcription factors NF-κB and AP-1 in PMA-differentiated THP-1 cells. Int Immunopharmacol 7:1488–1495. doi: 10.1016/j.intimp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Roger T, Out AT, Mukaida N, Matsushima K, Jansen MH, Lutter R. 1998. Enhanced AP-1 and NF-κB activities and stability of interleukin 8 (IL-8) transcripts are implicated in IL-8 mRNA superinduction in lung epithelial H292 cells. Biochem J 330:429–435. doi: 10.1042/bj3300429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hauf N, Charkraborty T. 2003. Suppression of NF-kappaB activation and proinflammatory cytokine expression by Shiga toxin-producing Escherichia coli. J Immunol 170:2074–2082. doi: 10.4049/jimmunol.170.4.2074. [DOI] [PubMed] [Google Scholar]
- 26.Ruchaud-Sparagano M, Maresca M, Kenny B. 2007. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell Microbiol 9:1909–1921. doi: 10.1111/j.1462-5822.2007.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angel P, Karin M. 1991. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1072:129–157. doi: 10.1016/0304-419X(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 28.Hershko DD, Robb BW, Luo G, Hasselgren P-O. 2002. Multiple transcription factors regulating the IL-6 gene are activated by cAMP in cultured Caco-2 cells. Am J Physiol Regul Integr Comp Physiol 283:R1140–R1148. doi: 10.1152/ajpregu.00161.2002. [DOI] [PubMed] [Google Scholar]
- 29.Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ, Hanson E, Eckmann L. 2008. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion forming enteric bacterial pathogens. J Immunol 180:6816–6826. doi: 10.4049/jimmunol.180.10.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Royan SV, Jones RM, Koutsouris A, Roxas JL, Falzari K, Weflen AW, Kim A, Bellmeyer A, Turner JR, Neish AS, Rhee K-J, Viswanathan VK, Hecht GA. 2010. Enteropathogenic E. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-κB activation. Mol Microbiol 78:1232–1245. doi: 10.1111/j.1365-2958.2010.07400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang D, Gho Y, Suh M, Kang C. 2002. Highly sensitive and fast protein detection with Coomassie brilliant blue in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Bull Korean Chem Soc 23:1511–1513. doi: 10.5012/bkcs.2002.23.11.1511. [DOI] [Google Scholar]
- 32.Ishihama Y, Rappsilber J, Mann M. 2006. Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J Proteome Res 5:988–994. doi: 10.1021/pr050385q. [DOI] [PubMed] [Google Scholar]
- 33.Rappsilber J, Mann M, Ishihama Y. 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 34.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 35.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. 2016. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 36.Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 37.Levine MM, Nalin DR, Hornick RB, Bergquist EJ, Waterman DH, Young CR, Sotman S, Rowe B. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet 311:1119–1122. doi: 10.1016/S0140-6736(78)90299-4. [DOI] [PubMed] [Google Scholar]
- 38.Zurawski DV, Mumy KL, Badea L, Prentice JA, Hartland EL, McCormick BA, Maurelli AT. 2008. The NleE/OspZ family of effector proteins is required for polymorphonuclear transepithelial migration, a characteristic shared by enteropathogenic Escherichia coli and Shigella flexneri infections. Infect Immun 76:369–379. doi: 10.1128/IAI.00684-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.