

Abstract
Viral infection of the central nervous system (CNS) can result in a multitude of responses including pathology, persistence or immune clearance. Lymphocytic choriomeningitis virus (LCMV) is a powerful model system to explore these potential outcomes of CNS infection due to the diversity of responses that can be achieved after viral inoculation. Several factors including tropism, timing, dose and variant of LCMV in combination with the development or suppression of the corresponding immune response dictates whether lethal meningitis, chronic infection or clearance of LCMV in the CNS will occur. Importantly, the functionality and positioning of the LCMV-specific CD8+ T cell response are critical in directing the subsequent outcome of CNS LCMV infection. Although a basic understanding of LCMV and immune interactions in the brain exists, the molecular machinery that shapes the balance between pathogenesis and clearance in the LCMV-infected CNS remains to be elucidated. This review covers the various outcomes of LCMV infection in the CNS and what is currently known about the impact of the virus itself versus the immune response in the development of disease or clearance.
Keywords: LCMV, Clone 13, Virus, Meningitis, Immunotherapy, Brain, Neurons, Astrocytes, Immunodeficient, Neonates, Growth Hormone, T cell, behavior, Review
2. INTRODUCTION
The parameters that dictate pathology versus clearance after CNS viral infection are relatively undefined. The CNS contains highly integrated populations of neurons and glial cells responsible for our livelihood, and immune infiltration has the potential to severely disrupt the function of this sensitive compartment. Consequently, through evolutionary pressures the CNS has become immunologically specialized and now possesses the ability to modulate (or suppress) leukocyte behavior in a manner that fundamentally differs from that observed in most peripheral tissues. This evolutionary acquisition, which is advantageous from the standpoint of host survival, likely facilitates the entry and replication of viruses within the CNS. A multitude of viruses have the capacity to gain access to the CNS and upon doing so can establish a chronic infection, drive immunopathology, or alter the function of residents cells, making clearance of the invading pathogen essential. Therefore, a fundamental knowledge of viral-immune interactions in the CNS is essential if we ultimately intend to therapeutically modulate CNS immune responses in humans to achieve viral clearance in the absence of immunopathology. LCMV infection of mice provides an outstanding model system to explore such interactions and extend the lessons learned to virus infections of humans.
3. GENERAL BACKGROUND ON LCMV
LCMV is a natural pathogen of both human and murine populations (1). Human transmission has been shown to occur in utero, upon transplantation with infected organs or upon contact with infected hamsters or laboratory animals (2–4). Congenital LCMV infection acts as an abortifacient and a fetal teratogen commonly manifested as chorioretinitis, hydrocephalus and microcephaly or macrocephaly (5–9). Adult infection with LCMV can lead to the development of fever, malaise, headaches, seizure and in some cases fatal meningitis (3, 10–13). Through the study of murine models of infection, a mechanistic understanding of LCMV induced immunopathology can be gained and applied to human disease. In addition, study of the LCMV model has served as a Rosetta stone for the field of viral immunology, resulting in the discovery of numerous generalizable findings that apply to a variety of viral and microbial infections in humans.
3.1. LCMV structure
LCMV is the prototypic member of the Arenaviridae family, which encompasses old and new world viruses including Lassa, Junin, Machupo and Tacaribe (14, 15). LCMV is a negative strand RNA virus that has two genome segments referred to as L and S. The L RNA segment is the larger portion of the genome with a size of 7.2kb compared to the 3.4kb size of the S RNA segment. A bidirectional coding strategy is used to synthesize the polymerase (L) and small RING finger protein Z from the L segment and the viral nucleoprotein (NP) and glycoprotein (GP) from the S segment (16–21). The GP protein is later post-translationally cleaved into GP1 and GP2, which are expressed on the envelope of the virus (22). GP1 mediates attachment of the virus to its cellular targets to initiate infection and thus determines tropism of LCMV. Therefore, mutations in this protein have the potential to alter viral targeting and influence the course of disease.
3.2. Advantages to the LCMV model system
LCMV has been used to make significant contributions to both the fields of virology and immunology, including seminal work detailing concepts such as immune tolerance, immunodominance, MHC restriction and the basis for viral persistence (23–29). Utilization of the LCMV model system provides some key advantages in our understanding of viral-immune interactions. One important benefit to the LCMV system is its great flexibility in the resultant outcome after infection. Depending on the strain, dose and route of LCMV infection, a wide variety of responses including viral clearance, immune suppression, viral persistence, hepatitis or fatal choriomeningitis can be induced (30–33). Another major advantage of the LCMV model system is the generation of experimental tools that can be used to trace and follow the anti-viral response. The ability to trace populations of LCMV-specific T cells gave way to identification of immuodominant epitopes that are observed after infection. Extensive mapping of the LCMV Armstrong (Arm) specific immune response in C57BL/6 mice revealed the generation of a polyclonal T cell response with defined MHC I and II restricted epitopes against both the glycoprotein (GP) and nucleoprotein (NP) of the virus (MHC I: Db GP33–41, Kb GP33–43, Db NP396–404, Db GP276–286, Db GP92–101, Kb GP118–125, Kb NP205–212; MHC II: I-Ab GP61–80, I-Ab NP309–328) thought to represent most of the LCMV Arm specific repertoire (34–36). Additionally, the creation of LCMV specific CD4+ and CD8+ T cell receptor (TCR) transgenics (referred to as SMARTA (37) and P14 (38), respectively) have advanced the field by providing traceable and transferable populations of virus specific T cells. The accumulation of such tools and their permutations allows for more detailed analyses of the immunopathogenesis observed following LCMV infection, which drives a variety of disease states.
4. LCMV INDUCED MENINGITIS
LCMV has the capacity to induce meningitis in both human and murine hosts (12, 13, 33). In adult immunocompetent mice, intracerebral (i.c.) injection of LCMV results in the development of an acute, fatal meningitis 6–8 days post infection (33, 39). Prior to death, mice demonstrate symptoms including ruffled fur, blepharitis, hunched posture and seizure. Notably, the ultimate death of the mouse is often associated with massive seizure induction. LCMV has been shown to localize and infect the ependyma, choroid plexus and meningeal regions of the brain, with relatively little virus detected in the brain parenchyma after i.c. infection (Figure 1) (31, 40, 41). Because LCMV is a non-cytopathic virus, the damage induced in the CNS is not caused by the virus, but rather is caused by the subsequent immune response. Accordingly, massive leukocytic infiltrates are found to accumulate in the meningeal and ventricular regions of the brain, which corresponds with the viral distribution (41–43). Infiltration of the brain parenchyma is also observed and may be critical for the disease process (44).
Figure 1.
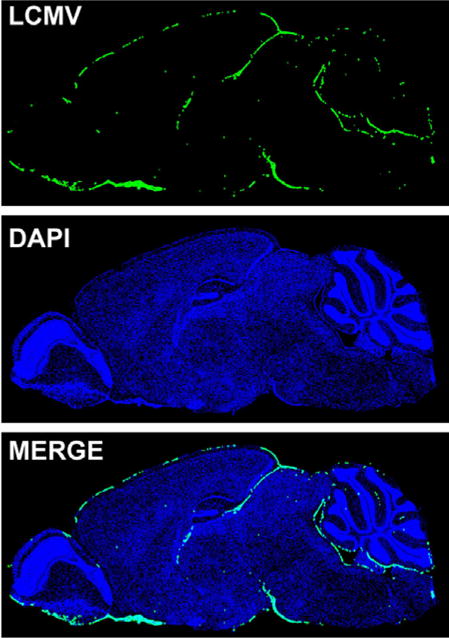
LCMV distribution during acute meningitis. The brain from an 8 week old C57BL/6 mouse infected with LCMV i.c. was harvested on day 6 post infection when severe seizures were apparent. Shown is the localization of LCMV (green) on a sagittal brain section. The virus was detected with a polyclonal anti-LCMV antibody and cell nuclei (blue) were stained with DAPI. Note that LCMV is distributed in the meninges with little to no infiltration into the brain parenchyma.
4.1. The role of CD8+ T cells and histocompatibility during meningitis
Induction of lethal LCMV induced meningitis is dependent on the generation of a productive immune response. T cells were directly implicated as key mediators of mortality when it was discovered that thymectomized mice had increased protection after i.c. infection with LCMV (45). Although the virus is administered i.c., approximately 90% of the inoculum is released into the blood and becomes available for the peripheral T cell priming that occurs over the next 3–5 days (42). Detailed T cell analyses in the cervical lymph node of LCMV infected BALB/c mice revealed significant activation marked by increased CD8+ T cell numbers starting on day 3 post infection and elevated levels of both CD44 and CD25 by day 6 post infection (46). Proliferative and cytotoxic capacities also increased progressively over time, corresponding to the development of a functional T cell response that preceded immunopathology (46).
Following priming activated T cells are then assumed to enter the circulation, traffic to the CNS and cause inflammation and disease. Accordingly, CD8+ T cells have been identified in and appear to be recruited to the cerebral spinal fluid (CSF) after LCMV infection (47, 48). CD8+ T cells were further defined as the essential T cell population needed for LCMV-induced meningitis by studies involving CD8+ T cell depletion (49), genetic knockouts (50–52), blocking peptides (53) and cytotoxic lymphocyte (CTL) transfer (43, 54–56). CD4+ T cells, B cells and NK cells are not essential for meningitis (49, 55, 57–59); however, in the absence of CD8+ T cells, the wasting disease that subsequently develops has been attributed to CD4+ T cells (50, 52). Disease induction by CD8+ T cells necessitates LCMV specificity and therefore cannot be mediated by non-specific bystander CD8+ T cells alone (43). Importantly, meningitis can be initiated in mice harboring a T cell repertoire with a low number naïve LCMV-specific CD8+ T cells. Intracerebral inoculation of ovalbumin CD8 TCR-tg mice (OT-I mice) (60, 61) with LCMV does not result in meningitis, but adoptive transfer of as few as 1,000 monospecific, naive DbGP33–41 CD8+ T cells (P14 cells) was sufficient to reconstitute disease in these mice (43). Therefore, LCMV-specific CD8+ T cells are absolutely required for the development of meningitis.
Supporting a role for CD8+ T cells in LCMV-induced meningitis, histocompatibility at the MHC class I locus was discovered by Zinkernagel and Doherty to be an essential requirement for pathology and disease following i.c. LCMV infection (56, 62, 63) – a finding worthy of the Nobel Prize. In fact, Doherty subsequently uncovered that maximal meningeal inflammation required that histocompatible MHC I be expressed on both bone marrow derived cells and radio-resistant cells; he presumed the latter to be endothelial cells (64). In vitro, LCMV infection enhanced MHC I expression on brain endothelial cells (65), suggesting an increased potential for CD8+ T cell engagement in vivo. Recently, the CNS vascular endothelium was shown to present a model antigen (Ag) to CD8+ T cells and allow for preferential entry of Ag-specific CD8+ T cells into the brain parenchyma (66). If this mechanism holds true in the context of meningitis, then vascular endothelial MHC I expression may be important for optimal entry of CD8+ T cells into the CNS. As LCMV specific CD8+ T cells are known to produce proinflammatory cytokines that can affect blood brain barrier integrity (67, 68), it is interesting to further theorize that cognate peptide-MHC I interactions on the vascular endothelium with LCMV-specific CD8+ T cells could stimulate cytokine release or actual cytolysis of the endothelium. These actions could then be directly responsible for the increased blood brain barrier permeability associated with CD8+ T cells during meningitis (69).
4.2. CD8+ T cell trafficking to the CNS during acute meningitis
Normally, T cell trafficking into various tissues is a multi-step process involving selectin mediated rolling, chemokine induced activation and integrin mediated adhesion on vascular endothelium, which is followed by extravasation (70). Although these steps are not completely defined for CNS T cell entry during LCMV-induced meningitis, some insights into the process do exist in the literature. Studies in CD43, an E-selectin ligand (71), deficient mice demonstrated decreased infiltration to the CNS (72) suggesting a role for this E-selectin in the trafficking events to the CNS. However, it was previously shown that entry of CD8+ T cells into this site after infection is actually an E/P selectin independent event (73). Therefore, during LCMV infection, the selectins used for CD8+ T cell rolling on the vascular endothelium of the CNS are still unclear and remain to be elucidated.
Several candidates for chemokine induced activation of integrins have been identified. By day 6 post infection when T cells are present in the CNS, mRNA levels of chemokines such as RANTES, IP-10, MIP-1α/β and MCP-1 are increased over what is observed on day 3 post-infection – a time point prior to T cell infiltration (74). Chemokine upregulation appears to be linked to IFNγ expression (75), which is interesting because IFNγ is not detected until later time points when T cells are present in the CNS. This suggests that the release of IFNγ by infiltrating CD8+ T cells may be an important step in the amplification of chemokine expression and T cell recruitment into the brain.
Chemokine receptors including CXCR3 and CCR5 are expressed on CD8+ T cells after LCMV infection (44, 75, 76). While CCR5 and CXCR3 are dispensable for initial T cell entry into the CSF of the LCMV infected mice (44, 77), the subsequent positioning of T cells in the brain parenchyma during LCMV-induced meningitis is controlled in part by interactions between CXCR3 and CXCL10 (44, 78). Mice genetically deficient in either the receptor (CXCR3) or the ligand (CXCL10) were found to be partially resistant to LCMV-induced meningitis, and this was attributed to a reduced number of CD8+ T cells found in the brain parenchyma (44, 78). The inability of researchers to identify requisite chemokine-chemokine receptor interactions for initial T cell entry into the brain of LCMV-infected mice might be due to the fact that the appropriate molecule (s) has not yet been studied or to functional redundancy in the system, which has the potential to obscure the relative importance of the previously mentioned chemokine/chemokine receptors. It is therefore of some importance to examine chemokine pathways using approaches that do not rely on genetic knockouts. Real time in vivo imaging of T cell extravasation in the presence of chemokine receptor blocking antibodies might provide a contemporary approach to address such an issue.
Expression of the integrins VLA-4, LFA-1 and CD11b on CD8+ T cells located in the secondary lymphoid organs and CSF after infection make them probable candidates for the last step of extravasation (75, 79–82). Correspondingly, the ligands for VLA-4 and LFA-1 (i.e., VCAM-1 and ICAM-1) are expressed on the vascular endothelium of the CNS during meningitis (79, 80, 83). Entry of CD8+ T cells into the brain is associated with massive infiltration of monocytes and non-specific CD8+ T cells, which is likely due to the aforementioned CD8+ T cell associated blood brain barrier activation and breakdown. Although many candidates for immune cell trafficking into the CNS have been identified, the multistep process for extravasation of the initial LCMV-specific CD8+ T cell population, which amplifies the magnitude of CNS immune infiltrate, requires further refinement.
4.3. T cell effector functions and meningitis
Despite the identification of CD8+ T cells as the critical population required for disease induction, the actual mechanism that results in mortality during LCMV infection is still unknown. After entry into the CNS, LCMV-specific CD8+ T cells co-localize in LCMV-infected regions of the brain. Direct in vivo engagement of infected target cells by LCMV-specific CTL is thought be involved in the pathogenic process, as immunological synapses between DbGP33–41 specific CTL and LCMV-infected targets were observed in symptomatic mice at the peak of disease (day 6 post-infection) (84). Productive CTL lysis of the LCMV infected cells is supported by the deposition of perforin onto the targets (84), the significant increase in the number of apoptotic cells (43) and the presence of LCMV NP circulating in the CSF (85). Interestingly, in CXCR3-deficient mice delayed disease, associated with poor parenchymal CTL recruitment, was observed despite the fact that infiltration of the CSF was unimpaired (44). Though it remains possible that death in LCMV-infected mice is caused by physical occlusion of the ventricular system or by increased intracranial pressure, the aforementioned data in CXCR3-deficient mice suggests a critical link between CTL localization and mortality in the LCMV model.
CTL have multiple pathways that can be used for target cell killing which include perforin-granzyme delivery, Fas/FasL interactions and cytokine production (e.g., TNF-α); however, elimination of individual pathways through genetic deficiency has minimally impacted disease progression and mortality in the LCMV model. It was originally thought that pathology and death in this model involved perforin-mediated lysis of target cells by CD8+ T cells. This was based upon the observation that perforin-deficient mice survived i.c. challenge with a viscerotropic strain of LCMV (86). Subsequent analyses of perforin-deficient mice infected with a neurotropic LCMV variant (Armstrong) revealed a delay in disease kinetics, but not protection from death, thus disproving that perforin was the sole effector molecule responsible for disease (87). Perforin-independent, granzyme B-mediated cytolysis and Fas/FasL interactions (88) have also been eliminated as being required for disease, as both granzyme B-deficient and Fas-deficient mice succumb to LCMV-induced meningitis ((89), unpublished results). Therefore, no evidence presently exists to support a direct link between a single contact-dependent CTL effector mechanism and the highly reproducible mortality observed in the model.
CTL also use cytokine release as an alternate means to lyse target cells. TNFα and IFNγ were potential candidates for disease progression based on their increased cerebral expression in symptomatic animals and their colocalization with infected regions (90–92). Depending on the time of administration, TNFα was shown to either exacerbate or alleviate meningitis in an adoptive transfer model of disease (93). However, three observations support that TNFα is not involved in disease pathogenesis: 1.) TNFα was not found in the CSF (94), 2.) mortality was observed despite intracranial neutralization of TNFα (95), and 3.) i.c. inoculation of TNFα deficient mice results in normal disease kinetics (unpublished results). IFNγ (96) are also dispensable for LCMV-induced meningitis, as deficient mice succumb to disease with kinetics similar to wild-type mice following i.c. challenge with neurotropic LCMV Arm. However, IFNγ deficiency does improve survival when mice are infected with a viscerotropic LCMV variant (96–98). This pattern of protection versus susceptibility based on the usage of a viscerotropic versus a neurotropic LCMV variant is likely due to the peripheral organs acting as an antigen sink that divert the CD8+ T cell away from the CNS (99). In conclusion, although CD8+ T cells are known to be crucial for disease, the mechanism (s) that mediate mortality during meningitis remain elusive.
5. LCMV AND NEONATAL INFECTION
In 1936 Traub made the seminal observation that a naturally occurring state of persistent LCMV infection could arise in most tissues without causing mortality (100). Later it was determined that LCMV infection either in utero or within 24 hours of birth leads to a life long carrier state marked by persistent viral infection rather than the clearance or death that is normally observed in adult mice (101–103). After inoculation, virus is disseminated globally throughout the body and is detected in major organs including the lung, liver, kidney, thymus, lymph nodes, pancreas and brain (104, 105). Within the brain parenchyma of the adult carrier mouse, the virus replicates almost exclusively in neurons (106, 107) (Figure 2). Chronic infection is established in part because LCMV replicates in the thymus, which leads to clonal deletion of LCMV-specific thymocytes (38). This was directly demonstrated by neonatal infection of LCMV-specific TCR tg mice, which resulted in the loss of cells in the CD4+CD8+ stage of thymic development and a diminished number of splenic CD8+ T cells (38). As a result of clonal deletion, the repertoire of T cells in the periphery of carrier mice has little to no cytolytic activity against LCMV and is unable to effectively purge the virus (108, 109). Notably, vaccination of carrier mice with an immunodominant CTL epitope for LCMV leads to the generation of a low affinity CTL response, albeit at an exceedingly low frequency, suggesting that thymic deletion of LCMV-specific CD8+ T cells is incomplete and that high affinity CD8+ T cell clones are eliminated in the thymus (110). Additionally, “cured” carrier mice after immune cell transfer (discussed below) can mount a functional endogenous CTL response that is able to clear virus upon re-challenge, suggesting a reversibility of tolerance once persistent antigen is removed (111).
Figure 2.
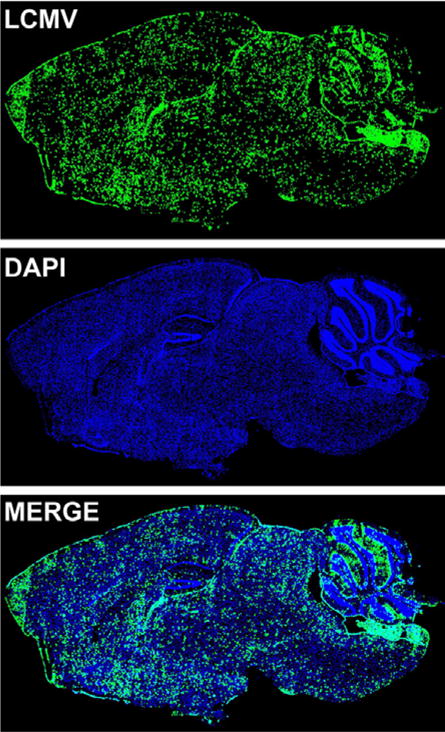
LCMV distribution in carrier mice infected in utero with LCMV. A brain from a C57BL/6 mouse infected with LCMV in utero via maternal vertical transmission was harvested at >8 weeks of age. Note that LCMV (LCMV) is localized throughout the brain parenchyma in a pattern reflecting neuronal infection. The meninges are also infected during this carrier state. Cell nuclei are shown in blue for anatomical purposes.
5.1. Immunotherapy to treat LCMV carrier mice
Induction of a carrier state in mice has served as a useful platform to study the potential of immunotherapy as an effective means for viral clearance. Initial work conducted by Mogens Volkert established that transfer of LCMV immune cells from donor mice into carrier recipients resulted in elimination of the virus (112). CD8+ T cells, H-2 compatibility and viral specificity, but not virus specific antibodies and CD4+ T cells, were all later defined as necessary parameters for successful immunotherapy (113–118). However, a role for B cells and CD4+ T cells may have been underestimated. In a system where high antibody production was observed, peripheral clearance of carrier mice persistently infected with LCMV WE was dependent on CD4+ T cells and B cells, but not CD8+ T cells (119). In the same study, immunotherapy of LCMV Arm carriers, which have lower antibody production after adoptive transfer compared to LCMV WE carriers, required CD8+ T cells, CD4+ T cells and B cells for viral elimination from tissues (119). Since B cells are critical for proper splenic architecture and resulting T cell responses (120), it is possible that this structural function is more important than antibody production in the context of successful clearance of carrier mice. This idea was substantiated in a study that used B cell deficient μMT mice to generate immune cells for immunotherapy (121). Adoptive transfer of LCMV immune cells from μMT-deficient mice resulted in decreased viral clearance in carrier mice infected with LCMV Arm. This defect was attributed to the impaired LCMV-specific CD4+ T cell responses observed in μMT donor mice (121). Further evaluation of CD4+ T cells following adoptive immunotherapy revealed that they are important for the long term maintenance of CD8+ T cell function (122–124). In fact, the extent of CD8+ T cell IFNγ and TNFα production directly correlated with the number of CD4 T cells co-transferred into recipient LCMV carrier mice (124). The numbers of CD4+ T cells, relative to CD8+ T cells, needed to provide sufficient help is surprisingly low. 350,000 CD8+ T cells and 7,000 CD4+ T cells (a ratio of 50:1) was defined as the minimum number of virus-specific memory cells required for successful immunotherapeutic clearance of LCMV Arm infected carrier mice (124). While CD8+ T cells are likely the predominant effector cell necessary for viral clearance in persistently infected mice, CD4+ T cells and B cells play an important accessory role by maximizing and sustaining the activities of the transferred CTL response.
One of the more striking features of carrier immunotherapy is the varied kinetics of viral clearance based on the location of viral sequestration (116, 118, 125, 126). Oldstone and colleagues demonstrated that LCMV was purged from peripheral tissues (e.g., serum, liver, lungs and spleen) by day 15 post-immunotherapy but persisted in the brain and kidney of carrier mice until around day 120 (116). Although initial studies of brain tissues after immunotherapy demonstrated a restriction of infiltrates to the leptomeninges with little to no inundation of the brain parenchyma (116, 126), a more sensitive assay revealed that GFP-tagged LCMV-specific CTL distribute evenly throughout the brain parenchyma within 8 days following immunotherapy (107). Interestingly, LCMV clearance from neurons in carrier mice occurs with minimal neuronal drop out, suggesting a noncytopathic mechanism of clearance (116, 126).
Because neurons express low to undetectable levels of MHC I, this is thought to be one mechanism that protects them from direct CTL-mediated lysis. Consequently, this protective mechanism might also give rise to the predilection of viruses to establish persistence in neurons. Reduced MHC I expression on neurons was first demonstrated in vitro using a neuronal cell line (127, 128). These cells also had a reduced ability to load MHC I with peptides (128). However, stimulation of the neuronal cell line by IFNγ resulted in upregulation of MHC I and subsequent lysis by CTL (127). Based on the hypothesis that neurons had a deficiency in MHC I expression, transgenic mice were generated to express H-2Db protein under the neuron-specific enolase promoter (NSE-Db mice). Adoptive transfer of LCMV-specific CTL in NSE-Db mice persistently infected with LCMV Arm resulted in CTL infiltration of the CNS, profound blood brain barrier breakdown, increased illness/death of carrier mice and faster CNS viral clearance in those that survived (129). These data suggest that neuronal MHC I expression is one bottleneck that restricts the speed with which CTL purge virus from neurons in LCMV carrier mice. This bottleneck is likely in place to minimize the amount of irreparable immune-mediated damage that can occur within the CNS.
In the absence of direct interactions between CTL and neurons, production of cytokines such as IFNα/β, IFNγ and TNFα may be important for viral clearance of the CNS. Each of these cytokines were shown be required for viral clearance following immunotherapy (107, 119, 122). A recent study examining mice 8 days post-immunotherapy revealed that a considerable number of antigen presenting cells (APCs) were recruited into the CNS of carrier mice as a direct consequence of the immunotherapy, and this APC recruitment correlated perfectly with the arrival of LCMV-specific CTL (107). Importantly, of the APC subsets only dendritic cells (not infiltrating macrophages or resident microglia) were able to stimulate LCMV-specific CTL to produce IFNγ and TNFα production, and immunological synapses between CTL and APCs were observed in situ (84). These data suggest that CNS-infiltrating DCs might serve as important accessory cells to support the activities of anti-viral CTL operating within the persistently infected CNS. These DCs could function either by promoting CTL survival/division or by fostering the production of antiviral cytokines to purge adjacently infected neurons. Thus, it is conceivable that direct engagement of neurons by CTL is not an absolute requirement for clearance.
5.2. Behavioral abnormalities in carrier mice
Although carrier mice do not display signs of neuronal loss during infection, they do exhibit signs of neuronal dysfunction manifested as abnormalities in behavior and learning (130–132). Specifically, persistently infected mice show reductions in spatial-temporal learning, assessed in a Y maze discriminated avoidance learning task, and decreases in explorative behavior in the context of novel environments (130–132). Expression of viral antigen is found in neurons localized in numerous regions of the brain including the neocortex, limbic system, hypothalamus, brain stem, thalamus, basal ganglia and the hippocampus - a region that is implicated in the development of memory and learning (104, 106). Recently, it was shown that persistent viral infection is associated with alterations in the host gene profile of the brain (132). Most of the genetic perturbations were downstream of type 1 interferon (132) - a cytokine known to be systemically elevated in LCMV carrier mice (133, 134). Because the carrier state is maintained in the absence of any major inflammation or necrosis in the brain parenchyma, it has been theorized that alterations in the expression of neuronal proteins are a direct result of the LCMV infection itself. Neurochemical abnormalities, manifested as hypersensitivity to cholinergic antagonists during learning and motor tests, and decreased choline acetyltransferase in neuroblastoma cells have been linked to LCMV infection (130, 131, 135). These data suggest that dysfunctional cholinergic responses might be responsible for the neurobehavioral defects observed in the carrier mice. In addition to neurochemical alterations, viral persistence was also associated with decreased neuronal plasticity. In vitro studies showed that LCMV prevented nerve growth factor (NGF) induced upregulation of growth-associated protein-43 (GAP-43) in PC12 cells (136). Importantly, these data were confirmed in vivo, as decreased expression of GAP-43 was observed in the hippocampus of LCMV carrier mice (136). Because GAP-43 has been implicated in neuronal plasticity (137), decreased expression in the hippocampus of carrier mice might also contribute to the virus-induced defects in learning capacity. These studies clearly indicate that LCMV has the capacity to significantly alter neuronal function in the absence of actual cytolysis.
5.3. LCMV and growth hormone deficiency
Neonatal infection of C3H/St mice results in the development of a growth hormone deficiency syndrome. This is characterized by growth retardation manifested as a decrease in body length and weight within weeks of birth (138). Disease induction is dependent on the strain of mouse and virus used. C3H/St mice were susceptible to disease, whereas both BALB/c and SWR/J mice were resistant (139, 140). The ability of LCMV variants to differentially infect growth hormone-producing cells localized in the anterior pituitary gland directly correlated with growth hormone deficiency during persistence (138, 141). LCMV Armstrong but not LCMV WE or Traub was able to induce disease (139). Growth hormone deficiency occurred in the absence of necrosis or inflammation in the pituitary gland, suggesting that LCMV altered cellular function rather than induce cell death (142). In support of this statement, LCMV was subsequently shown to selectively disrupt growth hormone production by interfering with the growth hormone transactivator factor GHF1 (Pit1) that binds to the growth hormone promoter (143). This interference resulted in decreased production of growth hormone mRNA and subsequent development of the growth hormone deficiency syndrome (144, 145). Therefore, the ability of LCMV to cause selective cellular dysfunction without lysis is not restricted to neurons and can be extended to other persistently infected cell populations.
6. LCMV AND ADULT INFECTION
In general, chronic LCMV infection in adult mice is favored under conditions where there is rapid viral replication and/or an impaired immune response. For example, mechanisms that allow for increased viral titers, including viral interferon insensitivity, higher replicative rates, increased inoculation dosage and suppression of the immune system can facilitate the transition from an acute to more chronic state of persistence (26, 146–149). Therefore, under permissive conditions LCMV can establish a state of persistence in adult mice.
6.1. LCMV clone 13 and chronic infection
The prototypic LCMV strain associated with chronic infection of immunocompetent adult mice is referred to as LCMV clone 13. This variant was isolated from mice persistently infected with Armstrong from birth (150). Genetic analysis of LCMV clone 13 compared to the parental Armstrong virus revealed only 2 genomic changes that led to alterations in the resultant amino acid sequence: a change from K to Q at position 1079 of the L segment encoding the polymerase and a change from F to L at position 260 of the S segment encoding the viral glycoprotein (28, 29, 151). Notably, the glycoprotein (F to L at position 260) mutation has been associated with both viral persistence and decreased CTL activity in addition to conferring an increased affinity for the LCMV receptor, α-dystroglycan (α-DG) (29, 152). Initial studies in carrier mice infected with LCMV Armstrong revealed that the F to L mutation at position 260 of the glycoprotein, which is observed in the clone 13 variant, appeared to occur preferentially in macrophages and peripheral tissues including the spleen, liver, lymph node, thymus, heart and lung within 2 months post infection (151, 153, 154). However, in the brain conversion from LCMV Armstrong to clone 13 occurred at lower levels (151), and in general, LCMV Armstrong was observed to out compete clone 13 for infection of neurons in vitro and brain tissue upon co-infection in vivo (154, 155). These data suggested that LCMV clone 13 was a more viscerotropic variant, whereas Armstrong was a more neurotropic variant.
After inoculation of adult mice with LCMV clone 13, virus is distributed throughout the body in areas including the serum, blood, spleen, liver, kidney and brain (28, 153, 154, 156, 157). In contrast to LCMV Armstrong infection, which is cleared within 2 weeks, LCMV clone 13 challenge results in persistent infection and diminished CTL responses (29, 152, 158, 159) in adult mice. Whereas clone 13 infection of neonates results in suppression of LCMV-specific responses, challenge of adult mice generates a generalized immune suppression that also diminishes cytotoxic responses to other viruses (158). This global inhibition is partially attributed to alterations in splenic architecture that occurs after clone 13 infection of adult but not neonatal mice (158). Further studies in adult mice demonstrated that enhanced viral infiltration into the white pulp of the spleen was associated with increased binding to α-DG (152) and that clone 13 also more effectively targeted fibroblastic reticular cells resulting in decreased conduit function of the spleen (160). The disruption in splenic function (158, 160), physical destruction of dendritic cells (161), and inhibition of dendritic cell differentiation and maturation (162, 163) are likely contributors to the immunosuppression observed during clone 13 infection. More recently, it has also been revealed that inhibitory molecule such as PD-1 (164) and IL-10 (165) are upregulated early following clone 13 infection and also contribute to the immunosuppressive milieu. In terms of CTL responses, clone 13 infection was shown to disrupt their dominance hierarchies and tissue distribution (157), induce deletion of immunodominant populations (159), and promote profound functional exhaustion, evidenced by a diminished capacity to produce IFNγ, TNFα, and IL-2 (157). Similar functional disruptions were also noted in the CD4 compartment (166, 167). However, despite this suppression in virus-specific immunity, systemic clone 13 clearance is eventually achieved in chronically infected mice.
6.2. LCMV clone 13 infection and clearance from the CNS
Intravenous inoculation of adult mice with LCMV clone 13 results in state of protracted viral clearance. In contrast to Armstrong, which is cleared acutely in 8–10 days, clone 13 distributes systemically and is not purged from most peripheral tissues (e.g. serum, spleen, lymph nodes, lung, and liver) until 40–80 days post-infection (156, 157). The time period preceding viral clearance is associated with profound immune exhaustion, during which LCMV-specific T cells show a diminished capacity to produce anti-viral cytokines and engage in cytolytic activities (157). Within the CNS clearance of LCMV clone 13 is delayed considerably and does not occur until day 200 post infection (156).
To gain novel insights into clone 13 infection of the CNS, our group recently studied clone 13 tropism and the responding CNS immune response to this variant following intravenous inoculation (156). At early time points post-infection, viral antigen was detected in the choroid plexus, meninges, ependymal cells and in regions surrounding the blood vasculature of the brain (Figure 3). Within a month, viral entry and dissemination throughout the brain parenchyma was achieved through astrocyte infection. Because astrocytes are a critical element of the blood brain barrier (BBB) (168), it is possible that the virus gains access from the blood stream into these cells through astrocytic foot processes that comprise the second layer of the BBB. Additionally, astrocytic expression of the LCMV receptor, α-dystroglycan, may increase the ability of clone 13 to readily gain access to this cell type (169, 170). Over time the virus moved throughout the astrocytic network, and complete inundation of the brain parenchyma was observed between days 30–60 post infection. In contrast, peak viral loads were achieved in the periphery by day 10 post infection. By 150 days post-infection, a remarkable shift in the distribution of parenchymal virus was observed. Viral antigen in astrocytes was reduced to an almost undetectable level and olfactory bulb neurons emerged as the last bastion supporting viral persistence. When LCMV clones were isolated from the brains of clone 13 infected mice at early and late stages of persistence, it was revealed that the leucine at position 260 of the viral glycoprotein, indicative of the clone 13 phenotype, was maintained throughout infection. Therefore, reversion or selection of a more neurotropic LCMV Armstrong virus was not a likely cause for the altered tropism observed between these two stages of chronic infection.
Figure 3.
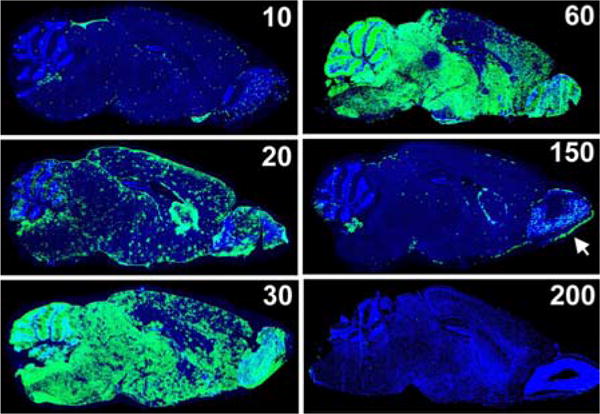
Localization of LCMV in the brain of clone 13 infected mice. The brains of C57BL/6 mice infected intravenously with LCMV clone 13 were harvested at time points spanning 10 to 200 days post infection. The distribution of LCMV (green) is shown on representative sagittal brain sections, and cell nuclei are shown in blue. On day 10 post-infection clone 13 is detected in the meninges, choroid plexus, ependyma as well as throughout the parenchyma in areas around blood vessels (note the punctuate pattern observed in the parenchyma). From days 20–60 post-infection, the virus inundates the brain parenchyma, spreading almost exclusively through the astrocytic network. Concurrent with reanimation of the peripheral T cell response clone 13 is purged from parenchymal astrocytes, and by day 150 the virus is found in its last refuge - olfactory bulb neurons as well as the meninges, ependyma and choroid plexus. Clone 13 is finally cleared from the brain by day 200 post infection.
Reflective of what is observed in the periphery, LCMV specific CD8+ T cells that reside in the CNS undergo a state of functional impairment after clone 13 infection. When analyzed ex vivo CNS-derived LCMV-specific CD8+ T cells showed signs of decreased IFNγ, TNFα, and IL-2 production by day 8 post-infection, but the functional impairment of CD8+ T cells was far more severe in peripheral tissues (e.g., spleen and liver). After day 60 post-infection, just prior to the initiation of viral clearance from astrocytes, LCMV-specific CD8+ T cells underwent a reanimation phase in both the periphery and CNS characterized by a resurgence of full cytokine production capacity. At this time increased leukocyte numbers were observed in the CNS, and diversification in the composition of the infiltrate was observed. In particular, a significant increase in the number of virus-specific CD4+ T cells and B cells was observed. These events coincided with the eventual clearance of clone 13 from the CNS and were initiated at a time when virus was purged almost entirely from peripheral tissues. These findings suggest that diversification of the CNS immune infiltrate is an important step in achieving viral clearance from this specialized compartment and that the timing of CNS clearance might be delayed by the peripheral “antigen sink” which spreads out the resources of the adaptive immune response (99). Once the peripheral viral burden is removed, the adaptive immune system can redirect all of its resources to CNS. Studies are underway to elucidate the exact components of CNS immune infiltrate responsible for purging clone 13 during this reanimation phase.
6.3. Chronic LCMV infection in immunocompromised mice
Chronic infection can also be established in adult immunocompromised mice after inoculation with LCMV Armstrong. Although intracranial injection of immunocompetent mice with LCMV normally results in acute lethal meningitis, a deficiency in the CD8+ T cell response enables the virus to establish persistence. For example, in CD3δ deficient mice, a delayed LCMV-specific CD8+ T cell response provides the virus enough time to switch tropism into neurons (51). The targeting of the virus to neurons changes the accessibility of LCMV peptide presentation because of low neuronal MHC I expression, and therefore the virus remains “hidden” even after a functional immune response is generated. Similarly, mice that mount a negligible LCMV-specific response due to forced expression of a CD8+ ovalbumin specific TCR (OT-I mice) (60, 61) also survive an intracranial challenge with LCMV (43). Initially, the anatomical viral distribution is confined to the meninges, ependyma and choroid plexus, as observed in wild type mice; however, by day 55 the localization changes and LCMV persists in neurons and astrocytes in addition to the meninges and ependyma. Importantly, transfer of as few as 1000 naive DbGP33–41 CD8+ T cells into OT-I mice prior to infection was sufficient to reconstitute the acute meningitis (43). These studies reflect two important points for LCMV viral persistence and disease. First, the immune response, particularly LCMV-specific CD8+ T cells, is necessary for clearance/immunopathology and thus a deficiency in this compartment can prevent disease and lead to uncontrolled infection. Second, a single monospecific CD8+ T cell population operating in sea of bystander T cells of an irrelevant specificity is sufficient drive an entire disease process (e.g., meningitis) with normal kinetics. These data suggest that bystander T cells are of minimal importance in mediating acute CNS diseases initiated by virus infection.
7. CONCLUSION
LCMV infection provides a powerful model system to study viral-immune cell interactions in the CNS because of the great diversity in observed outcomes. These outcomes range from severe immunopathology to chronic infection to viral clearance. Because LCMV is noncytolytic, the outcome of infection is largely influenced by two factors: 1.) viral tropism within the CNS, and 2.) the functionality of the adaptive immune system. In some instances, LCMV alone is sufficient to alter the function of the cells it infects, which can result in modifications in behavior and hormone regulation. However, the integrity of the CNS as well as the state of LCMV persistence within this compartment is usually dictated by the immune response that is mobilized to fight the infection. The most dramatic example of this is the fatal choriomeningitis that develops in response to intracerebral inoculation of immunocompetent mice with LCMV.
CD8+ T cells have emerged as key mediators of both pathology and clearance following LCMV infection. While acute lethal meningitis is caused by a robust LCMV-specific CD8+ T cell response, neonatal and adult persistence develops when CD8+ T cells are suppressed. This is observed when an immunosuppressive variant of LCMV is used (e.g. clone 13) or when immunologically compromised (or immature) mice are infected with LCMV. Once LCMV establishes persistence, it is possible to therapeutically purge the pathogen by supplementing the carrier mouse with virus-specific memory T cells. This remarkable therapeutic approach is also dependent on the presence of a functional CD8+ T cells, further emphasizing the importance of this cell population. Moreover, factors that aid in the induction and/or maintenance of virus-specific CTL, such as MHC and CD4+ T cell responses, also have a profound impact on the final outcome after infection. For example, adoptive immunotherapy is not successful unless CD8+ and CD4+ T cells cooperate to achieve clearance. Given the importance of CTL in the LCMV system as well as other viral models, we propose that it is important to gain additional insights into the behavior and function of these cells within the CNS. It is also of great importance to better understand how to therapeutically modulate CTL function, especially in patients that have an established CNS infection.
While peripheral responses have been extensively studied in the LCMV model, relatively little is known about viral and leukocyte behavior within the LCMV-infected CNS. There is a paucity of knowledge as to how and why LCMV tropism within the CNS shifts after infection, which effector mechanisms are critical for T cell clearance versus pathology, which molecules direct T cell trafficking into the LCMV-infected CNS, and whether LCMV-specific T cell priming (or restimulation) occurs within the CNS. The degree to which neurons and glia regulate virus-specific immune cells and the impact of immune-target interactions on the function the LCMV-infected CNS cells also remains to be elucidated. Addressing these unanswered questions in the LCMV model will provide important, fundamental information pertaining to CNS virus-immune interactions that can hopefully be extrapolated to states of viral infection in humans.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant AI070967-01 (to D.B. McGavern) and a grant from the Burroughs Wellcome Fund (to D.B McGavern). S.S. Kang is supported by NIH training grant NS041219-06.
References
- 1.Lledo L, Gegundez MI, Saz JV, Bahamontes N, Beltran M. Lymphocytic choriomeningitis virus infection in a province of Spain: analysis of sera from the general population and wild rodents. J Med Virol. 2003;70:273–5. doi: 10.1002/jmv.10389. [DOI] [PubMed] [Google Scholar]
- 2.Interim guidance for minimizing risk for human lymphocytic choriomeningitis virus infection associated with rodents. MMWR Morb Mortal Wkly Rep. 2005;54:747–9. [PubMed] [Google Scholar]
- 3.Rousseau MC, Saron MF, Brouqui P, Bourgeade A. Lymphocytic choriomeningitis virus in southern France: four case reports and a review of the literature. Eur J Epidemiol. 1997;13:817–23. doi: 10.1023/a:1007434521082. [DOI] [PubMed] [Google Scholar]
- 4.Rotbart HA. Viral meningitis. Semin Neurol. 2000;20:277–92. doi: 10.1055/s-2000-9427. [DOI] [PubMed] [Google Scholar]
- 5.Jamieson DJ, Kourtis AP, Bell M, Rasmussen SA. Lymphocytic choriomeningitis virus: an emerging obstetric pathogen? Am J Obstet Gynecol. 2006;194:1532–6. doi: 10.1016/j.ajog.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 6.Charrel RN, Retornaz K, Emonet S, Noel G, Chaumoitre K, Minodier P, Girard N, Garnier JM, de Lamballerie X. Acquired hydrocephalus caused by a variant lymphocytic choriomeningitis virus. Arch Intern Med. 2006;166:2044–6. doi: 10.1001/archinte.166.18.2044. [DOI] [PubMed] [Google Scholar]
- 7.Barton LL, Mets MB, Beauchamp CL. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am J Obstet Gynecol. 2002;187:1715–6. doi: 10.1067/mob.2002.126297. [DOI] [PubMed] [Google Scholar]
- 8.Wright R, Johnson D, Neumann M, Ksiazek TG, Rollin P, Keech RV, Bonthius DJ, Hitchon P, Grose CF, Bell WE, Bale JF., Jr Congenital lymphocytic choriomeningitis virus syndrome: a disease that mimics congenital toxoplasmosis or Cytomegalovirus infection. Pediatrics. 1997;100:E9. doi: 10.1542/peds.100.1.e9. [DOI] [PubMed] [Google Scholar]
- 9.Mets MB, Barton LL, Khan AS, Ksiazek TG. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am J Ophthalmol. 2000;130:209–15. doi: 10.1016/s0002-9394(00)00570-5. [DOI] [PubMed] [Google Scholar]
- 10.Barton LL, Hyndman NJ. Lymphocytic choriomeningitis virus: reemerging central nervous system pathogen. Pediatrics. 2000;105:E35. doi: 10.1542/peds.105.3.e35. [DOI] [PubMed] [Google Scholar]
- 11.Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, Comer JA, Guarner J, Paddock CD, DeMeo DL, Shieh WJ, Erickson BR, Bandy U, DeMaria A, Jr, Davis JP, Delmonico FL, Pavlin B, Likos A, Vincent MJ, Sealy TK, Goldsmith CS, Jernigan DB, Rollin PE, Packard MM, Patel M, Rowland C, Helfand RF, Nichol ST, Fishman JA, Ksiazek T, Zaki SR. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N Engl J Med. 2006;354:2235–49. doi: 10.1056/NEJMoa053240. [DOI] [PubMed] [Google Scholar]
- 12.Schanen A, Gallou G, Hincky JM, Saron MF. A rash, circulating anticoagulant, then meningitis. Lancet. 1998;351:1856. doi: 10.1016/S0140-6736(98)02207-7. [DOI] [PubMed] [Google Scholar]
- 13.Roebroek RM, Postma BH, Dijkstra UJ. Aseptic meningitis caused by the lymphocytic choriomeningitis virus. Clin Neurol Neurosurg. 1994;96:178–80. doi: 10.1016/0303-8467(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 14.Emonet S, Lemasson JJ, Gonzalez JP, de Lamballerie X, Charrel RN. Phylogeny and evolution of old world arenaviruses. Virology. 2006;350:251–7. doi: 10.1016/j.virol.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 15.Charrel RN, de Lamballerie X. Arenaviruses other than Lassa virus. Antiviral Res. 2003;57:89–100. doi: 10.1016/s0166-3542(02)00202-4. [DOI] [PubMed] [Google Scholar]
- 16.Riviere Y, Ahmed R, Southern PJ, Buchmeier MJ, Dutko FJ, Oldstone MB. The S RNA segment of lymphocytic choriomeningitis virus codes for the nucleoprotein and glycoproteins 1 and 2. J Virol. 1985;53:966–8. doi: 10.1128/jvi.53.3.966-968.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Southern PJ, Singh MK, Riviere Y, Jacoby DR, Buchmeier MJ, Oldstone MB. Molecular characterization of the genomic S RNA segment from lymphocytic choriomeningitis virus. Virology. 1987;157:145–55. doi: 10.1016/0042-6822(87)90323-0. [DOI] [PubMed] [Google Scholar]
- 18.Salvato M, Shimomaye E, Oldstone MB. The primary structure of the lymphocytic choriomeningitis virus L gene encodes a putative RNA polymerase. Virology. 1989;169:377–84. doi: 10.1016/0042-6822(89)90163-3. [DOI] [PubMed] [Google Scholar]
- 19.Salvato MS, Shimomaye EM. The completed sequence of lymphocytic choriomeningitis virus reveals a unique RNA structure and a gene for a zinc finger protein. Virology. 1989;173:1–10. doi: 10.1016/0042-6822(89)90216-x. [DOI] [PubMed] [Google Scholar]
- 20.Singh MK, Fuller-Pace FV, Buchmeier MJ, Southern PJ. Analysis of the genomic L RNA segment from lymphocytic choriomeningitis virus. Virology. 1987;161:448–56. doi: 10.1016/0042-6822(87)90138-3. [DOI] [PubMed] [Google Scholar]
- 21.Romanowski V, Matsuura Y, Bishop DH. Complete sequence of the S RNA of lymphocytic choriomeningitis virus (WE strain) compared to that of Pichinde arenavirus. Virus Res. 1985;3:101–14. doi: 10.1016/0168-1702(85)90001-2. [DOI] [PubMed] [Google Scholar]
- 22.Wright KE, Spiro RC, Burns JW, Buchmeier MJ. Post-translational processing of the glycoproteins of lymphocytic choriomeningitis virus. Virology. 1990;177:175–83. doi: 10.1016/0042-6822(90)90471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248:701–702. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
- 24.van der Most RG, Sette A, Oseroff C, Alexander J, Murali-Krishna K, Lau LL, Southwood S, Sidney J, Chesnut RW, Matloubian M, Ahmed R. Analysis of cytotoxic T cell responses to dominant and subdominant epitopes during acute and chronic lymphocytic choriomeningitis virus infection. J Immunol. 1996;157:5543–54. [PubMed] [Google Scholar]
- 25.Weidt G, Utermohlen O, Heukeshoven J, Lehmann-Grube F, Deppert W. Relationship among immunodominance of single CD8+ T cell epitopes, virus load, and kinetics of primary antiviral CTL response. J Immunol. 1998;160:2923–31. [PubMed] [Google Scholar]
- 26.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–61. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 27.Pircher H, Rohrer UH, Moskophidis D, Zinkernagel RM, Hengartner H. Lower receptor avidity required for thymic clonal deletion than for effector T-cell function. Nature. 1991;351:482–485. doi: 10.1038/351482a0. [DOI] [PubMed] [Google Scholar]
- 28.Matloubian M, Somasundaram T, Kolhekar SR, Selvakumar R, Ahmed R. Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J Exp Med. 1990;172:1043–8. doi: 10.1084/jem.172.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvato M, Borrow P, Shimomaye E, Oldstone MB. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J Virol. 1991;65:1863–9. doi: 10.1128/jvi.65.4.1863-1869.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchmeier MJ, Welsh RM, Dutko FJ, Oldstone MB. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv Immunol. 1980;30:275–331. doi: 10.1016/s0065-2776(08)60197-2. [DOI] [PubMed] [Google Scholar]
- 31.Cole GA, Gilden DH, Monjan AA, Nathanson N. Lymphocytic choriomeningitis virus: pathogenesis of acute central nervous system disease. Fed Proc. 1971;30:1831–41. [PubMed] [Google Scholar]
- 32.Borrow P, Oldstone MBA. Lymphocytic choriomeningitis virus. Lippincott-Raven Publishers; Philadelphia: 1997. [Google Scholar]
- 33.McGavern DB, Homann D, Oldstone MB. T cells in the central nervous system: the delicate balance between viral clearance and disease. J Infect Dis. 2002;186:S145–51. doi: 10.1086/344264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gairin JE, Mazarguil H, Hudrisier D, Oldstone MB. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J Virol. 1995;69:2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallimore A, Hombach J, Dumrese T, Rammensee HG, Zinkernagel RM, Hengartner H. A protective cytotoxic T cell response to a subdominant epitope is influenced by the stability of the MHC class I/peptide complex and the overall spectrum of viral peptides generated within infected cells. Eur J Immunol. 1998;28:3301–3311. doi: 10.1002/(SICI)1521-4141(199810)28:10<3301::AID-IMMU3301>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 36.van der Most RG, Murali-Krishna K, Whitton JL, Oseroff C, Alexander J, Southwood S, Sidney J, Chesnut RW, Sette A, Ahmed R. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology. 1998;240:158–167. doi: 10.1006/viro.1997.8934. [DOI] [PubMed] [Google Scholar]
- 37.Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol. 1998;28:390–400. doi: 10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 38.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–61. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 39.Allan JE, Dixon JE, Doherty PC. Nature of the inflammatory process in the central nervous system of mice infected with lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 1987;134:131–143. doi: 10.1007/978-3-642-71726-0_6. [DOI] [PubMed] [Google Scholar]
- 40.Doherty PC, Zinkernagel RM. T-cell-mediated immunopathology in viral infections. Transplant Rev. 1974;19:89–120. doi: 10.1111/j.1600-065x.1974.tb00129.x. [DOI] [PubMed] [Google Scholar]
- 41.Schwendemann G, Lohler J, Lehmann-Grube F. Evidence for cytotoxic T-lymphocyte-target cell interaction in brains of mice infected intracerebrally with lymphocytic choriomeningitis virus. Acta Neuropathol. 1983;61:183–95. doi: 10.1007/BF00691984. [DOI] [PubMed] [Google Scholar]
- 42.Mims CA. Intracerebral injections and the growth of viruses in the mouse brain. Br J Exp Pathol. 1960;41:52–59. [PMC free article] [PubMed] [Google Scholar]
- 43.McGavern DB, Truong P. Rebuilding an immune-mediated central nervous system disease: weighing the pathogenicity of antigen-specific versus bystander T cells. J Immunol. 2004;173:4779–90. doi: 10.4049/jimmunol.173.8.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christensen JE, Nansen A, Moos T, Lu B, Gerard C, Christensen JP, Thomsen AR. Efficient T-cell surveillance of the CNS requires expression of the CXC chemokine receptor 3. J Neurosci. 2004;24:4849–58. doi: 10.1523/JNEUROSCI.0123-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowe WP, Black PH, Levey RH. Protective effect of neonatal thymectomy on mouse LCM infection. Proc Soc Exp Biol. 1963;114:248–251. doi: 10.3181/00379727-114-28643. [DOI] [PubMed] [Google Scholar]
- 46.Lynch F, Doherty PC, Ceredig R. Phenotypic and functional analysis of the cellular response in regional lymphoid tissue during an acute virus infection. J Immunol. 1989;142:3592–8. [PubMed] [Google Scholar]
- 47.Zinkernagel RM, Doherty PC. Cytotoxic thymus-derived lymphocytes in cerebrospinal fluid of mice with lymphocytic choriomeningitis. J Exp Med. 1973;138:1266–9. doi: 10.1084/jem.138.5.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceredig R, Allan JE, Tabi Z, Lynch F, Doherty PC. Phenotypic analysis of the inflammatory exudate in murine lymphocytic choriomeningitis. J Exp Med. 1987;165:1539–51. doi: 10.1084/jem.165.6.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leist TP, Cobbold SP, Waldmann H, Aguet M, Zinkernagel RM. Functional analysis of T lymphocyte subsets in antiviral host defense. J Immunol. 1987;138:2278–81. [PubMed] [Google Scholar]
- 50.Fung-Leung WP, Kundig TM, Zinkernagel RM, Mak TW. Immune response against lymphocytic choriomeningitis virus infection in mice without CD8 expression. J Exp Med. 1991;174:1425–1429. doi: 10.1084/jem.174.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kappes DJ, Lawrence DM, Vaughn MM, Dave VP, Belman AR, Rall GF. Protection of CD3 delta knockout mice from lymphocytic choriomeningitis virus-induced immunopathology: implications for viral neuroinvasion. Virology. 2000;269:248–56. doi: 10.1006/viro.2000.0224. [DOI] [PubMed] [Google Scholar]
- 52.Quinn DG, Zajac AJ, Frelinger JA, Muller D. Transfer of lymphocytic choriomeningitis disease in beta 2-microglobulin-deficient mice by CD4+ T cells. Int Immunol. 1993;5:1193–8. doi: 10.1093/intimm/5.10.1193. [DOI] [PubMed] [Google Scholar]
- 53.Oldstone MB, von Herrath M, Lewicki H, Hudrisier D, Whitton JL, Gairin JE. Use of a high-affinity peptide that aborts MHC-restricted cytotoxic T lymphocyte activity against multiple viruses in vitro and virus-induced immunopathologic disease in vivo. Virology. 1999;256:246–57. doi: 10.1006/viro.1998.9593. [DOI] [PubMed] [Google Scholar]
- 54.Baenziger J, Hengartner H, Zinkernagel RM, Cole GA. Induction or prevention of immunopathological disease by cloned cytotoxic T cell lines specific for lymphocytic choriomeningitis virus. Eur J Immunol. 1986;16:387–93. doi: 10.1002/eji.1830160413. [DOI] [PubMed] [Google Scholar]
- 55.Dixon JE, Allan JE, Doherty PC. The acute inflammatory process in murine lymphocytic choriomeningitis is dependent on Lyt-2+ immune T cells. Cell Immunol. 1987;107:8–14. doi: 10.1016/0008-8749(87)90260-7. [DOI] [PubMed] [Google Scholar]
- 56.Doherty PC, Allan JE, Ceredig R. Contributions of host and donor T cells to the inflammatory process in murine lymphocytic choriomeningitis. Cell Immunol. 1988;116:475–81. doi: 10.1016/0008-8749(88)90246-8. [DOI] [PubMed] [Google Scholar]
- 57.Johnson ED, Monjan AA, Morse HC., 3rd Lack of B-cell participation in acute lymphocyte choriomeningitis disease of the central nervous system. Cell Immunol. 1978;36:143–50. doi: 10.1016/0008-8749(78)90257-5. [DOI] [PubMed] [Google Scholar]
- 58.Allan JE, Doherty PC. Natural killer cells contribute to inflammation but do not appear to be essential for the induction of clinical lymphocytic choriomeningitis. Scand J Immunol. 1986;24:153–62. doi: 10.1111/j.1365-3083.1986.tb02081.x. [DOI] [PubMed] [Google Scholar]
- 59.Allan JE, Doherty PC. Immune T cells can protect or induce fatal neurological disease in murine lymphocytic choriomeningitis. Cell Immunol. 1985;90:401–7. doi: 10.1016/0008-8749(85)90204-7. [DOI] [PubMed] [Google Scholar]
- 60.Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78:110–7. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- 61.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 62.Zinkernagel RM, Pfau CJ, Hengartner H, Althage A. Susceptibility to murine lymphocytic choriomeningitis maps to class I MHC genes–a model for MHC/disease associations. Nature. 1985;316:814–7. doi: 10.1038/316814a0. [DOI] [PubMed] [Google Scholar]
- 63.Doherty PC, Zinkernagel RM. H-2 compatibility is required for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. J Exp Med. 1975;141:502–7. doi: 10.1084/jem.141.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doherty PC, Allan JE. Role of the major histocompatibility complex in targeting effector T cells into a site of virus infection. Eur J Immunol. 1986;16:1237–42. doi: 10.1002/eji.1830161009. [DOI] [PubMed] [Google Scholar]
- 65.Gairin JE, Joly E, Oldstone MB. Persistent infection with lymphocytic choriomeningitis virus enhances expression of MHC class I glycoprotein on cultured mouse brain endothelial cells. J Immunol. 1991;146:3953–7. [PubMed] [Google Scholar]
- 66.Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007;204:2023–30. doi: 10.1084/jem.20070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol Dis. 2007;26:36–46. doi: 10.1016/j.nbd.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munoz-Fernandez MA, Fresno M. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog Neurobiol. 1998;56:307–40. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 69.Andersen IH, Marker O, Thomsen AR. Breakdown of blood-brain barrier function in the murine lymphocytic choriomeningitis virus infection mediated by virus-specific CD8+ T cells. J Neuroimmunol. 1991;31:155–63. doi: 10.1016/0165-5728(91)90021-x. [DOI] [PubMed] [Google Scholar]
- 70.Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm. 2006;113:477–85. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- 71.Matsumoto M, Atarashi K, Umemoto E, Furukawa Y, Shigeta A, Miyasaka M, Hirata T. CD43 functions as a ligand for E-Selectin on activated T cells. J Immunol. 2005;175:8042–50. doi: 10.4049/jimmunol.175.12.8042. [DOI] [PubMed] [Google Scholar]
- 72.Onami TM, Harrington LE, Williams MA, Galvan M, Larsen CP, Pearson TC, Manjunath N, Baum LG, Pearce BD, Ahmed R. Dynamic regulation of T cell immunity by CD43. J Immunol. 2002;168:6022–31. doi: 10.4049/jimmunol.168.12.6022. [DOI] [PubMed] [Google Scholar]
- 73.Bartholdy C, Marker O, Thomsen AR. Migration of activated CD8 (+) T lymphocytes to sites of viral infection does not require endothelial selectins. Blood. 2000;95:1362–9. [PubMed] [Google Scholar]
- 74.Asensio VC, Campbell IL. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol. 1997;71:7832–40. doi: 10.1128/jvi.71.10.7832-7840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nansen A, Marker O, Bartholdy C, Thomsen AR. CCR2+ and CCR5+ CD8+ T cells increase during viral infection and migrate to sites of infection. Eur J Immunol. 2000;30:1797–806. doi: 10.1002/1521-4141(200007)30:7<1797::AID-IMMU1797>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 76.de Lemos C, Christensen JE, Nansen A, Moos T, Lu B, Gerard C, Christensen JP, Thomsen AR. Opposing effects of CXCR3 and CCR5 deficiency on CD8+ T cell-mediated inflammation in the central nervous system of virus-infected mice. J Immunol. 2005;175:1767–75. doi: 10.4049/jimmunol.175.3.1767. [DOI] [PubMed] [Google Scholar]
- 77.Nansen A, Christensen JP, Andreasen SO, Bartholdy C, Christensen JE, Thomsen AR. The role of CC chemokine receptor 5 in antiviral immunity. Blood. 2002;99:1237–45. doi: 10.1182/blood.v99.4.1237. [DOI] [PubMed] [Google Scholar]
- 78.Christensen JE, de Lemos C, Moos T, Christensen JP, Thomsen AR. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol. 2006;176:4235–43. doi: 10.4049/jimmunol.176.7.4235. [DOI] [PubMed] [Google Scholar]
- 79.Christensen JP, Andersson EC, Scheynius A, Marker O, Thomsen AR. Alpha 4 integrin directs virus-activated CD8+ T cells to sites of infection. Journal of Immunology. 1995;154:5293–301. [PubMed] [Google Scholar]
- 80.Andersson EC, Christensen JP, Scheynius A, Marker O, Thomsen AR. Lymphocytic choriomeningitis virus infection is associated with long-standing perturbation of LFA-1 expression on CD8+ T cells. Scand J Immunol. 1995;42:110–8. doi: 10.1111/j.1365-3083.1995.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 81.Andersson EC, Christensen JP, Marker O, Thomsen AR. Changes in cell adhesion molecule expression on T cells associated with systemic virus infection. J Immunol. 1994;152:1237–45. [PubMed] [Google Scholar]
- 82.Nielsen HV, Christensen JP, Andersson EC, Marker O, Thomsen AR. Expression of type 3 complement receptor on activated CD8+ T cells facilitates homing to inflammatory sites. J Immunol. 1994;153:2021–8. [PubMed] [Google Scholar]
- 83.Marker O, Scheynius A, Christensen JP, Thomsen AR. Virus-activated T cells regulate expression of adhesion molecules on endothelial cells in sites of infection. J Neuroimmunol. 1995;62:35–42. doi: 10.1016/0165-5728(95)00099-n. [DOI] [PubMed] [Google Scholar]
- 84.McGavern DB, Christen U, Oldstone MB. Molecular anatomy of antigen-specific CD8 (+) T cell engagement and synapse formation in vivo. Nat Immunol. 2002;3:918–25. doi: 10.1038/ni843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kyburz D, Speiser DE, Battegay M, Hengartner H, Zinkernagel RM. Lysis of infected cells in vivo by antiviral cytolytic T cells demonstrated by release of cell internal viral proteins. Eur J Immunol. 1993;23:1540–5. doi: 10.1002/eji.1830230722. [DOI] [PubMed] [Google Scholar]
- 86.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 87.Storm P, Bartholdy C, Sorensen MR, Christensen JP, Thomsen AR. Perforin-deficient CD8+ T cells mediate fatal lymphocytic choriomeningitis despite impaired cytokine production. J Virol. 2006;80:1222–30. doi: 10.1128/JVI.80.3.1222-1230.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walsh CM, Matloubian M, Liu CC, Ueda R, Kurahara CG, Christensen JL, Huang MT, Young JD, Ahmed R, Clark WR. Immune function in mice lacking the perforin gene. Proc Nat Acad Sci. 1994;91:10854–8. doi: 10.1073/pnas.91.23.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zajac AJ, Dye JM, Quinn DG. Control of lymphocytic choriomeningitis virus infection in granzyme B deficient mice. Virology. 2003;305:1–9. doi: 10.1006/viro.2002.1754. [DOI] [PubMed] [Google Scholar]
- 90.Campbell IL, Hobbs MV, Kemper P, Oldstone MB. Cerebral expression of multiple cytokine genes in mice with lymphocytic choriomeningitis. J Immunol. 1994;152:716–23. [PubMed] [Google Scholar]
- 91.Frei K, Leist TP, Meager A, Gallo P, Leppert D, Zinkernagel RM, Fontana A. Production of B cell stimulatory factor-2 and interferon gamma in the central nervous system during viral meningitis and encephalitis. Evaluation in a murine model infection and in patients. J Exp Med. 1988;168:449–53. doi: 10.1084/jem.168.1.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gessner A, Drjupin R, Lohler J, Lother H, Lehmann-Grube F. IFN-gamma production in tissues of mice during acute infection with lymphocytic choriomeningitis virus. J Immunol. 1990;144:3160–5. [PubMed] [Google Scholar]
- 93.Doherty PC, Allan JE, Clark IA. Tumor necrosis factor inhibits the development of viral meningitis or induces rapid death depending on the severity of inflammation at time of administration. J Immunol. 1989;142:3576–80. [PubMed] [Google Scholar]
- 94.Leist TP, Frei K, Kam-Hansen S, Zinkernagel RM, Fontana A. Tumor necrosis factor alpha in cerebrospinal fluid during bacterial, but not viral, meningitis. Evaluation in murine model infections and in patients. J Exp Med. 1988;167:1743–8. doi: 10.1084/jem.167.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Leist TP, Zinkernagel RM. Treatment with antitumor necrosis factor alpha does not influence the immune pathological response against lymphocytic choriomeningitis virus. Cytokine. 1990;2:29–34. doi: 10.1016/1043-4666(90)90040-z. [DOI] [PubMed] [Google Scholar]
- 96.Nansen A, Christensen JP, Ropke C, Marker O, Scheynius A, Thomsen AR. Role of interferon-gamma in the pathogenesis of LCMV-induced meningitis: unimpaired leucocyte recruitment, but deficient macrophage activation in interferon-gamma knock-out mice. J Neuroimmunol. 1998;86:202–12. doi: 10.1016/s0165-5728(98)00055-1. [DOI] [PubMed] [Google Scholar]
- 97.Leist TP, Eppler M, Zinkernagel RM. Enhanced virus replication and inhibition of lymphocytic choriomeningitis virus disease in anti-gamma interferon-treated mice. J Virol. 1989;63:2813–9. doi: 10.1128/jvi.63.6.2813-2819.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pfau CJ, Gresser I, Hunt KD. Lethal role of interferon in lymphocytic choriomeningitis virus-induced encephalitis. J Gen Virol. 1983;64(Pt 8):1827–30. doi: 10.1099/0022-1317-64-8-1827. [DOI] [PubMed] [Google Scholar]
- 99.Thomsen AR, Volkert M, Marker O. The timing of the immune response in relation to virus growth determines the outcome of the LCM infection. Acta Pathol Microbiol Scand [C] 1979;87C:47–54. [PubMed] [Google Scholar]
- 100.Traub E. Persistence of lymphocytic choriomeningitis virus in immune animals and its relation to immunity. J Exp Med. 1936;63:847–861. doi: 10.1084/jem.63.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.King CC, Jamieson BD, Reddy K, Bali N, Concepcion RJ, Ahmed R. Viral infection of the thymus. J Virol. 1992;66:3155–60. doi: 10.1128/jvi.66.5.3155-3160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hotchin JE, Cinits M. Lymphocytic choriomeningitis infection of mice as a model for the study of latent virus infection. Can J Microbiol. 1958;4:149–63. doi: 10.1139/m58-016. [DOI] [PubMed] [Google Scholar]
- 103.Volkert M, Larsen JH. Studies On Immunological Tolerance To Lcm Virus. 5. The Induction Of Tolerance To The Virus. Acta Pathol Microbiol Scand. 1965;63:161–71. doi: 10.1111/apm.1965.63.2.161. [DOI] [PubMed] [Google Scholar]
- 104.Fazakerley JK, Southern P, Bloom F, Buchmeier MJ. High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: relevance to arenavirus disease and mechanisms of viral persistence. J Gen Virol. 1991;72:1611–25. doi: 10.1099/0022-1317-72-7-1611. [DOI] [PubMed] [Google Scholar]
- 105.Southern PJ, Blount P, Oldstone MB. Analysis of persistent virus infections by in situ hybridization to whole-mouse sections. Nature. 1984;312:555–8. doi: 10.1038/312555a0. [DOI] [PubMed] [Google Scholar]
- 106.Rodriguez M, Buchmeier MJ, Oldstone MB, Lampert PW. Ultrastructural localization of viral antigens in the CNS of mice persistently infected with lymphocytic choriomeningitis virus (LCMV) Am J Pathol. 1983;110:95–100. [PMC free article] [PubMed] [Google Scholar]
- 107.Lauterbach H, Zuniga EI, Truong P, Oldstone MB, McGavern DB. Adoptive immunotherapy induces CNS dendritic cell recruitment and antigen presentation during clearance of a persistent viral infection. J Exp Med. 2006;203:1963–75. doi: 10.1084/jem.20060039. Epub 2006 Jul 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cihak J, Lehmann-Grube F. Immunological tolerance to lymphocytic choriomeningitis virus in neonatally infected virus carrier mice: evidence supporting a clonal inactivation mechanism. Immunology. 1978;34:265–75. [PMC free article] [PubMed] [Google Scholar]
- 109.Marker O, Volkert M. Studies on cell-mediated immunity to lymphocytic choriomeningitis virus in mice. J Exp Med. 1973;137:1511–25. doi: 10.1084/jem.137.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.von Herrath MG, Berger DP, Homann D, Tishon T, Sette A, Oldstone MB. Vaccination to treat persistent viral infection. Virology. 2000;268:411–9. doi: 10.1006/viro.1999.0130. [DOI] [PubMed] [Google Scholar]
- 111.Jamieson BD, Ahmed R. T-cell tolerance: exposure to virus in utero does not cause a permanent deletion of specific T cells. Proc Natl Acad Sci U S A. 1988;85:2265–8. doi: 10.1073/pnas.85.7.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Volkert M. Studies on immunological tolerance to LCM virus. Treatment of virus carrier mice by adoptive immunization. Acta Path et Microbiol Scand. 1962;56:305–310. [PubMed] [Google Scholar]
- 113.Volkert M. Studies On Immunological Tolerance To Lcm Virus. 2. Treatment Of Virus Carrier Mice By Adoptive Immunization. Acta Pathol Microbiol Scand. 1963;57:465–87. [PubMed] [Google Scholar]
- 114.Volkert M, Larsen JH. Studies On Immunological Tolerance To Lcm Virus. 6. Immunity Conferred On Tolerant Mice By Immune Serum And By Grafts Of Homologous Lymphoid Cells. Acta Pathol Microbiol Scand. 1965;63:172–80. doi: 10.1111/apm.1965.63.2.172. [DOI] [PubMed] [Google Scholar]
- 115.Volkert M, Bro-Jorgensen K, Marker O. Persistent LCM virus infection in the mouse. Immunity and tolerance. Bull World Health Organ. 1975;52:471–8. [PMC free article] [PubMed] [Google Scholar]
- 116.Oldstone MB, Blount P, Southern PJ, Lampert PW. Cytoimmunotherapy for persistent virus infection reveals a unique clearance pattern from the central nervous system. Nature. 1986;321:239–43. doi: 10.1038/321239a0. [DOI] [PubMed] [Google Scholar]
- 117.Jamieson BD, Butler LD, Ahmed R. Effective clearance of a persistent viral infection requires cooperation between virus-specific Lyt2+ T cells and nonspecific bone marrow-derived cells. J Virol. 1987;61:3930–7. doi: 10.1128/jvi.61.12.3930-3937.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tishon A, Eddleston M, de la Torre JC, Oldstone MB. Cytotoxic T lymphocytes cleanse viral gene products from individually infected neurons and lymphocytes in mice persistently infected with lymphocytic choriomeningitis virus. Virology. 1993;197:463–7. doi: 10.1006/viro.1993.1613. [DOI] [PubMed] [Google Scholar]
- 119.Planz O, Ehl S, Furrer E, Horvath E, Brundler MA, Hengartner H, Zinkernagel RM. A critical role for neutralizing-antibody-producing B cells, CD4 (+) T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. Proc Natl Acad Sci USA. 1997;94:6874–9. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ngo VN, Cornall RJ, Cyster JG. Splenic T zone development is B cell dependent. J Exp Med. 2001;194:1649–60. doi: 10.1084/jem.194.11.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol. 1998;72:9208–16. doi: 10.1128/jvi.72.11.9208-9216.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tishon A, Lewicki H, Rall G, Von Herrath M, Oldstone MB. An essential role for type 1 interferon-gamma in terminating persistent viral infection. Virology. 1995;212:244–50. doi: 10.1006/viro.1995.1477. [DOI] [PubMed] [Google Scholar]
- 123.Hunziker L, Klenerman P, Zinkernagel RM, Ehl S. Exhaustion of cytotoxic T cells during adoptive immunotherapy of virus carrier mice can be prevented by B cells or CD4+ T cells. Eur J Immunol. 2002;32:374–82. doi: 10.1002/1521-4141(200202)32:2<374::AID-IMMU374>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 124.Berger DP, Homann D, Oldstone MB. Defining parameters for successful immunocytotherapy of persistent viral infection. Virology. 2000;266:257–263. doi: 10.1006/viro.1999.0074. [DOI] [PubMed] [Google Scholar]
- 125.Hoffsten PE, Oldstone MB, Dixon FJ. Immunopathology of adoptive immunization in mice chronically infected with lymphocytic choriomeningitis virus. Clin Immunol Immunopathol. 1977;7:44–52. doi: 10.1016/0090-1229(77)90028-9. [DOI] [PubMed] [Google Scholar]
- 126.Ahmed R, Jamieson BD, Porter DD. Immune therapy of a persistent and disseminated viral infection. J Virol. 1987;61:3920–9. doi: 10.1128/jvi.61.12.3920-3929.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Joly E, Mucke L, Oldstone MB. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 1991;253:1283–5. doi: 10.1126/science.1891717. [DOI] [PubMed] [Google Scholar]
- 128.Joly E, Oldstone MB. Neuronal cells are deficient in loading peptides onto MHC class I molecules. Neuron. 1992;8:1185–90. doi: 10.1016/0896-6273(92)90138-4. [DOI] [PubMed] [Google Scholar]
- 129.Rall GF, Mucke L, Oldstone MB. Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J Exp Med. 1995;182:1201–12. doi: 10.1084/jem.182.5.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gold LH, Brot MD, Polis I, Schroeder R, Tishon A, Torre J, Oldstone MBA, Koob GF. Behavioral effects of persistent lymphocytic choriomeningitis virus infection in mice. Behav Neural Biol. 1994;62:100–109. doi: 10.1016/s0163-1047(05)80031-7. [DOI] [PubMed] [Google Scholar]
- 131.Hotchin J, Seegal R. Virus-induced behavioral alteration of mice. Science. 1977;196:671–4. doi: 10.1126/science.854742. [DOI] [PubMed] [Google Scholar]
- 132.Kunz S, Rojek JM, Roberts AJ, McGavern DB, Oldstone MB, de la Torre JC. Altered central nervous system gene expression caused by congenitally acquired persistent infection with lymphocytic choriomeningitis virus. J Virol. 2006;80:9082–92. doi: 10.1128/JVI.00795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bukowski JF, Biron CA, Welsh RM. Elevated natural killer cell-mediated cytotoxicity, plasma interferon, and tumor cell rejection in mice persistently infected with lymphocytic choriomeningitis virus. J Immunol. 1983;131:991–6. [PubMed] [Google Scholar]
- 134.Saron MF, Riviere Y, Hovanessian AG, Guillon JC. Chronic production of interferon in carrier mice congenitally infected with lymphocytic choriomeningitis virus. Virology. 1982;117:253–6. doi: 10.1016/0042-6822(82)90524-4. [DOI] [PubMed] [Google Scholar]
- 135.Oldstone MB, Holmstoen J, Welsh RM., Jr Alterations of acetylcholine enzymes in neuroblastoma cells persistently infected with lymphocytic choriomeningitis virus. J Cell Physiol. 1977;91:459–72. doi: 10.1002/jcp.1040910316. [DOI] [PubMed] [Google Scholar]
- 136.de la Torre JC, Mallory M, Brot M, Gold L, Koob G, Oldstone MB, Masliah E. Viral persistence in neurons alters synaptic plasticity and cognitive functions without destruction of brain cells. Virology. 1996;220:508–15. doi: 10.1006/viro.1996.0340. [DOI] [PubMed] [Google Scholar]
- 137.Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- 138.Oldstone MB, Sinha YN, Blount P, Tishon A, Rodriguez M, von Wedel R, Lampert PW. Virus-induced alterations in homeostasis: alteration in differentiated functions of infected cells in vivo. Science. 1982;218:1125–7. doi: 10.1126/science.7146898. [DOI] [PubMed] [Google Scholar]
- 139.Oldstone MB, Ahmed R, Buchmeier MJ, Blount P, Tishon A. Perturbation of differentiated functions during viral infection in vivo. I. Relationship of lymphocytic choriomeningitis virus and host strains to growth hormone deficiency. Virology. 1985;142:158–74. doi: 10.1016/0042-6822(85)90430-1. [DOI] [PubMed] [Google Scholar]
- 140.Tishon A, Oldstone MB. Perturbation of differentiated functions during viral infection in vivo. In vivo relationship of host genes and lymphocytic choriomeningitis virus to growth hormone deficiency. Am J Pathol. 1990;137:965–9. [PMC free article] [PubMed] [Google Scholar]
- 141.Teng MN, Borrow P, Oldstone MB, de la Torre JC. A single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with the ability to cause growth hormone deficiency syndrome. J Virol. 1996;70:8438–43. doi: 10.1128/jvi.70.12.8438-8443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Oldstone MB, Rodriguez M, Daughaday WH, Lampert PW. Viral perturbation of endocrine function: disordered cell function leads to disturbed homeostasis and disease. Nature. 1984;307:278–81. doi: 10.1038/307278a0. [DOI] [PubMed] [Google Scholar]
- 143.de la Torre JC, Oldstone MB. Selective disruption of growth hormone transcription machinery by viral infection. Proc Natl Acad Sci U S A. 1992;89:9939–43. doi: 10.1073/pnas.89.20.9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Valsamakis A, Riviere Y, Oldstone MB. Perturbation of differentiated functions in vivo during persistent viral infection. III. Decreased growth hormone mRNA. Virology. 1987;156:214–20. doi: 10.1016/0042-6822(87)90400-4. [DOI] [PubMed] [Google Scholar]
- 145.Klavinskis LS, Oldstone MB. Lymphocytic choriomeningitis virus selectively alters differentiated but not housekeeping functions: block in expression of growth hormone gene is at the level of transcriptional initiation. Virology. 1989;168:232–5. doi: 10.1016/0042-6822(89)90262-6. [DOI] [PubMed] [Google Scholar]
- 146.Moskophidis D, Lechner F, Hengartner H, Zinkernagel RM. MHC class I and non-MHC-linked capacity for generating an anti-viral CTL response determines susceptibility to CTL exhaustion and establishment of virus persistence in mice. J Immunol. 1994;152:4976–83. [PubMed] [Google Scholar]
- 147.Moskophidis D, Battegay M, Bruendler MA, Laine E, Gresser I, Zinkernagel RM. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and to gamma interferon. J Virol. 1994;68:1951–5. doi: 10.1128/jvi.68.3.1951-1955.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Oldstone MB. Viral persistence: parameters, mechanisms and future predictions. Virology. 2006;344:111–8. doi: 10.1016/j.virol.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 149.Ou R, Zhou S, Huang L, Moskophidis D. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J Virol. 2001;75:8407–23. doi: 10.1128/JVI.75.18.8407-8423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MBA. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice: role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Villarete L, Somasundaram T, Ahmed R. Tissue-mediated selection of viral variants: correlation between glycoprotein mutation and growth in neuronal cells. J Virol. 1994;68:7490–6. doi: 10.1128/jvi.68.11.7490-7496.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Smelt SC, Borrow P, Kunz S, Cao W, Tishon A, Lewicki H, Campbell KP, Oldstone MB. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor alpha-dystroglycan correlate with viral tropism and disease kinetics. J Virol. 2001;75:448–57. doi: 10.1128/JVI.75.1.448-457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matloubian M, Kolhekar SR, Somasundarum T, Ahmed R. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J Virol. 1993;67:7340–7349. doi: 10.1128/jvi.67.12.7340-7349.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Evans CF, Borrow P, de la Torre JC, Oldstone MB. Virus-induced immunosuppression: kinetic analysis of the selection of a mutation associated with viral persistence. J Virol. 1994;68:7367–73. doi: 10.1128/jvi.68.11.7367-7373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dockter J, Evans CF, Tishon A, Oldstone MB. Competitive selection in vivo by a cell for one variant over another: implications for RNA virus quasispecies in vivo. J Virol. 1996;70:1799–803. doi: 10.1128/jvi.70.3.1799-1803.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lauterbach H, Truong P, McGavern DB. Clearance of an immunosuppressive virus from the CNS coincides with immune reanimation and diversification. Virol J. 2007;4:53. doi: 10.1186/1743-422X-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tishon A, Borrow P, Evans C, Oldstone MB. Virus-induced immunosuppression. 1. Age at infection relates to a selective or generalized defect. Virology. 1993;195:397–405. doi: 10.1006/viro.1993.1389. [DOI] [PubMed] [Google Scholar]
- 159.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–13. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, Larsen CP, Ahmed R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci U S A. 2007;104:15430–5. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Borrow P, Evans CF, Oldstone MB. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J Virol. 1995;69:1059–70. doi: 10.1128/jvi.69.2.1059-1070.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest. 2004;113:737–45. doi: 10.1172/JCI20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sevilla N, Kunz S, McGavern D, Oldstone MB. Infection of dendritic cells by lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 2003;276:125–44. doi: 10.1007/978-3-662-06508-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 165.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MBA. Interleukin-10 determines viral clearance or persistence in vivo: Antibody blockade of the IL-10 receptor restores T cell function and controls viral infection. Nat Med. 2006 doi: 10.1038/nm1492. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mothe BR, Stewart BS, Oseroff C, Bui HH, Stogiera S, Garcia Z, Dow C, Rodriguez-Carreno MP, Kotturi M, Pasquetto V, Botten J, Crotty S, Janssen E, Buchmeier MJ, Sette A. Chronic lymphocytic choriomeningitis virus infection actively down-regulates CD4+ T cell responses directed against a broad range of epitopes. J Immunol. 2007;179:1058–67. doi: 10.4049/jimmunol.179.2.1058. [DOI] [PubMed] [Google Scholar]
- 167.Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. 2005;79:10514–27. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 169.Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104:311–24. doi: 10.1016/s0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]
- 170.Yamamoto T, Kawaguchi M, Sakayori N, Muramatsu F, Morikawa S, Kato Y, Shibata N, Kobayashi M. Intracellular binding of fukutin and alpha-dystroglycan: relation to glycosylation of alpha-dystroglycan. Neurosci Res. 2006;56:391–9. doi: 10.1016/j.neures.2006.08.009. [DOI] [PubMed] [Google Scholar]