

Abstract
The objective of the study was to evaluate the epidemiology of patients with congenital myasthenic syndrome (CMS) in Israel. Targeted mutation analysis was performed based on the clinical symptoms and electrophysiological findings for known CMS. Additional specific tests were performed in patients of Iranian and/or Iraqi Jewish origin. All medical records were reviewed and clinical data, genetic mutations and outcomes were recorded. Forty-five patients with genetic mutations in known CMS genes from 35 families were identified. Mutations in RAPSN were identified in 13 kinships in Israel. The most common mutation was c.-38A>G detected in 8 patients of Iranian and/or Iraqi Jewish origin. Four different recessive mutations in COLQ were identified in 11 kinships, 10 of which were of Muslim-Arab descent. Mutations in CHRNE were identified in 7 kinships. Less commonly detected mutations were in CHRND, CHAT, GFPT1 and DOK7. In conclusion, mutations in RAPSN and COLQ are the most common causes of CMS in our cohort. Specific mutations in COLQ, RAPSN, and CHRNE occur in specific ethnic populations and should be taken into account when the diagnosis of a CMS is suspected.
Keywords: Congenital myasthenic syndrome (CMS), Genetic mutations, RAPSN, COLQ, CHRNE
1. Introduction
Congenital myasthenic syndromes (CMS) are a rare heterogeneous group of inherited neuromuscular disorders caused mainly by mutations in genes coding for proteins expressed at the neuromuscular junction (NMJ). The CMS are often classified as presynaptic, synaptic or postsynaptic based on the location of the disease protein or related to endplate development and maintenance [1].
In each CMS, the safety margin of the neuromuscular transmission is compromised by a different mechanism [1]. Recently, next generation sequencing (NGS) has identified new and ubiquitously expressed CMS genes that also reveal defects not confined to the NMJ [2]. To date, 20 CMS-causing genes have been identified [3].
CMS signs can manifest shortly after birth, while milder cases may remain undiagnosed until adolescence or even adulthood [4]. Minor myasthenic symptoms may be exacerbated by fever, infections or excitement. Typical clinical findings of the neonatal onset subtype include: feeding difficulties, poor suck and cry, choking spells, eyelid ptosis, as well as facial, bulbar, and generalized weakness. Congenital arthrogryposis multiplex (or congenital) may be present, as well as respiratory insufficiency with sudden apnea and cyanosis. Motor milestones may be delayed. Patients with onset later in childhood show abnormal muscle fatigability. Fluctuating eyelid ptosis and extraocular muscle weakness or paralysis can occur. Facial and bulbar weakness with nasal speech and difficulties in coughing and swallowing are infrequent in later childhood onset. Spinal deformity or muscle atrophy may also occur, sometimes with a characteristic ‘limb girdle’ pattern of weakness. CMS patients may be erroneously diagnosed as having acquired myasthenia gravis, myopathies or muscular dystrophies. Naturally, misdiagnosis leads to inappropriate treatment. Moreover, an accurate genetic diagnosis is important because therapy that benefits one type of CMS can be harmful in another. While next generation sequencing (NGS) methods are increasingly available, they are still expensive and the bioinformatics analysis can be time consuming. Israel’s population is characterized by ethnic heterogeneity, composed of Jews of many ethnic backgrounds, as well as Christians, Muslim Arabs, Druze, and Bedouins [5]. Here, we describe the genetic and clinical characteristics of Israeli CMS patients.
2. Patients and methods
Fifty-five patients suspected of CMS, based on clinical presentation and neurophysiological studies, were referred from four medical centers in Israel: Hadassah-Hebrew University Medical Center, Schneider Children’s Medical Center of Israel, Sourasky Medical Center and Wolfson Medical Center. The study was approved by the institution’s ethical committees.
Initial genetic evaluations were performed in accordance with patients’ clinical symptoms, and known CMS genes (each subunit of AChR, COLQ, CHAT, RAPSN and DOK7) were identified by Sanger sequencing. Molecular genetic analysis was mainly performed in Mayo Clinic laboratories, with patients of Iranian or Iraqi Jewish origin sent for confirmation of the c.-38A>G mutation in the promoter region of the RAPSN, in the molecular genetics laboratory in Wolfson Medical Center, Holon. In three patients, whole-exome sequencing was performed.
All medical records were reviewed as well as patients’ clinical data, electrophysiological studies, genetic mutation and recorded outcome. Forty-five CMS patients from 35 unrelated kinships with confirmed genetic mutations were included in the study. 10 patients were excluded as no genetic mutations identified in the sequenced genes constituted confirmation of the CMS diagnosis.
3. Results
3.1. Mutation frequencies
Overall, we found genetic mutations in 7 genes in our cohort. Mutations in RAPSN were the most prevalent among kinships in Israel (13 kinships, 37%), followed by mutations in collagenic tail peptide acetylcholinesterase (COLQ; 11 kinships, 31%) and the acetyl choline receptor (CHRNE; 7 kinships, 20%; Fig. 1).
Fig. 1.
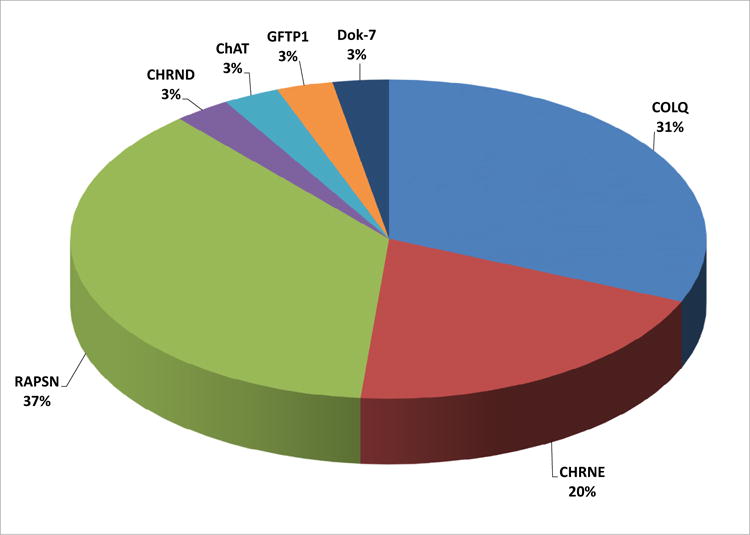
Percentage of identified genetic CMS mutations in the study by kinships. Mutations in RAPSN were the most prevalent among kinships in Israel (13 kinships, 37%), followed by mutations in COLQ (11 kinships, 31%) and CHRNE (7 kinships, 20%).
Within these kinships, COLQ mutations were identified in 16 patients, RAPSN mutations in 15 patients and CHRNE mutations were identified in 9 patients (Supplementary Table S1).
3.2. Characteristics of patients by affected genes
The characteristics of patient groups carrying mutations in the same genes are presented in Table 1.
Table 1.
Clinical and genetic data of patients with CMS.
| CMS gene/No. of patients | Mean age of onset | Consanguinity | Limb weakness | Ophthalmoplegia | Ptosis |
|---|---|---|---|---|---|
| COLQ n = 16 | Birth-infancy | 9/16 | 15/16 | 10/16 | 12/16 |
| CHRNE n = 9 | Birth-infancy | 6/9 | 8/9 | 9/9 | 9/9 |
| RAPSN n = 15 | Infancy | 1/15 | 1/15 | 0/15 | 15/15 |
| CHRND n = 2 | 2 y | 2/2 | 1/2 | 0/2 | 2/2 |
| CHAT n = 1 | Birth | 1/1 | 1/1 | 0/1 | 1/1 |
| GFPT1 n = 1 | 5 y | 0/1 | 1/1 | 0/1 | 0/1 |
| DOK7 n = 1 | 10 y | 1/1 | 1/1 | 0/1 | 1/1 |
3.2.1. Mutations in RAPSN
Patients with mutations in RAPSN were members of 13 distinct families. The most common mutation was the c.-210T>C (previously named c.-38A>G), identified in patients of Iranian and/or Iraqi Jewish origin [6,7]. The patients presented with typical ptosis, prognathism, and masticatory and facial weakness. The mean age of onset was in the first year of life. Only one patient was born to consanguineous parents.
A single patient in our cohort of European origin was heterozygous for the common p.Asn88Lys mutation [8]. This patient presented at birth with apnea, feeding problems, and hypotonia.
3.2.2. Mutations in COLQ
Patients with COLQ mutations were members of 11 distinct kinships. All but a single patient were of Arab Muslim origin. Four previously reported families also carried the p.Gly240* mutation [9]. One patient was heterozygous for p.Gly240* and p.Gly126Alafs*38. Two patients from distinct families were homozygous for p.Asn298Metfs*2 and one patient was homozygous for p.Pro265Alafs*37. Two patients from one kinship were homozygous for Arg410Trp. In 3 siblings of another family but of the same clan, the mutation was present at heterozygosity.
Symptoms of patients with mutations in COLQ consisted of ptosis, ophthalmoplegia and limb weakness, with variability among specific mutations. However, all patients harboring the Arg410Trp mutation had limb girdle and neck muscle weakness but no ptosis or ophthalmoplegia. The onset was at birth or during infancy and the consanguinity rate was 56%.
3.2.3. Mutations in CHRNE
Patients with CHRNE mutations were members of 7 families. Three patients from 3 families of Ashkenazi Jewish origin harbored the p.Asn452Glufs*4 mutation [10]; 1 patient was homozygous and 2 were heterozygous.
Three patients of Bedouin origin sharing a single kinship were heterozygous for c.246-11del4. This novel mutation, a deletion in intron 7 of CHRNE, weakens the polypyrimidine tract before exon 8 and predicts abnormal splicing. The patients (2 siblings and a maternal cousin) were born to consanguineous parents and presented at birth with ptosis, ophthalmoplegia, and facial weakness.
3.2.4. Other mutations
Less commonly detected mutations were in CHRND (n = 2), CHAT (n = 1), GFPT1 (n = 1) and DOK7 (n = 1).
4. Discussion
This is the first study presenting data on different CMS mutations in Israeli patients according to ethnicity. Israel’s population is particularly suited for genetic studies. It is unique in its multitude of clusters of different Jewish ethnicities, as well as Muslim communities and other subpopulations [5], each isolated by historical events, country of origin, and religion. In addition, the consanguinity rate in some subpopulations is high. We found that in our Israeli cohort, the largest kinship groups were patients with mutations in RAPSN (37%), COLQ (31%), and in CHRNE (20%). The relative frequency of genetic mutations in our cohort differs from other reported frequencies in the literature. In the Munich cohort of 680 CMS patients of mainly European origin, RAPSN mutations were found in only 15% of patients and COLQ in 12%, whereas 49% of all mutations were in CHRNE [11]. Similar frequencies were found in the Mayo Clinic cohort of 354 patients investigated between 1988 and 2014, where 51% had mutations in CHRNE, 14% in RAPSN, and 12% COLQ [3]. Some of our Israeli patients, whose genetic analyses were performed at the Mayo Clinic, were included in the latter large cohort.
The results of the study confirmed our initial assumption that our population will include a large proportion of carriers of the RAPSN c.-38A>G mutation, because it was the most prevalent mutation among previously-studied populations This mutation was found in a total of 12 kinships constituting 34% of all kinships in our population, and 92% of RAPSN mutations carrying kinships in our population.
In Israel, COLQ mutations were detected mainly in Arabs of Muslim origin. Only one patient with a COLQ mutation was of Jewish ethnicity, and this patient’s family has origins in Iraq. The most frequent COLQ mutations in the Israeli CMS patients were p.Gly240*, p.Pro265Alafs*37 and Arg410Trp, suggesting that these mutations are quite common in Middle-Eastern countries. To the best of our knowledge, the only group that reported on CMS in a non-Israeli Middle-Eastern population was Matlik and colleagues [12]. They described a Syrian family with CMS and identified a novel COLQ missense mutation p.Ile337Thr [12]. None of our patients carried this mutation.
Of the COLQ mutations, the Arg410Trp mutation segregated with Muslim-Arab families originating from a northern village in Israel. These patients had limb girdle and neck muscle weakness but no ptosis or ophthalmoplegia compared to other patients with COLQ mutations. Patients 14, 15 and 16 were compound heterozygotes for Arg410Trp/p.Ile265Tyrfs*27 and resided in another village in northern Israel. Interestingly, patient 10, the first cousin of patients 14 to 16, harbored a completely different mutation of COLQ – p.Pro265Alafs*37 mutation. This mutation causes a frameshift after codon 262, predicting 36 missense codons followed by a stop codon. The mutation spares 85% of the collagen domain but truncates COLQ and causes loss of function [13]. Even though COLQ mutations were diagnosed in only 11 compared to 13 kinships with RAPSN mutations, there were 16 patients with COLQ mutations compared to 15 with RAPSN mutations due to more siblings with the same mutations and larger families in the COLQ families.
Defects in CHRNE accounted for 20% of our population, which is much less than the estimated frequency of 50% in the literature [3,11]. The p.Asn452Glufs*4 mutation detected in 3 Ashkenazi Jewish patients is a known founder mutation in patients originating from North Africa, Spain, Portugal and Brazil [14–16]. Considering that CMS is uncommon among Ashkenazi Jews, it is possible that these patients are representatives of a small subgroup of the Ashkenazi Jewish descendent population that has acquired this mutation owing to historical events of migration. An enticing hypothesis is that this mutation was common among Jews that were deported from Spain in the 15th century. Based on our results, we suggest adding Ashkenazi Jewish origin to the list of patients from North Africa, Spain, Portugal and Brazil for screening for this mutation at an early stage in the diagnosis of CMS.
In our cohort, we found only a single patient with a mutation in DOK7 (patient 45) compared to the 10% found in previously-described cohorts. This patient of Muslim-Arab origin presented with limb girdle weakness and was homozygous for the p.Lys320Serfs*136mutation. This mutation was also reported in a large Muslim-Arab kindred from a town in Wadi Ara, Israel. In that family, symptoms presented at birth and the muscle weakness mimicked congenital muscular dystrophy or limb girdle muscular dystrophy [17–19]. Our patient’s symptoms started in late childhood and were consistent with limb girdle weakness.
Our findings reflect the distinct genetic and ethnic diversity of the population in Israel. In our cohort, rare mutations were more common than in previously published studies, probably due to the high consanguinity rate in certain subgroups of the Israeli population. We propose a diagnostic strategy based on phenotypic clues, ethnic origin and geographical residency. A focused, knowledge-based diagnostic approach can assist in genetic counseling. Moreover, most CMS are treatable and choosing the appropriate drug is dependent on the underlying molecular defect.
Although the majority of individuals with CMS benefit from acetylcholine esterase (AChE) inhibitors (pyridostigmine), some CMS subtypes such as endplate acetylcholinesterase deficiency (EP AChE deficiency) due to mutations in COLQ, slow-channel CMS (SCCMS), or DOK7-related CMS are refractory to, or deteriorate with, the use of AChE inhibitors [20–23]. Ephedrine and Albuterol [24] are alternative treatment options for CMS subtypes caused by mutations in COLQ and DOK7 as well as the epsilon subunit mutation of AChR [25]. Quinidine and fluoxetine are treatment for individuals with genetically defined slow-channel CMS [20].
In accordance with our aim to facilitate diagnosis without use of NGS, only patients with confirmed CMS mutations are included in this study. We excluded 10 patients with CMS with no identifiable defect in known CMS genes. Other genes responsible for CMS in Israeli patients still await discovery and for these patients, NGS will be needed to discover the disease genes.
Based on our results, our recommendation for physicians in our region suspecting CMS is to consider the patient’s specific combination of clinical symptoms, origin and geographic location and use this information for targeted mutation analyses. This information may also be useful for physicians encountering suspected CMS in patients of Middle-Eastern origins in other parts of the world.
Supplementary Material
Acknowledgments
Mutation analysis performed in Dr. Andrew Engel’s laboratory was supported by NIH Grant NS6277.
Appendix: Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.nmd.2016.11.014.
References
- 1.Engel AG. Current status of the congenital myasthenic syndromes. Neuromuscul Disord. 2012;22:99–111. doi: 10.1016/j.nmd.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodríguez Cruz PM, Palace J, Beeson D. Inherited disorders of the neuromuscular junction: an update. J Neurol. 2014;261:2234–43. doi: 10.1007/s00415-014-7520-7. [DOI] [PubMed] [Google Scholar]
- 3.Engel AG, Shen X-M, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015;14:420–34. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzoni PJ, Scola RH, Kay CSK, Werneck LC. Congenital myasthenic syndrome: a brief review. Pediatr Neurol. 2012;46:141–8. doi: 10.1016/j.pediatrneurol.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Rosner G, Rosner S, Orr-Urtreger A. Genetic testing in Israel: an overview. Annu Rev Genomics Hum Genet. 2009;10:175–92. doi: 10.1146/annurev.genom.030308.111406. [DOI] [PubMed] [Google Scholar]
- 6.Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum Mol Genet. 2003;12:739–48. doi: 10.1093/hmg/ddg089. [DOI] [PubMed] [Google Scholar]
- 7.Goldhammer Y, Blatt I, Sadeh M, Goodman RM. Congenital myasthenia associated with facial malformations in Iraqi and Iranian Jews. A new genetic syndrome. Brain. 1990;113(Pt 5):1291–306. doi: 10.1093/brain/113.5.1291. [DOI] [PubMed] [Google Scholar]
- 8.Müller JS, Mildner G, Müller-Felber W, et al. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology. 2003;60:1805–10. doi: 10.1212/01.wnl.0000072262.14931.80. [DOI] [PubMed] [Google Scholar]
- 9.Shapira YA, Sadeh ME, Bergtraum MP, et al. Three novel COLQ mutations and variation of phenotypic expressivity due to G240X. Neurology. 2002;58:603–9. doi: 10.1212/wnl.58.4.603. [DOI] [PubMed] [Google Scholar]
- 10.Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RC. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann Neurol. 1996;40:810–17. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- 11.Abicht A, Dusl M, Gallenmüller C, et al. Phenotype-guided gene-after-gene sequencing in diagnostic practice: a study of 680 patients. Hum Mutat. 2012;33:1474–84. doi: 10.1002/humu.22130. [DOI] [PubMed] [Google Scholar]
- 12.Matlik HN, Milhem RM, Saadeldin IY, Al-Jaibeji HS, Al-Gazali L, Ali BR. Clinical and molecular analysis of a novel COLQ missense mutation causing congenital myasthenic syndrome in a Syrian family. Pediatr Neurol. 2014;51:165–9. doi: 10.1016/j.pediatrneurol.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Ohno K, Brengman J, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci USA. 1998;95:9654–9. doi: 10.1073/pnas.95.16.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abicht A, Stucka R, Karcagi V, et al. A common mutation (epsilon1267delG) in congenital myasthenic patients of Gypsy ethnic origin. Neurology. 1999;53:1564–9. doi: 10.1212/wnl.53.7.1564. [DOI] [PubMed] [Google Scholar]
- 15.Richard P, Gaudon K, Haddad H, et al. The CHRNE 1293insG founder mutation is a frequent cause of congenital myasthenia in North Africa. Neurology. 2008;71:1967–72. doi: 10.1212/01.wnl.0000336921.51639.0b. [DOI] [PubMed] [Google Scholar]
- 16.Mihaylova V, Scola RH, Gervini B, et al. Molecular characterisation of congenital myasthenic syndromes in Southern Brazil. J Neurol Neurosurg Psychiatry. 2010;81:973–7. doi: 10.1136/jnnp.2009.177816. [DOI] [PubMed] [Google Scholar]
- 17.Mahjneh I, Vannelli G, Bushby K, Marconi GP. A large inbred Palestinian family with two forms of muscular dystrophy. Neuromuscul Disord. 1992;2:277–83. doi: 10.1016/0960-8966(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 18.Mahjneh I, Bushby K, Anderson L, et al. Merosin-positive congenital muscular dystrophy: a large inbred family. Neuropediatrics. 1999;30:22–8. doi: 10.1055/s-2007-973452. [DOI] [PubMed] [Google Scholar]
- 19.Mahjneh I, Lochmüller H, Muntoni F, Abicht A. DOK7 limb-girdle myasthenic syndrome mimicking congenital muscular dystrophy. Neuromuscul Disord. 2013;23:36–42. doi: 10.1016/j.nmd.2012.06.355. [DOI] [PubMed] [Google Scholar]
- 20.Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252–7. doi: 10.1016/j.nurt.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palace J, Lashley D, Newsom-Davis J, et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130:1507–15. doi: 10.1093/brain/awm072. [DOI] [PubMed] [Google Scholar]
- 22.Kinali M, Beeson D, Pitt MC, et al. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol. 2008;201–202:6–12. doi: 10.1016/j.jneuroim.2008.06.026.. [DOI] [PubMed] [Google Scholar]
- 23.Schara U, Lochmüller H. Therapeutic strategies in congenital myasthenic syndromes. Neurotherapeutics. 2008;5:542–7. doi: 10.1016/j.nurt.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liewluck T, Selcen D, Engel AG. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok-7 myasthenia. Muscle Nerve. 2011;44:789–94. doi: 10.1002/mus.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadeh M, Shen X-M, Engel AG. Beneficial effect of albuterol in congenital myasthenic syndrome with epsilon-subunit mutations. Muscle Nerve. 2011;44:289–91. doi: 10.1002/mus.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.