

Abstract
Background
Rho kinases (ROCKs) contribute to allergic airways disease. ROCKs also play a role in lymphocyte proliferation and migration.
Objective
To determine the role of ROCK2 acting within CD4+ cells in allergic airways responses.
Methods
ROCK2 haploinsufficient (ROCK2+/-) and wildtype mice were sensitized with ovalbumin (OVA). ROCK2+/- mice then received either CD4+ cells from ROCK2 sufficient OVA TCR transgenic (OT-II) mice or saline i.v. 48 hours before challenge with aerosolized OVA. Wildtype mice received saline before challenge. Allergic airway responses were measured 48 hours after the last challenge. Allergic airways responses were also assessed in mice lacking ROCK2 only in CD4+ cells (ROCK2CD4Cre mice) versus control (CD4-Cre and ROCK2flox/flox) mice.
Results
OVA-induced increases in bronchoalveolar lavage lymphocytes, eosinophils, IL-13, IL-5, and eotaxin were reduced in ROCK2+/- versus wildtype mice, as were airway hyperresponsiveness and mucous hypersecretion. In ROCK2+/- mice, adoptive transfer with CD4+ cells from OT-II mice restored effects of OVA on lymphocytes, eosinophils, IL-13, IL-5, and mucous hypersecretion to wildtype levels, whereas eotaxin and airway hyperrresponsiveness were not affected. ROCK2 inhibitors reduced IL-13-induced release of eotaxin from airway smooth muscle (ASM), similar to effects of these inhibitors on ASM contractility. Despite the ability of adoptive transfer to restore allergic airways inflammation in ROCK2 insufficient mice, allergic inflammation was not different in ROCK2CD4Cre versus control mice.
Conclusion
ROCK2 contributes to allergic airways responses likely via effects within ASM cells and within non lymphocyte cells involved in lymphocyte activation and migration into the airways.
Introduction
The Rho-associated coiled-coil forming kinases (ROCKs) are serine/threonine kinases that are targets of the Rho family of small GTP-binding proteins [1, 2]. Multiple lines of evidence support the hypothesis that ROCK proteins are involved in the pathogenesis of allergic asthma. Airway narrowing caused by airway smooth muscle (ASM) contraction is a canonical feature of asthma, and ROCKs participate in smooth muscle contraction via their ability to inhibit myosin light chain phosphatase (MLCP) [3-5]. ROCKs can also participate in cell migration, including the migration of T lymphocytes and eosinophils [6-9], cells that infiltrate the airways in allergic asthma. ROCKs are activated by GTP binding to RhoA, and RhoA expression increases in bronchi of allergen sensitized and challenged rats and guinea pigs [10, 11]. Furthermore, many of the inflammatory mediators released upon allergen challenge are capable of activating RhoA [12]. Indeed, in ovalbumin (OVA) sensitized mice, OVA aerosol challenge results in ROCK activation in the lungs that peaks about 4 hours post challenge and resolves over the next 48 hours [13]. Importantly, the ROCK inhibitors, Y27632 and fasudil, attenuate airway hyperresponsiveness (AHR), eosinophilic airway inflammation, Th2 cytokine expression, and mucous hypersecretion in lungs of OVA sensitized and challenged animals [14-17]. Similarly, ROCK insufficient mice also have reduced responses to allergen challenge [13]. However, the cell types in which ROCK acts to induce these effects have not been established.
Actions of ROCK within Th2 lymphocytes could account for the observed reductions in allergic airway responses after ROCK inhibition or in animals with ROCK insufficiency. In allergen sensitized and challenged mice, the type 2 cytokine, IL-13, is required for AHR, airway eosinophilia, and mucous cell hyperplasia [18, 19], and ROCK inhibitors reduce pulmonary expression of IL-13 after allergen sensitization and challenge [17]. Inhibiting ROCK also prevents lymphocyte chemotaxis induced by SDF-1, CCL19 and CCL20 [20-22] and lymphocyte proliferation induced by concavalin A or anti-CD3 [23]. Likewise, in co-cultures of dendritic cells with T-lymphocytes from mice that are transgenic for a T-cell receptor (TCR) that recognizes OVA peptide (OT-II mice), ROCK inhibition decreases the lymphocyte proliferation observed upon addition of OVA [13]. ROCK inhibitors also inhibit cytokine release by peripheral blood T cells stimulated with ConA [24].
Two ROCK isoforms have been identified: ROCK1 and ROCK2 [25, 26]. The two isoforms have similar but not identical substrate specificities. There are also differences in their cellular expression, their intracellular localization, and their regulation [25, 27-31]. These differences suggest that ROCK1 and ROCK2 may have non redundant functions in regulating airway responses. Indeed, ROCK2+/- mice have reduced allergic airway responses even when ROCK1 is fully functional, indicating that ROCK2 has a role not fulfilled by ROCK1 [13]. Similarly, ROCK2 does not compensate for loss of ROCK1 [13]: ROCK1+/- mice have reduced allergic airway responses even when ROCK2 is fully functional. Here we focused on the role of ROCK2.
The purpose of this study was to examine the hypothesis that ROCK2 activation within lymphocytes contributes to allergen-induced AHR, eosinophilia, and mucous cell hyperplasia. Most ROCK2-/- mice die either in gestation or early postnatally [32]. However, ROCK2+/- mice are viable. Lungs and other tissues from ROCK2+/- mice express about 50% of wildtype ROCK2 levels without changes in ROCK1 [13, 33]. We assessed allergic airway responses in WT and ROCK2+/- mice that had been sensitized with OVA and challenged with either PBS or OVA aerosol. To examine the role of ROCK2 within lymphocytes, prior to OVA aerosol challenge ROCK2+/- mice were administered (i.v.) either saline or splenic T lymphocytes derived from OT-II mice. We reasoned that if the reduced allergic airway responses observed in ROCK2+/- mice was the result of loss of lymphocytes in the airways consequent to the effects of ROCK2 in lymphocyte migration, proliferation, or cytokine production, then these functional deficits would be overcome if we provided ROCK2 sufficient lymphocytes that could respond to OVA. In contrast, if the role of ROCK2 was primarily the result of its actions in other cell types, no change in allergic airways responses would occur even if ROCK2+/- mice were provided with ROCK2 sufficient lymphocytes. We also crossed ROCK2 floxed mice (ROCK2flox/flox) [34] with mice expressing Cre recombinase under the influence of a CD4 promoter (CD4-Cre mice) to generate mice that lacked ROCK2 only in CD4 cells (ROCK2CD4Cre). Allergic airways responses were then assessed in ROCK2CD4Cre mice and their controls (ROCK2flox/flox and CD4-Cre mice).
Methods
Animals
The Harvard Medical Area Standing Committee on Animals approved these studies. ROCK2+/- mice were generated as previously described [33] and were on a C57BL/6 background. Gender-matched littermates were used as wildtype (WT) controls. OT-II and CD4-Cre mice were purchased from The Jackson Laboratories (Bar Harbor, ME). ROCK2flox/flox mice were generated as previously described [34]. ROCK2CD4Cre mice were obtained by breeding ROCK2flox/flox with CD4-Cre mice.
Protocol
Two cohorts of male mice were used. In the first cohort, 4-5 week old WT and ROCK2+/- mice were sensitized to ovalbumin (OVA, Grade V, Sigma-Aldrich Co., St. Louis, MO) by i.p. injection with alum (Imject, Pierce) on days 0 and 14 as previously described [35, 36](see Fig.1A for schematic). On day 26, ROCK2+/- mice received an i.v. injection of either saline or CD4+ cells from OT-II mice, as described below. WT mice received an injection of saline. On days 28 through 30, mice were challenged for 30 min with an aerosol of either PBS or 1% OVA, as previously described [13]. 48 h after the last challenge, mice were anesthetized and instrumented for the measurement of airway responsiveness. Mice were then euthanized with an overdose of sodium pentobarbital, blood was harvested by cardiac puncture for the preparation of serum, bronchoalveolar lavage (BAL) was performed, and the lungs were excised. One lung was fixed for subsequent histological assessment of mucous cell hyperplasia. The other lung was placed in RNAlater (Qiagen, Valencia, CA) for subsequent preparation of RNA and qRT-PCR. In the second cohort (Fig.1B), the same protocol was used to sensitize, challenge, and evaluate ROCK2flox/flox, CD4-Cre, and ROCK2CD4Cre mice, except that no injections were given on day 26, and no histology was performed. Instead, in these mice, one lung was used for flow cytometry to evaluate Th2 and Th17 cells.
Figure 1.

Schematic diagram of the experimental protocols used in this study.
CD4 cell preparation for adoptive transfer
To obtain CD4+ cells for adoptive transfer, spleens of OT-II mice were harvested, splenocytes isolated, and CD4+ lymphocytes enriched by negative selection using a pan antibody panel conjugated to magnetic beads (Dynabeads, Life Technologies, Carlsbad, CA USA). The purity of the cell population was determined by flow cytometry after blocking of Fc receptor and surface staining for CD45-PE/Cy7 (30-F11), CD3-APC-Cy7 (17A2), and CD4-AF488 (GK1.5). All antibodies were purchased from Biolegend. Using this process, we enriched the splenocyte from approximately 20% to over 70% CD4+ lymphocytes (Fig.S1).
Adoptive Transfer of CD4+ T cells from OT-II mice
ROCK2+/- mice were treated i.v. before the initiation of OVA challenge with either saline or 106 CD4+ cells from mice expressing a TCRαβ transgene for OVA peptide (OT-II mice). The number of CD4 cells was chosen based on its ability to restore the reduced BAL lymphocytes observed in allergen challenged ROCK2+/- mice to levels observed in similarly challenged WT mice (Fig.2A).
Figure 2.

Regulation of allergic inflammation by ROCK2. Bronchoalveolar lavage (BAL) lymphocytes (A), IL-13 (B), IL-5 (C), eosinophils (D), eotaxin (E), and mast-cell tryptase 1 (MCPT1) (a marker of mast-cell degranulation) (F) were assessed in WT and ROCK2+/- mice that were sensitized with OVA and challenged with either aerosolized OVA or PBS. WT mice were treated with saline and ROCK2+/- mice were treated with either saline (SAL) or splenic CD4+ cells from OT-II mice (CD4+) as described in Fig.1A. BAL was performed 48 h after the last challenge. Results are mean ± SEM of data from 8-19 mice per group. * p<0.05 versus PBS in mice with same genotype and treatment; + p<0.05 versus WT mice; and # p<0.05 versus ROCK2+/- mice treated with saline.
Bronchoalveolar Lavage (BAL)
BAL was performed and total and differential cell counts evaluated as previously described [37-39]. BAL supernatants were stored at -80°C and subsequently analyzed by ELISA for IL-5, IL-13, eotaxin (R&D Systems Inc., Minneapolis, MN) and mast cell tryptase 1 (eBiosciences, San Diego, CA). Serum was prepared and analyzed for OVA-specific IgE by ELISA (Cayman, Ann Arbor, MI). For the IL-5 ELISA, BAL fluid was concentrated 5-10 fold by centrifugation through a 3kDa exclusion filter for 20 minutes as recommended by the vendor (Amicon Ultra-0.5mL 3kDa, Millipore, Billerica, USA). Values expressed are those in the original BAL fluid.
Measurement of pulmonary mechanics and airway responsiveness
Mice were anesthetized with xylazine (7 mg/kg) and sodium pentobarbital (50 mg/kg) and instrumented for the measurement of pulmonary mechanics and airway responsiveness to aerosolized methacholine by the forced oscillation technique, as previously described [39].
Histopathologic assessment
Lung sections were used to quantify goblet cell hyperplasia by determining the number of PAS+ cells in each airway. In cohort 1 (Fig.1A), after BAL, the right mainstem bronchus was clamped and the right lung was removed for subsequent preparation of RNA. The left lung was inflated in situ at 20 cm of water with 4% paraformaldehyde and then removed for paraffin embedding, sectioning, and staining for periodic acid-Schiff (PAS) to evaluate goblet cell hyperplasia. Goblet cell hyperplasia was quantified by determining the number of PAS+ cells in each airway (3-6 airways per mouse) and dividing by the perimeter of the airway measured at the basal membrane. The images were captured with a high definition AxyoCam camera (Zeiss, Jena, Germany) coupled to an optical microscope (Olympus, Japan).
RNA Extraction and Real-Time PCR
The left lung was harvested and RNA prepared and reverse transcribed for real time PCR as previously described [40]. Primers for murine Muc5ac and 18S ribosomal RNA were previously described [36]. Muc5ac mRNA abundance relative to 18S was calculated based on the ΔΔCT method. For each set of primers, melting curve analysis yielded a single peak consistent with one PCR product.
Effect of ROCK inhibition on eotaxin release from human airway smooth muscle (ASM) cells
Primary human ASM cells from 2 different healthy donors (courtesy of Dr. Reynold Panettieri Jr., University of Pennsylvania) were grown until confluent, then washed with PBS, serum deprived, and hormone supplemented as previously described [41]. 24 h later, cells were treated with the pan-ROCK inhibitor Y-27632 (Calbiochem, San Diego, CA) or a ROCK2 specific inhibitor SR-3677 (Tocris, Bristol, UK)[42, 43]. 30 minutes later, cells were challenged with IL-13 (eBiosciences) or with PBS. 24 h later, cell supernatants were collected and assayed for eotaxin by ELISA (R&D Systems).
Flow cytometry
In mice in cohort 2 (Fig.1B), lung cell suspensions were obtained, stained for surface markers (CD45 and CD4) and intracellular IL-13 and IL-17, followed by flow-cytometric analysis. Briefly, the lungs were excised, and the right lung placed in medium containing monensin (Golgi Stop, BD Biosciences, NJ). Single cell suspensions were prepared and stimulated with PMA and ionomycin as previously described [40]. Fixed and permeabilized cells had their Fc receptors blocked (FcX, Biolegend) and were stained as follows: CD45-PE/Cy7 (30F11), CD4-AF488 (GK1.5), CD3-APC/Cy7 (17A2), IL-17A-AF647 (TC11-18H10.1), IL-13-AF647 (eBio13A, Biolegend), or with isotype control (all antibodies from Biolegend unless noted). Cells were counted by flow cytometry (Canto-II, BD Biosciences, NJ), and analyzed with FlowJo software (FlowJo, Ashland, OR). Cells were gated for CD45+ cells, followed by gating for CD3 and CD4. CD45+CD3+CD4+ cells were then gated for IL-17A or IL-13.
Statistical Analysis
BAL and flow-cytometry cell data were log transformed for statistical analysis. Data were analyzed by ANOVA or factorial ANOVA using STATISTICA software (StatSoft®; Tulsa, OK). Fisher's LSD test was used for post hoc comparisons. A p value < 0.05 was considered statistically significant.
Results
Adoptive transfer of OVA-specific CD4+ cells rescues attenuated allergic airway inflammation in ROCK2+/- mice
Compared to WT mice, ROCK2+/- mice had attenuated inflammatory responses to allergen: BAL lymphocytes, IL-5, IL-13, eosinophils, and eotaxin were each lower in ROCK2+/- mice treated with saline than in WT treated with saline (Fig. 2A-E). In contrast, BAL MCPT1, a marker of mast cell degranulation, was unaffected by ROCK2 insufficiency (Fig.2F). Reductions in airway inflammation in ROCK2+/- mice were not the result of effects of ROCK2 on allergic sensitization since there was no statistically significant difference in OVA-specific IgE in ROCK2+/- versus WT mice (Fig.S2) (note that all mice were OVA sensitized).
Adoptive transfer of CD4+ cells from OT-II mice into ROCK2+/- mice was used to reverse the effects of ROCK2 insufficiency on OVA-induced recruitment of lymphocytes to the lungs. Indeed, BAL lymphocytes were no longer different in WT mice versus ROCK2+/- mice that had received OVA-specific CD4+ cells (Fig.2A). Adoptive transfer also reversed the effects of ROCK2 insufficiency on BAL IL-13 and IL-5, and eosinophils: in ROCK2+/- mice that received CD4+ T cells, OVA-induced increases in BAL IL-13, IL-5 and eosinophils were no longer different from WT mice (Fig.2B-D).
BAL cells were also assessed in some additional OVA-sensitized and challenged WT mice that were administered OT-II cells i.v. before OVA challenge, and compared to cohort-matched WT mice administered i.v. saline before challenge. Whereas adoptive transfer of ROCK2 sufficient lymphocytes to ROCK2 insufficient mice increased both BAL lymphocytes and eosinophils (Fig.2A,D), the same adoptive transfer was without effect in WT mice that were already sufficient for ROCK2 (Figure S3A,B), indicating that in WT mice, simply increasing the number of circulating lymphocytes did not result in additional BAL lymphocytes, or any subsequent effects on BAL eosinophils.
IL-13 is an important stimulus for release of eotaxin in allergic airways [41, 44, 45]. Nevertheless, despite the ability of adoptive transfer of CD4+ cells to restore BAL IL-13 to normal WT levels in ROCK2+/- mice (Fig.2B), BAL eotaxin was not restored (Fig. 2E). These data could be the result of ROCK2 acting within the cells that produce eotaxin. ASM cells produce eotaxin in response to IL-13 [41, 46], and are one important source of eotaxin in asthmatic airways [47, 48]. Consequently, we examined the impact of a chemical inhibitor of ROCK2 on IL-13-induced eotaxin release from human ASM cells. As previously described [41], IL-13 (100 ng/ml) increased eotaxin release from ASM cells (Fig.3). Pre-treatment with the ROCK2 specific inhibitor, SR3677 [42], caused a dose-dependent decrease in IL-13-stimulated eotaxin release (Fig. 3A), indicating that ROCK2 is required for eotaxin release from ASM cells. Similar results were obtained with the pan ROCK inhibitor, Y-27632 [49](Fig.3B). To our knowledge, this is first report of a role for ROCK2 in eotaxin release from any cell type. Hence, it is conceivable that ROCK2 also contributes to the release of eotaxin from other cell types in the airways that are important for producing this chemokine, for example, airway epithelial cells.
Figure 3.

IL-13-induced eotaxin release from human airway smooth muscle (ASM) cells requires ROCK2. Human ASM cells from 2 different donors were grown to confluency, serum deprived and insulin and transferrin supplemented for 24 h, and then treated with increasing concentrations of either (A) the ROCK2 specific inhibitor, SR3677, or (B) the pan ROCK inhibitor, Y-27632, for 30 minutes before challenge with IL-13 (100 ng/ml) for 24 hrs. Eotaxin in the supernatant was measured by ELISA and is expressed as a % of the eotaxin measured in the IL-13 treated cells that did not receive ROCK inhibitor. Results are mean ± SEM of n=4-8 per treatment. *p<0.05 vs untreated cells; # p<0.05 vs IL-13 only treated cells. The ELISA assay was conducted at least in duplicate for each donor and at each concentration. Data were normalized to eotaxin levels of IL-13 treated cells that did not receive ROCK inhibitors.
OVA-specific CD4+ cells rescue attenuated mucous cell hyperplasia in ROCK2+/- mice
ROCK2 insufficiency reduced the mucous cell hyperplasia that was induced by OVA challenge (Fig.4): compared to OVA-challenged WT mice, OVA challenged ROCK2+/- mice treated with saline had fewer PAS+ cells in the airway epithelium (Fig.4A) and lower pulmonary Muc5a mRNA abundance (Fig.4B), consistent with previous observations [13]. ROCK2 acting within epithelial cells to promote the processes that allow for airway epithelial cell differentiation to goblet cells could account for this phenomenon. However, adoptive transfer of CD4+ cells from ROCK2 sufficient OT-II mice reversed the effects of ROCK2 insufficiency on mucous cell hyperplasia (Fig.4). IL-13 is known to promote mucous cell hyperplasia and Muc5ac mRNA expression [50], and reduced BAL IL-13 in ROCK2+/- was also reversed after adoptive transfer of ROCK2 sufficient CD4+ cells (Fig. 2B), suggesting that ROCK2 within airway epithelial cells is not essential for mucous cell hyperplasia.
Figure 4.

Regulation of goblet cell hyperplasia by ROCK2. (A) PAS+ cell density (expressed as the number of PAS+ cells per 100 um of basement membrane) quantified morphometrically and (B) pulmonary mRNA abundance of Muc5ac and in WT and ROCK2+/- mice that were sensitized with OVA and challenged with either aerosolized OVA or PBS. WT mice were treated with saline and ROCK2+/- mice were treated with either saline (SAL) or splenic CD4+ cells from OT-II mice (CD4+) as described in Fig.1A. Results are mean ± SEM of 3 PBS challenged mice per group and 6-7 OVA challenged mice per group. * p<0.05 versus PBS in mice with same genotype and treatment and # p<0.05 versus WT.
OVA-specific CD4+ cells do not rescue attenuated OVA-induced AHR in ROCK2+/- mice
Compared to WT mice challenged with PBS, WT mice challenged with OVA developed AHR: methacholine-induced changes in each of the pulmonary mechanical parameters examined (Rn (Newtonian resistance), G (tissue resistance) and H (tissue elastance)) were greater in OVA- challenged than PBS-challenged WT mice (Fig.5). Airway responsiveness was significantly reduced in ROCK2+/- versus WT mice challenged with OVA and treated with saline (Fig 5), indicating that ROCK2 is required for OVA-induced AHR, as previously described [13]. In contrast to the effects of adoptive transfer on allergic airway inflammation (Fig.2), we observed no effect of adoptive transfer of CD4+ cells on OVA-induced AHR in ROCK2+/- mice: airway responsiveness was not different in OVA challenged ROCK2+/- mice treated with saline versus those treated with CD4+ cells (Fig.5).
Figure 5.

Attenuation of allergen-induced AHR in ROCK2+/- in mice is independent of CD4+ cells. (A) newtonian resistance - Rn, a measure of central airways response (B) the coefficient of lung tissue damping - G, and (C) the coefficient of lung tissue elastance - H. Both G and H are indices of changes in the lung periphery including airway closure. Results are mean ± SEM of data from 8-21 mice per group. * p<0.05 versus PBS in mice with same genotype and treatment; + p<0.05 versus WT mice; and # p<0.05 versus ROCK2+/- mice treated with saline.
Genetic deletion of ROCK2 in CD4+ cells
The data from the experiments above using adoptive transfer of CD4 cells (Fig.2) suggested that ROCK2 acting within CD4+ cells was required for OVA-induced eosinophil recruitment, Th2 cytokine production, and mucous hypersecretion but not AHR. To confirm such a role for ROCK2, we genetically depleted ROCK2 specifically in CD4+ cells by crossing ROCK2flox/flox mice with mice expressing Cre recombinase under control of a CD4 promoter (CD4-Cre mice). Interestingly, OVA-induced increases in BAL lymphocytes, and IL-13 were not different in ROCK2CD4cre versus control mice (ROCK2flox/flox and CD4Cre mice) (Fig.6A,B), nor was there any difference in the number of pulmonary IL-13+CD4+ (Th2) cells (Fig. 6C) in ROCK2CD4cre versus ROCK2flox/flox mice). Genetic deletion of ROCK2 in CD4 cells also failed to affect BAL eosinophils (Fig.6D) and to prevent allergen-induced AHR (Fig. 6E).
Figure 6.

Genetic ablation of ROCK2 in CD4 cells does not alter allergen-induced inflammation or AHR. (A) BAL lymphocytes, (B) BAL IL-13, (C) pulmonary IL-13+ Th2 cells, (D) BAL eosinophils, (E) airway responsiveness assessed using changes in G, and (F) pulmonary Th17 cells (CD45+CD3+CD4+IL17+ cells). Airway responsiveness assessed using Rn and H is found in Figure E3. Results are mean ± SEM of data from n=5-12 mice per group, *p<0.05 vs PBS challenge and # p<0.05 vs control mice. Control mice in all studies except flow cytometry were a combination of ROCK2flox/flox and CD4-Cre mice. In flow-cytometric analysis control group was composed only by ROCK2flox/flox mice.
It has been previously established that ROCK2 is required for induction of Th17 cells in response to other stimuli [51, 52]. Hence, to confirm functional ROCK2 depletion within CD4+ cells in the lungs of ROCK2CD4cre mice, we examined pulmonary Th17 cells. Compared to ROCK2flox/flox mice, pulmonary IL-17A+CD4+ cells were substantially reduced in both PBS and OVA challenged ROCK2CD4cre mice (Fig.6F).
Discussion
Our data indicate an important role for ROCK2 in allergic airways disease: OVA-induced cellular inflammation, type 2 cytokine production, mucous hypersecretion, and AHR were each substantially reduced in ROCK2+/- versus WT mice treated with saline (Figs. 2,4,5), consistent with previous reports [13, 17]. The pan ROCK inhibitors, fasudil and Y-27632, also attenuate these outcomes in allergen challenged mice [16, 17, 53]. Together, the data indicate a key role for ROCK2 in allergen- induced eosinophil recruitment, mucous cell hyperplasia, and AHR. Data from this study allow us to begin to determine the cellular site of action of ROCK2 in mediating these events, as described below.
Administration of CD4+ cells from ROCK2 sufficient OT-II mice restored BAL lymphocytes and their type 2 cytokines, IL-13 and IL-5, in ROCK2+/- mice (Fig.2). Administration of these CD4+ cells also restored eosinophilic inflammation and mucous cell hyperplasia (Fig.2,4), consistent with the known role for IL-5 as an eosinophil survival factor [54] and the requirement for IL-13 for allergen induced mucous cell hyperplasia [45]. Nevertheless, genetically depleting ROCK2 only in CD4+ cells had no effect on OVA-induced increases in BAL lymphocytes, or associated changes in IL-13 expression or eosinophil migration (Fig.6). How can these observations be reconciled? First, we cannot rule out the possibility that the observed effects of adoptive transfer were the result not of CD4+ cells, but of some other spleen cells transferred with them, since the cells transferred were not 100% CD4 cells (Figure S1). Second, the transendothelial migration of lymphocytes from the blood into tissues occurs in a manner that reportedly involves ROCK-dependent signaling events in both the lymphocytes and the endothelial cells [6, 55]. As described above, we observed impaired OVA-induced lymphocyte recruitment to the lungs only when both endothelial cells and lymphocytes had impaired ROCK2 expression (data summarized in Fig.7). Thus, it is may be that normal expression and function of ROCK2 in either lymphocytes or in pulmonary vascular endothelial cells is sufficient for transmigration of lymphocytes into the lungs: though reduced compared to normal, lymphocyte/endothelial cell adhesion is still sufficiently strong in mice expressing normal ROCK2 in either cell type to permit normal lymphocyte transmigration (Fig.7). Additional experiments in endothelial cell specific ROCK2-/- mice will be required to address this possibility.
Figure 7.
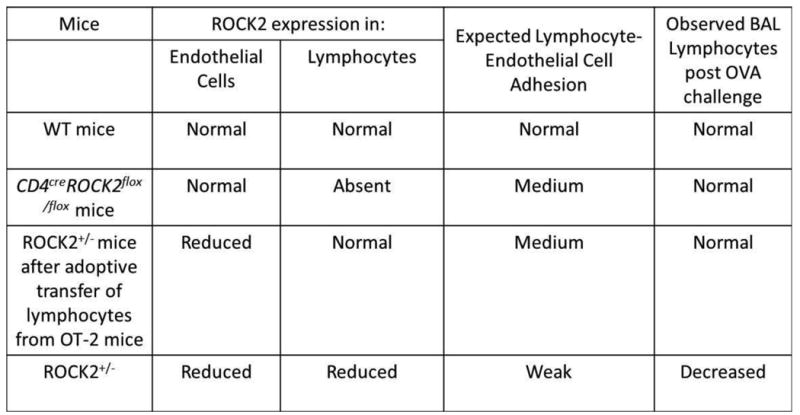
Summary of effects of various deletions and manipulations on BAL lymphocytes.
Others have reported that ROCK contributes to T cell migration and proliferation [13, 20-23]. Hence, we were somewhat surprised by the lack of effect of CD4+ cell specific ROCK2 deficiency on OVA-induced lymphocyte recruitment and IL-13 expression (Fig.6). However, the known effects of ROCK within lymphocytes [6, 13, 20-23] are largely based on experiments with ROCK inhibitors like Y-27632, that block both ROCK1 and ROCK2. Taken together with our observations (Fig.6), the data suggest that effects of ROCK within lymphocytes are mediated by ROCK1 rather than ROCK2.
ROCK2 also contributed to OVA-induced AHR (Fig.5). In mice, IL-13 is required for allergen-induced AHR [56-58] and BAL IL-13 was also reduced in ROCK2+/- versus WT mice challenged with OVA (Fig.2B). However, returning BAL IL-13 to normal WT levels by administration of CD4+ cells to ROCK2+/- mice (Fig.2B) did not reverse the attenuated AHR in these mice (Fig.5). These data suggest that reduced BAL IL-13 in ROCK2+/- mice was not the cause of their attenuated AHR. Instead, actions of ROCK2 within ASM cells likely account for this phenomenon. Agonists like methacholine increase intracellular Ca++ in ASM causing myosin light chain (MLC) phosphorylation, an event required for ASM contraction. ROCKs phosphorylate the myosin binding subunit of MLCP, thereby inhibiting it [59]. MLCP dephosphorylates MLC[59]. By inhibiting MLCP, ROCK prolongs MLC phosphphorylation and promotes smooth muscle contraction. Consequently, ROCK inhibitors or genetic insufficiency in ROCK1 or ROCK2 attenuate contractile responses in tracheal rings stimulated with cholinergic agonists [60, 61]. Similarly, the ability of IL-17A to augment ROCK2 expression contributes to the ability of IL-17A to augment contractile responses of ASM [62]. The observation that ROCK2 specific inhibitors attenuate contractile agonist-induced force generation in cultured human ASM cells [63] also indicates that ROCK2 in particular can promote ASM contraction. Thus, following allergen challenge, ROCK2 acting within ASM cells likely contributes not only to eotaxin release but also to AHR. Experiments with smooth muscle specific ROCK2 deficient mice will ultimately be needed to directly test this hypothesis.
Although we did not observe any significant attenuation of airway eosinophilia, BAL type 2 cytokines, or AHR in mice lacking ROCK2 only in CD4 cells (Fig.6), Th17 cells were markedly reduced in these mice (Fig.6F). ROCK2 phosphorylates IRF4, a transcription factor required for the production of IL-17 and is required for the induction of Th17 cells [51]. Hence, our data (Fig.6F) confirm functional depletion of ROCK2 within CD4+ cells in the lungs of these mice. This attenuation of Th17 cells without changes in the major features of allergic airways disease (Fig.6) is consistent with data indicating that IL-17A is not required for allergic airways disease in this model [64]. The observation that ROCK2CD4cre mice had marked reductions in Th17 cells is consistent with the observation that ROCK2 specific inhibitor, KD-025, attenuates the progression of collagen-induced arthritis in mice, a Th17 dependent process [52]. KD-025 also reduces IL-17 production in human T cells without affecting IFNγ production [52]. Together with our data indicating no effect of CD4+ specific ROCK2 deficiency on IL-13 expression (Fig.6), the data also indicate that Th17 cells are more dependent upon ROCK2 than other CD4+ cells (Th1 and Th2). Whether ROCK2 plays a role in IL-17A expression by other cells, like γδ T cells and innate lymphoid cells type 3 (ILC3), remains to be established.
In summary, our data indicate that ROCK2 is a potential therapeutic target for allergic asthma: allergen-induced inflammation, goblet cell hyperplasia, and AHR were each attenuated in ROCK2+/- versus WT mice. ROCK2 in CD4+ T cells does not appear to be the required for allergic airways responses. Instead, ASM cells and endothelial cells necessary for transvascular migration of lymphocytes, appear to be the primary ROCK2 effector cells contributing to allergic airways responses. The pan ROCK inhibitor, fasudil, is currently in clinical trials in the US for targeting a number of diseases (see ClinicalTrials.gov NCT00120718, NCT00670202, NCT00498615) but it may not be feasible for treatment of asthma since concurrent inhibition of both ROCK isoforms may produce undesirable effects such as hypotension [65]. However, selective ROCK2 inhibitors (e.g. KD-025) are also in development [52]. Greater understanding of the site and mechanism of ROCK2 may permit the use of ROCK2 selective inhibitors for reduction of the symptoms of allergic asthma when conventional asthma therapies have failed. Finally, current asthma treatment poorly controls mucous hypersecretion [66], and targeting ROCK2 may be beneficial in decreasing the mucus hypersecretion in asthmatics.
Supplementary Material
Figure S1. Representative flow cytometric data from splenocytes (CD4+ cells) before enrichment and after enrichment with negative selection using magnetic beads.
Figure S2. OVA-specific IgE levels measured by ELISA in serum from OVA sensitized WT and ROCK2+/- mice that were challenged with either aerosolized OVA or PBS. WT mice were treated with saline and ROCK2+/- mice were treated with either saline (SAL) or splenic CD4+ cells from OT-II mice (CD4+) as described in Fig.1A. Blood was harvested 48 h after the last challenge. Results are mean ± SEM of data from 8-10 mice per group. No statistical significance was observed among groups after one-way ANOVA.
Figure S3. Bronchoalveolar lavage (BAL) lymphocytes (A), and eosinophils (B), and airway responsiveness assessed using Newtonian resistance (Rn) or the coefficients of lung tissue damping (G) or elastance (H) (panels C,D, and E, respectively) in WT mice that were sensitized and challenged with aerosolized OVA. These mice were treated with saline (SAL) or splenic CD4+ cells from OT-II mice (CD4+) as described in Fig.1A. Results are mean ± SEM of data from 7-9 mice per group.
Figure S4. Newtonian resistance (Rn) and tissue elastance (H) from ROCK2CD4Cre and control mice sensitized and challenged with OVA or PBS. Results are mean ± SEM of data from 5-12 mice per group. ROCK2flox/flox and CD4-Cre data were combined as a control group. Factorial ANOVA, * p<0.05 versus PBS in mice with same genotype.
Acknowledgments
This study was supported by National Institute of Health grants: HL091933, P30ES000002, and HL052233. JAM was supported by F32 ES022556. The authors would also like to thank the Vermont Lung Center (VLC) at University of Vermont, and Drs. Charles Irvin and Matthew Poynter at VLC for use of instrumentation for morphometric analysis. VLC is supported by NIH IDeA COBRE P30GM103532. The authors also acknowledge the valuable input from Drs. Jennifer Mitchel and Jin-Ah Park, MIPS, Harvard T.H. Chan School of Public Health.
References
- 1.Leung T, et al. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16(10):5313–27. doi: 10.1128/mcb.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsui T, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15(9):2208–16. [PMC free article] [PubMed] [Google Scholar]
- 3.Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522(Pt 2):177–85. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang H, et al. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. American journal of physiology, Cell physiology. 2005;289(2):C352–60. doi: 10.1152/ajpcell.00111.2005. [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeldt HM, et al. Sphingosine-1-phosphate stimulates contraction of human airway smooth muscle cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17(13):1789–99. doi: 10.1096/fj.02-0836com. [DOI] [PubMed] [Google Scholar]
- 6.Smith A, et al. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. Journal of cell science. 2003;116(Pt 15):3123–33. doi: 10.1242/jcs.00606. [DOI] [PubMed] [Google Scholar]
- 7.Alblas J, et al. Activation of Rhoa and ROCK are essential for detachment of migrating leukocytes. Mol Biol Cell. 2001;12(7):2137–45. doi: 10.1091/mbc.12.7.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Worthylake RA, et al. RhoA is required for monocyte tail retraction during transendothelial migration. The Journal of cell biology. 2001;154(1):147–60. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adachi T, et al. The functional role of rho and rho-associated coiled-coil forming protein kinase in eotaxin signaling of eosinophils. J Immunol. 2001;167(8):4609–15. doi: 10.4049/jimmunol.167.8.4609. [DOI] [PubMed] [Google Scholar]
- 10.Schaafsma D, et al. Allergic sensitization enhances the contribution of Rho-kinase to airway smooth muscle contraction. Br J Pharmacol. 2004;143(4):477–84. doi: 10.1038/sj.bjp.0705903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiba Y, Misawa M. The role of RhoA-mediated Ca2+ sensitization of bronchial smooth muscle contraction in airway hyperresponsiveness. J Smooth Muscle Res. 2004;40(4-5):155–67. doi: 10.1540/jsmr.40.155. [DOI] [PubMed] [Google Scholar]
- 12.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–58. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 13.Zhu M, et al. Role of Rho kinase isoforms in murine allergic airway responses. Eur Respir J. 2011;38(4):841–50. doi: 10.1183/09031936.00125010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henry PJ, et al. Inhibitors of prostaglandin transport and metabolism augment protease-activated receptor-2-mediated increases in prostaglandin E2 levels and smooth muscle relaxation in mouse isolated trachea. J Pharmacol Exp Ther. 2005;314(3):995–1001. doi: 10.1124/jpet.105.086124. [DOI] [PubMed] [Google Scholar]
- 15.Schaafsma D, et al. Inhalation of the Rho-kinase inhibitor Y-27632 reverses allergen-induced airway hyperresponsiveness after the early and late asthmatic reaction. Respir Res. 2006;7:121. doi: 10.1186/1465-9921-7-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaafsma D, et al. The inhaled Rho kinase inhibitor Y-27632 protects against allergen-induced acute bronchoconstriction, airway hyperresponsiveness, and inflammation. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L214–9. doi: 10.1152/ajplung.00498.2007. [DOI] [PubMed] [Google Scholar]
- 17.Taki F, et al. Effects of Rho-kinase inactivation on eosinophilia and hyper-reactivity in murine airways by allergen challenges. Clin Exp Allergy. 2007;37(4):599–607. doi: 10.1111/j.1365-2222.2007.02693.x. [DOI] [PubMed] [Google Scholar]
- 18.Grunig G, et al. Requirement for IL-13 independently of IL-4 in experimental asthma [see comments] Science. 1998;282(5397):2261–3. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wills-Karp M, et al. Interleukin-13: central mediator of allergic asthma [see comments] Science. 1998;282(5397):2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 20.Vicente-Manzanares M, et al. A role for the Rho-p160 Rho coiled-coil kinase axis in the chemokine stromal cell-derived factor-1alpha-induced lymphocyte actomyosin and microtubular organization and chemotaxis. J Immunol. 2002;168(1):400–10. doi: 10.4049/jimmunol.168.1.400. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, et al. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin II. Proc Natl Acad Sci U S A. 2013;110(29):11976–81. doi: 10.1073/pnas.1306180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bardi G, Niggli V, Loetscher P. Rho kinase is required for CCR7-mediated polarization and chemotaxis of T lymphocytes. FEBS letters. 2003;542(1-3):79–83. doi: 10.1016/s0014-5793(03)00351-x. [DOI] [PubMed] [Google Scholar]
- 23.Tharaux PL, et al. Rho kinase promotes alloimmune responses by regulating the proliferation and structure of T cells. Journal of immunology. 2003;171(1):96–105. doi: 10.4049/jimmunol.171.1.96. [DOI] [PubMed] [Google Scholar]
- 24.Aihara M, et al. Effect of Y-27632 on release of cytokines from peripheral T cells in asthmatic patients and normal subjects. Int Immunopharmacol. 2004;4(4):557–61. doi: 10.1016/j.intimp.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa O, et al. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392(2):189–93. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 26.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4(6):446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 27.Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170(3):443–53. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneko T, et al. Identification of calponin as a novel substrate of Rho-kinase. Biochem Biophys Res Commun. 2000;273(1):110–6. doi: 10.1006/bbrc.2000.2901. [DOI] [PubMed] [Google Scholar]
- 29.Yoneda A, et al. Fibronectin matrix assembly requires distinct contributions from Rho kinases I and -II. Mol Biol Cell. 2007;18(1):66–75. doi: 10.1091/mbc.E06-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sapet C, et al. Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2. Blood. 2006;108(6):1868–76. doi: 10.1182/blood-2006-04-014175. [DOI] [PubMed] [Google Scholar]
- 31.Zhang YM, et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. Faseb J. 2006;20(7):916–25. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 32.Thumkeo D, et al. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol Cell Biol. 2003;23(14):5043–55. doi: 10.1128/MCB.23.14.5043-5055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noma K, et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118(5):1632–44. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okamoto R, et al. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013;27(4):1439–49. doi: 10.1096/fj.12-217018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams AS, et al. Role of the adiponectin binding protein, T-cadherin (cdh13), in allergic airways responses in mice. PLoS One. 2012;7(7):e41088. doi: 10.1371/journal.pone.0041088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verbout NG, et al. Impact of adiponectin overexpression on allergic airways responses in mice. J Allergy (Cairo) 2013;2013:349520. doi: 10.1155/2013/349520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston RA, et al. Type I interleukin-1 receptor is required for pulmonary responses to subacute ozone exposure in mice. Am J Respir Cell Mol Biol. 2007;37(4):477–84. doi: 10.1165/rcmb.2006-0315OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu M, et al. Role of TNFR1 in the innate airway hyperresponsiveness of obese mice. J Appl Physiol (1985) 2012;113(9):1476–85. doi: 10.1152/japplphysiol.00588.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams AS, et al. Obesity and airway responsiveness: Role of TNFR2. Pulm Pharmacol Ther. 2012 doi: 10.1016/j.pupt.2012.05.001. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kasahara DI, et al. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol. 2012;188(9):4558–67. doi: 10.4049/jimmunol.1102363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore PE, et al. IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L847–53. doi: 10.1152/ajplung.00245.2001. [DOI] [PubMed] [Google Scholar]
- 42.Feng Y, et al. Discovery of substituted 4-(pyrazol-4-yl)-phenylbenzodioxane-2-carboxamides as potent and highly selective Rho kinase (ROCK-II) inhibitors. J Med Chem. 2008;51(21):6642–5. doi: 10.1021/jm800986w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herskowitz JH, et al. Pharmacologic inhibition of ROCK2 suppresses amyloid-beta production in an Alzheimer's disease mouse model. J Neurosci. 2013;33(49):19086–98. doi: 10.1523/JNEUROSCI.2508-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, et al. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J Immunol. 1999;162(5):2477–87. [PubMed] [Google Scholar]
- 45.Zhu Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103(6):779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirst SJ, et al. Selective induction of eotaxin release by interleukin-13 or interleukin-4 in human airway smooth muscle cells is synergistic with interleukin-1beta and is mediated by the interleukin-4 receptor alpha-chain. Am J Respir Crit Care Med. 2002;165(8):1161–71. doi: 10.1164/ajrccm.165.8.2107158. [DOI] [PubMed] [Google Scholar]
- 47.Kuperman DA, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8(8):885–9. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 48.Ghaffar O, et al. Constitutive and cytokine-stimulated expression of eotaxin by human airway smooth muscle cells. Am J Respir Crit Care Med. 1999;159(6):1933–42. doi: 10.1164/ajrccm.159.6.9805039. [DOI] [PubMed] [Google Scholar]
- 49.Uehata M, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 50.Zuhdi Alimam M, et al. Muc-5/5ac mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways. Am J Respir Cell Mol Biol. 2000;22(3):253–60. doi: 10.1165/ajrcmb.22.3.3768. [DOI] [PubMed] [Google Scholar]
- 51.Biswas PS, et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest. 2010;120(9):3280–95. doi: 10.1172/JCI42856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zanin-Zhorov A, et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A. 2014;111(47):16814–9. doi: 10.1073/pnas.1414189111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Righetti RF, et al. Effects of Rho-kinase inhibition in lung tissue with chronic inflammation. Respir Physiol Neurobiol. 2014;192:134–46. doi: 10.1016/j.resp.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 54.Yamaguchi Y, et al. Analysis of the survival of mature human eosinophils: interleukin-5 prevents apoptosis in mature human eosinophils. Blood. 1991;78(10):2542–7. [PubMed] [Google Scholar]
- 55.Li H, et al. ROCK inhibitor fasudil attenuated high glucose-induced MCP-1 and VCAM-1 expression and monocyte-endothelial cell adhesion. Cardiovasc Diabetol. 2012;11:65. doi: 10.1186/1475-2840-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hamelmann E, et al. Antiinterleukin-5 antibody prevents airway hyperresponsiveness in a murine model of airway sensitization. Am J Respir Crit Care Med. 1997;155(3):819–25. doi: 10.1164/ajrccm.155.3.9117011. [DOI] [PubMed] [Google Scholar]
- 57.Walter DM, et al. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J Immunol. 2001;167(8):4668–75. doi: 10.4049/jimmunol.167.8.4668. [DOI] [PubMed] [Google Scholar]
- 58.Venkayya R, et al. The Th2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am J Respir Cell Mol Biol. 2002;26(2):202–8. doi: 10.1165/ajrcmb.26.2.4600. [DOI] [PubMed] [Google Scholar]
- 59.Shi J, Wei L. Rho Kinases in Cardiovascular Physiology and Pathophysiology: The Effect of Fasudil. J Cardiovasc Pharmacol. 2013;62(4):341–354. doi: 10.1097/FJC.0b013e3182a3718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaafsma D, et al. Differential Rho-kinase dependency of full and partial muscarinic receptor agonists in airway smooth muscle contraction. Br J Pharmacol. 2006;147(7):737–43. doi: 10.1038/sj.bjp.0706665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kasahara DI, et al. Abrogation of Airway Hyperresponsiveness but not Inflammation by Rho kinase Insufficiency. Clin Exp Allergy. 2014 doi: 10.1111/cea.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kudo M, et al. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012;18(4):547–54. doi: 10.1038/nm.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kasahara DI, et al. ROCK Insufficiency Attenuates Ozone-Induced Airway Hyperresponsiveness in Mice. Am J Physiol Lung Cell Mol Physiol. 2015:ajplung 00372 2014. doi: 10.1152/ajplung.00372.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzukawa M, et al. Epithelial cell-derived IL-25, but not Th17 cell-derived IL-17 or IL-17F, is crucial for murine asthma. J Immunol. 2012;189(7):3641–52. doi: 10.4049/jimmunol.1200461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerthoffer WT, Solway J, Camoretti-Mercado B. Emerging targets for novel therapy of asthma. Curr Opin Pharmacol. 2013;13(3):324–30. doi: 10.1016/j.coph.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gras D, et al. Bronchial epithelium as a target for innovative treatments in asthma. Pharmacol Ther. 2013;140(3):290–305. doi: 10.1016/j.pharmthera.2013.07.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative flow cytometric data from splenocytes (CD4+ cells) before enrichment and after enrichment with negative selection using magnetic beads.
Figure S2. OVA-specific IgE levels measured by ELISA in serum from OVA sensitized WT and ROCK2+/- mice that were challenged with either aerosolized OVA or PBS. WT mice were treated with saline and ROCK2+/- mice were treated with either saline (SAL) or splenic CD4+ cells from OT-II mice (CD4+) as described in Fig.1A. Blood was harvested 48 h after the last challenge. Results are mean ± SEM of data from 8-10 mice per group. No statistical significance was observed among groups after one-way ANOVA.
Figure S3. Bronchoalveolar lavage (BAL) lymphocytes (A), and eosinophils (B), and airway responsiveness assessed using Newtonian resistance (Rn) or the coefficients of lung tissue damping (G) or elastance (H) (panels C,D, and E, respectively) in WT mice that were sensitized and challenged with aerosolized OVA. These mice were treated with saline (SAL) or splenic CD4+ cells from OT-II mice (CD4+) as described in Fig.1A. Results are mean ± SEM of data from 7-9 mice per group.
Figure S4. Newtonian resistance (Rn) and tissue elastance (H) from ROCK2CD4Cre and control mice sensitized and challenged with OVA or PBS. Results are mean ± SEM of data from 5-12 mice per group. ROCK2flox/flox and CD4-Cre data were combined as a control group. Factorial ANOVA, * p<0.05 versus PBS in mice with same genotype.