

Abstract
Zika virus is an emerging mosquito‐borne pathogen that is associated with Guillain–Barré syndrome in adults and microcephaly and other neurological defects in newborns. Despite being declared an international emergency by the World Health Organization, comparatively little is known about its biology. Here, we investigate the strategies employed by the virus to suppress the host antiviral response. We observe that once established, Zika virus infection is impervious to interferon treatment suggesting that the virus deploys effective countermeasures to host cell defences. This is confirmed by experiments showing that Zika virus infection impairs the induction of type‐I interferon as well as downstream interferon‐stimulated genes. Multiple viral proteins affect these processes. Virus‐mediated degradation of STAT2 acts to reduce type‐I and type‐III interferon‐mediated signaling. Further, the NS5 of Zika virus binds to STAT2, and its expression is correlated with STAT2 degradation by the proteasome. Together, our findings provide key insights into how Zika virus blocks cellular defense systems. This in turn is important for understanding pathogenesis and may aid in designing antiviral therapies.
Keywords: flavivirus, interferon response, NS5, STAT2, Zika virus
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Zika virus (ZIKV) is an emerging arbovirus belonging to the family Flaviviridae and is transmitted by mosquitoes of the Aedes spp 1. The virus was originally identified in Uganda in 1947, but the first major reported outbreak of this pathogen was not until 2007 in the Micronesian island of Yap 2. Over the next seven years, outbreaks were reported in other Pacific islands 3. In 2015, the largest Zika pandemic to date began in Brazil 4 and has since rapidly spread throughout the Americas infecting nearly two million people. Although most Zika cases in Brazil were asymptomatic or manifested as a flu‐like illness, there was a very large increase in the incidence of Guillain–Barré syndrome 5 and neurological birth defects including microcephaly in newborns 6. It is now generally accepted that ZIKV is a teratogenic agent that crosses the placental barrier and inflicts severe neurological damage in developing foetuses 7, 8. In addition to transmission by mosquito bites, ZIKV can be spread by sexual contact 9, 10.
The interferon (IFN) system plays a crucial role in combating viral infections in mammalian cells. The genetic material from incoming RNA viruses is detected by toll‐like receptors present on the cell surface and in endosomal compartments, whereas double‐strand RNA (dsRNA) from intracellular viral replication is detected by the cytoplasmic RNA sensors RIG‐I and MDA5 11. This leads to activation of transcription factors such as interferon regulatory factor 3 (IRF3) and NF‐κB resulting in IFN production. In the IFN signaling (effector) phase, secreted IFN binds to cognate receptors and initiates a signaling cascade that results in transcription of IFN‐stimulated genes (ISGs), which encode proteins that inhibit virus replication and spread. Productive replication of flaviviruses is linked with their ability to block IFN induction and signaling pathways. For example, Dengue (DENV) and West Nile viruses, which are close relatives to ZIKV, have evolved diverse strategies to block the IFN response 12. The protective role of the IFN system against flaviviruses is evident from the observation that unlike wild‐type mice, type‐I IFN signaling‐deficient mice are highly susceptible to infection by both ZIKV 13 and DENV 14, 15.
In this study, we investigated the strategies used by ZIKV to suppress host innate immune defenses in order to establish productive infection. We observed that ZIKV‐infected cells failed to mount a robust immune response, particularly in the early phase of infection. Although ZIKV replication was significantly inhibited in cells that were primed with the dsRNA analogue, poly(I:C) or by pre‐treatment with IFNs, once established, ZIKV replication was impervious to either treatment. This suggests that the virus employs effective countermeasures to the innate immune response, and indeed, we observed strong suppression of IFN and ISG expression in ZIKV‐infected cells. Viral proteins NS1, NS4A, and NS5 were identified as key suppressors of type‐I IFN induction. Moreover, we observed a significant reduction in type‐I and type‐III IFN signaling in ZIKV‐infected cells as well as degradation of the antiviral transcriptional activator STAT2, a key component of these signaling pathways. Further analysis identified proteasomal degradation as the mechanism by which STAT2 is depleted in ZIKV‐infected cells. Finally, we identified ZIKV NS5 protein as a potent inhibitor of IFN signaling that acts by inducing degradation of STAT2. A direct interaction between ZIKV NS5 and STAT2 was detected, which was primarily mediated by the methyltransferase (MTase) domain of NS5. These data suggest that ZIKV employs multiple mechanisms to block the host IFN response.
Results and Discussion
ZIKV inhibits the host cell IFN response
To establish an environment that is conducive to replication, many viruses block the host cell IFN response through multiple mechanisms. For example, DENV inhibits the RIG‐I‐mediated responses by blocking both induction and downstream signaling of type‐I IFN, a process that involves the viral proteins NS2B‐3, NS4A, NS4B, and NS5 16, 17. As a first step toward understanding how ZIKV affects the IFN system, we measured the induction of Ifnb and the downstream antiviral gene Ifit1 by qRT–PCR at various times post‐infection. During the early phase of ZIKV infection, there was delayed induction of Ifnb and Ifit1 and peak expression of both genes was not observed until 24–48 h (Fig EV1). This was not due to an inability of cells to mount a rapid response as it can be seen that poly(I:C) treatment induced robust expression of these genes by 12 h. The delayed IFN response in ZIKV‐infected cells could be due to an inability of the cells to efficiently detect viral RNA and/or an active suppression of the RNA‐mediated antiviral signaling pathway by ZIKV. To address these potential scenarios, we pre‐treated cells with poly(I:C) to activate the innate immune response and then examined the effect on ZIKV infection. As shown in Fig 1A, virus replication was reduced by > 80% in treated cells, indicating that at early stages of infection, the cells can mount a strong antiviral response. However, addition of poly(I:C) to cells that were pre‐infected with ZIKV had little effect on replication (Fig 1A), suggesting that the virus efficiently blocks dsRNA‐stimulated IFN response.
Figure EV1. The IFN response is delayed during ZIKV infection.
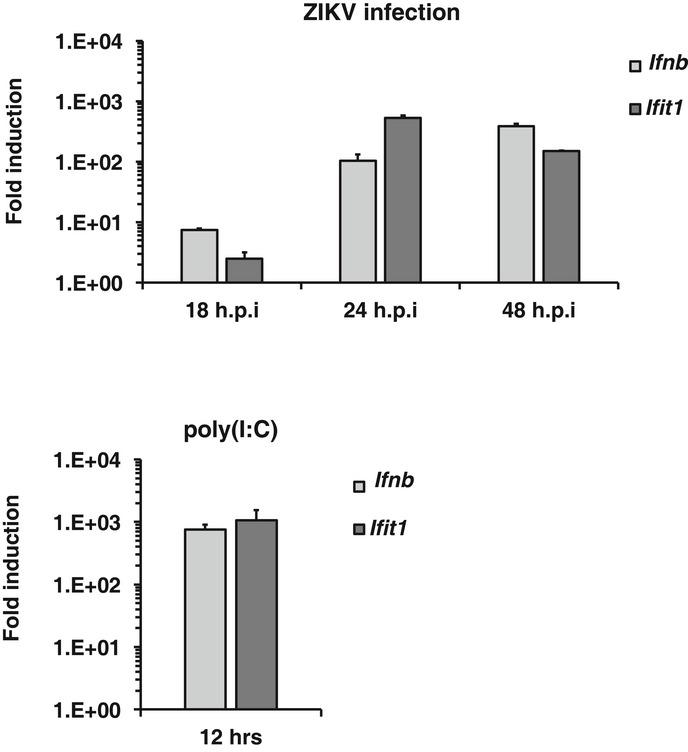
A549 cells were infected with ZIKV (MOI = 2) for 18, 24 and 48 h. At each time point, relative levels of Ifnb and Ifit1 mRNA (compared to mock‐infected cells) were determined by qRT–PCR (upper panel). As a positive control for IFN induction, cells were transfected with 2 μg of poly(I:C) for 12 h (lower panel). Levels of Ifnb and Ifit1 mRNA (compared to untreated cells) are shown. Values are expressed as mean ± standard error. N = 3.
Figure 1. ZIKV inhibits induction of the IFN response.
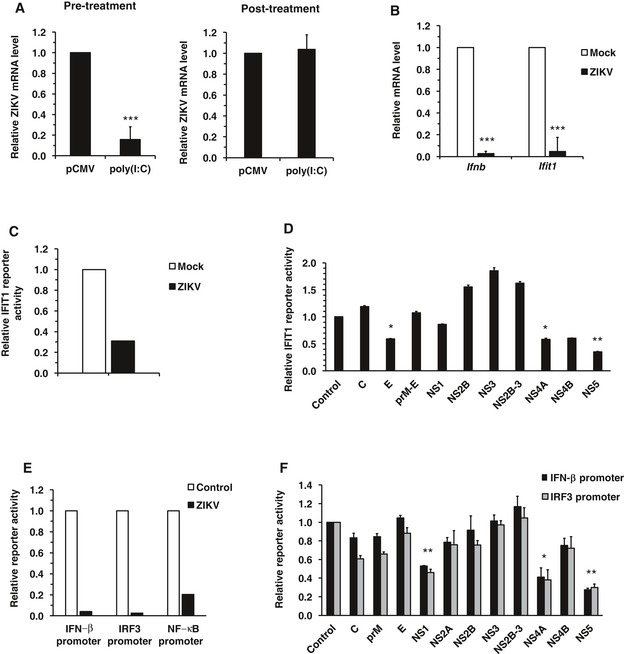
- A549 cells were transfected with 2 μg of poly(I:C) for 6 h and then infected with ZIKV (MOI = 3) for 18 h (pre‐treatment) or infected with ZIKV first for 6 h and then transfected with poly(I:C) for 12 h (post‐treatment). Levels of ZIKV genomic RNA were determined by qRT–PCR. Values are expressed as mean ± standard error. ***P < 0.001 (Student's t‐test), N = 3.
- A549 cells infected with ZIKV (MOI = 3) for 6 h were transfected with 2 μg poly(I:C) for 12 h after which levels of Ifnb and Ifit1 mRNA were determined by qRT–PCR. Values are expressed as mean ± standard error. ***P < 0.001 (Student's t‐test), N = 3.
- A549 cells were infected with ZIKV (MOI = 5) for 16 h and then transfected with an IFIT1 promoter‐driven firefly luciferase plasmid (pGL3B/561) and a constitutively active Renilla luciferase construct (pRL‐TK), as well as 1 μg of poly(I:C). Eight hours later, cell lysates were harvested and subjected to luciferase assay. Values are expressed as mean of two independent experiments, N = 2.
- HEK293T cells were transfected with plasmids encoding each of the 10 ZIKV proteins. Sixteen hours later, they were transfected with an IFIT1 promoter‐driven firefly luciferase plasmid (pGL3B/561) and a constitutively active Renilla luciferase construct (pRL‐TK), as well as 1 μg of poly(I:C). Eight hours later, cell lysates were harvested and subjected to luciferase assay. C, capsid; E, envelope. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test), N = 4.
- A549 cells were infected with ZIKV (MOI = 5) for 16 h and then transfected with the indicated promoter‐driven firefly luciferase plasmids and a constitutively active Renilla luciferase construct (pRL‐TK), as well as 1 μg of poly(I:C). Eight hours later, cell lysates were harvested and subjected to luciferase assay. Values are expressed as mean of two independent experiments, N = 2.
- HEK293T cells were transfected with plasmids encoding individual ZIKV proteins, the indicated promoter‐driven firefly luciferase plasmids and a constitutively active Renilla luciferase construct (pRL‐TK), as well as 0.4 μg of poly(I:C). Twenty‐four hours later, cell lysates were harvested and subjected to luciferase assay. C, capsid; E, envelope. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test); N = 3.
To further investigate whether ZIKV modulates the induction and/or effector phase of the IFN response, we quantified poly(I:C)‐induced expression of Ifnb and Ifit1 in ZIKV‐infected and mock‐treated cells. As shown in Fig 1B, ZIKV infection strongly inhibited expression of Ifnb and Ifit1 indicating a suppression of both induction and signaling of IFNs. Consistent with these data, the activity of the IFIT1 promoter, which has both IRF3‐binding sites and interferon‐sensitive response element (ISRE), was reduced in ZIKV‐infected cells (Fig 1C). To determine which viral component(s) is responsible for suppressing the IFN system, we expressed individual ZIKV proteins and examined their effects on dsRNA‐induced IFN response by measuring activity of the IFIT1 promoter. Our data showed that IFIT1 promoter activity was affected by multiple ZIKV proteins, with the strongest suppression (~70%) observed in cells expressing NS5 and a moderate inhibition (40%) in those expressing E or NS4A (Fig 1D). To further understand how ZIKV inhibits the induction of type‐I IFNs, we examined the effect of viral infection on IFN‐β, IRF3, and NF‐κB reporter activities. These experiments revealed that ZIKV infection efficiently blocked poly(I:C)‐stimulated expression of all three reporters (Fig 1E). To identify the viral component(s) responsible for suppressing type‐I IFN production, we measured the activities of these reporters in the presence of individual ZIKV proteins. The expression of NS1, NS4A, and NS5 significantly inhibited the activities of both IFN‐β and IRF3 reporters (Fig 1F) while a moderate reduction in NF‐κB activity was observed in cells expressing NS5 (Fig EV2). Taken together, these data indicate that ZIKV is very efficient at blocking both the induction and effector phases of the type‐I IFN response, a process that is mediated by the viral proteins NS1, NS4A, and NS5.
Figure EV2. ZIKV NS5 reduces NF‐κB reporter activity.
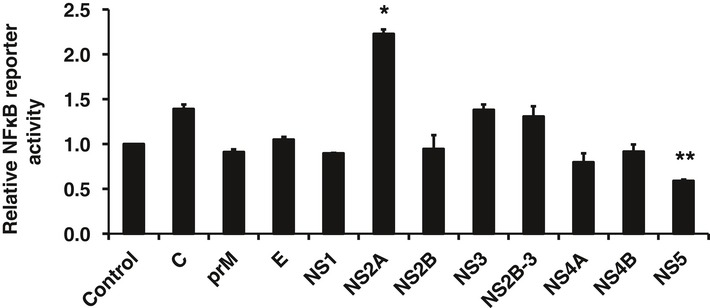
HEK293T cells were transfected with plasmids encoding different ZIKV proteins. Sixteen hours later, they were transfected with an NF‐κB promoter‐driven firefly luciferase plasmid (pNF‐κB‐luciferase) and a constitutively active Renilla luciferase construct (pRL‐TK), as well as 1 μg of poly(I:C). Twenty‐four hours later, cell lysates were harvested and subjected to luciferase assay. C, capsid; E, envelope. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test); N = 3.
ZIKV blocks IFN signaling
Recent studies in mouse models suggest that the effector phase of IFN response is important for controlling ZIKV infection and associated pathogenesis 13, 18. Here, we further investigated the effect of ZIKV infection on the IFN effector phase and the key viral components involved. To determine how sensitive ZIKV is to IFN‐mediated antiviral signaling, A549 cells were treated with IFN‐α, IFN‐γ, or IFN‐λ prior to infection and then replication was assessed. Whereas ZIKV replication was moderately affected by IFN‐λ, the virus was much more sensitive to pre‐treatment with IFN‐α or IFN‐γ (Fig 2A). These results are consistent with previous observations with ZIKV 19 and other flaviviruses such as DENV 20 indicating that in order to maintain a productive infection, the virus must employ strategies to block IFN‐dependent antiviral defense. To address the question of whether ZIKV actively suppresses IFN signaling, we infected cells with ZIKV followed by treatment with different IFNs. Data in Fig 2A show that viral replication was largely unaffected when IFNs were administered after productive ZIKV infection was already established. These results are consistent with data in Fig 2B showing that IFN‐induced expression of the ISG Ifit1 is substantially reduced by ZIKV infection. Interestingly, Ifit1 induction by IFN‐α and IFN‐λ, but not IFN‐γ, was suppressed in ZIKV‐infected cells (Fig 2B). This indicates that type‐I and type‐III IFN signaling but not type‐II signaling pathways are blocked by ZIKV. To further investigate this, we measured the IFN‐α and IFN‐γ signaling through ISRE‐ and gamma‐activated sequence (GAS) promoters, respectively, in ZIKV‐infected cells. We observed that IFN‐dependent ISRE signaling but not GAS signaling was reduced by ZIKV (Fig 2C). To determine which ZIKV protein mediates the subversion of type‐I IFN signaling, we expressed individual ZIKV proteins in HEK293T cells and examined their effects on the ISRE promoter activity. As shown in Fig 2D, expression of NS5 protein strongly reduced (90%) ISRE‐dependent IFN signaling. In summary, these data indicate that ZIKV interferes with the effector phase of the IFN response, a process mediated in large part by NS5 protein.
Figure 2. ZIKV infection blocks IFN signaling.
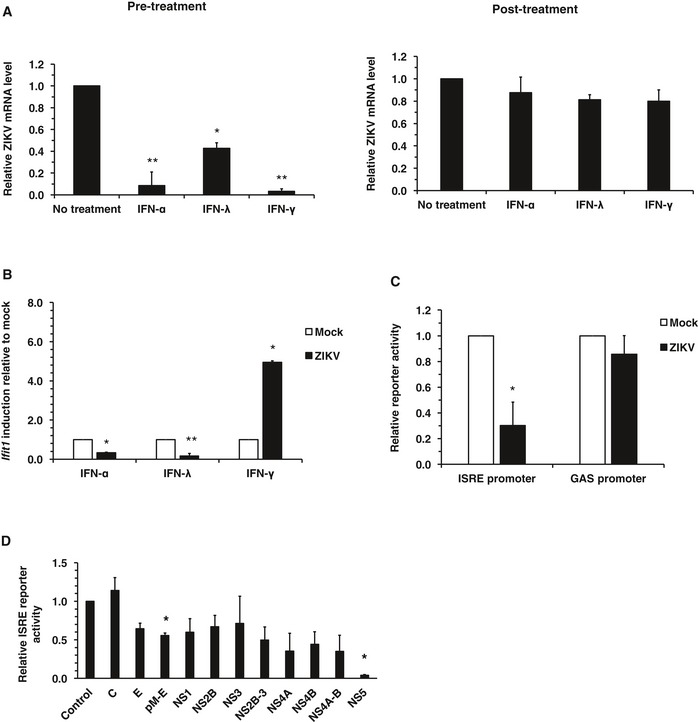
- A549 cells were treated with IFN‐α (100 U/ml), IFN‐λ (200 ng/ml), or IFN–γ (10 U/ml) for 6 h after which they were infected with ZIKV (MOI = 3) for 18 h (pre‐treatment). Alternatively, cells were first infected with ZIKV for 6 h followed by treatment with IFN for 12 h (post‐treatment). Total RNA was isolated, and levels of ZIKV genomic RNA were determined by qRT–PCR. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test); N = 3.
- A549 cells were infected with ZIKV (MOI = 3) for 6 h prior to addition of IFN‐α (100 U/ml), IFN‐λ (200 ng/ml), or IFN‐γ (10 U/ml) for 12 h. Total RNA was isolated, and levels of Ifit1 RNA were determined by qRT–PCR. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test); N = 3.
- A549 cells were infected with ZIKV (MOI = 5) for 6 h and then transfected with ISRE or GAS promoter‐driven firefly luciferase together with a constitutively active Renilla luciferase construct. Sixteen hours later, cells were treated with IFN‐α (100 U/ml) or IFN‐γ (10 U/ml) for 2 h. Cells lysates were harvested, and relative luciferase activity from ISRE and GAS promoters were determined. Values are expressed as mean ± standard error. *P < 0.05 (Student's t‐test); N = 3.
- HEK293T cells were transfected with ZIKV protein expression constructs together with an ISRE promoter‐driven firefly luciferase plasmid (pGL3B/561) and a constitutively active Renilla luciferase construct (pRL‐TK). After 24 h, they were treated with IFN‐α (100 U/ml) for 10 h. Cell lysates were then harvested and subjected to luciferase assays. C, capsid; E, envelope. Values are expressed as mean ± standard error. *P < 0.05 (Student's t‐test); N = 3.
ZIKV infection induces degradation of human STAT2
The transcriptional activator STAT2 is a critical component of antiviral defense systems that acts downstream of type‐I/III IFN receptor activation. Previous studies have shown that STAT2 is targeted by several viruses including respiratory syncytial virus 21, 22, Nipah virus 23, 24, and the closely related DENV 25. Initially, we examined whether levels of STAT2 protein were affected by ZIKV infection. Immunoblotting revealed that within 24 h, STAT2 protein levels were almost completely abolished in ZIKV‐infected cells, while STAT1 levels remained unchanged (Fig 3A). The fact that STAT1 was unaltered in ZIKV‐infected cells is consistent with our earlier data showing that type‐II IFN signaling is not significantly disrupted by ZIKV infection (Fig 2C). Analyses by confocal microscopy confirmed the immunoblot data showing that STAT2 was absent from ZIKV‐infected A549 cells (Fig 3C), whereas STAT1 was unaffected (Fig EV3A). The same phenotype was observed in primary human fetal astrocytes (Fig 3C), and it is tempting to speculate that the virus deploys this mechanism to undermine host IFN response in the fetal brain. Next, we examined the mechanism by which ZIKV induces STAT2 degradation. ZIKV‐infected cells were treated with inhibitors of the proteasome (MG132 and epoxomicin) or lysosomes (bafilomycin‐A1). STAT2 levels were rescued by treatment with both MG132 and epoxomicin (Fig 3B) but not bafilomycin‐A1 (Fig EV3B), indicating that ZIKV infection results in proteasome‐dependent STAT2 degradation.
Figure 3. ZIKV infection causes degradation of human STAT2.
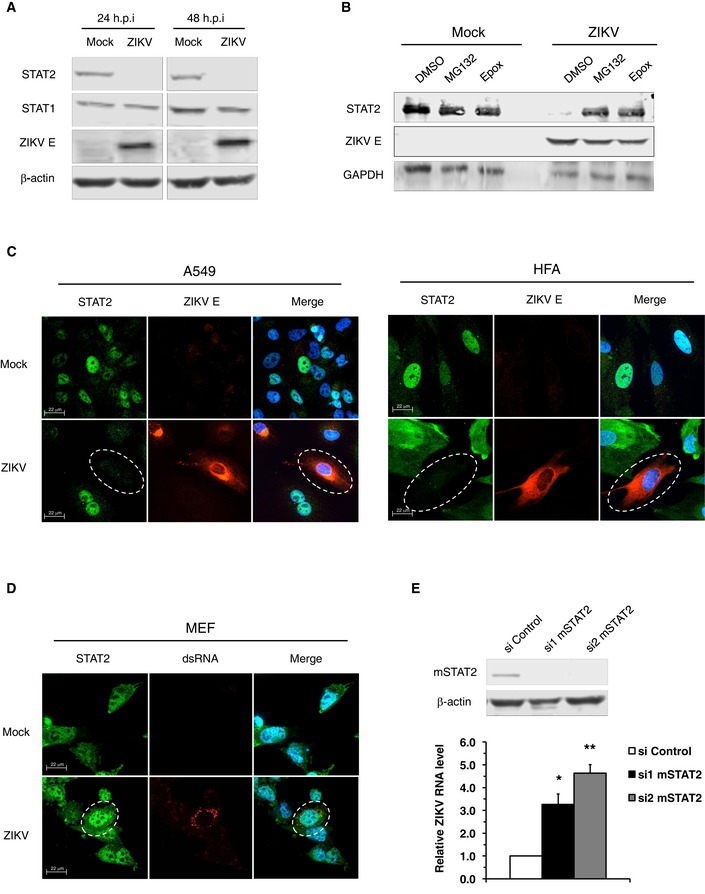
- A549 cells were infected with ZIKV (MOI = 5) for 24–48 h after which cell lysates were processed by SDS–PAGE and immunoblotting. Representative panels are shown. The experiment was repeated three times.
- A549 cells were infected with ZIKV (MOI = 5) for 24 h and then treated with DMSO, MG132 (20 μM), or epoxomicin (Epox; 400 nM) for 12 h. Cell lysates were processed by SDS–PAGE and immunoblotting. Representative panels are shown. The experiment was repeated three times.
- A549 cells (MOI = 2) or human primary fetal astrocytes (HFA) (MOI = 5) were infected with ZIKV for 24 and 48 h, respectively, after which IFN‐α (100 U/ml) was added for 2 h. Cells were then fixed and processed for indirect immunofluorescence. Images were acquired on a spinning disk confocal microscope with a 40× objective. Dashed line white circles indicated ZIKV‐infected cells. Representative panels are shown. The experiment was repeated two times.
- Mouse embryonic fibroblasts (MEF) were infected with ZIKV (MOI = 5) for 48 h after which IFN‐α (200 U/ml) was added for 2 h. Samples were processed for immunofluorescence assay as described in panel (C). Dashed line white circles indicated ZIKV‐infected cells. Representative panels are shown. The experiment was repeated two times.
- STAT2 levels in MEFs were decreased by transfection with siRNAs for 48 h after which the cells were infected with ZIKV (MOI = 5) for another 48 h. The silencing efficiency was determined by immunoblotting. Representative panels are shown. The experiment was repeated three times. The ZIKV replication was measured by qRT–PCR. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test); N = 3.
Figure EV3. ZIKV infection does not affect STAT1 levels.
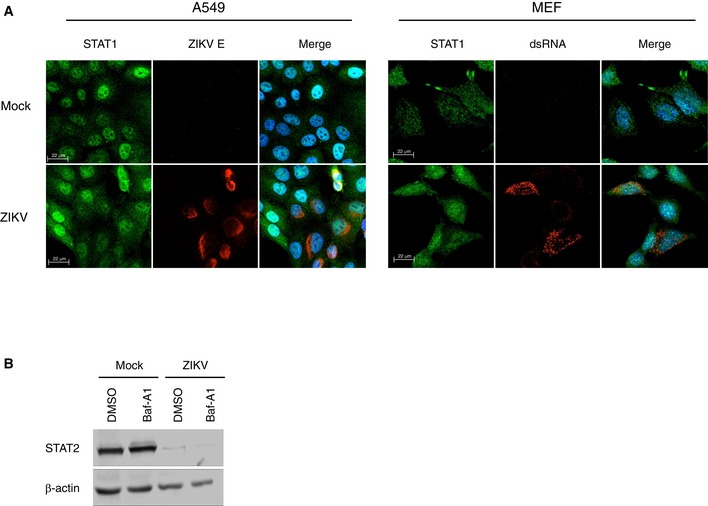
- A549 cells or mouse embryonic fibroblasts (MEFs) were infected with ZIKV (MOI = 2) for 24 and 48 h after which IFN‐α (100 U/ml) was added for 2 h. Cells were then fixed and processed for indirect immunofluorescence. Representative panels are shown. The experiment was repeated two times.
- A549 cells were infected with ZIKV (MOI = 5) for 24 h and then treated with DMSO or bafilomycin‐A1 (Baf‐A1) (400 nM) for 12 h. Cell lysates were processed by SDS–PAGE and immunoblotting. Representative panels are shown. The experiment was repeated three times.
Recent studies demonstrated that ZIKV is not lethal for wild‐type mice, whereas those lacking type‐I IFN receptors (AG129, Ifnar1 −/−) are highly susceptible to the virus 13, 18. Based on our data, we surmised that one of the limiting factors for productive infection in wild‐type mice could be the inability of ZIKV to induce STAT2 degradation in murine cells. Consistent with this hypothesis, we observed that ZIKV infection of mouse embryonic fibroblasts (MEFs) did not result in loss of STAT2 (Fig 3D). Moreover, transient knockdown of STAT2 in MEFs enhanced ZIKV replication by up to fivefold (Fig 3E). This suggests that in addition to IFNAR−/−, mice deficient in STAT2 may serve as an animal model for ZIKV infection. Collectively, these results suggest that targeting of STAT2 for degradation is a key mechanism by which ZIKV blocks IFN signaling in human cells.
ZIKV NS5 protein interacts with human STAT2 and induces its degradation
To identify the viral component(s) that are required for STAT2 degradation, we expressed individual ZIKV proteins in A549 cells and examined their effects on the levels of STAT2. STAT2 levels were dramatically reduced in cells expressing NS5, indicating a role for this protein in ZIKV‐induced degradation of STAT2 (Fig EV4). This result is consistent with our earlier observation that NS5 strongly suppresses IFN‐mediated ISRE signaling (Fig 2D). Previous studies have shown that NS5 of DENV also causes loss of STAT2 16, 26. However, unlike DENV NS5, ZIKV NS5‐mediated degradation of STAT2 did not require cotranslational endoproteolytic processing (Fig EV4). To further investigate the nature of NS5‐dependent degradation of STAT2, levels of STAT2 were compared in cells expressing each of the two functional domains of NS5, methyl transferase (MTase) and polymerase (RdRP). While STAT2 degradation was negligible in cells expressing the RdRP domain, levels of STAT2 were significantly lower in many cells expressing the MTase domain (Fig 4A and B). Methyltransferase activity was not required for STAT2 degradation as evidenced by the fact that levels of the protein were lower in cells expressing the S56A mutant (Fig 4A and B). Finally, unlike DENV NS5 16, the first 10 amino acid residues of ZIKV NS5 are not important for degradation of STAT2 (Fig 4A and B).
Figure EV4. Expression of ZIKV NS5 induces degradation of STAT2.
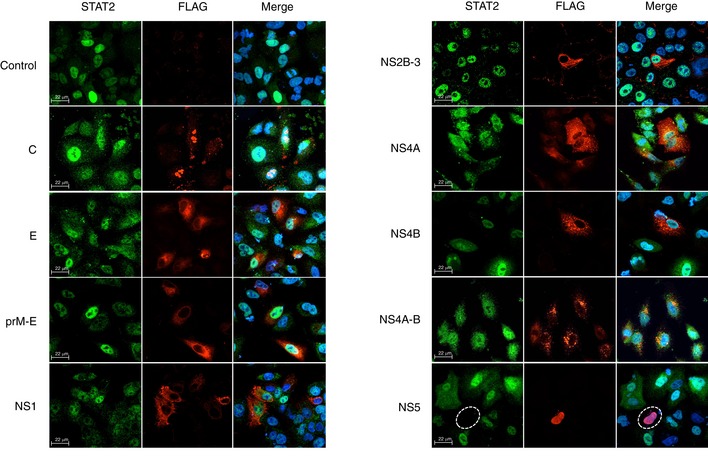
A549 cells were transfected with control plasmid pcDNA 3.1 or plasmids encoding FLAG‐tagged ZIKV proteins. At 48 h, post‐transfection cells were treated with IFN‐α (100 U/ml) for 2 h and then processed for indirect immunofluorescence. Images were acquired on a spinning disk confocal microscope with a 40× objective. NS5‐positive cells are indicated by dashed white circles. Representative panels are shown. The experiment was repeated three times.
Figure 4. NS5 protein interacts with human STAT2 and induces its degradation.
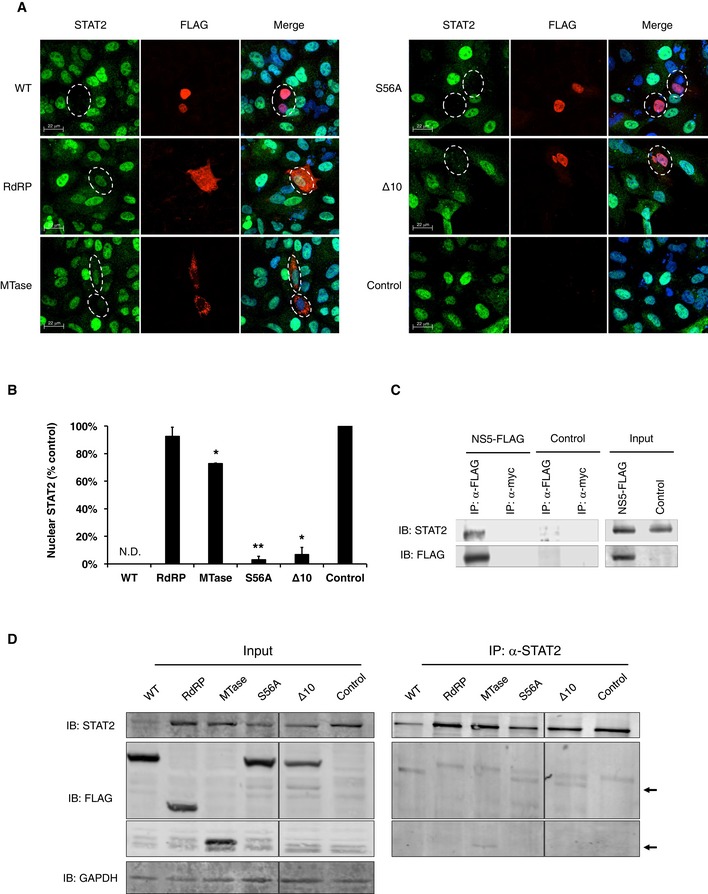
- A549 cells were transfected with control plasmid pcDNA 3.1 or plasmids encoding FLAG‐tagged WT or mutant NS5 proteins. At 48 h, post‐transfection cells were treated with IFN‐α (100 U/ml) for 2 h and then processed for indirect immunofluorescence. Images were acquired on a spinning disk confocal microscope with a 40× objective. NS5‐positive cells are indicated by dashed white circles. Representative panels are shown. The experiment was repeated three times.
- Quantification of nuclear STAT2 was performed using Volocity image analyses software. A minimum of 20 cells were counted for each sample. N.D., not detected. Values are expressed as mean ± standard error. *P < 0.05, **P < 0.01 (Student's t‐test); N = 3.
- A549 cells were transfected with plasmids encoding FLAG‐tagged ZIKV NS5 or FLAG alone for 24 h and then treated with epoxomicin (400 nM) for 24 h. Cells were harvested and processed for immunoprecipitation (IP) using a mouse anti‐FLAG or anti‐myc antibody followed by SDS–PAGE and immunoblotting. Representative panels are shown. The experiment was repeated three times.
- A549 cells were transfected with plasmids encoding FLAG‐tagged WT, mutant NS5 proteins, or FLAG alone for 48 h. Cells were harvested and processed for immunoprecipitation (IP) using rabbit anti‐STAT2 followed by SDS–PAGE and immunoblotting. Arrows indicate co‐immunoprecipitated WT NS5 and NS5 mutants that are in a complex with STAT2. Representative panels are shown. The experiment was repeated two times.
Next, we examined whether NS5 of ZIKV binds STAT2 by performing co‐immunoprecipitation in cells expressing FLAG‐tagged NS5. We observed stable interaction between NS5 and STAT2 (Fig 4C), a process that may be required for the eventual degradation of STAT2. We further mapped this interaction to the MTase domain (Fig 4D), which is consistent with the finding that expression of this domain alone decreases levels of STAT2 protein. However, the MTase domain was not as effective as full‐length NS5 at inducing degradation of STAT2 indicating that other regions of the protein may contribute to this process. Together, our data indicate that a major mechanism by which ZIKV disrupts IFN signaling is through NS5‐mediated binding and degradation of STAT2.
Recent studies in mouse models 13, 18 and primary trophoblasts 27 point to a vital role for the IFN system in curtailing ZIKV infection. Here, we show that ZIKV employs multiple strategies to block antiviral signaling in host cells. This includes interfering with the production of type‐I IFNs as well as downstream signaling. Specifically, ZIKV inhibits the induction of type‐I IFNs by suppressing the IRF3 and NF‐κB signaling, which is mediated by NS1, NS4A, and NS5. We also showed that ZIKV efficiently blocks IFN signaling by inducing proteasome‐mediated degradation of STAT2. We demonstrated that NS5 is the key viral determinant for STAT2 degradation and it interacts with STAT2 through its MTase domain. We are further investigating the mechanisms by which this pathogen interferes with antiviral signaling and it is hoped that the resulting information may be exploited for design of vaccines or compounds that block ZIKV replication.
Materials and Methods
Cell culture and virus infection
A549 cells, Vero cells, HEK293T cells, and mouse embryonic fibroblasts (MEFs) from the American Type Culture Collection (Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 100 U/ml penicillin and streptomycin, 1 mM HEPES (Gibco), 2 mM glutamine (Gibco), 10% heat‐inactivated fetal bovine serum (FBS; Gibco) at 37°C in 5% CO2. Primary human fetal astrocytes (HFAs) were prepared as previously described 28 from 15‐ to 19‐week aborted fetuses with written consent approved under the protocol 1,420 by the University of Alberta Human Research Ethics Board (Biomedical). The Zika virus (strain PLCal_ZIKV) was kindly provided by Dr. David Safronetz at the Public Health Agency of Canada. Virus manipulations were performed according to level‐2 containment procedures. Virus stocks were generated and then titrated (by plaque assay) using Vero cells.
Plasmids and transfection
ZIKV prM, NS2A, NS2B, NS3, NS2B‐3, NS4A, NS4B, NS4A‐B, and NS5 were generated by polymerase chain reaction (PCR) using a partial clone of strain H/PF/2013 ZIKV (A. Kumar, T.C. Hobman, unpublished data) as the template and cloned into between NheI and XhoI restriction sites in pCDNA 3.1 plasmid. ZIKV capsid and envelope were generated by PCR using cDNA generated from total RNA extracted from ZIKV (strain PLCal_ZIKV)‐infected A549 cells. The cDNAs were subcloned between NheI and XhoI restriction sites in pCDNA 3.1. All primer pairs used in PCR are tagged with a FLAG epitope at the C‐terminus (Table EV1). The pCDNA 3.1 prM‐E and NS1‐FLAG was commercially synthesized based on the sequence from ZIKV (strain H/PF/2013). To ensure proper topology of the viral proteins, prM‐E construct was generated with the membrane anchor of capsid, envelope with the last transmembrane segment of prM, NS1 with the last transmembrane segment of envelope protein, and NS4B with the 2K fragment. NS5 mutants were generated by PCR using pCDNA3.1‐NS5‐FLAG as template and primer pairs described in Table EV1. For indirect immunofluorescence analysis in A549 cells and MEFs, transfection of the appropriate expression plasmids was performed using TransIT‐LT1 (Mirus). For luciferase reporter assays in A549 or HEK293T cells, plasmid transfection was performed using Lipofectamine 2000 (Invitrogen). poly(I:C) (Sigma‐Aldrich) was transfected into cells using TransIT‐LT1.
Antibodies and compounds
The following antibodies were purchased from the indicated sources. Rabbit anti‐STAT2 (human, sc‐476), rabbit anti‐STAT2 (mouse, sc‐950), and rabbit anti‐STAT1 (sc‐346) from Santa Cruz; mouse anti‐FLAG (F3165) and mouse anti‐β‐actin (a3853) from Sigma‐Aldrich; mouse monoclonal anti‐double‐strand RNA J2 (10010200) from Scicons, Hungary; rabbit anti‐GAPDH (ab9485), Abcam; mouse antiflavivirus E protein (4G2) (MAB10216) from Millipore 19; mouse anti‐myc (9E10) (ATCC CRL−1729) from ATCC. The proteasome inhibitors MG132 and epoxomicin and the lysosome inhibitor bafilomycin‐A1 were purchased from Sigma‐Aldrich.
Immunoprecipitation
A549 cells were transfected with pCDNA NS5‐FLAG, NS5 mutants, or pCDNA‐FLAG with Lipofectamine 2000 (Life Technologies) for indicated time periods with treatments described. The cells were pelleted and resuspended in IP buffer (137 mM NaCl, 50 mM Tris pH 7.5, 1% NP‐40, 1 mM NaF, 1 mM DTT, and protease inhibitor cocktail (Roche)). The supernatant was clarified by centrifugation at 16,000 g for 15 min. Aliquots of the cell lysate were incubated overnight with the appropriate antibody at 4°C followed by incubation with Protein G beads for 2 h. After washes with IP buffer, SDS sample buffer was added to the beads and boiled and the proteins resolved by SDS–PAGE.
Immunoblotting
Total cell lysates from A549 cells, MEFs, or HEK293T cells were collected at designated time points post‐infection, post‐silencing, or post‐transfection and were washed three times with phosphate‐buffered saline (PBS) before lysing with 2× SDS sample buffer with or without β‐mercaptoethanol (2%). The samples were incubated at 98°C for 10 min to denature proteins. Proteins were separated by SDS–PAGE and transferred to polyvinylidene difluoride membranes for immunoblotting, as described previously 2.
Confocal microscopy
A549 cells, HFAs or MEFs on coverslips were fixed for 15 min at room temperature with 4% electron microscopy grade paraformaldehyde (Electron Microscope Sciences) in PBS. Samples were then washed three times with PBS and incubated in blocking buffer (0.2% Triton X‐100 [VWR Internationals] and 3% bovine serum albumin [BSA; Sigma‐Aldrich] in PBS) at room temperature for 1 h. Incubations with primary antibodies in blocking buffer were carried out at room temperature for 1 h, followed by three washes in wash buffer (0.3% BSA and 0.02% Triton X‐100 in PBS). Samples were then incubated with the indicated secondary antibodies in blocking buffer for 1 h at room temperature, followed by three washes in wash buffer. The indicated secondary antibodies (Invitrogen) were used at 1:1,000 dilutions in blocking buffer. Prior to mounting, samples were incubated with DAPI (4′,6‐diamidino‐2‐phenylindole; Sigma‐Aldrich) (1 μg/ml) for 30 min at room temperature before washing. Coverslips were mounted on microscope slides using Prolong Gold anti‐fade mounting reagent (Life Technologies). Images were acquired using an Olympus IX‐81 spinning disk confocal microscope equipped with a 40×/1.42‐numerical‐aperture oil PlanApo N objective. Images were analyzed using Volocity 6.2.1 software (PerkinElmer).
Quantitative real‐time PCR (qRT–PCR)
Total RNA from A549 cells or MEFs was isolated using the RNA NucleoSpin Kit (Macherey Nagel) and reverse‐transcribed using random primers (Invitrogen) and ImProm‐II reverse transcriptase (Promega) at 42°C for 1.5 h. The resulting cDNAs were mixed with the appropriate primers (Integrated DNA Technologies) and the PerfecTa SYBR green SuperMix with Low ROX (Quanta Biosciences) and amplified for 40 cycles (30 s at 94°C, 40 s at 55°C and 20 s at 68°C) in a Stratagene Mx3005P qRT–PCR machine. The gene targets and primers used are listed in Table EV2. The ΔCT values were calculated using Actb mRNA as the internal control. The ΔΔC T values were determined using control samples as the reference value. Relative levels of mRNAs were calculated using the formulas 2(−ΔΔCT).
Luciferase reporter assay
A549 cells and HEK293T cells were transfected with the following promoter reporter (firefly luciferase) constructs IFIT1: pGL3B/561 (gift from Ganes Sen), IFN‐β: p125‐luc and IRF3: p55‐CIB‐Luc (provided by T. Taniguchi, University of Tokyo, Japan), NF‐κB: pNF‐kB‐Luc (Stratagene), ISRE: pGL4 ISRE (Promega), or GAS: pGAS‐Luc plasmid (Stratagene) along with the Renilla luciferase: pRL‐TK (Promega) as a transfection control. The cells were harvested at indicated time points by washing the cells once with PBS and then lysing in 200 μl Luciferase Lysis buffer (0.1% [v/v] Triton X‐100; 25 mM glycylglycine [pH 7.8]; 15 mM MgSO4; 4 mM EGTA; and 1 mM dithiothreitol), and the samples were stored at −80°C. For luciferase assays, the samples were thawed and aliquoted in duplicates for both firefly and Renilla luciferase assays. The firefly luciferase substrate D‐luciferin (Gold Biotechnology, USA) was prepared at a final concentration of 70 μM in luciferase assay buffer (25 mM glycylglycine [pH 7.8], 15 mM K2PO4, [pH 7.8], 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, and 2 mM ATP), added to samples and incubated in the absence of light for 5 min. Luciferase activity measured using an Illuminator plate reader (BioTek). For Renilla luciferase measurements, the substrate coelenterazine (Gold Biotechnology, USA) was prepared at a final concentration of 1.4 μM in luciferase assay buffer (25 mM glycylglycine [pH 7.8], 15 mM K2PO4 [pH 7.8], 15 mM MgSO4, and 4 mM EGTA), added to samples and luciferase activity was measured using an Illuminator plate reader (BioTek).
siRNA silencing
MEFs were transfected with the indicated siRNAs (Table EV3) using RNAiMAX (Invitrogen). Forty‐eight hours later, cells were mock‐infected or infected with ZIKV (MOI = 5) for 48 h. Total cell lysates or total RNA were isolated and processed for Western blot and qRT–PCR, respectively.
Author contributions
AK, SH, and TCH conceived the research. AK, SH, and TCH drafted the manuscript. AK, SH, AMA, DL, VM, and CO carried out the experiments. AK and SH created the figures. WB and CP generated the HFAs. All authors discussed the results and contributed to the revision of the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Acknowledgements
The authors thank Eileen Reklow and Cheyneille Oliver for technical support. This work was funded by the grants from the Canadian Institutes of Health Research (CIHR), the Li Ka Shing Institute of Virology and the Women & Children's Health Research Institute to T.C.H. A.K. and D.L. are funded by postdoctoral fellowships from the Alberta Innovates‐Health Solutions. A.A. is the recipient of a Doctoral scholarship from CIHR. T.C.H. and C.P. hold Canada Research Chair awards.
EMBO Reports (2016) 17: 1766–1775
References
- 1. Musso D, Gubler DJ (2016) Zika Virus. Clin Microbiol Rev 29: 487–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C et al (2009) Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360: 2536–2543 [DOI] [PubMed] [Google Scholar]
- 3. Musso D, Cao‐Lormeau VM, Gubler DJ (2015) Zika virus: following the path of dengue and chikungunya? Lancet 386: 243–244 [DOI] [PubMed] [Google Scholar]
- 4. Zanluca C, de Melo VC, Mosimann AL, Dos Santos GI, Dos Santos CN, Luz K (2015) First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz 110: 569–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith DW, Mackenzie J (2016) Zika virus and Guillain‐Barre syndrome: another viral cause to add to the list. Lancet 387: 1486–1488 [DOI] [PubMed] [Google Scholar]
- 6. Rodrigues LC (2016) Microcephaly and Zika virus infection. Lancet 387: 2070–2072 [DOI] [PubMed] [Google Scholar]
- 7. Mlakar J, Korva M, Tul N, Popovic M, Poljsak‐Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V et al (2016) Zika virus associated with microcephaly. N Engl J Med 374: 951–958 [DOI] [PubMed] [Google Scholar]
- 8. Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM (2016) Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol 47: 6–7 [DOI] [PubMed] [Google Scholar]
- 9. D'Ortenzio E, Matheron S, de Lamballerie X, Hubert B, Piorkowski G, Maquart M, Descamps D, Damond F, Yazdanpanah Y, Leparc‐Goffart I (2016) Evidence of Sexual Transmission of Zika Virus. N Engl J Med 374: 2195–2198 [DOI] [PubMed] [Google Scholar]
- 10. Deckard DT, Chung WM, Brooks JT, Smith JC, Woldai S, Hennessey M, Kwit N, Mead P (2016) Male‐to‐Male Sexual Transmission of Zika Virus ‐ Texas, January 2016. MMWR Morb Mortal Wkly Rep 65: 372–374 [DOI] [PubMed] [Google Scholar]
- 11. Jensen S, Thomsen AR (2012) Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86: 2900–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ye J, Zhu B, Fu ZF, Chen H, Cao S (2013) Immune evasion strategies of flaviviruses. Vaccine 31: 461–471 [DOI] [PubMed] [Google Scholar]
- 13. Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS (2016) A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 19: 720–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnson AJ, Roehrig JT (1999) New mouse model for dengue virus vaccine testing. J Virol 73: 783–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prestwood TR, Morar MM, Zellweger RM, Miller R, May MM, Yauch LE, Lada SM, Shresta S (2012) Gamma interferon (IFN‐gamma) receptor restricts systemic dengue virus replication and prevents paralysis in IFN‐alpha/beta receptor‐deficient mice. J Virol 86: 12561–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ashour J, Laurent‐Rolle M, Shi PY, Garcia‐Sastre A (2009) NS5 of dengue virus mediates STAT2 binding and degradation. J Virol 83: 5408–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dalrymple NA, Cimica V, Mackow ER (2015) Dengue Virus NS Proteins Inhibit RIG‐I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon‐Regulating Virulence Determinant. MBio 6: e00553‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, Langsjoen RM, Paessler S, Vasilakis N, Weaver SC (2016) Characterization of a Novel Murine Model to Study Zika Virus. Am J Trop Med Hyg 94: 1362–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera‐Lecoin M, Surasombatpattana P, Talignani L, Thomas F et al (2015) Biology of Zika Virus Infection in Human Skin Cells. J Virol 89: 8880–8896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diamond MS, Harris E (2001) Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR‐independent mechanism. Virology 289: 297–311 [DOI] [PubMed] [Google Scholar]
- 21. Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M et al (2007) Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin‐Cullin E3 ligase. J Virol 81: 3428–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu X, Zheng J, Zheng K, Hou Y, Zhao F, Zhao D (2014) Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology 57: 65–73 [DOI] [PubMed] [Google Scholar]
- 23. Rodriguez JJ, Cruz CD, Horvath CM (2004) Identification of the nuclear export signal and STAT‐binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol 78: 5358–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodriguez JJ, Parisien JP, Horvath CM (2002) Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol 76: 11476–11483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones M, Davidson A, Hibbert L, Gruenwald P, Schlaak J, Ball S, Foster GR, Jacobs M (2005) Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J Virol 79: 5414–5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mazzon M, Jones M, Davidson A, Chain B, Jacobs M (2009) Dengue virus NS5 inhibits interferon‐alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J Infect Dis 200: 1261–1270 [DOI] [PubMed] [Google Scholar]
- 27. Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB (2016) Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 19: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vivithanaporn P, Asahchop EL, Acharjee S, Baker GB, Power C (2016) HIV protease inhibitors disrupt astrocytic glutamate transporter function and neurobehavioral performance. AIDS 30: 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File