

Abstract
Factors that influence the charging of protein ions formed by electrospray ionization from aqueous solutions in which proteins have native structures and function were investigated. Protein ions ranging in molecular weight from 12.3 to 79.7 kDa and pI values from 5.4 to 9.6 were formed from different solutions and reacted with volatile bases of gas-phase basicities higher than that of ammonia in the cell of a Fourier-transform ion cyclotron resonance mass spectrometer. The charge-state distribution of cytochrome c ions formed from aqueous ammonium or potassium acetate is the same. Moreover, ions formed from these two solutions do not undergo proton transfer to 2-fluoropyridine, which is 8 kcal/mol more basic than ammonia. These results provide compelling evidence that proton-transfer between ammonia and protein ions does not limit protein ion charge in native electrospray ionization. Both circular dichroism and ion mobility measurements indicate that there are differences in conformation of proteins in pure water and aqueous ammonium acetate, and these differences can account for the difference in the extent of charging and proton-transfer reactivities of protein ions formed from these solutions. The extent of proton-transfer of the protein ions with higher gas-phase basicity bases trends with how closely the protein ions are charged to the value predicted by the Rayleigh limit for spherical water droplets approximately the same size as the proteins. These results indicate that droplet charge limits protein ion charge in native mass spectrometry and are consistent with these ions being formed by the charged residue mechanism.
Graphical abstract
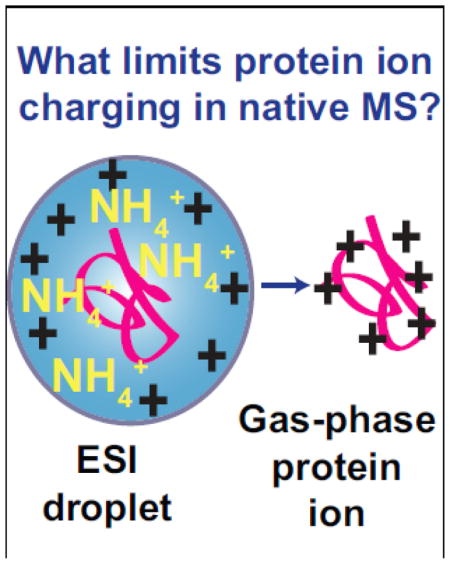
Introduction
Many factors affect the extent of charging of intact gas-phase macromolecular ions formed by electrospray ionization (ESI) [1–19], but the conformation of a molecule in solution is one of the most significant [3–5]. Broad distributions of highly charged protein ions are formed from solutions in which proteins are denatured, such as water/methanol/acid solutions. In contrast, narrower distributions of lower charge states are formed from buffered aqueous solutions in which proteins are in native or native-like conformations [3]. Other factors, such as surface tension of the ESI droplet [6, 7], instrumental parameters [3–5, 8], supercharging methods [7, 9–13], and gaseous reactions with acid or base vapors [16–19] can also affect the extent of charging of protein ions formed by ESI. The maximum extent of charging of denatured and supercharged protein ions can be limited by the proton-transfer reactivity of the ion relative to that of the electrospray solvent or other molecules in solution [6, 18, 19].
Factors that limit the extent of charging of protein ions formed from buffered aqueous solutions by ESI are debated [15, 20–27], and several mechanisms for ion formation of protein ions from buffered aqueous solutions have been proposed [23, 28–30]. In the charged residue mechanism (CRM) for ion formation, multiply charged gas-phase ions are formed by solvent evaporation in the late stages of droplet lifetime [28]. De la Mora [15] and others [7, 31, 32] showed that the extent of charging of globular macromolecular ions formed from aqueous solutions is close to the number of charges predicted by the Rayleigh limit (ZR) for a water droplet that is approximately the same size as the macromolecule. The maximum charge of globular protein and dendrimer ions from 6 to 1400 kDa formed from aqueous solutions was between 65 to 110 % of ZR and proportional to the square root of the molecular weight, suggesting the formation of globular ions from 6 to 1400 kDa follows the CRM [15]. The maximum charge of polyethylene glycol (PEG) ions with molecular weights greater than 50 kDa exceeded ZR, suggesting that these ions are formed from nonspherical droplets that fail to undergo Coulombic fissions because the PEG backbone is highly charged [15]. In the chain ejection model (CEM), folded native protein ions are formed by the CRM, but highly charged unfolded protein ions are ejected from the ESI droplet before complete desolvation occurs [20, 33–36].
Salts, or buffers commonly used in native ESI mass spectrometry, can also affect the extent of charging on protein ions formed from aqueous solutions [22, 37–40]. In the combined charged residue-field emission model (CCRFEM) for ion formation, small ions residing at the droplet surface evaporate from the droplet at a rate determined by the electric field strength at the droplet surface and ion solvation energies, and macromolecules located in the interior of the droplet ionize by the CRM [29]. In the CCRFEM, buffer ions with low solvation energies evaporate, carrying away charge from the ESI droplet, such that less charge remains on the macromolecules in the droplet after solvent evaporation [29]. Recently, Allen et al. [31] reported that protein ions formed from buffered aqueous ESI solutions with molecular weights less than 130 kDa were charged less than ZR, but protein ions with molecular weights greater than 130 kDa were charged closer to ZR. The authors suggested that this is consistent with charge-carrier emission of buffer ions limiting the charge of protein ions with molecular weights less than 130 kDa formed from buffered aqueous ESI solutions [31].
It has also been proposed that the extent of charging of proteins from native solutions is limited by proton-transfer reactivity between protein ions and commonly added salts, such as ammonium acetate or ammonium bicarbonate, which are extensively used as buffers in aqueous ESI solutions [2, 23, 26, 41–49]. Kebarle and coworkers suggested that charging of protein ions formed by ESI from aqueous ammonium salt solutions is limited by proton-transfer between the protein ion and ammonium or ammonia at the surface of the ESI droplet in the final stages of solvent evaporation [23, 41, 47]. In this mechanism, NH3 formed in the last stages of the ESI process can accept a proton from the protein resulting in less highly charged ions [41].
Adding basic molecules directly to aqueous ESI solutions can result in lower protein ion charging [49, 50] either as a result of proton-transfer reactions or competition for protons in the ionization process. Protein ions formed from aqueous solutions can be denatured by exposing the ESI droplet to gaseous acids or bases which increase the charging as a result of the change in protein conformation [16, 17]. The proton-transfer reactivities of protein ions with volatile bases, formed by ESI from solutions in which the proteins are denatured, have been investigated experimentally [14, 18, 51–56] and modeled computationally [14, 19, 26, 41, 47, 56]. Proton-transfer reactions between protein ions and volatile basic molecules show that the apparent gas-phase basicity of high charge state ions is lower than that of low charge state ions [14, 18, 52, 56], and that proton-transfer rates between protein ions and basic molecules depend on temperature [51, 57, 58]. Conformation also affects the proton-transfer reactivity of protein ions [14, 52, 56]. The apparent gas-phase basicity of disulfide-intact lysozyme ions formed from solutions in which the protein conformation is compact is lower than that of disulfide-reduced lysozyme ions of the same charge state formed from solutions in which the protein is denatured [56].
Here, we investigate factors that limit the extent of charging of protein ions formed from buffered aqueous solutions using proton-transfer reactions with volatile molecules more basic than ammonia, circular dichroism and ion mobility mass spectrometry. Cytochrome c has nearly identical secondary structure in solutions of aqueous ammonium acetate or potassium acetate, and the charge-state distributions of cytochrome c ions formed from these solutions are similar. Moreover, the protein ions formed from these solutions do not react with 2-fluoropyridine which is 8 kcal/mol more basic NH3. These results show that charging of protein ions in native mass spectrometry is not limited by the presence of NH4+ or NH3 in solution. The extent of the proton-transfer reactivities of protein ions formed from native aqueous solutions depends on conformation and how closely the ions are charged to ZR. These results indicate that the charge on the ESI droplets limits the extent of charging of the molecular ions of proteins in native mass spectrometry.
Experimental
Proton-transfer reactions
Mass spectral data were acquired using a 9.4 T Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer that is described elsewhere [59]. Protein ions were formed from aqueous ammonium acetate, potassium acetate or pure water solutions by nanoelectrospray ionization using borosilicate capillaries (1.0 mm o.d./0.78 mm i.d., Sutter Instruments, Novato, CA, USA) that were pulled to a tip i.d. of 0.8 μm or 1.6 μm with a Flaming/Brown micropipette puller (Model P-87, Sutter Instruments, Novato, CA, USA). Tip diameters were measured with a scanning electron microscope (Hitachi TM-1000 SEM, Schaumburg, IL, USA) at the Electron Microscope Laboratory at UC Berkeley. Nanoelectrospray was initiated by applying a potential of about +0.7 to 1.2 kV to a 0.127 mm diameter platinum wire inserted into the capillary and in contact with the sample solution.
Volatile bases were degassed using several freeze-pump-thaw cycles and introduced into the mass spectrometer through a sapphire leak valve to a vacuum chamber pressure of 1.0 × 10−8 Torr (base pressure is ~3 × 10−10 Torr) measured using an ion gauge located remotely from the ion cell. The ion gauge was not calibrated to the pressure of the volatile basic molecules in the ion cell. Rate constants of cytochrome c ions formed from water/methanol/acetic acid and reacted with pyridine are within 35–85% of those reported by Schnier et al. [14]. Protein ions were reacted in the cell with 2-fluoropyridine (2-FP), pyridine, diethylamine (DEA) and dipropylamine (DPA) for up to 120 s.
Lyophilized protein powders of equine cytochrome c, bovine carbonic anhydrase, jack bean concanavalin A, human holo-transferrin, equine myoglobin, 2-FP, DEA, DPA, ammonium acetate, potassium acetate and sodium acetate are from Sigma (St. Louis, MO) and pyridine is from Fisher Scientific (Waltham, MA). Protein solutions were prepared at 10 μM in Millipore Milli-Q water, 10 mM ammonium acetate, 10 mM potassium acetate, or 10 mM sodium acetate, except for holo-transferrin where 200 mM ammonium acetate was used to maintain sufficient ion signal.
Ion mobility mass spectrometry
Arrival time distributions and mass spectral data were acquired using a Waters Synapt G2 (Waters, Milford, MA, USA) that is located at University of California, San Francisco. The traveling wave ion mobility cell was operated with a constant wave velocity of 800 m/s, wave height of 40 V, helium flow rate of 180 mL/min, and IMS (N2) flow rate of 90 mL/min. The time of flight mass analyzer was operated in sensitivity mode (“V”). Calculated collision cross sections were obtained from the arrival time distributions using the procedure described by Bush et al. [60]. Cytochrome c, ubiquitin, bovine serum albumin, avidin, β-lactoglobulin, and concanavalin A were used as calibrant ions. Arrival times were assigned as the center of the full-width at half-maximum for the arrival time distribution for each ion. Average collision cross sections, CCSav, were calculated as an average weighted sum of the collision cross sections for each charge-state distribution.
The charge-state distributions of protein ions obtained with the FT-ICR and Synapt mass spectrometers are similar, except for carbonic anhydrase ions formed from pure water. Charge states up to the 19+ and 15+ were observed with these respective instruments. The higher charging with the FT-ICR mass spectrometer is likely due to more droplet heating in this instrument which can result in some unfolding of the protein in the droplet prior to ion formation [10].
Circular dichroism
Circular dichroism (CD) data were acquired using a Jasco Model 815 spectropolarimeter (JASCO, Inc., Easton, MD, USA). Wavelength scans from 190 to 260 nm at 20 °C were acquired for solutions containing 10 μM cytochrome c in pure water, 10 mM ammonium acetate, potassium acetate, and sodium acetate.
Results and Discussion
Effects of protein conformation on protein ion charging
The average charge of cytochrome c and carbonic anhydrase ions formed from pure water is higher than that from aqueous ammonium acetate (Table 1). The charge-state distribution of carbonic anhydrase ions formed from pure water is bimodal (Supplemental Figure 1), with a small distribution of higher charge state ions indicative of partially unfolded structure [3]. The conformation of proteins in aqueous solution can be affected by the presence of salts [61, 62]. To determine if these differences in charging from pure water versus aqueous ammonium salt solutions are due to salts in solution affecting the conformation of the proteins, the α-helical and β-strand content of cytochrome c and carbonic anhydrase was probed using circular dichroism (CD). CD spectra of 10 μM cytochrome c and carbonic anhydrase in pure water, 10 mM ammonium acetate, and 10 mM potassium acetate from 200 to 260 nm are shown in Figure 1a and 1b. K+ was chosen because NH4+ and K+ have similar ionic radii [63], activity coefficients [64], and Gibbs solvation free energies [63]. The CD spectrum of carbonic anhydrase in pure water has a band at 204 nm that is absent in the CD spectra of carbonic anhydrase with ammonium or potassium acetate (Figure 1a). This band corresponds to a molar ellipticity contribution from tryptophan residues that couple with other aromatic residues [65]. This band in the spectrum of carbonic anhydrase in water but not in ammonium or potassium acetate indicates that the conformation is different in water when these two salts are not present. The CD spectra of cytochrome c in pure water, ammonium acetate and potassium acetate have unresolved bands at 208 and 222 nm (Figure 1b). These bands are characteristic of a predominately α-helical protein [65]. The ellipticity at 222 nm of cytochrome c in pure water is slightly more negative than that of cytochrome c in aqueous potassium or ammonium acetate solutions. The ellipticity of a partially denatured protein can become more negative at 222 nm as a result of the loss of positive ellipticity contributions of aromatic side chains [65]. The CD spectra of carbonic anhydrase and cytochrome c in aqueous ammonium and potassium acetate solutions are very similar (Figure 1a–b), indicating that secondary structure of the protein in these solutions is also similar. These CD measurements indicate that the presence of salts in solution affects the secondary structure of carbonic anhydrase and to a lesser extent, cytochrome c, compared to that in pure water, and the secondary structure of these two proteins is similar in ammonium and potassium acetate.
Table 1.
Molecular weight, pI [75], number of basic residues, maximum charge (Zmax), average charge (Zav), extent of proton-transfer with DPA, and effective density values for each protein. Ions formed from 10 mM aqueous ammonium acetate solution unless otherwise noted
| molecular weight (kDa) | pI | number of basic residues | Zmax | Zav | reaction with DPA (120 s) | Effective Density (g/cm3) | ||
|---|---|---|---|---|---|---|---|---|
| −ΔZmax | −ΔZav | |||||||
| cytochrome c | 12.3 | 9.6 | 24 | 8 | 7.1 | 1.0 ± 0.0 | 1.1 ± 0.1 | 0.50 |
| cytochrome c (water) | 12.3 | 9.6 | 24 | 12 | 8.1 | 3.0 ± 0.0 | 1.8 ± 0.1 | 0.44 |
| myoglobin | 17.6 | 7.4 | 32 | 9 | 8.0 | 0.3 ± 0.5 | 0.6 ± 0.3 | 0.52 |
| carbonic anhydrase | 29.1 | 6.8 | 43 | 12 | 9.8 | 0.7 ± 0.5 | 0.4 ± 0.1 | 0.74 |
| carbonic anhydrase (water) | 29.1 | 6.8 | 43 | 19 | 12.9 | 4.3 ± 2.0 | 2.8 ± 0.1 | 0.69 |
| concanavalin A dimer | 51.7 | 6.0 | 48 | 17 | 15.1 | 1.3 ± 0.5 | 1.5 ± 0.5 | 0.58 |
| holo-transferrin (200 mM ammonium acetate) | 79.7 | 5.4 | 64 | 21 | 19.2 | 1.7 ± 0.5 | 1.4 ± 0.3 | 0.60 |
Figure 1.

(a) CD spectra of 10 μM carbonic anhydrase and (b) 10 μM cytochrome c in pure water (solid blue line), 10 mM ammonium acetate (green dotted line), and 10 mM potassium acetate (purple dashed line). (c) Collision cross sections of 10 μM cytochrome c ions (pink) and carbonic anhydrase ions (blue) and formed from 10 mM ammonium acetate (square, diamond, respectively) or pure water (triangle, circle, respectively) as a function of charge state. (d) Arrival time distributions of cytochrome c 8+ ions and carbonic anhydrase 12+ ions formed from water (blue) or 10 mM ammonium acetate (green)
The protein conformation in solution can also affect the resulting conformations of ions produced by ESI from these solutions [66–68]. Ion mobility experiments were performed to investigate if the gas-phase conformations of carbonic anhydrase and cytochrome c ions formed from pure water and aqueous ammonium acetate differ. The collision cross sections of carbonic anhydrase and cytochrome c ions formed from pure water and aqueous 10 mM ammonium acetate as a function of charge state were obtained using traveling wave ion mobility mass spectrometry and are shown in Figure 1c. The collision cross sections for the same charge state ions formed from pure water or ammonium acetate solutions are indistinguishable within experimental error with the exception of 12+ charge state of carbonic anhydrase. For this charge state, the cross section is slightly larger when this ion is formed from water compared to that formed from ammonium acetate (Figure 1d). The more extended conformers of the 12+ charge state of carbonic anhydrase ions formed from pure water are more abundant compared to those formed from ammonium acetate (Figure 1d).
The collisional cross sections of the 6+ through 8+ charge states of cytochrome c ions formed from 10 mM aqueous ammonium acetate and pure water are indistinguishable, except for the relative abundances of the the two conformers of the 8+ charge state (Figure 1d). The compact conformer of 8+ cytochrome c is slightly more abundant from 10 mM ammonium acetate than from pure water (Figure 1d). Higher charge states of cytochrome c and carbonic anhydrase ions that are formed from pure water have larger collision cross sections than the lower charge states that are formed from both ammonium acetate and pure water (Figure 1c). These ion mobility measurements show that the conformations of cytochrome c and carbonic anhydrase ions formed from pure water can be different than the conformation of these ions formed from 10 mM ammonium acetate. The CD measurements show that cytochrome c and carbonic anhydrase have different secondary structure in pure water and ammonium acetate, and this produces a difference in the gas-phase conformations. These results show that the difference in charging of cytochrome c and carbonic anhydrase ions formed from pure water and 10 mM ammonium acetate is likely due to differences in conformations of the proteins in these solutions.
Gas-phase proton-transfer reactions between protein ions and volatile bases
In order to determine if proton transfer reactivity between NH3 and proteins affects the charge of protein ions formed from aqueous ammonium acetate solutions [23, 46, 47], proton-transfer experiments for five proteins ranging in molecular weight from 12.3 to 79.7 kDa and pI values of 5.4 to 9.6 were performed with volatile bases with gas-phase basicities (GB) that are higher than that of NH3 (196 kcal/mol). The charge-state distribution of each protein (cytochrome c, myoglobin, carbonic anhydrase, concanavalin A dimer, holo-transferrin) did not change when these ions were reacted with 2-fluoropyridine (2-FP) (GB = 204 kcal/mol) for 120 s at 1.0 × 10−8 Torr. Lower charge states of these five proteins were formed by proton transfer to pyridine, diethylamine (DEA), and dipropylamine (DPA), which have GB values of GB = 215, 221, 225 kcal/mol, respectively [69]. For example, there is no change in the charge-state distribution of concanavalin A dimer ions after reaction with 2-FP for 120 s (Figure 2). However, reaction of these ions with pyridine (120 s) results in a decrease in relative abundance of the 16+ charge state and increase in the relative abundances of the 14+ and 13+ (Figure 2). The absence of any proton-transfer between the five proteins and 2-FP (204 kcal/mol) indicates that no proton-transfer reaction with NH3 (196 kcal/mol) would occur under these same conditions. These results show that the apparent GB values for all the protein ions formed directly from aqueous ammonium acetate are more than 8 kcal/mol higher than the GB of NH3.
Figure 2.

Mass spectra of concanavalin A dimer ions formed from 10 mM ammonium acetate after reaction with either 2-fluoropyridine or pyridine for (a) 0 and (b) 120 s
The concentration of the bases in these experiments is very low and is substantially lower than the corresponding concentration of ammonium in solution and gaseous ammonia in the ESI interface, although the long reaction times used in these experiment at least partially compensates for this difference. To evaluate if ammonium or ammonia affect protein charging from aqueous buffered ammonium acetate solutions, cytochrome c ions were formed from either 10 mM ammonium acetate or 10 mM potassium acetate. Potassium has a similar ionic radius [63], activity coefficient [64], and Gibbs solvation free energy [63] to ammonium but potassium cannot undergo proton transfer reactions with protein ions. In addition, secondary structure of cytochrome c in ammonium and potassium acetate solutions is very similar (Figure 1a and 1b). The charge-state distributions of cytochrome c ions from these solutions are the same (Figure 3, top). This result provides compelling evidence that the presence of NH4+ or NH3 in solution does not affect the charging of proteins in native mass spectrometry. Moreover, the charge-state distributions of ions formed from either aqueous ammonium acetate or potassium acetate solutions do not change upon reaction with 2-FP (Figure 3). This demonstrates that the apparent gas-phase basicity of proteins is greater than the gas-phase basicity of NH3 whether or not NH3 or NH4+ is present. Acetate and water can potentially proton-transfer with proteins. Different anions can affect protein charging from solutions in which proteins are denatured [70] or solutions in which proteins have native structures [71]. In these experiments, acetate and water are present in both the ammonium and potassium acetate solutions, and therefore should not affect our conclusion about NH4+ or NH3 not affecting protein charge. These results clearly show that proton-transfer between proteins and NH3 does not affect the charge of ions formed from ammonium acetate solutions and that proton transfer to ammonia does not limit the charging of proteins in native mass spectrometry.
Figure 3.

Mass spectra of 10 μM cytochrome c ions formed from 10 mM ammonium acetate and 10 mM potassium acetate reacted with 2-fluoropyrine for 0 and 120 s
Charging of Protein Ions and the Rayleigh Limit
To determine if the charging of the protein ions under these conditions is close to ZR [7, 15, 31, 32], the maximum and average charge of the protein ions, Zmax and Zav, respectively, were compared to ZR for water droplets of comparable sizes to the proteins. ZR was calculated by approximating protein ions as spheres with a density of 1 g/cm3 [7, 15, 31, 32]. For protein ions formed from aqueous ammonium acetate, Zav is between 73.2 and 86.5 % of ZR and Zmax is between 85.7 and 95.0 % of ZR, which is within the range of previously reported values [15, 31, 32]. However, Zav and Zmax for protein ions formed from pure water are higher than the values for ions formed from aqueous ammonium acetate. Zav for ions formed from pure water is between 93.6 and 96.1 % of ZR and Zmax is between 137 and 142 % of ZR.
To determine if the difference in the conformation of the protein in pure water compared to in aqueous ammonium acetate can account for Zmax greater than ZR for protein ions formed from pure water, ZR(CCS) was calculated by approximating the protein ions as spheres with densities obtained from average collision cross sections, CCSav. CCSav values were calculated as an average weighted sum of the collision cross sections for each charge-state distribution from the solution from which the ions were formed. ZR(CCS) takes into account, in part, conformational differences of protein ions formed from pure water and aqueous ammonium acetate because protein ions with larger collision cross sections have lower effective densities. The average effective density of protein ions formed from aqueous ammonium acetate is 0.58 g/cm3, which is comparable to previously reported values [60]. However, the effective densities of cytochrome c and carbonic anhydrase ions formed from pure water are slightly lower than those of ions formed from aqueous ammonium acetate (Table 1).
ZR, ZR(CCS), Zmax and Zav for protein ions formed from pure water and aqueous ammonium acetate are shown as a function of molecular weight (Supplemental Figure 2) and normalized to ZR (Figure 4). Zmax and Zav for protein ions formed from ammonium acetate increase with the square root of molecular weight (Supplemental Figure 2), consistent with previously reported results [15, 31, 32]. ZR(CCS) is greater than ZR by an average of 41 ± 14 % (Figure 4), consistent with protein ions formed from pure water that are partially elongated and not spherical. Zmax for protein ions formed from pure water is between 85 and 110 % of ZR(CCS) and Zav is between 56 and 75 % of ZR(CCS). The results are consistent with the higher charging for the ions formed from pure water being a result of their less compact structures compared to those formed from ammonium acetate.
Figure 4.

Zmax (purple square), Zav (pink triangles), ZR(CCS) (blue circles) for protein ions formed from aqueous ammonium acetate (filled) or pure water (open) relative to ZR (green line) as a function of molecular weight. ZR represents the maximum number of charges on a protein predicted using the Rayleigh limit for a droplet the same size as a spherical protein with a density of 1.0 g/cm3. ZR(CCS) represents the maximum number of charges predicted using the Rayleigh limit for a spherical droplet with the same radius as the CCSav for the protein ions.
Recently, Allen et al. investigated the effects of charge carrier emission of buffer ions on the charging of proteins ions formed from buffered aqueous ESI solutions by comparing Zav of protein ions to ZR [31]. The authors calculated ZR by approximating the ions as spherical with densities of 0.6, 1.0, and 1.2 g/cm3. For a density of 1.0 g/cm3, Zav of protein cations with molecular weights between 5.8 and 468 kDa was 60–90 % of ZR [31]. Zav of protein cations with molecular weights less than 130 kDa were only up to 83 % of ZR, but Zav of ions greater than 130 kDa were closer to ZR [31]. The authors suggest that this shows a molecular weight dependence of protein ions charging to ZR that is consistent with charge-carrier emissions of buffer ions at critical field strengths limiting the charge of protein ions [31]. Here, the molecular weight range of proteins investigated is much smaller, only 12.3 to 79.7 kDa, but there is no trend between protein molecular weight and charging of protein ions to ZR within this molecular weight range. (Figure 4). Zmax of the protein ions formed from aqueous ammonium acetate is close to ZR, ranging from 86 to 95 % ZR. The extent of charging is very close to that expected from the charged residue mechanism, given that factors, such as conformation, can affect charging in this simplistic assumption in determining ZR. Our results suggest that if charge-carrier emission from aqueous ammonium acetate solutions occurs, it does not significantly affect the charge of the protein ions in native mass spectrometry.
Factors that affect the relative proton-transfer reactivities of protein ions
The relative proton-transfer reactivities of the five proteins with basic molecules was investigated by comparing the change in charge-state distributions when the ions are reacted with DPA (Table 1). The relative extent of proton-transfer between the five proteins investigated and DPA does not trend with the protein pI, number of basic residues, or the fraction of basic residues in the protein (Table 1, Figure 5b–e). The extent of proton-transfer between protein ions and DPA is greater for ions formed from pure water than from aqueous ammonium acetate (Table 1). The proton-transfer reactivity of a protein ion is affected by both the number of charges and the ion conformation [56, 72]. The ions from pure water are more highly charged than those formed from aqueous ammonium acetate, which can increase proton-transfer reactivity but are also less compact, which can decrease proton-transfer reactivity. Some partial collapse of the more highly charged, less compact ions may also occur in the gas phase [73] which would result in higher proton transfer reactivity. The time scale of the ion mobility measurements and the proton transfer reactivity experiments differ significantly, and any change in conformation in the latter may not be reflected in the ion mobility measurements.
Figure 5.

(a) The percent Zmax (circle) and Zav (square) of ZR, for protein ions formed from aqueous ammonium acetate (filled markers) or pure water (open markers) prior to reaction with a base v. the decrease in Zmax (circle) and Zav (square) after 120 s reaction with DPA. (b) fraction of basic residues, (c) molecular weight (d) number of basic residues (e) isoelectric point (pI) for each protein as a function of the decrease in Zmax (circle) and Zav (square) after 120 s reaction with DPA. (i) corresponds to cytochrome c, (ii) myoglobin, (iii) carbonic anhydrase, (iv) concanavalin A dimer and (v) holo-transferrin ions, respectively.
The extent of proton-transfer of protein ions with DPA trends with how closely the ions are charged to ZR. Figure 5a shows the absolute decrease in protein ion charge, ΔZmax and ΔZav, upon reaction with DPA (120 s) as a function of percent Zmax and Zav of ZR for the protein ions before reaction. Protein ions with the greatest Z/ZR value undergo the most proton-transfer with DPA (Figure 5a). This suggests that how closely protein ions are charged to ZR affects the proton-transfer reactivities of the ions.
Conclusions
Factors that limit the charge of protein ions formed by ESI from aqueous solutions by ESI were investigated. The charge-state distributions of cytochrome c ions formed from aqueous potassium acetate or ammonium acetate are the same. Potassium has a similar Gibbs solvation free energy, activity coefficient, and ionic radius to ammonium, so any ion evaporation that occurs should be similar for both ions. However, potassium cannot undergo proton transfer reactions with proteins. The similar charging of cytochrome c ions formed from potassium or ammonium acetate solutions shows that the presence of NH4+ or NH3 does not affect charging in native mass spectrometry. Protein ions from these two solutions do not proton-transfer with 2-fluoropyridine, which has a gas-phase basicity that is 8 kcal/mol higher than that of ammonia, demonstrating that the apparent gas-phase basicity of protein ions formed in native mass spectrometry is greater than that of ammonia.
Protein ions formed from aqueous ammonium acetate solutions are charged closely to the values predicted by the Rayleigh limit for spherical water droplets similar in size to the proteins. The average charge of protein ions formed from aqueous ammonium acetate is between 73 and 87 % of ZR, which is within the range of previously reported values. The maximum charge of protein ions formed from aqueous ammonium acetate is between 86 to 95 % of ZR. The maximum charge of the protein ions is close to ZR, suggesting the charge-carrier emission process in the combined charged residue-field emission model does not significantly limit the charging of these protein ions.
The extent of proton-transfer between protein ions formed from aqueous solutions and molecules with much higher gas-phase basicities depends on several factors. Protein ions formed from pure water are more highly charged with less compact conformations than those formed from aqueous ammonium acetate and undergo more proton transfer with basic molecules. The extent of proton-transfer between protein ions with more basic molecules is related to how closely the protein ions are charged to ZR for spherical water droplets of comparable size to the proteins. These results are consistent with the charged residue mechanism in which ESI droplet charge limits the charge of protein ions formed from buffered aqueous solutions, when the proteins have compact folded conformations. This knowledge of the factors limiting the charging of protein ions formed from buffered aqueous solutions forms a foundation for efforts to join mass spectrometry with other solution techniques, such as high-throughput X-ray scattering, to reduce gaps between cloning library technologies and the throughput capacity of analytical platforms to characterize proteins [74].
Supplementary Material
Supplemental Figure 1 Mass spectrum of 10 μM carbonic anhydrase formed from pure water (FT-ICR).
Supplemental Figure 2 Zmax (purple square), Zav (pink triangles) for protein ions formed from aqueous ammonium acetate (filled) or pure water (open) as a function of molecular weight. Green line represents ZR, the maximum number of charges on a protein predicted using the Rayleigh limit for a droplet the same size as a spherical protein with a density of 1.0 g/cm3. ZR(CCS) (blue circles) represents the maximum number of charges predicted using the Rayleigh limit for a spherical droplet with the same radius as the CCSav for the protein ions.
Supplemental Table 1 Collisional cross sections (CCS) of cytochrome c and carbonic anhydrase ions formed from pure water or 10 mM aqueous ammonium acetate.
Acknowledgments
The authors thank the National Institutes of Health (Grant no. R01GM097357) for financial support and the Sandler-Moore Mass Spectrometry Core Facility at UCSF for use of the Synapt G2. John A. Tainer is partly supported by a Robert A. Welch Chemistry Chair, the Cancer Prevention and Research Institute of Texas, and the University of Texas System Science and Technology Acquisition and Retention. The authors also thank Department of Energy, Office of Basic Energy Sciences, Integrated Diffraction Analysis Technologies (IDAT) program at Lawrence Berkeley National Lab for use of the Jasco 815 spectropolarimeter and Dr. Catherine Going for helpful discussions.
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization–principles and practice. Mass Spectrom Rev. 1990;9:37–70. [Google Scholar]
- 2.Kebarle P, Verkerk UH. Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom Rev. 2009;28:898–917. doi: 10.1002/mas.20247. [DOI] [PubMed] [Google Scholar]
- 3.Chowdhury SK, Katta V, Chait BT. Probing conformational changes in proteins by mass spectrometry. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 4.Dobo A, Kaltashov IA. Detection of multiple protein conformational ensembles in solution via deconvolution of charge-state distributions in ESI MS. Anal Chem. 2001;73:4763–4773. doi: 10.1021/ac010713f. [DOI] [PubMed] [Google Scholar]
- 5.Loo JA, Loo RRO, Udseth HR, Edmonds CG, Smith RD. Solvent-induced conformational changes of polypeptides probed by electrospray-ionization mass spectrometry. Rapid Commun Mass Spectrom. 1991;5:101–105. doi: 10.1002/rcm.1290050303. [DOI] [PubMed] [Google Scholar]
- 6.Iavarone AT, Jurchen JC, Williams ER. Effects of solvent on the maximum charge state and charge state distribution of protein ions produced by electrospray ionization. J Am Soc Mass Spectrom. 2000;11:976–985. doi: 10.1016/S1044-0305(00)00169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iavarone AT, Williams ER. Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J Am Chem Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson BA. Declustering and fragmentation of protein ions from an electrospray ion source. J Am Soc Mass Spectrom. 1997;8:1053–1058. [Google Scholar]
- 9.Iavarone AT, Jurchen JC, Williams ER. Supercharged protein and peptide ions formed by electrospray ionization. Anal Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sterling HJ, Cassou CA, Susa AC, Williams ER. Electrothermal supercharging of proteins in native electrospray ionization. Anal Chem. 2012;84:3795–3801. doi: 10.1021/ac300468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassou CA, Sterling HJ, Susa AC, Williams ER. Electrothermal supercharging in mass spectrometry and tandem mass spectrometry of native proteins. Anal Chem. 2013;85:138–146. doi: 10.1021/ac302256d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teo CA, Donald WA. Solution additives for supercharging proteins beyond the theoretical maximum proton-transfer limit in electrospray ionization mass spectrometry. Anal Chem. 2014;86:4455–4462. doi: 10.1021/ac500304r. [DOI] [PubMed] [Google Scholar]
- 13.Lomeli SH, Peng IX, Yin S, Loo RRO, Loo JA. New reagents for increasing ESI multiple charging of proteins and protein complexes. J Am Soc Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnier PD, Gross DS, Williams ER. Electrostatic forces and dielectric polarizability of multiply protonated gas-phase cytochrome c ions probed by ion/molecule chemistry. J Am Chem Soc. 1995;117:6747–6757. [Google Scholar]
- 15.de la Mora JF. Electrospray ionization of large multiply charged species proceeds via Dole’s charged residue mechanism. Anal Chim Acta. 2000;406:93–104. [Google Scholar]
- 16.Kharlamova A, Prentice BM, Huang TY, McLuckey SA. Electrospray droplet exposure to gaseous acids for the manipulation of protein charge state distributions. Anal Chem. 2010;82:7422–7429. doi: 10.1021/ac101578q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kharlamova A, McLuckey SA. Negative Electrospray Droplet Exposure to Gaseous Bases for the Manipulation of Protein Charge State Distributions. Anal Chem. 2011;83:431–437. doi: 10.1021/ac1027319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams ER. Proton transfer reactivity of large multiply charged ions. J Mass Spectrom. 1996;31:831–842. doi: 10.1002/(SICI)1096-9888(199608)31:8<831::AID-JMS392>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnier PD, Gross DS, Williams ER. On the maximum charge state and proton transfer reactivity of peptide and protein ions formed by electrospray ionization. J Am Soc Mass Spectrom. 1995;6:1086–1097. doi: 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 20.Ahadi E, Konermann L. Modeling the behavior of coarse-grained polymer chains in charged water droplets: implications for the mechanism of electrospray ionization. J Phys Chem B. 2012;116:104–112. doi: 10.1021/jp209344z. [DOI] [PubMed] [Google Scholar]
- 21.Wang G, Cole RB. Charged residue versus ion evaporation for formation of alkali metal halide cluster ions in ESI. Anal Chim Acta. 2000;406:53–65. [Google Scholar]
- 22.Wang G, Cole RB. Effect of solution ionic strength on analyte charge state distributions in positive and negative ion electrospray mass spectrometry. Anal Chem. 1994;66:3702–3708. [Google Scholar]
- 23.Felitsyn N, Peschke M, Kebarle P. Origin and number of charges observed on multiply-protonated native proteins produced by ESI. Int J Mass Spectrom. 2002;219:39–62. [Google Scholar]
- 24.Samalikova M, Grandori R. Protein Charge-State Distributions in Electrospray-Ionization Mass Spectrometry Do Not Appear To Be Limited by the Surface Tension of the Solvent. J Am Chem Soc. 2003;125:13352–13353. doi: 10.1021/ja037000u. [DOI] [PubMed] [Google Scholar]
- 25.Samalikova M, Grandori R. Testing the role of solvent surface tension in protein ionization by electrospray. J Mass Spectrom. 2005;40:503–510. doi: 10.1002/jms.821. [DOI] [PubMed] [Google Scholar]
- 26.Marchese R, Grandori R, Carloni P, Raugei S. A computational model for protein ionization by electrospray based on gas-phase basicity. J Am Soc Mass Spectrom. 2012;23:1903–1910. doi: 10.1007/s13361-012-0449-0. [DOI] [PubMed] [Google Scholar]
- 27.Spencer EAC, Ly T, Julian RR. Formation of the serine octamer: Ion evaporation or charge residue? Int J Mass Spectrom. 2008;270:166–172. [Google Scholar]
- 28.Dole M, Mack LL, Hines RL, Mobley RC. Molecular Beams of Macroions. J Chem Phys. 1968;49:2240–2449. [Google Scholar]
- 29.Hogan CJ, Carroll JA, Rohrs HW, Biswas P, Gross ML. Combined Charged Residue-Field Emission Model of macromolecular electrospray ionization. Anal Chem. 2009;81:369–377. doi: 10.1021/ac8016532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metwally H, McAllister RG, Konermann L. Exploring the Mechanism of Salt-Induced Signal Suppression in Protein Electrospray Mass Spectrometry Using Experiments and Molecular Dynamics Simulations. Anal Chem. 2015;87:2434–2442. doi: 10.1021/ac5044016. [DOI] [PubMed] [Google Scholar]
- 31.Allen SJ, Schwartz AM, Bush MF. Effects of polarity on the structures and charge States of native-like proteins and protein complexes in the gas phase. Anal Chem. 2013;85:12055–12061. doi: 10.1021/ac403139d. [DOI] [PubMed] [Google Scholar]
- 32.Heck AJR, Van Den Heuvel RHH. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom Rev. 2004;23:368–389. doi: 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- 33.Konermann L, Ahadi E, Rodriguez AD, Vahidi S. Unraveling the mechanism of electrospray ionization. Anal Chem. 2013;85:2–9. doi: 10.1021/ac302789c. [DOI] [PubMed] [Google Scholar]
- 34.Konermann L, Rodriguez AD, Liu J. On the formation of highly charged gaseous ions from unfolded proteins by electrospray ionization. Anal Chem. 2012;84:6798–6804. doi: 10.1021/ac301298g. [DOI] [PubMed] [Google Scholar]
- 35.Yue X, Vahidi S, Konermann L. Insights into the mechanism of protein electrospray ionization from salt adduction measurements. J Am Soc Mass Spectrom. 2014;25:1322–1331. doi: 10.1007/s13361-014-0905-0. [DOI] [PubMed] [Google Scholar]
- 36.Vahidi S, Stocks BB, Konermann L. Partially disordered proteins studied by ion mobility-mass spectrometry: implications for the preservation of solution phase structure in the gas phase. Anal Chem. 2013;85:10471–10478. doi: 10.1021/ac402490r. [DOI] [PubMed] [Google Scholar]
- 37.Flick TG, Williams ER. Supercharging with trivalent metal ions in native mass spectrometry. J Am Soc Mass Spectrom. 2012;23:1885–1895. doi: 10.1007/s13361-012-0463-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu MM, Rempel DL, Zhao J, Giblin DE, Gross ML. Probing Ca2+-induced conformational changes in porcine calmodulin by H/D exchange and ESI-MS: Effect of cations and ionic strength. Biochemistry. 2003;42:15388–15397. doi: 10.1021/bi035188o. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Håkansson K. Divalent metal ion-peptide interactions probed by electron capture dissociation of trications. J Am Soc Mass Spectrom. 2006;17:1731–1741. doi: 10.1016/j.jasms.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 40.Ly T, Julian RR. Protein-metal interactions of calmodulin and α-synuclein monitored by selective noncovalent adduct protein probing mass spectrometry. J Am Soc Mass Spectrom. 2008;19:1663–1672. doi: 10.1016/j.jasms.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 41.Verkerk UH, Peschke M, Kebarle P. Effect of buffer cations and of H3O+ on the charge states of native proteins. Significance to determinations of stability constants of protein complexes. J Mass Spectrom. 2003;38:618–631. doi: 10.1002/jms.475. [DOI] [PubMed] [Google Scholar]
- 42.Prakash H, Kansara BT, Mazumdar S. Effects of salts on the charge-state distribution and the structural basis of the most-intense charge-state of the gaseous protein ions produced by electrospray ionization. Int J Mass Spectrom. 2010;289:84–91. [Google Scholar]
- 43.Grandori R. Origin of the conformation dependence of protein charge-state distributions in electrospray ionization mass spectrometry. J Mass Spectrom. 2003;38:11–15. doi: 10.1002/jms.390. [DOI] [PubMed] [Google Scholar]
- 44.Watt SJ, Sheil M, Beck JL, Prosselkov P, Otting G, Dixon NE. Effect of protein stabilization on charge state distribution in positive and negative ion electrospray ionization mass spectra. J Am Soc Mass Spectrom. 2007;18:1605–1611. doi: 10.1016/j.jasms.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Kebarle P. A brief overview of the present status of the mechanisms involved in electrospray mass spectrometry. J Mass Spectrom. 2000;35:804–817. doi: 10.1002/1096-9888(200007)35:7<804::AID-JMS22>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 46.Peschke M, Verkerk UH, Kebarle P. Features of the ESI mechanism that affect the observation of multiply charged noncovalent protein complexes and the determination of the association constant by the titration method. J Am Soc Mass Spectrom. 2004;15:1424–1434. doi: 10.1016/j.jasms.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Peschke M, Blades A, Kebarle P. Charged states of proteins. Reactions of doubly protonated alkyldiamines with NH3: Solvation or deprotonation Extension of two proton cases to multiply protonated globular proteins observed in the gas phase. J Am Chem Soc. 2002;124:11519–11530. doi: 10.1021/ja012591e. [DOI] [PubMed] [Google Scholar]
- 48.Hiraoka K, Asakawa Y, Kawashima Y, Okazaki S, Nakamura M, Yamamoto Y, Takamizawa A. The effect of the presence of foreign salts on the formation of gaseous ions for electrospray and laser spray. Rapid Commun Mass Spectrom. 2004;18:2437–2442. doi: 10.1002/rcm.1644. [DOI] [PubMed] [Google Scholar]
- 49.Catalina MI, Van Den Heuvel RHH, van Duijn E, Heck AJR. Decharging of globular proteins and protein complexes in electrospray. Chem Eur J. 2005;11:960–968. doi: 10.1002/chem.200400395. [DOI] [PubMed] [Google Scholar]
- 50.Le Blanc JCY, Wang J, Guevremont R, Siu KWM. Electrospray mass spectra of protein cations formed in basic solutions. Org Mass Spectrom. 1994;29:587–593. [Google Scholar]
- 51.Loo RRO, Smith RD. Proton transfer reactions of multiply charged peptide and protein cations and anions. J Mass Spectrom. 1995;30:339–347. [Google Scholar]
- 52.Cassady CJ, Wronka J, Kruppa GH, Laukien FH, Hettich R. Deprotonation reactions of multiply protonated ubiquitin ions. Rapid Commun Mass Spectrom. 1994;8:394–400. doi: 10.1002/rcm.1290080511. [DOI] [PubMed] [Google Scholar]
- 53.Winger BE, Light-Wahl KJ, Smith RD. Gas-phase proton transfer reactions involving multiply charged cytochrome c ions and water under thermal conditions. J Am Soc Mass Spectrom. 1992;3:624–630. doi: 10.1016/1044-0305(92)85003-3. [DOI] [PubMed] [Google Scholar]
- 54.Valentine SJ, Counterman AE, Clemmer DE. Conformer-dependent proton-transfer reactions of ubiquitin ions. J Am Soc Mass Spectrom. 1997;8:954–961. [Google Scholar]
- 55.McLuckey SA, Van Berkel GJ, Glish GL. Reactions of dimethylamine with multiply charged ions of cytochrome c. J Am Chem Soc. 1990;112:5668–5670. [Google Scholar]
- 56.Gross DS, Schnier PD, Rodriguez-Cruz SE, Fagerquist CK, Williams ER. Conformations and folding of lysozyme ions in vacuo. Proc Natl Acad Sci USA. 1996;93:3143–3148. doi: 10.1073/pnas.93.7.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogorzalek Loo RR, Smith RD. Investigation of the Gas-Phase Structure of Electrosprayed Proteins Using Ion-Molecule Reactions. J Am Soc Mass Spectrom. 1994;5:207–220. doi: 10.1016/1044-0305(94)85011-9. [DOI] [PubMed] [Google Scholar]
- 58.Ogorzalek Loo RR, Udseth HR, Smith RD. A new approach for the study of gas-phase ion-ion reactions using electrospray ionization. J Am Soc Mass Spectrom. 1992;3:695–705. doi: 10.1016/1044-0305(92)87082-A. [DOI] [PubMed] [Google Scholar]
- 59.Jurchen JC, Williams ER. Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J Am Chem Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal Chem. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 61.Baldwin RL. How Hofmeister ion interactions affect protein stability. Biophysical Journal. 1996;71:2056–2063. doi: 10.1016/S0006-3495(96)79404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goto Y, Fink AL. Conformational states of beta-lactamase: molten-globule states at acidic and alkaline pH with high salt. Biochemistry. 1989;28:945–952. doi: 10.1021/bi00429a004. [DOI] [PubMed] [Google Scholar]
- 63.Marcus Y. Thermodynamics of solvation of ions. Part 5 Gibbs free energy of hydration at 298.15 K. Faraday Trans. 1991;87:2995–2999. [Google Scholar]
- 64.Kielland J. Individual Activity Coefficients of Ions in Aqueous Solutions. J Am Chem Soc. 1937;59:1675–1678. [Google Scholar]
- 65.Fasman GD, editor. Circular Dichroism and the Conformational Analysis of Biomolecules. Springer Science and Business Media; Boston, MA: 2013. [Google Scholar]
- 66.Shi L, Holliday AE, Khanal N, Russell DH, Clemmer DE. Configurationally-Coupled Protonation of Polyproline-7. J Am Chem Soc. 2015;137:8680–8683. doi: 10.1021/jacs.5b04287. [DOI] [PubMed] [Google Scholar]
- 67.Hernández H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 68.Han L, Hyung SJ, Mayers JJS, Ruotolo BT. Bound anions differentially stabilize multiprotein complexes in the absence of bulk solvent. J Am Chem Soc. 2011;133:11358–11367. doi: 10.1021/ja203527a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hunter E, Lias SG. Evaluated gas phase basicities and proton affinities of molecules: an update. J Phys Chem Ref Data. 1998;27:413–656. [Google Scholar]
- 70.Mirza UA, Chait BT. Effects of Anions on the Positive Ion Electrospray Ionization Mass Spectra of Peptides and Proteins. Anal Chem. 1994;66:2898–2904. doi: 10.1021/ac00090a017. [DOI] [PubMed] [Google Scholar]
- 71.Flick TG, Cassou CA, Chang TM, Williams ER. Solution additives that desalt protein ions in native mass spectrometry. Anal Chem. 2012;84:7511–7517. doi: 10.1021/ac301629s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loo RRO, Loo JA, Udseth HR, Fulton JL, Smith RD. Protein structural effects in gas phase ion/molecule reactions with diethylamine. Rapid Commun Mass Spectrom. 1992;6:159–165. doi: 10.1002/rcm.1290060302. [DOI] [PubMed] [Google Scholar]
- 73.Bornschein RE, Hyung SJ, Ruotolo BT. Ion mobility-mass spectrometry reveals conformational changes in charge reduced multiprotein complexes. J Am Soc Mass Spectrom. 2011;22:1690–1698. doi: 10.1007/s13361-011-0204-y. [DOI] [PubMed] [Google Scholar]
- 74.Brunette TJ, Parmeggiani F, Huang PS, Bhabha G, Ekiert DC, Tsutakawa SE, Hura GL, Tainer JA, Baker D. Exploring the repeat protein universe through computational protein design. Nature. 2015;528:580–584. doi: 10.1038/nature16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Mass spectrum of 10 μM carbonic anhydrase formed from pure water (FT-ICR).
Supplemental Figure 2 Zmax (purple square), Zav (pink triangles) for protein ions formed from aqueous ammonium acetate (filled) or pure water (open) as a function of molecular weight. Green line represents ZR, the maximum number of charges on a protein predicted using the Rayleigh limit for a droplet the same size as a spherical protein with a density of 1.0 g/cm3. ZR(CCS) (blue circles) represents the maximum number of charges predicted using the Rayleigh limit for a spherical droplet with the same radius as the CCSav for the protein ions.
Supplemental Table 1 Collisional cross sections (CCS) of cytochrome c and carbonic anhydrase ions formed from pure water or 10 mM aqueous ammonium acetate.