

Abstract
Repairing double-strand breaks (DSBs) is particularly challenging in pericentromeric heterochromatin, where the abundance of repeated sequences exacerbates the risk of ectopic recombination and chromosome rearrangements. Recent studies in Drosophila cells revealed that faithful homologous recombination repair of heterochromatic DSBs relies on the relocalization of DSBs to the nuclear periphery before Rad51 recruitment. Here we summarize the exciting progress in understanding this pathway, including conserved responses in mammalian cells and surprising similarities with mechanisms available in yeast to deal with DSBs in other sites that are difficult to repair, including other repeated sequences. We will also point out some of the most important open questions in the field and emerging evidence suggesting that deregulating these pathways might have dramatic consequences for human health.
Keywords: Heterochromatin, homologous recombination, genome stability, nuclear architecture, DSB repair, Drosophila
Unique mechanisms regulate homologous recombination repair in heterochromatin
Double-strand break (DSB) repair in pericentromeric heterochromatin (hereafter ‘heterochromatin’, See Glossary) is particularly challenging because of the abundance of repeated DNA sequences prone to non-allelic (ectopic) recombination [1, 2]. In Drosophila, about half of these sequences consist of simple ‘satellite’ DNA repeats, predominantly tandem 5-base pair sequences, repeated for hundreds of kilobases to megabases, while the rest are composed of scrambled clusters of transposable elements and about 250 isolated genes [3–5]. Pericentromeric heterochromatin occupies nearly 30% of fly and human genomes [3–5], and is typically enriched for ‘silent’ chromatin marks (e.g., H3K9me2,3 and its associated heterochromatin protein 1, or HP1), but it is absent in budding yeast. Notably, while these sequences are late replicating in Drosophila and mammalian cells [6, 7], they are functionally and structurally distinct from late replicating silent regions associated with the lamina (lamina-associated domains, or LADs) [8–10] and, in contrast to those, they are not usually distributed along the nuclear periphery (see for example [2, 11–14]). Heterochromatin is likely maintained in multicellular eukaryotes for its critical roles in centromere function [15–17], meiotic pairing [18, 19], and sister chromatid cohesion [20, 21], but how these large stretches of highly repeated DNA sequences are safely repaired is just starting to emerge.
Repeated sequences associated with different chromosomes can engage in ectopic recombination during DSB repair, leading to chromosome rearrangements and widespread genome instability [1, 2]. Homologous recombination (HR) starts when DSBs are resected to form single-stranded DNA (ssDNA) filaments, which invade ‘donor’ homologous sequences used as templates for DNA synthesis and repair [22]. In single copy sequences, a unique donor is present on the sister chromatid or the homologous chromosome, and repair is largely ‘error free’ [22]. In heterochromatin, however, the availability of thousands to millions of potential donor sequences can initiate unequal sister chromatid exchanges or inter-chromosomal recombination, leading to deletions, duplications, translocations, and formation of dicentric or acentric chromosomes [1, 23–25]. Despite this danger, HR is a primary pathway used to repair heterochromatic DSBs in both Drosophila and mammalian cells [12, 14, 25–28], and specialized mechanisms have evolved to enable ‘safe’ HR repair in heterochromatin while preventing aberrant recombination.
Major progress in understanding heterochromatin repair pathways comes from recent studies in Drosophila cells, where the organization of all pericentromeric regions in one distinct nuclear domain facilitates the study of nuclear dynamics during repair [12, 29]. These studies revealed that early repair steps occur inside the heterochromatin domain [12, 30], but later steps only occur after a dynamic expansion of the heterochromatin domain [12] and a striking relocalization of repair sites to the nuclear periphery [12, 25, 27, 30, 31]. This regulation of repair in space and time relies on a temporary block of HR progression inside the heterochromatin domain, and the restart of repair after relocalization [12, 25, 28]. Relocalization likely promotes safe HR while preventing aberrant recombination by isolating the DSBs and their templates (on the homologous chromosome or the sister chromatid) away from ectopic sequences before strand invasion [2]. Remarkable similarities to this pathway have been described in mouse cells [2, 14, 32, 33], where heterochromatin is organized in several nuclear domains called ‘chromocenters’, revealing highly conserved strategies for heterochromatin repair.
Here, we review the current understanding of the molecular mechanisms involved, with a focus on discoveries in Drosophila cells and conserved pathways in mouse cells. We highlight the importance of SUMOylation and nuclear architecture in regulating HR progression in heterochromatin, and point out interesting similarities between this response and previously identified mechanisms that repair other repeated DNA sequences (rDNA and telomeres) or other ‘difficult’ regions of the genome across different organisms. Finally, we propose how deregulation of these pathways might impact genome stability in health and in disease.
Silencing components and SUMOylation block HR progression inside the heterochromatin domain
Analyses of protein recruitment to repair foci suggest that the initial steps of the DNA damage response occur with high efficiency in heterochromatin. Specifically, DSB induction in heterochromatin results in checkpoint-dependent phosphorylation of H2A variants in Drosophila and mouse heterochromatin, resulting in γH2Av and γH2AX foci, respectively [2, 12, 14, 33] (Figure 1). The γH2Av-binding protein Mdc1/Mu2 (Mediator of DNA damage checkpoint 1) is also recruited to these sites within minutes from DSB formation in Drosophila cells [12, 27, 30]. Interestingly, in Drosophila cells foci of ATRIP (ATR interacting protein) and TopBP1 (Topoisomerase II Binding Protein 1), which are recruited to resected DSBs, appear brighter and form faster in heterochromatin than in euchromatin [12], suggesting that resection and/or ATRIP-TopBP1 recruitment is particularly efficient in heterochromatin [2, 12, 14, 27, 33] (Figure 1). These observations reversed the previous assumption that heterochromatin compaction and silencing present a barrier to initiating repair responses and support the interesting possibility that early repair steps (e.g., resection) are enhanced inside the domain. However, Rad51 and Rad54 are not recruited to heterochromatic DSBs until after relocalization [12, 14, 25, 28], suggesting that Rad51 loading and strand invasion are initially halted in heterochromatin.
Figure 1 (Key Figure). Model for the pathway that relocalizes heterochromatic DSBs to the nuclear periphery for continuing recombinational repair.

A) Schematic view of Drosophila chromosomes showing the position and extension of pericentromeric heterochromatin (adapted from [29]). Cellular (B) and molecular (C) views show that when DSBs form in Drosophila heterochromatin (in yellow), early damage responses efficiently occur inside the domain. These likely include DSB detection, checkpoint activation, resection, and the recruitment of Smc5/6 and SUMO E3 ligases (i.e., Nse2/Qjt, Nse2/Cerv and dPIAS). SUMOylation of unknown targets blocks HR progression and ectopic recombination. SUMOylated proteins also recruit the STUbL protein Dgrn, and this might be sufficient to induce the relocalization to nuclear pores and INMPs at the nuclear periphery. The RENi protein dRad60 associates with STUbL at the nuclear periphery, promoting STUbL-mediated ubiquitination of SUMOylated targets, removal of the block to HR progression, Rad51 recruitment, and ‘safe’ HR repair. This removal of the block might rely on proteasome-mediated degradation of ubiquitinated target (as shown). Alternatively, these targets might become active after ubiquitination or de-SUMOylation (not shown). This model also predicts that sister chromatids or homologous chromosomes (black line) relocalize in concert with the damaged site to provide homologous templates for repair completion.
In Drosophila, this temporary block to HR progression requires the heterochromatin-specific components Su(var)3–9 (Suppressor of variegation 3–9), SetDB1 (SET Domain Bifurcated 1) and HP1a [12], revealing the importance of heterochromatin silencing in the regulation of DSB repair. Downstream from these components, the block is mediated by Smc5/6 (Structural Maintenance of Chromosomes 5/6 complex) and SUMOylation [12, 25, 28], with three participating SUMO-E3 ligases: dPIAS (protein inhibitor of activated STAT), and the Smc5/6 complex subunits Nse2/Qjt (Quijote) and Nse2/Cerv (Cervantes) [12, 25, 28] (Figure 1, Table 1). Removing this block results in abnormal recruitment of Rad51 inside the domain and aberrant recombination, leading to the formation of recombination-dependent heterochromatic DNA filaments between dividing cells and chromosome rearrangements [12, 23–25, 28]. This suggests the importance of the block to Rad51 recruitment inside the heterochromatin domain to prevent aberrant recombination between heterochromatic repeated sequences.
Table 1. Main repair components responsible for heterochromatin repair in Drosophila with corresponding homologous proteins in S. cerevisiae and mammalian cells.
See text for details.
| Function in DSB repair | S.cerevisiae | D. melanogaster | Mammals |
|---|---|---|---|
| Heterochromatin-associated protein | - | HP1a/Su(var)205 | HP1α, HP1β, HP1γ |
| H3K9me2/3 methyltransferases | - | Su(var)3–9 SetDB1/Egg |
Suv39H1, Suv39H2 SetDB1, SetDB2 |
| Smc subunits of the Smc5/6 complex | Smc5/6 | Smc5/6 | Smc5/6 |
| E3 SUMO ligase of the Smc5/6 complex | Mms21 | Qjt, Cerv | Nse2 |
| PIAS-family E3 SUMO ligases | Siz1, Siz2 | dPIAS/Su(var)2–10 | PIAS1, PIAS2 PIAS3, PIAS4 |
| SUMO-targeted Ub ligase (STUbL) | Slx5/8 | Dgrn | Rnf4 |
| RENi protein | Esc2 | dRad60 | Nip45 |
| DSB anchoring sub-component of the nuclear pore complex | Nup84 | Nup107 | Nup107 |
| SUN-family inner nuclear membrane ‘anchoring’ proteins | Mps3 | Koi, Spag4 | Sun1-3, Sun5, Spag4 |
| Checkpoint kinases | Mec1 Tel1 |
ATR/Mei-41 ATM/Tefu |
ATR ATM |
In addition to blocking HR progression, SUMOylation is required for relocalizing heterochromatic DSBs to the nuclear periphery in Drosophila [12, 25]. The underlying mechanism is still unclear, but the association of SUMOylated proteins with the SUMO-targeted ubiquitin ligase (STUbL) Dgrn (Degringolade, the Drosophila homolog of yeast Slx5/8 and human Rnf4; Table 1) might be sufficient to trigger this movement. Accordingly, Dgrn is recruited to heterochromatic DSBs before their departure from the domain [28], and is required for relocalization [25]. Importantly, Dgrn is required for relocalizing heterochromatic DSBs, but not for blocking HR recruitment inside the domain [25]. This reveals that the block to HR progression and relocalization of heterochromatic DSBs are two genetically separable pathways, with SUMOylation controlling both, but Dgrn being responsible only for relocalization. The Smc5/6 complex is also required for heterochromatin repair in mouse cells, where it blocks the recruitment of the non-homologous end-joining (NHEJ) component Ku80 and promotes DSB relocalization [14], revealing a conserved role of Smc5/6 in the maintenance of heterochromatin stability (Figure 2). However, the loss of HP1 proteins and Smc5/6 leads to abnormal formation of Rad51 foci inside the heterochromatin domain in Drosophila [12, 25, 28] but not in mouse cells [14], suggesting that distinct or redundant mechanisms contribute to blocking HR progression in mammalian heterochromatin.
Figure 2. Comparison between Drosophila and mouse heterochromatin repair pathways.

In both Drosophila and mouse cells, DSBs leave the heterochromatin domain during HR repair. Resection and the Smc5/6 complex are required for relocalization, and Rad51 is recruited after relocalization. Strand invasion components (including Rad51) mediate relocalization likely by stabilizing repair sites outside the heterochromatin domain. Mouse Smc5/6 also blocks NHEJ inside the domain. Heterochromatin expansion occurs during relocalization in both systems (not shown), potentially contributing to DSB signaling and repair and/or dynamics. In Drosophila cells, but not in mouse cells, Smc5/6 and HP1a are sufficient to block HR progression inside the heterochromatin domain, thus preventing ectopic recombination between heterochromatic sequences. Further, NHEJ is available as an alternative pathway for heterochromatin repair in G1; this pathway occurs without relocalization in mouse cells, but might require relocalization in Drosophila cells. Finally, Drosophila heterochromatic DSBs relocalize to the nuclear periphery to continue repair, while the final destination of this movement in mouse cells is still unclear. Whether STUbL and RENi proteins participate in heterochromatin repair in mouse cells is also unknown.
Together, these studies identified heterochromatin silencing proteins and SUMOylation as central components required for blocking Rad51 recruitment inside the heterochromatin domain, and for preventing aberrant recombination between repeated sequences. SUMOylation might also work in concert with STUbL proteins to trigger relocalization of DSBs to the nuclear periphery, providing a critical regulation of heterochromatin repair in space and time.
Heterochromatic DSBs move to the nuclear periphery to complete ‘safe’ HR repair
In Drosophila cells, heterochromatic DSBs relocalize to nuclear pores or inner nuclear membrane proteins (INMPs) at the nuclear periphery before recruiting Rad51 and continuing HR repair [25]. Specifically, the nuclear pore ‘outer ring’ Nup107–160 complex and the INMPs of the SUN family, Koi (Klaroid) and Spag4 (Sperm-associated antigen 4) independently anchor heterochromatic DSBs after relocalization [25] (Figure 1, Table 1).
In addition to providing anchoring functions, thus keeping DSBs away from the heterochromatin domain during strand invasion, the nuclear periphery seems to play an active role in repair progression. In fact, ATRIP focus disassembly and Rad51 recruitment, which reflect HR progression [12], occur only after DSBs have moved to the nuclear periphery [25]. This progression is impaired in the absence of nuclear periphery anchoring components, revealing that anchoring to the periphery is not only concomitant with, but also a prerequisite for the continuation of HR [25].
What promotes the continuation of HR repair at the nuclear periphery is unclear, but the STUbL Dgrn and its partner, the RENi (Rad60-Esc2-Nip45) family protein dRad60, might play a central role in this repair step. Dgrn and dRad60 are highly enriched at the nuclear periphery [25], where they colocalize with both nuclear pores and INMPs [25]. Unlike Dgrn, dRad60 is not recruited to heterochromatic DSBs before relocalization [28], suggesting a specific function of dRad60 at later repair steps. While the functions of RENi proteins are still poorly understood, it has been proposed that they might promote the activity of STUbLs on their targets [34–36]. Thus, dRad60 might facilitate the Dgrn-dependent ubiquitination of SUMOylated targets at the nuclear periphery, relieving the block to HR progression specifically in this location.
Based on previous studies of the role of STUbL proteins in HR repair, ubiquitination might promote proteasome-mediated degradation of components that block HR progression [37–41], or the activation of these components to continue repair [42]. The SUMO-peptidase Ulp1 (Ubiquitin-like-specific protease 1) is also enriched at nuclear pores, potentially providing an alternative pathway for removal of the block [43–45]. Finally, Dgrn and dRad60 likely contribute to ‘docking’ DSBs at the nuclear periphery via their direct interaction with the Smc5/6 complex after DSB relocalization [25]. Understanding the role(s) of STUbL-RENi proteins, the potential involvement of SUMO-proteases, and the interplay between these proteins and other repair and nuclear periphery components, remain important open questions in the field.
Notably, ‘safe’ HR progression at the nuclear periphery requires the presence of donor sequences, suggesting that homologous templates relocalize together with the broken site to the nuclear periphery. While the mechanisms that maintain an association between damage sites and their templates are still unknown, they might include cohesins [46–49] and proteins required for mitotic pairing of homologous chromosomes in flies [50, 51]. Accordingly, both homologous chromosomes and sister chromatids are used as templates for HR repair of Drosophila heterochromatin, although with a preference for the sister chromatid [27].
Loss of anchoring at the nuclear periphery leads to DSBs that explore more space in the nucleus and eventually return inside the heterochromatin domain without completing repair [25]. While there is risk for ectopic recombination associated with resected DSBs, defective anchoring is not sufficient to induce Rad51 recruitment inside the heterochromatin domain or aberrant recombination between heterochromatic sequences in the short-term [25]. This is likely because the Nse2 and dPIAS-dependent protection of the domain is still functional in the absence of anchoring components [12, 25, 28]. However, repair defects resulting from defective anchoring have dramatic consequences for the cells, leading to radiation sensitivity, accumulation of micronuclei, changes in the number of satellites, and widespread chromosome rearrangements [25]. Micronuclei likely result from extra chromosomal circles or acentric chromosome fragments [1, 23, 52], while changes in satellite numbers might be a consequence of intra-chromosomal recombination [1]. Chromosomal abnormalities are mostly characterized by centromere fusions and loss of entire chromosomes and arms, as expected for heterochromatin repair defects [24, 25]. Together, these phenotypes are a consequence of inaccurate or lack of repair at heterochromatic sequences, revealing the importance of the relocalization pathway in the completion of faithful heterochromatin repair and genome integrity.
Nuclear architecture and dynamics participate in HR repair of other repetitive sequences
In addition to regulating heterochromatin repair, nuclear architecture and dynamics play essential roles in repair and stability of other types of repeated sequences, as revealed by numerous studies from yeast to mammalian cells in the past decade (see [53–58] for other recent reviews on this topic). Understanding the differences and similarities across different damage sites and model systems, and the regulatory mechanisms involved in various contexts, is an exciting new frontier in the genome stability field.
Three major types of dynamics have been identified during HR repair. First, mobilization of repair sites is a common response during inter-homolog recombination [59–61], and likely reflects the exploration of the nuclear space during ‘homology search’ (reviewed in [62]). Second, undamaged chromatin also becomes overall more dynamic, albeit to a lesser extent compared to damaged sites [60, 63–65]. This might be the result of a global relaxation of the chromatin [66, 67], a phenomenon possibly linked to heterochromatin expansion [12, 14, 32] and that might facilitate chromatin accessibility to repair components as well as DSB relocalization. Third, repair sites relocalize to specific subnuclear compartments when the lesion occurs in DNA regions that are intrinsically difficult to repair such as at repeated sequences [2, 12, 14, 25, 28, 68–70], collapsed forks [37, 69], eroded telomeres [45, 71], subtelomeric regions [72, 73], or persistent/unrepairable DSBs (e.g., in the absence of a donor sequence for HR repair) [37, 72, 74–78] (Figure 3). In these contexts, relocalization might be required to avoid aberrant recombination with ectopic repeated sequences [12, 25, 68, 69] and/or promote alternative repair pathways when repair is stalled [37, 45, 71, 75, 76, 78] (recently reviewed in [53, 54]).
Figure 3. Overview of relocalization pathways and signaling mechanisms.
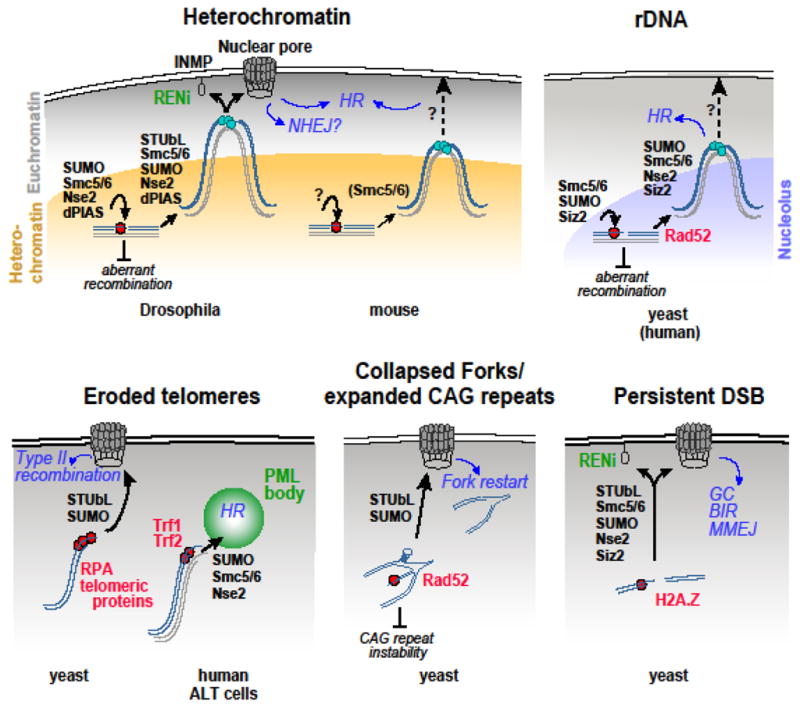
The models show the molecular mechanisms responsible for relocalizing DSBs to sub-nuclear domains to continue repair, including the current understanding of the final destination of these movements, regulatory components of the SUMOylation pathway involved (black, bold), and repair outcomes (blue, italics). The potential role of SUMOylation/relocalization in genome stability is also shown (black, italics). Light blue circles indicate Rad51 recruitment and strand invasion. Question marks point to some of the questions highlighted in the main text. Whether SUMOylation contributes to the spatial and temporal regulation of rDNA repair in human cells is still unclear. GC: Gene conversion. BIR: Break-induced replication. MMEJ: Microhomology-mediated end joining.
The molecular mechanisms required for heterochromatin repair share striking similarities with those responding to DSBs in regions that are difficult to repair (Figure 3, Table 1). For example, pioneering studies in yeast revealed that DSBs in ribosomal DNA (rDNA) repeats move to outside the nucleoli to complete HR repair, and this requires both Smc5/6 and SUMOylation by Siz2 (a dPIAS homologue) [68]. Given the abundance of repeated sequences, rDNA presents similar repair challenges as pericentromeric heterochromatin in multicellular eukaryotes. Further, yeast ‘unrepairable’ DSBs move to nuclear pores or the SUN-family INMP Mps3 (a Koi and Spag4 homolog) [37, 74–78]. This relocalization relies on the STUbL-RENi proteins Slx5/8-Esc2, Smc5/6, and SUMOylation by Nse2 and Siz2 [37, 74, 75, 78]. Yeast eroded telomeres and expanded CAG repeats prone to fork collapse also relocalize to nuclear pores in a STUbL- and SUMOylation-dependent manner [45, 69, 71]. Interestingly, eroded telomeres and damaged CAG repeats do not seem to rely on Mps3 for repair [45, 69], suggesting that different nuclear periphery sites are specialized for dealing with distinct types of damage and/or DNA sequences. The similarity between these relocalization pathways identified in yeast and those participating in Drosophila heterochromatin repair is particularly surprising, given that budding yeast lacks pericentromeric heterochromatin and the ‘silent’ histone marks and associated proteins required to relocalize heterochromatic DSBs [12]. However, the existence of these mechanisms in yeast suggests that they originated early in the evolution, and they have evolved to deal with the complexity of repairing the long stretches of highly repeated sequences that characterize the pericentromeric regions of multicellular eukaryotes.
While DSBs in different repeated DNA sequences typically move to a new nuclear location to continue repair, the final destination of this movement might not always be the nuclear periphery. DSBs induced in mouse heterochromatin with ion irradiation or Cas9 seem to move for a relatively short distance, reaching the periphery of the chromocenters before recruiting Rad51 [14, 33] (Figure 2). Similarly, rDNA of yeast and mammalian cells seem to remain proximal to the nucleolus during HR progression [68, 70] (Figure 3). Whether these sites also associate (even if only transiently) with the nuclear periphery has not been investigated. Live imaging and tracking of repair foci will be necessary to establish whether association with the nuclear periphery occurs, and/or whether alternative subnuclear structures provide anchoring functions.
For example, PML (promyelocytic leukemia) bodies have been proposed as alternative subnuclear domains regulating HR progression in mammalian cells [79] (Figure 3). Specifically, in human ALT cells the alternative HR repair pathway required for telomere elongation occurs after relocalization of telomeres to ALT-associated PML bodies (APBs) and requires Smc5/6 and SUMOylation of telomeric proteins by Nse2 [80]. Given the similarity between this regulation and the mechanisms of targeting to the nuclear periphery, it has been proposed that PML bodies provide functional environments for repair similar to those available at nuclear pores and INMPs in yeast and Drosophila [79]. Alternative sites for HR repair might be particularly important to limit the distance traveled and the possibility of ectopic recombination and translocation [81] in large nuclei, like in mammalian cells, but more studies are required to test these hypotheses.
Together, these studies revealed a critical role for nuclear dynamics and nuclear architecture in DSB repair of repeated DNA sequences that are at high risk for aberrant recombination. Conversely, DSBs in single-copy sequences repaired by NHEJ or HR with the sister chromatid do not seem to undergo long-range movements [37, 82]. Also, NHEJ in heterochromatic repeated sequences does not trigger relocalization in mouse cells [14]. However, a dynamic response has been suggested during heterochromatin repair by NHEJ in Drosophila tissues [27], and more studies are required to understand these potential differences. Determining how different signaling mechanisms direct repair sites to distinct sub-nuclear domains in order to promote ‘safe’ or ‘alternative’ repair pathways, and how they selectively target some DSBs but not others, are central questions in the fields of DNA repair and nuclear dynamics.
Signaling pathways and mechanisms responsible for relocalization of heterochromatic DSBs and other repetitive sequences
SUMOylation is likely a major signal responsible for targeting repair sites to the nuclear periphery, for both heterochromatic DSBs in Drosophila [12, 25, 28], and lesions in DNA regions that are intrinsically difficult to repair in yeast [37, 45, 68, 69, 74, 75, 78] (Figure 3). Interestingly, artificial tethering of polySUMO tails or the STUbL Slx5 to undamaged chromatin in yeast is sufficient to induce the relocalization of these sites to nuclear pores [78], suggesting the tantalizing possibility that STUbL recruitment to polySUMOylated proteins is a universal signal for initiating long-range movements.
Elegant work in yeast also revealed a major influence of the cell cycle on SUMOylation and the type of anchoring site utilized at the nuclear periphery. In G1 or S/G2 cells, persistent DSBs are poly-SUMOylated by the coordinated action of Nse2 and Siz2 and targeted to nuclear pores through the STUbL Slx5/8 [78]. In S/G2, however, mono-SUMOylation by Smc5/6-Nse2 is sufficient to target damage sites to the nuclear periphery, and relocalization preferentially ends at the SUN domain protein Mps3 [78]. Chromatin remodelers also affect the final destination of the movement, with Ino80 and Swr1 preferentially targeting DSBs to Mps3 and nuclear pores, respectively [76]. While the mechanisms responsible for these differences are still under investigation, these results reveal distinct targeting mechanisms for different nuclear periphery sites, and the surprising influence of the cell cycle phase on the extent of SUMOylation, chromatin remodelers involved, and the destination of the movement (reviewed in [56]). The significance of this differential targeting might relate to the availability of different repair pathways at distinct nuclear periphery sites to recover lesions that are difficult to repair, depending on whether the sister chromatid is available or not. In fact, at least in yeast, targeting to SUN domain proteins protects from recombination, while targeting to nuclear pores facilitates HR progression [75, 76]. More studies are required to understand how common the cell-cycle-dependent regulation is in different organisms, and the extent to which the cell cycle, chromatin remodelers, and nuclear periphery anchoring influence heterochromatin repair. The presence of two Nse2 and two Mps3 paralogs in Drosophila potentially adds additional levels of regulation to the system [25, 28], and unraveling these pathways is another exciting challenge for future studies.
A central question is what proteins are targeted by SUMOylation to trigger relocalization, and important studies in yeast and human ALT cells have identified at least some of these regulators (Figure 3). Relocalization and repair of damaged rDNA and expanded CAG repeats depends on Rad52 SUMOylation [68, 69], while H2AZ SUMOylation participates in targeting persistent DSBs to nuclear pores [74, 76]. Relocalization of eroded telomeres to the nuclear pore relies on SUMOylation of telomeric components and RPA [45, 80]. Because a large number of proteins are SUMOylated during the DNA damage response (see for example [36, 83–85]), it is likely that more than one component contributes to signaling DSBs for relocalization, and an interesting possibility is that different targets are specialized for distinct damage sites and relocalization destinations. Further, given that SUMOylation is a common response during DSB repair, a threshold of SUMOylation might also need to be reached to trigger relocalization, perhaps as a result of persistent signaling at DSBs that are difficult to repair. What components are targeted for the spatial and temporal regulation of heterochromatin repair is still a major unanswered question, but possible targets include histones [74, 76, 86], RPA (Replication protein A) [38, 45, 87, 88], Mdc1/Mu2 [38], Smc5/6 subunits [36, 83], Blm (Bloom syndrome protein) [89, 90], and other repair [36, 69, 83, 84] and chromatin [41, 91] components.
Other regulatory mechanisms participating in the mobilization of DSBs include checkpoint kinases [11, 12, 91], resection proteins [12, 14], and strand invasion components [12]. In addition to being required for relocalizing different types of DSBs to the nuclear periphery [37, 45, 74], checkpoint kinases also contribute to the movement of repair sites during inter-homologous HR repair [59, 60], and the global increase in chromatin mobility [64] in yeast. These functions are mediated, at least in part, by chromatin remodelers [64, 76, 92]. Similarly, resection and strand invasion proteins participate in repair dynamics in different contexts [59, 60, 69, 74–76]. Thus, these pathways likely provide signals or means for DSB mobilization regardless of the purpose and the final destination of the movement.
In agreement with this idea, Drosophila checkpoint kinases ATM (Ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3-related) are required for both global heterochromatin expansion and relocalization of DSBs [12]. Heterochromatin expansion likely reflects a relaxation of the chromatin and contributes to relocalization by enabling more exploration of the nuclear space before nuclear periphery anchoring. Accordingly, the peak of heterochromatin expansion corresponds to the time when most relocalization occurs [12], and RNAi depletion of components required for expansion also affects DSB relocalization [12]. However, notably, we identified conditions that impair relocalization of heterochromatic DSBs without affecting global heterochromatin expansion (i.e., Nse2/Qjt RNAi [28]), revealing that global chromatin expansion is not sufficient to induce relocalization of heterochromatic DSBs.
In heterochromatin, checkpoint kinases might also be required to loosen the chromatin locally by reducing the association of silencing components with repair sites and facilitating repair progression or the ‘looping’ of damaged sites to outside of the domain. Accordingly, mouse ATM phosphorylates the HP1-interacting protein Kap1 (KRAB-associated protein-1), thus reducing the strength of Kap1 interaction with damaged heterochromatin, and promoting the release of the chromatin modifier Chd3 (Chromodomain Helicase DNA binding protein 3), chromatin relaxation, and heterochromatin repair [11, 91]. Checkpoint kinases also regulate chromatin relaxation in heterochromatin through the recruitment of ISWI-class chromatin remodelers [93]. Interestingly, HP1β is targeted by Casein Kinase to facilitate HP1β release from damage sites, suggesting redundant targets and effectors in this response [32]. However, whether these chromatin changes contribute to mobilizing heterochromatic DSBs or are required for other repair steps is still unclear. The local reduction of HP1a signals at heterochromatic repair sites occurs after relocalization in Drosophila cells, suggesting that changes to the local heterochromatin structure have late functions in repair (e.g., during Rad51 recruitment or strand invasion) [12]. In agreement with this idea, blocking Kap1 phosphorylation in mouse cells impairs heterochromatin repair but does not affect the relocalization of heterochromatic DSBs [14].
It has been suggested that resection of heterochromatic DSBs might contribute to triggering relocalization, such as via RPA recruitment and SUMOylation [12, 14], while strand invasion components (e.g., Brca2, Rad51 and Rad54) ‘trap’ repair sites outside the heterochromatin domain after relocalization. However, more studies are needed to determine how checkpoint components, resection proteins, chromatin remodelers and strand invasion proteins contribute to heterochromatin repair dynamics, and the potential role of local chromatin changes in the spatial and temporal regulation of heterochromatin repair.
Finally, a major open question in the field is whether the movements of repair sites are driven by motor proteins or rely on increased exploration of the nucleus via Brownian motion followed by anchoring to nuclear structures. Homology search during HR with the homologous chromosome in yeast is largely characterized by constrained Brownian motion [59, 60]. However, surprisingly, Rad51-dependent homology search during ALT repair of telomeres displays rapid directional movements [61], suggesting active forces are involved. Intriguingly, the movement of DSBs and unprotected telomeres in mouse cells is dependent on microtubules and SUN/KASH proteins of the LINC complex spanning the nuclear envelope [65]. While these movements have been mostly correlated with pathological NHEJ events [65], the LINC complex also facilitates the dynamics and HR repair of persistent DSBs in S. pombe [77], and Kinesin-14 mediates the transient interaction of telomeric DSBs with the nuclear pores for break-induced replication (BIR) in S. cerevisiae [73], suggesting broader functions of microtubule-driven motions in DSB repair. Understanding how commonly those forces contribute to directional movements of repair sites and whether similar activities participate in heterochromatin repair dynamics, is another exciting direction for future studies.
Heterochromatin instability in human disease
One of the main discoveries of studies so far is that silencing components, Smc5/6 and SUMOylation are essential for preventing aberrant recombination among repeated sequences [12, 23–25, 28], while promoting faithful HR repair of DSBs in heterochromatin [11, 12, 25]. Nuclear architecture components also play central roles in heterochromatin stability, by enabling the completion of heterochromatic repair in a ‘safe’ environment that averts aberrant recombination and chromosome rearrangements [25].
Strikingly, desilencing of heterochromatic repeats is a common response during early tumorigenesis [94, 95], and HP1 is frequently deregulated in cancer cells [96–98], suggesting heterochromatin de-silencing as a major trigger for aberrant recombination and cancer progression. Consistent with this idea, mouse mutants in heterochromatin silencing components and in the SUMO E3 ligase Nse2 display high cancer incidence [99, 100], and defective heterochromatin repair has been implicated in the cancer-prone syndrome Ataxia-telangiectasia [11]. Further, BRCA1 (breast cancer 1) is required for maintaining heterochromatin silencing, suggesting heterochromatin deprotection as a source of genome instability in breast cancer [95]. Nuclear pore components are also frequently deregulated in cancer cells [101], further strengthening the idea that heterochromatin repair dysfunction contributes to cancer progression (see [101–103] for recent reviews on this topic).
Dramatic changes in heterochromatin structure and organization are also typically observed during aging (reviewed in [104, 105]). Pericentromeric heterochromatin loses both H3K9me3 and HP1 proteins in older flies and human cells, leading to the abnormal expression of satellite sequences [106–108]. This is potentially linked to an overall reduction of silencing components in older cells and/or their sequestration in genomic region that acquire heterochromatin-like structure, such as the senescence-associated heterochromatin foci (SAHF) observed in human cell cultures [105, 109]. Loss of pericentric heterochromatin silencing is potentially a driving force for age-dependent genome instability and cell lethality. Accordingly, cells from older individuals or progeria patients are characterized by loss of heterochromatin marks and higher levels of DNA damage [106, 107]; human cell models of the premature aging Werner Syndrome display the loss of silencing marks in heterochromatin [110]; and mutation of heterochromatin silencing components reduces the lifespan in flies [24, 108]. Defective HR repair in older organisms might further aggravate the consequences of losing heterochromatin protection [111–114]. Interestingly, moderate overexpression of HP1a in flies results in lifespan extension [108], suggesting HP1a loss in older animals as a critical factor in aging. However, more studies are required to establish whether HP1a deregulation contributes to aging by affecting heterochromatin repair. Deregulation of nuclear pores during physiological aging provides other potential sources of heterochromatin repair defects [115].
Finally, loss of heterochromatin silencing and de-repression of satellite sequences has been suggested as a source of neurodegeneration in tauopathies such as Alzheimer’s disease [116]. Whether these aging-related disorders depend on heterochromatin repair defects is also unknown. Understanding the role of silencing and nuclear architecture in heterochromatin repair is essential for understanding how deregulation of these pathways contributes to cancer and other aging-related diseases, including neurological disorders.
Concluding Remarks
This is an exciting time for heterochromatin repair. The tools are now in place for in-depth characterization of the molecular mechanisms involved, including the role of nuclear architecture and dynamics in repair and maintenance of heterochromatic repeated sequences. The emerging picture is that relocalization of DSBs away from the heterochromatin domain before strand invasion enables safe HR repair of heterochromatic DNA sequences. This likely facilitates the interaction of resected filaments with homologous chromosomes or sister chromatids relocalized in concert with the lesion, while at the same time preventing strand invasion of ectopic sequences associated with other pericentromeric regions inside the heterochromatin domain. SUMOylation is central to this pathway, given that it regulates repair progression in coordination with relocalization, guaranteeing that strand invasion only occurs after relocalization. Other central components required for this mechanism to work include the STUbL-RENi proteins, and anchoring structures at the nuclear periphery. More studies are required to establish the conservation of these pathways across different organisms and cell types, the influence of cell cycle phases, the SUMOylation targets, the pathways for repair restart at the nuclear periphery, the forces responsible for relocalization, and the chromatin changes responsible for these dynamics. Identifying the molecular mechanisms involved in this complex pathway is a necessary step to determine how deregulation of these responses contributes to cancer, aging, and aging-related human disorders, potentially revolutionizing approaches to disease prevention and treatments (See Outstanding Questions Box).
Outstanding Questions Box.
What mechanisms facilitate early repair steps inside the heterochromatin domain?
What proteins are SUMOylated to halt HR progression and promote the relocalization of heterochromatic DSBs to the nuclear periphery?
How do SUMO-targeted ubiquitin ligases and SUMO proteases contribute to heterochromatin repair progression at the nuclear periphery?
What regulates the balance between HR and NHEJ pathways in heterochromatin?
How does the cell cycle influence heterochromatin repair dynamics and repair pathway choices?
Is relocalization of heterochromatic DSBs driven by active forces or Brownian motion followed by anchoring structures?
How do checkpoint kinases, resection components, and strand invasion proteins contribute to heterochromatin repair dynamics?
How do local and global chromatin changes contribute to heterochromatin repair?
What mechanisms regulate anchoring to nuclear pores versus INMPs, and do these different locations impact the outcomes of heterochromatin repair?
Is heterochromatin repair completed at the nuclear periphery or are DSBs released from the periphery after strand invasion?
Does heterochromatin repair continue at the nuclear periphery in all organisms, or are alternative anchoring structures used in different organisms?
How is the heterochromatin nuclear structure and epigenetic composition reestablished after repair completion?
What human diseases are specifically driven by heterochromatin repair defects?
Trends Box.
Heterochromatic DSB repair is spatially and temporally regulated by SUMOylation and SUMO-targeted ubiquitin ligases (STUbLs).
DSBs leave the heterochromatin domain before recruiting Rad51 and continuing HR repair in Drosophila and mouse cells, revealing conserved repair pathways.
SUMOylation blocks HR progression inside the heterochromatin domain, thus preventing aberrant recombination among repeated sequences on different chromosomes.
‘Safe’ HR repair of heterochromatic DSBs continues at nuclear pores and inner nuclear membrane proteins at the nuclear periphery in Drosophila cells.
The mechanisms regulating heterochromatin repair in space and time show striking similarities with pathways available in yeast to deal with other DSBs that are difficult to repair, including in rDNA, collapsed forks, and eroded telomeres.
Heterochromatin instability is emerging as a potential driving force for tumorigenesis and other aging-related human disorders.
Acknowledgments
This work was supported by the University of Southern California (USC) Gold Family Fellowship and the USC Research Enhancement Fellowship to T.R., NIH R21ES021541, NIH R01GM117376, The Rose Hills Foundation, and the E. Mallinckrodt Jr. Foundation to I.C. We would like to thank the Chiolo Lab for helpful discussions, and S. Keagy, C. Freudenreich, and L. Delabaere for insightful comments on the manuscript.
Glossary
- ALT cells
human cells that use the recombination-dependent ALT (alternative lengthening of telomere) pathway to extend telomere length in the absence of telomerase.
- BIR (break-induced replication)
HR pathway that repairs DSBs when homology is restricted to one end. It establishes a unidirectional replication fork that copies the donor template until the chromosome end.
- Cohesions
sister chromatid pairing maintained by cohesins.
- Collapsed forks
replication fork that lost the ability to replicate the DNA, for example as a result of replisome dissociation or DSB formation at stalled fork.
- Chromocenter
a cluster of pericentromeric heterochromatin from different chromosomes visible as DAPI-bright region during interphase in mouse cells.
- Extra-chromosomal circles
circular DNA fragments stably maintained in the cells and largely derived from intrachromosomal homologous recombination among tandem repeated sequences.
- Euchromatin
generally used to indicate the gene-rich portion of the genome frequently enriched for active histone marks.
- GC (gene conversion)
most common pathway responsible for HR repair, in which both ends of the break interact with homologous templates for repair synthesis.
- Heterochromatin
generally used to indicate the gene-poor portion of the genome that remain condensed in interphase and is largely composed of high levels of repeated DNA sequences and ‘silent’ histone marks.
- Heterochromatin expansion
~20% increase in volume occupied by the heterochromatin domain (in Drosophila) and chromocenter (in mouse cells) during the DSB response.
- Homologous mitotic (somatic) pairing
pairing between homologous chromosomes in interphase of mitotic cells, commonly observed in Drosophila.
- Inner nuclear membrane proteins
transmembrane proteins embedded in the internal membrane of the nuclear envelope.
- KASH protein
outer nuclear membrane component of a LINC complex characterized by a KASH (Klarsicht, ANC-1, Syne Homology) protein domain.
- Kinesins
motor proteins required for molecule and organelle transport along microtubules.
- Ku80
subunit of the ku70/80 complex involved in NHEJ repair of DSBs.
- LINC complex
complex formed by the interaction of SUN and KASH family proteins at the nuclear envelope, which can transfer forces from the cytoskeleton to the nuclear interior.
- Micronuclei
small nuclei that form when a chromosome or a chromosome fragment is not incorporated in the main nucleus during cell division, usually resulting from genome instability.
- MMEJ (microhomology-mediated end joining)
alternative NHEJ (Alt-NHEJ) pathway that relies on 5–25 base pair micro-homology to align the broken ends before re-joining.
- NHEJ (non-homologous end joining)
mechanism that repairs DSBs by direct ligation of the broken ends. It is largely homology-independent.
- Nuclear Pore
large multimeric protein complex that forms channels across the nuclear envelope to enable regulated molecule transport. It also functions as a chromatin anchoring structure for DNA transcription and repair.
- Nucleolus
nuclear domain responsible for ribosome synthesis and assembly, which contains rDNA, transcribed rRNA, and ribosomal proteins.
- Pericentromeric heterochromatin
the largest contiguous stretches of heterochromatin in multi-cellular eukaryotes. Predominantly made of highly repeated DNA sequences surrounding the centromeres.
- PML bodies
nuclear punctate structures containing the protein PML (romyelocytic Leukemia) and other components, implicated in telomere lengthening and DNA repair in mammalian cells.
- rDNA (ribosomal DNA)
~150–200 copies of genes codifying rRNA, repeated in tandem.
- Resection
one of the earliest steps of HR repair resulting in the formation of ssDNA at DSB sites to initiate strand invasion.
- Rad51
homologous recombination protein that forms a nucleofilament by coating the ssDNA of resected DSBs. It mediates homology search and strand invasion.
- Rad54
homologous recombination protein that promotes Rad51 dissociation after strand invasion, facilitating DNA synthesis and HR repair progression.
- Satellite DNA
large array of tandemly repeated DNA sequences representing one of the main constituents of pericentromeric heterochromatin.
- SUMOylation
post-translational protein modification that modifies protein function through the covalent attachment of SUMO (Small Ubiquitin-like Modifier) peptides.
- SUN protein
Inner nuclear membrane component of the LINC complex containing a SUN (Sad1p, UNC-84) protein domain.
- Telomere erosion
progressive shortening of telomeres occurring as a result of DNA replication cycles. In the absence of telomerase or alternative lengthening processes, telomere erosion eventually triggers a damage response.
- Taupathies
neurodegenerative diseases (e.g., Alzheimer’s disease and Huntington’s disease), characterized by the pathological aggregation of tau protein in the human brain.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peng JC, Karpen GH. Epigenetic regulation of heterochromatic DNA stability. Curr Opin Genet Dev. 2008;18(2):204–11. doi: 10.1016/j.gde.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiolo I, Tang J, Georgescu W, Costes SV. Nuclear dynamics of radiation-induced foci in euchromatin and heterochromatin. Mutat Res. 2013;750(1–2):56–66. doi: 10.1016/j.mrfmmm.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, Villasante A, Dimitri P, Karpen GH, Celniker SE. Sequence Finishing and Mapping of Drosophila melanogaster Heterochromatin. Science. 2007;316(5831):1625–1628. doi: 10.1126/science.1139816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, Parker SC, Gu T, Kundaje A, Riddle NC, Bishop E, Egelhofer TA, Hu SS, Alekseyenko AA, Rechtsteiner A, Asker D, Belsky JA, Bowman SK, Chen QB, Chen RA, Day DS, Dong Y, Dose AC, Duan X, Epstein CB, Ercan S, Feingold EA, Ferrari F, Garrigues JM, Gehlenborg N, Good PJ, Haseley P, He D, Herrmann M, Hoffman MM, Jeffers TE, Kharchenko PV, Kolasinska-Zwierz P, Kotwaliwale CV, Kumar N, Langley SA, Larschan EN, Latorre I, Libbrecht MW, Lin X, Park R, Pazin MJ, Pham HN, Plachetka A, Qin B, Schwartz YB, Shoresh N, Stempor P, Vielle A, Wang C, Whittle CM, Xue H, Kingston RE, Kim JH, Bernstein BE, Dernburg AF, Pirrotta V, Kuroda MI, Noble WS, Tullius TD, Kellis M, MacAlpine DM, Strome S, Elgin SC, Liu XS, Lieb JD, Ahringer J, Karpen GH, Park PJ. Comparative analysis of metazoan chromatin organization. Nature. 2014;512(7515):449–52. doi: 10.1038/nature13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, Booth BW, Pfeiffer BD, George RA, Svirskas R, Krzywinski M, Schein J, Accardo MC, Damia E, Messina G, Mendez-Lago M, de Pablos B, Demakova OV, Andreyeva EN, Boldyreva LV, Marra M, Carvalho AB, Dimitri P, Villasante A, Zhimulev IF, Rubin GM, Karpen GH, Celniker SE. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 2015;25(3):445–58. doi: 10.1101/gr.185579.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol. 1992;116(5):1095–110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schubeler D, Scalzo D, Kooperberg C, van Steensel B, Delrow J, Groudine M. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat Genet. 2002;32(3):438–42. doi: 10.1038/ng1005. [DOI] [PubMed] [Google Scholar]
- 8.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453(7197):948–51. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 9.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38(4):603–13. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148(3):458–72. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31(2):167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 12.Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144(5):732–44. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tjong H, Li W, Kalhor R, Dai C, Hao S, Gong K, Zhou Y, Li H, Zhou XJ, Le Gros MA, Larabell CA, Chen L, Alber F. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc Natl Acad Sci U S A. 2016;113(12):E1663–72. doi: 10.1073/pnas.1512577113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsouroula K, Furst A, Rogier M, Heyer V, Maglott-Roth A, Ferrand A, Reina-San-Martin B, Soutoglou E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol Cell. 2016;63(2):293–305. doi: 10.1016/j.molcel.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Murphy TD, Karpen GH. Localization of centromere function in a Drosophila minichromosome. Cell. 1995;82(4):599–609. doi: 10.1016/0092-8674(95)90032-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, Willard HF. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat Genet. 1997;15(4):345–55. doi: 10.1038/ng0497-345. [DOI] [PubMed] [Google Scholar]
- 17.Sun X, Wahlstrom J, Karpen G. Molecular structure of a functional Drosophila centromere. Cell. 1997;91(7):1007–19. doi: 10.1016/s0092-8674(00)80491-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dernburg AF, Sedat JW, Hawley RS. Direct evidence of a role for heterochromatin in meiotic chromosome segregation. Cell. 1996;86(1):135–46. doi: 10.1016/s0092-8674(00)80084-7. [DOI] [PubMed] [Google Scholar]
- 19.Karpen GH, Le MH, Le H. Centric heterochromatin and the efficiency of achiasmate disjunction in Drosophila female meiosis. Science. 1996;273(5271):118–22. doi: 10.1126/science.273.5271.118. [DOI] [PubMed] [Google Scholar]
- 20.Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. Requirement of heterochromatin for cohesion at centromeres. Science. 2001;294(5551):2539–42. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- 21.Hahn M, Dambacher S, Dulev S, Kuznetsova AY, Eck S, Worz S, Sadic D, Schulte M, Mallm JP, Maiser A, Debs P, von Melchner H, Leonhardt H, Schermelleh L, Rohr K, Rippe K, Storchova Z, Schotta G. Suv4–20h2 mediates chromatin compaction and is important for cohesin recruitment to heterochromatin. Genes Dev. 2013;27(8):859–72. doi: 10.1101/gad.210377.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kowalczykowski SC. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb Perspect Biol. 2015;7(11) doi: 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng JC, Karpen GH. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol. 2007;9(1):25–35. doi: 10.1038/ncb1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng JC, Karpen GH. Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS Genet. 2009;5(3):e1000435. doi: 10.1371/journal.pgen.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, Chiolo I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol. 2015;17(11):1401–11. doi: 10.1038/ncb3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28(21):3413–27. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janssen A, Breuer GA, Brinkman EK, van der Meulen AI, Borden SV, van Steensel B, Bindra RS, LaRocque JR, Karpen GH. A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev. 2016;30(14):1645–57. doi: 10.1101/gad.283028.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryu T, Bonner M, Chiolo I. Cervantes and Quijote protect heterochromatin from aberrant recombination and lead the way to the nuclear periphery. Nucleus. 2016 doi: 10.1080/19491034.2016.1239683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, Peach SE, Shanower G, Zheng H, Kuroda MI, Pirrotta V, Park PJ, Elgin SC, Karpen GH. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 2011;21(2):147–63. doi: 10.1101/gr.110098.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dronamraju R, Mason JM. MU2 and HP1a regulate the recognition of double strand breaks in Drosophila melanogaster. PLoS ONE. 2011;6(9):e25439. doi: 10.1371/journal.pone.0025439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterson CL. The ins and outs of heterochromatic DNA repair. Dev Cell. 2011;20(3):285–7. doi: 10.1016/j.devcel.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453(7195):682–6. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 33.Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, Lobrich M, Taucher-Scholz G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011;39(15):6489–99. doi: 10.1093/nar/gkr230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prudden J, Perry JJP, Arvai AS, Tainer JA, Boddy MN. Molecular mimicry of SUMO promotes DNA repair. Nat Struct Mol Biol. 2009;16(5):509–16. doi: 10.1038/nsmb.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekiyama N, Arita K, Ikeda Y, Hashiguchi K, Ariyoshi M, Tochio H, Saitoh H, Shirakawa M. Structural basis for regulation of poly-SUMO chain by a SUMO-like domain of Nip45. Proteins. 2010;78(6):1491–502. doi: 10.1002/prot.22667. [DOI] [PubMed] [Google Scholar]
- 36.Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, Zhou H. Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet. 2013;9(8):e1003670. doi: 10.1371/journal.pgen.1003670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science. 2008;322(5901):597–602. doi: 10.1126/science.1162790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012;26(11):1179–95. doi: 10.1101/gad.188284.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo K, Zhang H, Wang L, Yuan J, Lou Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012;31(13):3008–19. doi: 10.1038/emboj.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin Y, Seifert A, Chua JS, Maure JF, Golebiowski F, Hay RT. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 2012;26(11):1196–208. doi: 10.1101/gad.189274.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo CY, Li X, Kong XQ, Luo C, Chang CC, Chung Y, Shih HM, Li KK, Ann DK. An arginine-rich motif of ring finger protein 4 (RNF4) oversees the recruitment and degradation of the phosphorylated and SUMOylated Kruppel-associated box domain-associated protein 1 (KAP1)/TRIM28 protein during genotoxic stress. J Biol Chem. 2014;289(30):20757–72. doi: 10.1074/jbc.M114.555672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci Signal. 2012;5(253):ra88. doi: 10.1126/scisignal.2003485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith M, Bhaskar V, Fernandez J, Courey AJ. Drosophila Ulp1, a nuclear pore-associated SUMO protease, prevents accumulation of cytoplasmic SUMO conjugates. J Biol Chem. 2004;279(42):43805–14. doi: 10.1074/jbc.M404942200. [DOI] [PubMed] [Google Scholar]
- 44.Palancade B, Liu X, Garcia-Rubio M, Aguilera A, Zhao X, Doye V. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol Biol Cell. 2007;18(8):2912–23. doi: 10.1091/mbc.E07-02-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Churikov D, Charifi F, Eckert-Boulet N, Silva S, Simon MN, Lisby M, Geli V. SUMO-Dependent Relocalization of Eroded Telomeres to Nuclear Pore Complexes Controls Telomere Recombination. Cell Rep. 2016;15(6):1242–53. doi: 10.1016/j.celrep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Kim JS, Krasieva TB, LaMorte V, Taylor AM, Yokomori K. Specific recruitment of human cohesin to laser-induced DNA damage. J Biol Chem. 2002;277(47):45149–53. doi: 10.1074/jbc.M209123200. [DOI] [PubMed] [Google Scholar]
- 47.Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16(6):1003–15. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 48.Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16(6):991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 49.Oum JH, Seong C, Kwon Y, Ji JH, Sid A, Ramakrishnan S, Ira G, Malkova A, Sung P, Lee SE, Shim EY. RSC facilitates Rad59-dependent homologous recombination between sister chromatids by promoting cohesin loading at DNA double-strand breaks. Mol Cell Biol. 2011;31(19):3924–37. doi: 10.1128/MCB.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKee BD. Homologous pairing and chromosome dynamics in meiosis and mitosis. Biochim Biophys Acta. 2004;1677(1–3):165–80. doi: 10.1016/j.bbaexp.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 51.Joyce EF, Williams BR, Xie T, Wu CT. Identification of genes that promote or antagonize somatic homolog pairing using a high-throughput FISH-based screen. PLoS Genet. 2012;8(5):e1002667. doi: 10.1371/journal.pgen.1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26(1):125–32. doi: 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- 53.Geli V, Lisby M. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays. 2015;37(12):1287–92. doi: 10.1002/bies.201500084. [DOI] [PubMed] [Google Scholar]
- 54.Freudenreich CH, Su XA. Relocalization of DNA Lesions to the Nuclear Pore Complex. FEMS Yeast Res. 2016;16(8):fow095. doi: 10.1093/femsyr/fow095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harding SM, Greenberg RA. Choreographing the Double Strand Break Response: Ubiquitin and SUMO Control of Nuclear Architecture. Front Genet. 2016;7:103. doi: 10.3389/fgene.2016.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horigome C, Gasser SM. SUMO wrestles breaks to the nuclear ring’s edge. Cell Cycle. 2016:1–3. doi: 10.1080/15384101.2016.1216904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalousi A, Soutoglou E. Nuclear compartmentalization of DNA repair. Curr Opin Genet Dev. 2016;37:148–57. doi: 10.1016/j.gde.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 58.Seeber A, Gasser SM. Chromatin organization and dynamics in double-strand break repair. Curr Opin Genet Dev. 2016;43:9–16. doi: 10.1016/j.gde.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Dion V, Kalck V, Horigome C, Towbin BD, Gasser SM. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol. 2012;14(5):502–9. doi: 10.1038/ncb2465. [DOI] [PubMed] [Google Scholar]
- 60.Mine-Hattab J, Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol. 2012;14(5):510–7. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- 61.Cho NW, Dilley RL, Lampson MA, Greenberg RA. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014;159(1):108–21. doi: 10.1016/j.cell.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dion V, Gasser SM. Chromatin movement in the maintenance of genome stability. Cell. 2013;152(6):1355–64. doi: 10.1016/j.cell.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 63.Krawczyk PM, Borovski T, Stap J, Cijsouw T, ten Cate R, Medema JP, Kanaar R, Franken NA, Aten JA. Chromatin mobility is increased at sites of DNA double-strand breaks. J Cell Sci. 2012;125(Pt 9):2127–33. doi: 10.1242/jcs.089847. [DOI] [PubMed] [Google Scholar]
- 64.Seeber A, Dion V, Gasser SM. Checkpoint kinases and the INO80 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev. 2013;27(18):1999–2008. doi: 10.1101/gad.222992.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell. 2015;163(4):880–93. doi: 10.1016/j.cell.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172(6):823–34. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8(8):870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 68.Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragón L, Lisby M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol. 2007:923–31. doi: 10.1038/ncb1619. [DOI] [PubMed] [Google Scholar]
- 69.Su XA, Dion V, Gasser SM, Freudenreich CH. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 2015;29(10):1006–17. doi: 10.1101/gad.256404.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Sluis M, McStay B. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015;29(11):1151–63. doi: 10.1101/gad.260703.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Géli V, Lisby M. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat Cell Biol. 2009;11(8):980–7. doi: 10.1038/ncb1910. [DOI] [PubMed] [Google Scholar]
- 72.Therizols P, Fairhead C, Cabal GG, Genovesio A, Olivo-Marin JC, Dujon B, Fabre E. Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J Cell Biol. 2006;172(2):189–99. doi: 10.1083/jcb.200505159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chung DK, Chan JN, Strecker J, Zhang W, Ebrahimi-Ardebili S, Lu T, Abraham KJ, Durocher D, Mekhail K. Perinuclear tethers license telomeric DSBs for a broad kinesin- and NPC-dependent DNA repair process. Nat Commun. 2015;6:7742. doi: 10.1038/ncomms8742. [DOI] [PubMed] [Google Scholar]
- 74.Kalocsay M, Hiller NJ, Jentsch S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell. 2009;33(3):335–43. doi: 10.1016/j.molcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009;23(8):912–27. doi: 10.1101/gad.1782209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM. SWR1 and INO80 Chromatin Remodelers Contribute to DNA Double-Strand Break Perinuclear Anchorage Site Choice. Mol Cell. 2014;55(4):626–39. doi: 10.1016/j.molcel.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 77.Swartz RK, Rodriguez EC, King MC. A role for nuclear envelope-bridging complexes in homology-directed repair. Mol Biol Cell. 2014;25(16):2461–71. doi: 10.1091/mbc.E13-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, Gasser SM. PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev. 2016;30(8):931–45. doi: 10.1101/gad.277665.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagai S, Davoodi N, Gasser SM. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011;21(3):474–85. doi: 10.1038/cr.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Potts PR, Yu H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 2007;14(7):581–90. doi: 10.1038/nsmb1259. [DOI] [PubMed] [Google Scholar]
- 81.Roukos V, Burgess RC, Misteli T. Generation of cell-based systems to visualize chromosome damage and translocations in living cells. Nat Protoc. 2014;9(10):2476–92. doi: 10.1038/nprot.2014.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007:675–82. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, Zhao X. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell. 2012;45(3):422–32. doi: 10.1016/j.molcel.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Psakhye I, Jentsch S. Protein Group Modification and Synergy in the SUMO Pathway as Exemplified in DNA Repair. Cell. 2012:1–14. doi: 10.1016/j.cell.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 85.Thu YM, Van Riper SK, Higgins L, Zhang T, Becker JR, Markowski TW, Nguyen HD, Griffin TJ, Bielinsky AK. Slx5/Slx8 Promotes Replication Stress Tolerance by Facilitating Mitotic Progression. Cell Rep. 2016;15(6):1254–65. doi: 10.1016/j.celrep.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Groocock LM, Nie M, Prudden J, Moiani D, Wang T, Cheltsov A, Rambo RP, Arvai AS, Hitomi C, Tainer JA, Luger K, Perry JJ, Lazzerini-Denchi E, Boddy MN. RNF4 interacts with both SUMO and nucleosomes to promote the DNA damage response. EMBO Rep. 2014;15(5):601–8. doi: 10.1002/embr.201338369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burgess RC, Rahman S, Lisby M, Rothstein R, Zhao X. The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol Cell Biol. 2007;27(17):6153–62. doi: 10.1128/MCB.00787-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chung I, Zhao X. DNA break-induced sumoylation is enabled by collaboration between a SUMO ligase and the ssDNA-binding complex RPA. Genes Dev. 2015;29(15):1593–8. doi: 10.1101/gad.265058.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eladad S, Ye TZ, Hu P, Leversha M, Beresten S, Matunis MJ, Ellis NA. Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum Mol Genet. 2005;14(10):1351–65. doi: 10.1093/hmg/ddi145. [DOI] [PubMed] [Google Scholar]
- 90.Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, Enomoto T, Ohta K, Foiani M. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell. 2006;127(3):509–22. doi: 10.1016/j.cell.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 91.Goodarzi AA, Kurka T, Jeggo PA. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat Struct Mol Biol. 2011;18(7):831–9. doi: 10.1038/nsmb.2077. [DOI] [PubMed] [Google Scholar]
- 92.Neumann FR, Dion V, Gehlen LR, Tsai-Pflugfelder M, Schmid R, Taddei A, Gasser SM. Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev. 2012;26(4):369–83. doi: 10.1101/gad.176156.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klement K, Luijsterburg MS, Pinder JB, Cena CS, Del Nero V, Wintersinger CM, Dellaire G, van Attikum H, Goodarzi AA. Opposing ISWI- and CHD-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J Cell Biol. 2014;207(6):717–33. doi: 10.1083/jcb.201405077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, Rivera MN, Bardeesy N, Maheswaran S, Haber DA. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331(6017):593–6. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477(7363):179–84. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kirschmann DA, Lininger RA, Gardner LM, Seftor EA, Odero VA, Ainsztein AM, Earnshaw WC, Wallrath LL, Hendrix MJ. Down-regulation of HP1Hsalpha expression is associated with the metastatic phenotype in breast cancer. Cancer Res. 2000;60(13):3359–63. [PubMed] [Google Scholar]
- 97.De Lange R, Burtscher H, Jarsch M, Weidle UH. Identification of metastasis-associated genes by transcriptional profiling of metastatic versus non-metastatic colon cancer cell lines. Anticancer Res. 2001;21(4A):2329–39. [PubMed] [Google Scholar]
- 98.De Koning L, Savignoni A, Boumendil C, Rehman H, Asselain B, Sastre-Garau X, Almouzni G. Heterochromatin protein 1alpha: a hallmark of cell proliferation relevant to clinical oncology. EMBO Mol Med. 2009;1(3):178–91. doi: 10.1002/emmm.200900022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107(3):323–37. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 100.Jacome A, Gutierrez-Martinez P, Schiavoni F, Tenaglia E, Martinez P, Rodriguez-Acebes S, Lecona E, Murga M, Mendez J, Blasco MA, Fernandez-Capetillo O. NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J. 2015;34(21):2604–19. doi: 10.15252/embj.201591829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Simon DN, Rout MP. Cancer and the nuclear pore complex. Adv Exp Med Biol. 2014;773:285–307. doi: 10.1007/978-1-4899-8032-8_13. [DOI] [PubMed] [Google Scholar]
- 102.Carone DM, Lawrence JB. Heterochromatin instability in cancer: from the Barr body to satellites and the nuclear periphery. Semin Cancer Biol. 2013;23(2):99–108. doi: 10.1016/j.semcancer.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saksouk N, Simboeck E, Dejardin J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin. 2015;8:3. doi: 10.1186/1756-8935-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol. 2007;8(9):692–702. doi: 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- 105.Criscione SW, Teo YV, Neretti N. The Chromatin Landscape of Cellular Senescence. Trends Genet. 2016;32(11):751–761. doi: 10.1016/j.tig.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312(5776):1059–63. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103(23):8703–8. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Larson K, Yan SJ, Tsurumi A, Liu J, Zhou J, Gaur K, Guo D, Eickbush TH, Li WX. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012;8(1):e1002473. doi: 10.1371/journal.pgen.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 110.Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A, Yuan T, Yang J, Li Y, Shi L, Guan D, Pan H, Duan S, Ding Z, Li M, Yi F, Bai R, Wang Y, Chen C, Yang F, Li X, Wang Z, Aizawa E, Goebl A, Soligalla RD, Reddy P, Esteban CR, Tang F, Liu GH, Belmonte JC. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348(6239):1160–3. doi: 10.1126/science.aaa1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hendricks CA, Almeida KH, Stitt MS, Jonnalagadda VS, Rugo RE, Kerrison GF, Engelward BP. Spontaneous mitotic homologous recombination at an enhanced yellow fluorescent protein (EYFP) cDNA direct repeat in transgenic mice. Proc Natl Acad Sci U S A. 2003;100(11):6325–30. doi: 10.1073/pnas.1232231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.White RR, Sung P, Vestal CG, Benedetto G, Cornelio N, Richardson C. Double-strand break repair by interchromosomal recombination: an in vivo repair mechanism utilized by multiple somatic tissues in mammals. PLoS One. 2013;8(12):e84379. doi: 10.1371/journal.pone.0084379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sukup-Jackson MR, Kiraly O, Kay JE, Na L, Rowland EA, Winther KE, Chow DN, Kimoto T, Matsuguchi T, Jonnalagadda VS, Maklakova VI, Singh VR, Wadduwage DN, Rajapakse J, So PT, Collier LS, Engelward BP. Rosa26-GFP direct repeat (RaDR-GFP) mice reveal tissue- and age-dependence of homologous recombination in mammals in vivo. PLoS Genet. 2014;10(6):e1004299. doi: 10.1371/journal.pgen.1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Delabaere L, Ertl HA, Massey DJ, Hofley CM, Sohail F, Bienenstock EJ, Sebastian H, Chiolo I, LaRocque JR. Aging impairs double-strand break repair by homologous recombination in Drosophila germ cells. Aging Cell. 2016 doi: 10.1111/acel.12556.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136(2):284–95. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frost B, Hemberg M, Lewis J, Feany MB. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17(3):357–66. doi: 10.1038/nn.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]