

Abstract
Key points
Spinocerebellar ataxia type 6 (SCA6) is a midlife‐onset neurodegenerative disease caused by a CACNA1A mutation; CACNA1A is also implicated in cerebellar development.
We have previously shown that when disease symptoms are present in midlife in SCA684Q/84Q mice, cerebellar Purkinje cells spike with reduced rate and precision. In contrast, we find that during postnatal development (P10–13), SCA684Q/84Q Purkinje cells spike with elevated rate and precision.
Although surplus climbing fibres are linked to ataxia in other mouse models, we found surplus climbing fibre inputs on developing (P10–13) SCA684Q/84Q Purkinje cells when motor deficits were not detected.
Developmental alterations were transient and were no longer observed in weanling (P21–24) SCA684Q/84Q Purkinje cells.
Our results suggest that changes in the developing cerebellar circuit can occur without detectable motor abnormalities, and that changes in cerebellar development may not necessarily persist into adulthood.
Abstract
Although some neurodegenerative diseases are caused by mutations in genes that are known to regulate neuronal development, surprisingly, patients may not present disease symptoms until adulthood. Spinocerebellar ataxia type 6 (SCA6) is one such midlife‐onset disorder in which the mutated gene, CACNA1A, is implicated in cerebellar development. We wondered whether changes were observed in the developing cerebellum in SCA6 prior to the detection of motor deficits. To address this question, we used a transgenic mouse with a hyper‐expanded triplet repeat (SCA684Q/84Q) that displays late‐onset motor deficits at 7 months, and measured cerebellar Purkinje cell synaptic and intrinsic properties during postnatal development. We found that firing rate and precision were enhanced during postnatal development in P10–13 SCA684Q/84Q Purkinje cells, and observed surplus multiple climbing fibre innervation without changes in inhibitory input or dendritic structure during development. Although excess multiple climbing fibre innervation has been associated with ataxic symptoms in several adult transgenic mice, we observed no detectable changes in cerebellar‐related motor behaviour in developing SCA684Q/84Q mice. Interestingly, we found that developmental alterations were transient, as both Purkinje cell firing properties and climbing fibre innervation from weanling‐aged (P21–24) SCA684Q/84Q mice were indistinguishable from litter‐matched control mice. Our results demonstrate that significant alterations in neuronal circuit development may be observed without any detectable behavioural read‐out, and that early changes in brain development may not necessarily persist into adulthood in midlife‐onset diseases.
Keywords: ataxia, cellular electrophysiology, cerebellum, development
Key points
Spinocerebellar ataxia type 6 (SCA6) is a midlife‐onset neurodegenerative disease caused by a CACNA1A mutation; CACNA1A is also implicated in cerebellar development.
We have previously shown that when disease symptoms are present in midlife in SCA684Q/84Q mice, cerebellar Purkinje cells spike with reduced rate and precision. In contrast, we find that during postnatal development (P10–13), SCA684Q/84Q Purkinje cells spike with elevated rate and precision.
Although surplus climbing fibres are linked to ataxia in other mouse models, we found surplus climbing fibre inputs on developing (P10–13) SCA684Q/84Q Purkinje cells when motor deficits were not detected.
Developmental alterations were transient and were no longer observed in weanling (P21–24) SCA684Q/84Q Purkinje cells.
Our results suggest that changes in the developing cerebellar circuit can occur without detectable motor abnormalities, and that changes in cerebellar development may not necessarily persist into adulthood.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CF
climbing fibre
- CF‐EPSC
climbing fibre‐mediated excitatory postsynaptic current
- CV
coefficient of variation
- EPSC
excitatory postsynaptic current
- mIPSC
miniature inhibitory postsynaptic current
- SCA6
spinocerebellar ataxia type 6
- VGluT2
vesicular glutamate transporter type 2
Introduction
Spinocerebellar ataxia type 6 (SCA6) is an autosomal dominant disease caused by a CAG‐repeat expansion mutation in the CACNA1A gene encoding the pore‐forming α1A‐subunit of voltage‐dependent P/Q‐type calcium channels (Zhuchenko et al. 1997) that are highly expressed in cerebellar Purkinje cells (Hillman et al. 1991; Westenbroek et al. 1995). The CACNA1A gene also produces a cleaved C‐terminal (CT) gene product, α1ACT, that is observed in human SCA6 patients and transgenic mouse models, and is located in both the cytosol and nucleus (Ishikawa et al. 2001; Mark et al. 2015) where it may act as a transcriptional regulator and influence cerebellar development (Du et al. 2013). SCA6 is characterized by progressive motor symptoms including ataxia and occulomotor abnormalities; symptoms typically appear in midlife, although the age of onset can vary widely, from late teens to old age (Yabe et al. 1998). Purkinje cell degeneration is a prominent postmortem feature in SCA6 patients (Yang et al. 2000) and is observed in several mouse models of SCA6 (Unno et al. 2012; Jayabal et al. 2015; Mark et al. 2015), although disease onset precedes Purkinje cell loss in some mouse models (Unno et al. 2012; Jayabal et al. 2015).
P/Q channel expression can be detected just a few days after birth in cerebellar Purkinje cells (Indriati et al. 2013), and both the P/Q channel (Miyazaki et al. 2004; Hashimoto et al. 2011) and the α1ACT gene product (Du et al. 2013) have been shown to be involved in Purkinje cell development. Thus, it is surprising that motor symptoms in SCA6 patients manifest only at mid‐life (Matsumura et al. 1997; van de Warrenburg et al. 2002; Ashizawa et al. 2013), not earlier when the protein is first expressed, as might be predicted. Specifically, both P/Q channels (Miyazaki et al. 2004; Hashimoto et al. 2011) and the α1ACT gene product (Du et al. 2013) have been shown to be involved in the maturation of climbing fibre innervation onto Purkinje cells. Additionally, P/Q channels regulate Purkinje cell firing precision in adult mice (Womack et al. 2004), although to our knowledge, the role of P/Q channels has not been studied in the developing SCA6 cerebellum. We wondered whether brain development might be altered in the SCA6 brain, despite the absence of motor symptoms, since it is possible that developmental changes may produce an altered mature cerebellar microcircuit that is predisposed to later pathophysiology.
To address whether developmental alterations are observed in the SCA6 cerebellum, we used electrophysiology and two‐photon imaging in a transgenic SCA6 knock‐in mouse model (SCA684Q) that has a hyper‐expanded 84‐Q tract in the humanized P/Q channel locus (Watase et al. 2008). Both homozygous and heterozygous SCA684Q mice develops motor coordination problems as adults (Watase et al. 2008; Jayabal et al. 2015), and we have recently shown that a common cerebellar Purkinje cell firing deficit is observed in both mice at ages when ataxia is observed, and that rescuing this firing deficit in homozygous SCA684Q/84Q mice improves motor coordination (Jayabal et al. 2016). These results suggest that homozygous SCA684Q/84Q mice are both a good and a practical model for SCA6, which we used to address whether changes were present during postnatal neuronal development. We observed changes in the action potential firing rate and precision firing, as well as in climbing fibre innervation, but not in inhibitory drive or dendritic structure of Purkinje cells from developing (P10–13) SCA684Q/84Q mice. No differences in the behavioural read‐out of these circuit alterations were detected in developing (P10–13) SCA684Q/84Q mice, as predicted by the absence of symptoms in children with the SCA6 mutation. Interestingly, the synaptic and cellular changes observed in developing SCA684Q/84Q mice were transient, and these properties were indistinguishable from WT Purkinje cells from weanling (P21–24) mice, an age when climbing fibre and Purkinje cell firing properties have largely matured (McKay & Turner, 2005; van Welie et al. 2011; Hashimoto & Kano, 2013). Our results demonstrate that developmental perturbations do not necessarily cause detectable behavioural abnormalities, and that changes in circuit development may be transient.
Methods
Ethical approval
Breeding and animal procedures were approved by the McGill Animal Care Committee and were in accordance with the rules and regulations set in place by the Canadian Council on Animal Care.
Animals
Transgenic mice harbouring a humanized 84‐CAG repeat tract knocked into the mouse Cacna1a locus were obtained from The Jackson Laboratory (Bar Harbor, ME, USA; strain: B6.129S7‐Cacna1atm3Hzo/J; stock number: 008683). Heterozygous mice were bred to obtain litter‐matched homozygous (SCA684Q/84Q) and wild‐type (WT) control mice that were used in the experiments.
Acute cerebellar slice preparation
Mice were first deeply anaesthetized with isoflurane and checked for foot‐paw reflex. Mice were then decapitated and their brains were extracted from the skull and rapidly placed in ice‐cold artificial cerebrospinal fluid (ACSF; in mm: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 20 glucose, bubbled with 95% O2–5% CO2 to maintain pH at 7.3; osmolarity 323 mOsm1) (Watt et al. 2009). Parasagittal slices 250 μm thick of cerebellar vermis were cut using a Leica VT 1000S vibrating blade microtome and were incubated in ACSF at 37°C for ∼30–45 min and were then incubated at room temperature before being used in experiments (Watt et al. 2009). All chemicals were procured from Sigma‐Aldrich (Oakville, ON, Canada).
Electrophysiology
Since a posterior–anterior gradient has been observed for climbing fibre innervation (Sugihara, 2005), and membrane properties differ across lobules in mature Purkinje cells (Kim et al. 2012), we have performed all our recordings in anterior lobule III, in order to minimize cross‐lobule variability that might otherwise confound our results obtained during development. Extracellular recordings were acquired with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) in voltage‐clamp mode at 33–34°C, by visually identifying Purkinje cell somata using an upright microscope (Scientifica, Uckfield, UK). On‐line data acquisition and off‐line data analysis were performed using custom‐designed IGOR Pro acquisition and data analysis software (Wavemetrics, Portland, OR, USA).
For climbing fibre measurements, whole‐cell recordings were made from Purkinje cells at 31–33°C. The internal solution was composed of (in mm): 60 CsCl, 10 potassium d‐gluconate, 20 tetraethylammonium (TEA)‐Cl, 20 BAPTA, 4 MgCl2, 4 ATP, 30 Hepes and 0.2% w/v biocytin (pH 7.3, adjusted with CsOH; osmolality 290 mosmol kg–1). Stimulation pipettes were filled with ACSF and were visually positioned in the granule cell layer ∼50 μm away from the Purkinje cell soma. Purkinje cells were voltage clamped at −60 mV and were briefly depolarized to −20 mV when stimuli (duration, 1 ms; amplitude, 0–50 mV) were applied at 20 s inter‐sweep intervals. Climbing fibres were evoked by parametrically increasing the stimulation intensity from 0 to 50 mV; the number of climbing fibres evoked was estimated by counting the number of all‐or‐nothing steps that were evoked in the resulting postsynaptic current (Fig. 3 A) In a subset of experiments, we stimulated climbing fibres with two pulses, with a 100 ms inter‐stimulus interval in order to examine the paired‐pulse ratio at the climbing fibre synapse (Fig. 3 B), which is known to display paired‐pulse depression (Hashimoto & Kano, 1998).
Figure 3. All‐or‐nothing climbing fibres display paired‐pulse depression.
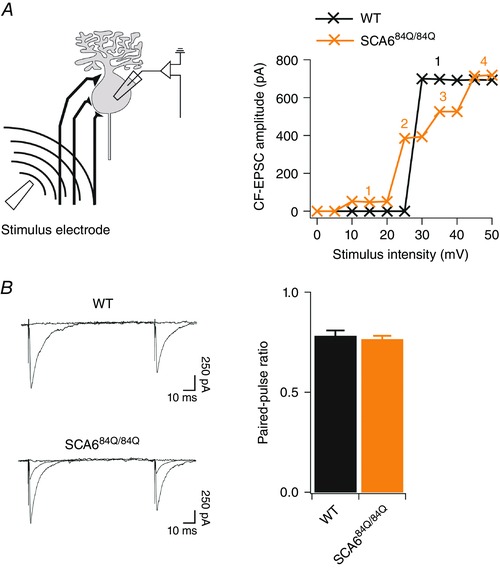
A, left, schematic diagram showing recording configuration. A stimulation electrode was placed in the granule cell layer (∼50 μm from Purkinje cell layer) and climbing fibres were recruited by increasing the stimulation intensity (0–50 mV) parametrically while recording intracellularly from a nearby Purkinje cell. Right, example plots showing evoked EPSC amplitude over the range of stimulus intensity used. WT example (black) shows 1 all‐or‐nothing step in the EPSC response over a range of stimuli, while SCA684Q/84Q example cell (orange) has 4 discrete all‐or‐nothing steps in the EPSC response over the same range of stimulus intensity. B, left, sample recordings from WT (top) and overlaid sample recordings of two climbing fibres in SCA684Q/84Q (bottom) Purkinje cells showing similar degrees of paired‐pulse depression at the climbing fibre synapse in both genotypes. Right, the paired‐pulse ratio, or EPSC2 amplitude/EPSC1 amplitude, is similar in WT (black) and SCA684Q/84Q (orange) Purkinje cells (WT: PPR = 0.78 ± 0.03, n = 4; SCA684Q/84Q: PPR = 0.77 ± 0.02, n = 9; not significantly different, P = 0.59).
For recording miniature inhibitory postsynaptic currents (mIPSCs) from Purkinje cells, whole‐cell recordings were made using internal solution containing (in mm): 60 CsCl, 10 potassium d‐gluconate, 20 TEA‐Cl, 20 BAPTA, 4 MgCl2, 4 ATP, 30 Hepes, 0.2% w/v biocytin (pH 7.3, adjusted with CsOH; osmolality 290 mosmol kg–1). Spontaneous mIPSCs were recorded in ACSF containing 10 μm 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX), 0.2 μm tetrodotoxin (TTX), and 50 μm 2R‐amino‐5‐phosphonovaleric acid (APV) from visually identified Purkinje cells that were voltage clamped at −60 mV.
Purkinje cell morphology
After recording of climbing fibre‐mediated excitatory postsynaptic currents (CF‐EPSCs) or mIPSCs, filled Purkinje cells were imaged using a custom‐built two‐photon microscope (in 1 μm optical sections) with a Ti:Sapphire laser (MaiTai; SpectraPhysics, Santa Clara, CA, USA) tuned to 775 nm. Image acquisition was done using ScanImage (Pologruto et al. 2003) running in Matlab (Mathworks, Natick, MA, USA). Purkinje cells and their dendrites were reconstructed manually using Neurolucida software (MBF Biosciences, Williston, VT, USA). Sholl analysis on Purkinje cells was performed using 1 μm concentric circles surrounding the soma as previously described using ImageJ (Ferreira et al. 2014).
Immunohistochemistry
Mice were transcardially perfused with 4% paraformaldehyde (EMS, Hatfield, PA, USA) and brains were extracted and incubated at 4°C in paraformaldehyde for 24 h, then transferred to phosphate‐buffered saline (PBS) with 0.05% sodium azide. Brains were sliced on a Leica Vibratome 3000 plus vibrating blade microtome into 100 μm thick coronal or sagittal slices. Rabbit anti‐calbindin (D‐28k; Swant, Marly, Switzerland; cat. no.: CB38), and mouse anti‐vesicular glutamate transporter type 2 (VGluT2) (Millipore, Temecula, CA, USA; cat. no.: MAB5504) primary antibodies were added to blocking solution PBS with 0.4% triton‐X, 0.05% sodium azide, and 5% BSA) at a dilution of 1:1000 and 1:300, respectively, and the slices were incubated at room temperature on a rotary shaker at 70 r.p.m. for 72 h. After removal of the remaining antibody with three to five washes with blocking solution, the slices were then incubated for 90 min at room temperature with Alexa‐594 tagged donkey anti‐rabbit antibody (1:1000; Jackson ImmunoResearch, West Grove, PA, USA; cat. no.: 711‐585‐152) and Alexa‐488 tagged donkey anti‐mouse antibody (1:300; Jackson ImmunoResearch, cat. no.: 715‐545‐150). To avoid cross reactivity, Fab fragment donkey anti‐mouse IgG was used (Jackson ImmunoResearch, cat. no.: 715‐007‐003). Sections were then rinsed and immediately mounted onto slides with Prolong Gold Antifade (Invitrogen) and stored in the dark at 4°C. Slices were imaged with a custom‐built two‐photon microscope with a Ti:Sapphire laser (MaiTai; SpectraPhysics) tuned to 775 nm. Image acquisition was done using ScanImage (Pologruto et al. 2003) running in Matlab, and all image acquisition and analysis was done blinded to genotype.
Behaviour
To assess cerebellar‐related behaviour in developing mice (P10), we conducted righting reflex, negative geotaxis and a wire‐hanging assay as described previously (Heyser, 2004; Yu et al. 2008; Takahashi et al. 2009; Markvartova et al. 2010; Mocholi et al. 2011; Zhang et al. 2014). For the righting reflex assay, each mouse was placed on its back and the time it took the mouse to right itself was measured (Yu et al. 2008; Mocholi et al. 2011; Zhang et al. 2014). For the negative geotaxis assay, each mouse was placed on a 45 deg wooden inclined plane with its body pointed towards the base of the platform (Mocholi et al. 2011; Zhang et al. 2014) and the time it took to turn around and face the top of the platform was measured. For the wire hanging test, we measured how long it took for a mouse to fall that was holding on to a thin wire (diameter 1 mm) suspended over a cushioned surface (Takahashi et al. 2009; Markvartova et al. 2010).
To assess motor coordination in weanlings (P21), we used a Rotarod (Stoelting Europe, Dublin, Ireland) as previously described (Watase et al. 2008; Jayabal et al. 2015), since we found that this was the most robust assay for detecting motor changes in older SCA684Q/84Q mice (Jayabal et al. 2015). Mice were acclimatized for an hour in the experimental room before conducting the assay. Mice were then placed on an accelerating rotating rod (from 4 to 40 r.p.m. over 5 min, followed by up to 5 min at 40 r.p.m.), and the latency for mice to fall was noted. The experiments were conducted for one to four trials per day daily from day (D) 1 to D4.
Data analysis
We used custom‐designed IGOR Pro software for data analysis of spike timing and regularity, and for synaptic excitatory postsynaptic current (EPSC) and mIPSC properties. The spike timing precision was quantified using the coefficient of variation (CV) or CV2. CV was calculated using the formula CV = σISI/μISI, where σISI and μISI are the standard deviation and mean, respectively, of the inter‐spike interval (ISI). Additionally, we used CV2, which measures short‐scale regularity in spike trains, using the formula (Holt et al. 1996):
The number of climbing fibres making synapses onto Purkinje cells was estimated by counting the number of discrete climbing fibre all‐or‐nothing evoked EPSC steps as previously described (Hashimoto & Kano, 2003). The paired‐pulse ratio was measured as the EPSC2 amplitude/EPSC1 amplitude. The disparity index, a measure of the coefficient of the variation of the evoked EPSC amplitudes, was computed to see if climbing fibres are differentially strengthened in transgenic mice, according to the formula (Hashimoto & Kano, 2003):
where SD is the standard deviation and M is the mean individual climbing fibre response:
The disparity ratio, an additional measure of the difference between the amplitude of climbing fibre EPSCs, was also computed, according to the formula (Hashimoto & Kano, 2003):
A is the amplitude of an individual EPSC, AN is the largest EPSC amplitude and n is the number of individual EPSCs observed.
Statistics
Data are reported as mean ± SEM. To compare the distributions of the number of climbing fibres per Purkinje cell, we used the Mann–Whitney U test. For all other analyses, comparisons were made using Student's unpaired two‐tailed t test. Statistical comparisons were made in IGOR Pro software with the level of significance (α) set at P < 0.05.
Results
Developmental perturbation of Purkinje cell output in SCA684Q/84Q mouse cerebellum
Purkinje cell firing abnormalities have been shown to occur at the time of disease onset in SCA684Q/84Q mice (Jayabal et al. 2016), and during later stages of disease in a separate SCA6 mouse model (Mark et al. 2015). To address whether Purkinje cell firing properties were affected during development, we recorded extracellular spontaneous Purkinje cell activity in acute sagittal cerebellar slices from P10–13 WT and SCA684Q/84Q mice, since the cerebellar circuit at this age is still undergoing profound developmental changes (McKay & Turner, 2005; van Welie et al. 2011; Hashimoto & Kano, 2013; White & Sillitoe, 2013). We found that developing SCA684Q/84Q Purkinje cells fired with elevated firing rates and enhanced precision of spike timing (Fig. 1 A and B). Firing rates were on average higher than age‐matched WT (mean WT frequency: 78.5 ± 4.0 Hz; n = 33; mean SCA684Q/84Q frequency: 97.6 ± 5.9 Hz, n = 34; significantly different, P = 0.009; Fig. 1 C), in contrast to the reduced firing rates and precision that was observed at 7 months old when motor phenotype is observed (Jayabal et al. 2016), or in a separate SCA6 model when ataxia was present (Mark et al. 2015). Interestingly, the CV was not found to be significantly different between SCA684Q/84Q and WT Purkinje cells (WT: CV = 0.25 ± 0.02, n = 33; SCA684Q/84Q: CV = 0.21 ± 0.01, n = 34, not significantly different from WT, P = 0.09). However, at this age both WT and SCA684Q/84Q Purkinje cells displayed relatively regular firing interspersed with occasional pauses, which has been shown to elevate CV even when cells are otherwise firing regularly (Holt et al. 1996; Shin et al. 2007). Thus, using an alternative measure of regularity, CV2, which captures spiking regularity better than CV in these conditions (Holt et al. 1996; Shin et al. 2007), we found that SCA684Q/84Q Purkinje cells displayed more regular firing with enhanced firing precision in comparison to WT litter‐matched controls (WT: CV2 = 0.24 ± 0.01, n = 33; SCA684Q/84Q: CV2 = 0.19 ± 0.009, n = 34; P = 0.0006; Fig. 1 D).
Figure 1. Enhanced Purkinje cell firing precision and rate in developing SCA684Q/84Q mice.
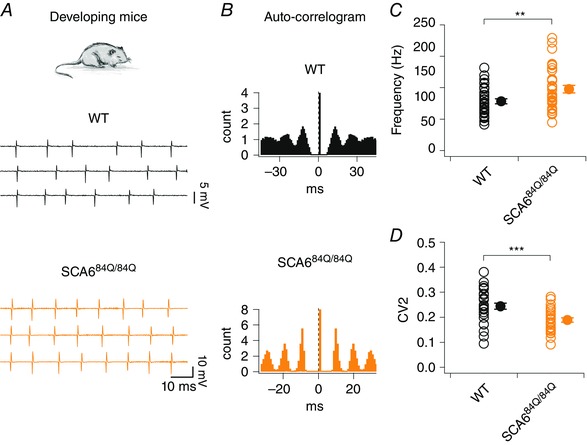
A, top, experiments were performed in P10–13 developing WT and SCA684Q/84Q mice. Bottom, sample traces from representative WT (middle, black) and SCA684Q/84Q (bottom, orange) Purkinje cells. WT cells fire with lower precision and frequency at this age than in adult (compare with Fig. 6 A and C), while developing SCA684Q/84Q Purkinje cells show enhanced firing rate and precision. B, corresponding auto‐correlograms highlight the lack of precision firing in the WT cell (top; with less distinct peaks) in contrast to the highly precise spike timings observed in SCA684Q/84Q Purkinje cell (bottom; with more distinct peaks). C, SCA684Q/84Q Purkinje cells fire action potentials at higher rates compared to WT cells at this developmental stage (P = 0.009). D, Purkinje cell firing precision is enhanced in SCA684Q/84Q Purkinje cells compared to WT as reflected by a lower CV2 of inter‐spike intervals (WT: CV2 = 0.24 ± 0.01, n = 33 cells; SCA684Q/84Q: CV2 = 0.19 ± 0.009, n = 34; significantly different, P = 0.00061). Comparisons made by Student's t test; ** P < 0.01; ***P< 0.001.
We wondered whether the higher firing rates seen in SCA684Q/84Q Purkinje cells caused enhanced spike‐timing precision, because shorter inter‐spike intervals allow for less variance in spike timing. To address this, we compared CV2 in both WT and SCA684Q/84Q Purkinje cells firing below 120 Hz, since the majority of WT cells had firing rates in this range. Using this subset of lower firing rate cells (WT<120Hz: mean frequency = 76.8 ± 3.8 Hz, n = 32; SCA684Q/84Q <120Hz: mean frequency = 82.2 ± 3.8 Hz, n = 26; not significantly different from WT, P = 0.32), we found that the precision of the spiking in SCA684Q/84Q Purkinje cells remained significantly enhanced even when firing rates had been matched (WT<120Hz: CV2 = 0.24 ± 0.012, n = 32; SCA684Q/84Q <120Hz: CV2 = 0.19 ± 0.009, n = 26; significantly different, P = 0.004). Thus, the enhanced precision of Purkinje cell firing from SCA684Q/84Q mice was not an epiphenomenon arising from enhanced firing rate, but reflected a real change in the pattern of spiking.
Since Purkinje cell firing rates increase during development (Gruol et al. 1992; McKay & Turner, 2005; Watt et al. 2009; Arancillo et al. 2015), we wondered whether changes observed in firing precision in SCA684Q/84Q Purkinje cells might arise due to subtle differences in ages sampled across genotype. We observed no significant differences in Purkinje cell age from our action potential recordings (WT: animal age = P11.5 ± 0.17, n = 33; SCA684Q/84Q: animal age = P11.2 ± 0.18, n = 34; not significantly different, P = 0.21). However, to address this in more detail, we subgrouped our data into P10–11 and P12–13 time windows. Interestingly, SCA684Q/84Q Purkinje cells had no significant differences in firing frequency at both P10–11 and P12–13 (P10–11 WT: mean frequency = 73.5 ± 4.9 Hz, n = 17; P10–11 SCA684Q/84Q: mean frequency = 94.1 ± 8.9 Hz, n = 18; not significantly different, P = 0.053; and P12–13 WT: mean frequency = 83.9 ± 6.5 Hz, n = 16; P12–13 SCA684Q/84Q: mean frequency = 101.7 ± 7.6 Hz, n = 16; not significantly different, P = 0.072), likely to be due to the smaller sample sizes. However, in spite of small sample size, SCA684Q/84Q Purkinje cells displayed significant reductions in CV2 at both P10–11 and P12–13 (P10–11: WT: CV2 = 0.24 ± 0.01, n = 17; SCA684Q/84Q: CV2 = 0.18 ± 0.01, n = 18; P = 0.0036; P12–13: WT: CV2 = 0.25 ± 0.02, n = 16; SCA684Q/84Q: CV2 = 0.20 ± 0.01, n = 16; P = 0.041), suggesting that the enhanced firing precision in SCA684Q/84Q Purkinje cells persists over the entire P10–13 developmental time window.
Purkinje cell dendritic morphology is unaltered in developing SCA684Q/84Q mice
Elevated firing rates in Purkinje cells are associated with cellular maturation (Gruol et al. 1992; McKay & Turner, 2005; Watt et al. 2009; Arancillo et al. 2015), suggesting that SCA684Q/84Q mice might develop more rapidly than WT. To examine this possibility, we looked at a common read‐out of cellular maturity in Purkinje cells: the complexity of the dendritic arbour, since Purkinje cell dendrites undergo a dramatic period of growth over the second and third postnatal weeks of development in rodents (McKay & Turner, 2005). Using Alexa 594 to fill neurons through a patch pipette, we captured two‐photon image stacks of Purkinje cell somata and dendrites from lobule III of developing (P10–13) WT and SCA684Q/84Q mice (Fig. 2 A), and used Sholl analysis to assess the size and complexity of Purkinje cell dendrites (Fig. 2 B) (Ferreira et al. 2014). We found no differences in the examined measures of dendritic complexity, including the number of primary dendrites (Fig. 2 C; branching index), the enclosing radius (Fig. 2 C), the convex hull area (Fig. 2 C), and the number of branch points (Fig. 2 C). These results suggest that dendritic maturation – a proxy for the stage of maturation of Purkinje cells – is not affected in SCA684Q/84Q mice in spite of enhanced firing properties, and suggests that Purkinje cells are not maturing more rapidly in SCA684Q/84Q mice.
Figure 2. Normal dendritic morphology observed in developing SCA684Q/84Q Purkinje cells.
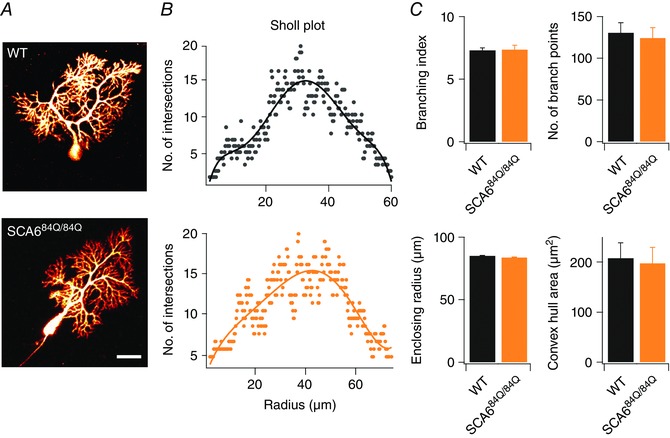
A, representative maximal intensity projection of 2‐photon stacks of Alexa‐594 filled Purkinje cells from P11 WT (top) and P10 SCA684Q/84Q mice (bottom). Scale bar, 20 μm. B, linear Sholl plots from representative Purkinje cells from WT (top, black) and SCA684Q/84Q Purkinje cells (bottom, orange) are similar. C, no significant differences were observed in several measurements of dendritic structure including branching index (WT: 7.32 ± 0.19, n = 9; SCA684Q/84Q: 7.38 ± 0.33, n = 9; not significantly different, P = 0.87), number of branch points (WT: 130.78 ± 11.92, n = 9; SCA684Q/84Q: 124.56 ± 12.25, n = 9; not significantly different, P = 0.72), enclosing radius (WT: 85.24 ± 2.01, n = 9; SCA684Q/84Q: 83.85 ± 1.55, n = 9; not significantly different, P = 0.59), and the convex hull area (WT: 208.78 ± 31.69, n = 9; SCA684Q/84Q: 83.85 ± 1.55, n = 9; not significantly different, P = 0.81). n = 9 for WT and n = 9 for SCA684Q/84Q Purkinje cells; comparisons made with Student's t test.
Selective alterations in Purkinje cell synaptic inputs in developing SCA684Q/84Q mice
Purkinje cells are initially innervated by multiple climbing fibres (>5) in the first postnatal week of development in mice, and then undergo a developmental refinement in which one climbing fibre synapse is strengthened while others are weakened and eventually eliminated, leaving the majority of Purkinje cells mono‐innervated by a single climbing fibre by the third postnatal week of development (Hashimoto & Kano, 2003). Since surplus climbing fibres have been associated with ataxia (Offermanns et al. 1997; Ebner et al. 2013) and P/Q channel knock‐out mice show changes in climbing fibre elimination (Miyazaki et al. 2004), we wondered whether climbing fibre development is altered in SCA684Q/84Q mice in development.
We made whole‐cell recordings in Purkinje cells in acute cerebellar slices from P10–13 WT and SCA684Q/84Q mice and elicited all‐or‐nothing climbing fibre responses (Fig. 3 A; see Methods for details). Climbing fibre synapses normally show paired‐pulse depression when stimulated with short interstimulus intervals (Hashimoto & Kano, 1998). We found that both WT and SCA684Q/84Q Purkinje cells displayed similar levels of paired‐pulse depression (P = 0.59; Fig. 3 B), which supports that we were recording from climbing fibre synapses, and suggests that presynaptic release properties were likely to be normal at SCA684Q/84Q climbing fibre synapses (Hashimoto & Kano, 1998). At this developmental age (P10–13), synapse elimination was well underway in WT cerebellum (Hashimoto & Kano, 2003), and the majority of WT Purkinje cells were innervated by one or two climbing fibres as previously described (Fig. 4 A) (Hashimoto & Kano, 2003). We found that 7/22 or 31.8% of P10–13 Purkinje cells were innervated by one climbing fibre, 12/22 or 54.5% by two climbing fibres, and 3/22 or 13.6% by three climbing fibres (Fig. 4 B). However, using the same stimulation range in SCA684Q/84Q mouse slices to evoke climbing fibre EPSC responses in Purkinje cells (e.g. Fig. 4 A shows 4 discrete climbing fibres), we found that only 1/21 or 4.8% of SCA684Q/84Q Purkinje cells were innervated by one climbing fibre, 11/21 or 52.4% were innervated by two climbing fibres, 8/21 or 38.1% were innervated by three, and 1/21 or 4.8% were innervated by four or more climbing fibres (Fig. 4 B). This distribution of climbing fibre innervation is significantly different in SCA684Q/84Q mice compared to WT (Mann–Whitney U test, P = 0.0063; Fig. 4 B). Interestingly, there were no significant differences in synaptic properties of evoked synaptic currents, including rise time, peak and decay time constants, suggesting that the composition of the receptors at these synapses is not significantly altered in SCA684Q/84Q mice (Table 1). Additionally, we saw no differences in the disparity index and ratio of weak and strong climbing fibres in multiply innervated Purkinje cells, suggesting that the process of synapse elimination, although apparently delayed, followed the normal stages of refinement in other respects (Table 1).
Figure 4. Persistent multiple climbing fibre innervation onto Purkinje cells of SCA684Q/84Q mice observed during the second postnatal week of development.
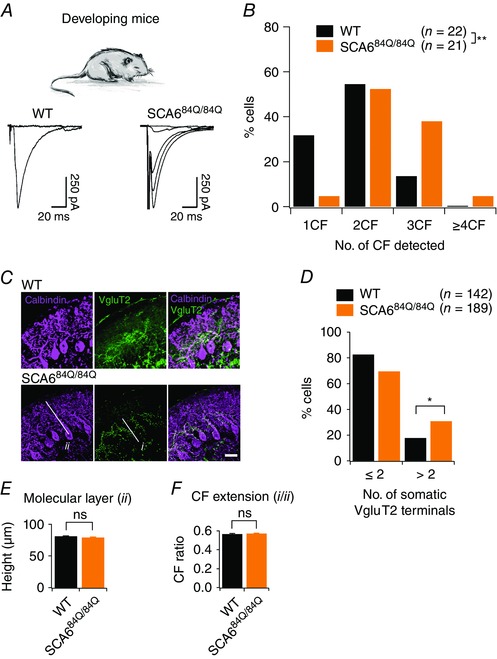
A, top, experiments conducted in P10–13 developing WT and SCA684Q/84Q mice. Bottom, example traces showing climbing fibre innervation from P11 WT mice (left, a maximum of 1 climbing fibre was elicited with extracellular stimulation) and from P12 SCA684Q/84Q mice (right, 4 all‐or‐nothing climbing fibres were elicited with extracellular stimulation). B, histogram showing distribution of Purkinje cells that are innervated by 1, 2, 3, or more climbing fibres (CF) from WT (black) and SCA684Q/84Q (orange) mice. Distribution is significantly shifted to Purkinje cells with higher numbers of climbing fibres at this age in SCA684Q/84Q mice (significantly different, Mann–Whitney U test, P = 0.0063). No other significant differences in EPSC properties were observed (see Table 1). C, representative images show immunocytochemistry labelling of calbindin (purple) and the transporter VGluT2 that is enriched at climbing fibre synapses (green) in P10 WT (top) and SCA684Q/84Q cerebellar slices. D, bar graph showing the number of Purkinje cells with somatic VGluT2 puncta for WT (black) and SCA684Q/84Q (orange) mice. More SCA684Q/84Q Purkinje cells display higher numbers of somatic VGluT2 puncta than WT (significantly different, Student's t test, P = 0.01). E and F, changes in the height of the molecular layer (E; indicated as ii in image in C; WT: 80.81 ± 0.98, n = 3 mice; SCA684Q/84Q: 78.89 ± 1.18, n = 4 mice, not significantly different, Student's t test, P = 0.21) and climbing fibre (CF) extension, the extent of the molecular layer innervated by climbing fibres (F; i/ii in image in C; WT: 0.56 ± 0.01, n = 3 mice; SCA684Q/84Q: 0.57 ± 0.01, n = 4 mice, not significantly different, Student's t test, P = 0.61) are unchanged in SCA684Q/84Q mice, arguing that multiple innervation arises due to the retention of somatic synapses. *P < 0.05; **P < 0.001.
Table 1.
Synaptic properties for climbing fibre (CF) EPSCs and mIPSCs recorded from Purkinje cells in developing (P10–13) mice
| Condition | |||
|---|---|---|---|
| Developing (P10–13) Purkinje cell synaptic properties | WT | SCA684Q/84Q | P |
| CF‐EPSC rise time (ms) | 0.43 ± 0.01 | 0.42 ± 0.03 | 0.56 |
| CF‐EPSC τ decay (ms) | 10.20 ± 0.03 | 10.13 ± 0.10 | 0.39 |
| CF max. amplitude (nA) | 1.71 ± 0.04 | 1.80 ± 0.21 | 0.75 |
| CF disparity index | 0.88 ± 0.04 | 0.91 ± 0.13 | 0.82 |
| CF disparity ratio | 0.46 ± 0.05 | 0.65 ± 0.21 | 0.43 |
| n for CF data (subset of cells analysed for disparity)a | 22 (15) | 21 (20) | |
| mIPSC rise time (ms) | 1.05 ± 0.02 | 1.03 ± 0.03 | 0.65 |
| mIPSC τ decay (ms) | 5.13 ± 0.04 | 5.13 ± 0.04 | 0.96 |
| mIPSC Frequency (Hz) | 6.10 ± 0.60 | 6.57 ± 0.54 | 0.57 |
| mIPSC Amplitude (pA) | 31.10 ± 2.65 | 32.49 ± 3.42 | 0.75 |
| n for mIPSC data | 8 | 8 | |
Mean ± SEM is shown for each condition, and the P value for a Student's t test comparison is shown. aOnly the subset of multiply innervated climbing fibres can be analysed for the disparity index and ratio; n for this analysis is indicated in the parentheses.
Since climbing fibre development is age dependent (Hashimoto & Kano, 2003), we wondered whether we were biasing our data by grouping Purkinje cells from P10–13 mice together. We observed no significant differences in the average ages of the recorded Purkinje cells, arguing that there is no bias in age across genotype (WT: mean age = P11.50 ± 0.19, n = 22; SCA684Q/84Q: mean age = P11.52 ± 0.24, n = 21; not significantly different, P = 0.46). To look in more detail at age‐dependent changes in climbing fibre innervation, we compared our data for P10–11 and P12–13. For P10–11, we found that 2/11 or 18% of WT Purkinje cells were mono‐innervated, 7/11 or 64% were innervated by two climbing fibres, and 2/11 or 18% were innervated by three climbing fibres. This distribution was not significantly different for SCA684Q/84Q Purkinje cells, where 1/9 or 13% of cells were mono‐innervated, 3/9 or 38% were innervated by two climbing fibres, and 5/9 or 63% were innervated by three climbing fibres (Mann–Whitney U test, P = 0.15). Interestingly, the distribution of climbing fibre innervation patterns was significantly different in P12–13 Purkinje cells. For WT mice, 5/11 or 45% of Purkinje cells were mono‐innervated, 5/11 or 45% of cells were innervated by two climbing fibres, and 1/11 or 9% of cells were innervated by three climbing fibres. For P12–13 SCA684Q/84Q mice, 0/12 or 0% were mono‐innervated, 8/12 or 67% were innervated by two climbing fibres, 2/12 or 25% were innervated by three climbing fibres, and 1/12 or 8% were innervated by four or more. These distributions are significantly different (Mann–Whitney U test, P = 0.01), suggesting that differences we observe may arise in particular from differences in later stage (P12–13) climbing fibre elimination.
To complement our electrophysiological estimate of climbing fibre innervation in developing (P10–13) SCA684Q/84Q mice, we used immunocytochemistry to look for morphological changes in climbing fibre innervation (Fig. 4 C). We measured the number of vesicular glutamate transporter type 2 (VGluT2) puncta on the soma of Purkinje cells in developing SCA684Q/84Q mice and found that Purkinje cells with more than two somatic boutons were more common in SCA684Q/84Q mice than WT (Fig. 4 D; P = 0.01). We measured the molecular layer height (ii in Fig. 4 C and E) and climbing fibre extension (extent of Purkinje cell dendrite that had VGluT2 staining; i/ii in Fig. 4 C and F where i denotes VGluT2 height in the molecular layer) in developing SCA684Q/84Q mice, and found that they were not different from WT (P = 0.61). Taken together, our electrophysiology and immunocytochemistry results suggest that P10–13 SCA684Q/84Q Purkinje cells retain surplus somatically located multiple climbing fibres, in comparison to WT.
Climbing fibre elimination on Purkinje cells is regulated by the development of inhibitory synapses on Purkinje cell somata from basket cells (Nakayama et al. 2012). We wondered whether the observed delay in Purkinje cell climbing fibre synapse elimination in developing (P10–13) SCA684Q/84Q mice reflects a change in inhibitory innervation of Purkinje cells. To address this, we isolated inhibitory mIPSCs, reflecting inhibition from basket, stellate and Purkinje cells (Fig. 5 A) (Orduz & Llano, 2007; Watt et al. 2009). We found that both mIPSC frequency and mIPSC amplitude were similar in SCA684Q/84Q and WT Purkinje cells (Fig. 5 B). Furthermore, we observed no differences between WT and SCA684Q/84Q mIPSCs synaptic properties such as rise time and decay time constant (Fig. 5 B and Table 1), suggesting that inhibitory synapse number, location and receptor composition at synapses were not different in developing SCA684Q/84Q mice. This suggests that the surplus climbing fibre innervation we observed in developing SCA684Q/84Q mice was not due to changes in the development of inhibitory input onto Purkinje cells.
Figure 5. Inhibitory input onto Purkinje cells appears normal in developing (P10–13) SCA684Q/84Q mice.

A, representative mIPSC traces from P10 WT (top, black) and SCA684Q/84Q (bottom, orange) Purkinje cells are shown on the left. Average mIPSCs are shown on the right. B, no significant differences are observed between mIPSC properties including rise time, frequency, decay time constant τ, and amplitude from SCA684Q/84Q (orange) and WT (black) Purkinje cells (see Table 1). Not significantly different (P > 0.05), Student's t test; n = 8 for both WT and SCA684Q/84Q Purkinje cells.
No apparent motor deficit in developing SCA684Q/84Q mice
Since changes in firing rate (Hourez et al. 2011), firing precision (e.g. Walter et al. 2006; Alviña & Khodakhah, 2010; Mark et al. 2015; Jayabal et al. 2016), and climbing fibre innervation (e.g. Offermanns et al. 1997; Shuvaev et al. 2011) have previously been associated with motor deficits, we wondered whether the changes we observed in developing SCA684Q/84Q mice were associated with impaired motor function. We were unable to use a Rotarod to assay motor coordination in P10 mice because their eyes were not yet open. Instead, we utilized three behavioural assays that have been associated with impaired cerebellar function in developing mice: righting reflex, negative geotaxis and a wire‐hanging test (Fig. 6) (Takahashi et al. 2009; Mocholi et al. 2011; Van Leuven et al. 2013; Zhang et al. 2014). Both WT and SCA684Q/84Q mice were able to right themselves rapidly when placed on their backs (n = 28 for WT; n = 30 for SCA684Q/84Q mice; P = 0.67; Fig. 6 A), and showed similar negative geotaxis behaviour (n = 28 for WT; n = 30 for SCA684Q/84Q mice; P = 0.62; Fig. 6 B). Furthermore, both WT and SCA684Q/84Q mice showed similar latency to fall on the wire‐hanging test (n = 19 for WT; n = 21 for SCA684Q/84Q mice; P = 0.77; Fig. 5 C). Although there may be subtle motor deficits in SCA684Q/84Q mice that could be detected with more sensitive behavioural assays, our results suggest that the developmental changes observed in SCA684Q/84Q mice do not produce significant changes in motor coordination, which is consistent with human SCA6.
Figure 6. Normal cerebellar‐related behaviour is observed in developing (P10–13) SCA684Q/84Q mice.
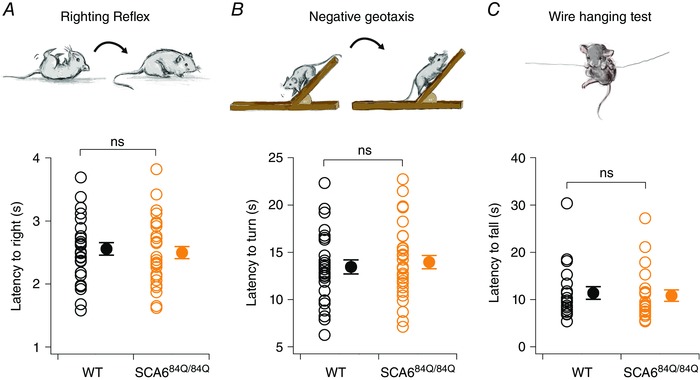
We used three simple motor behaviour assays to evaluate cerebellar‐related behaviour during postnatal development since more standard assays like Rotarod cannot be used before eye opening. A, no changes were observed in the righting reflex, and both SCA684Q/84Q and WT mice took the same time to right when placed on their backs (time to right, WT: 2.6 ± 0.1 s, n = 28, black; SCA684Q/84Q: 2.5 ± 0.1 s, n = 30, orange; not significantly different, P = 0.67). B, a negative geotaxis assay was also used to assay cerebellar‐related behaviour. We observed no significant differences in the latency to turn so that their snout faces up when placed facing down on a shallow incline for WT and SCA684Q/84Q mice (time to turn, WT: 13.5 ± 0.7 s, n = 28; SCA684Q/84Q: 14.0 ± 0.7 s, n = 30; not significantly different, P = 0.62). C, we also used a wire‐hanging test to assay motor strength and cerebellar‐related behaviour. Both WT and SCA684Q/84Q mice showed similar latencies to fall from a wire (latency to fall, WT: 11.4 ± 1.4 s, n = 19; SCA684Q/84Q: 10.9 ± 1.2 s, n = 21; not significantly different, P = 0.77). Comparisons made by Student's t test; ns: P > 0.05.
Recovery of circuit properties in weanling SCA684Q/84Q mouse cerebellum
Our finding that cerebellar development was altered in SCA684Q/84Q mice without concomitant motor deficits during development leads to the question of how long these alterations persist. Do the cerebellar microcircuit alterations persist throughout the lifetime prior to disease onset, for instance, without producing a behavioural output? We addressed this question using weanling (P21–24) mice, an age at which much of the developmental transitions for climbing fibre and firing properties have matured (McKay & Turner, 2005; Watt et al. 2009; Hashimoto & Kano, 2013). We measured spontaneous firing properties from WT and SCA684Q/84Q P21–24 Purkinje cells (Fig. 7 A) in cerebellar slices and found that firing rate was not significantly different (mean P21–24 WT frequency: 86.6 ± 8.3 Hz, n = 35; mean P21–24 SCA684Q/84Q frequency = 76.8 ± 6.1, n = 37; not significantly different, P = 0.34; Fig. 7 A and C). Both WT and SCA684Q/84Q Purkinje cells from weanling mice fire action potentials with high precision, reflected in both the distinct peaks of the autocorrelograms (Fig. 7 B), and the low CV and CV2 of interspike intervals, which was also found to not be significantly different across genotypes (WT: CV = 0.20 ± 0.04, n = 35; SCA684Q/84Q: CV = 0.19 ± 0.04, n = 37; not significantly different, P = 0.89, data not shown; WT: CV2 = 0.14 ± 0.01, n = 35; SCA684Q/84Q: CV2 = 0.16 ± 0.01, n = 37; not significantly different, P = 0.32; Fig. 7 D). These results suggest that Purkinje cell firing properties were only transiently perturbed during development and later recovered so that SCA684Q/84Q Purkinje cell firing was restored to WT levels when the cerebellar microcircuit matures.
Figure 7. Purkinje cell firing properties recovered in weanling SCA684Q/84Q mice.
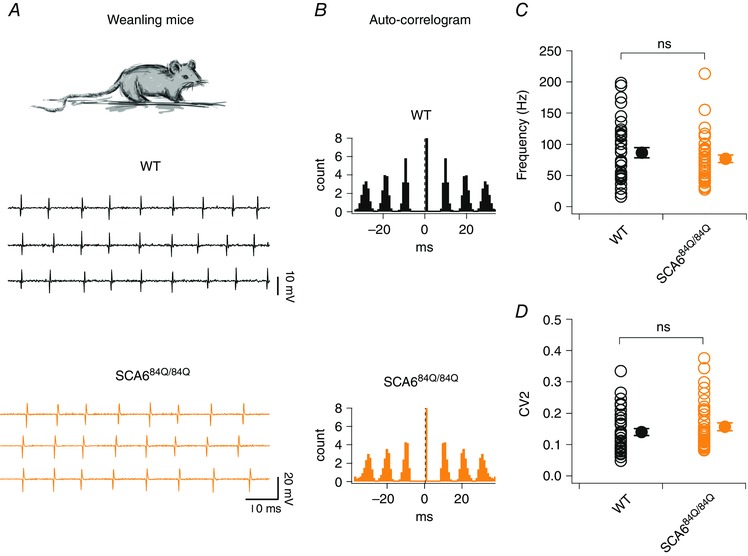
A, top, recordings were made from Purkinje cells from mouse cerebellar slices from weanlings (P21–24). Sample recordings from WT (middle, black) and SCA684Q/84Q Purkinje cells (bottom, orange). B, auto‐correlograms from spike trains from representative neurons show that both WT (top, black) and SCA684Q/84Q (orange, bottom) Purkinje cells display highly regular firing patterns in weanlings. C, no significant changes are observed in the firing frequencies in WT and SCA684Q/84Q Purkinje cells (P = 0.34). D, spike regularity, as measured by CV2, is unchanged in WT and SCA684Q/84Q Purkinje cells (P = 0.32). Comparisons made by Student's t test; ns: P > 0.05.
Interestingly, although firing frequency has been reported to increase during development (Gruol et al. 1992; McKay & Turner, 2005; Watt et al. 2009; Arancillo et al. 2015), we did not observe a statistically significant increase in WT firing frequency comparing our developmental (P10–13) and weanling (P21–24) data (P = 0.38, compare Figs 1 C and 7 C). However, Purkinje cells from WT weanlings showed significantly enhanced firing precision compared to earlier in development (at P10–13), as measured by CV2 (P < 0.0001, compare Figs 1 D and 7 D), which is consistent with a recent developmental study of Purkinje cell simple spiking regularity in vivo (Arancillo et al. 2015).
Since persistent multiple climbing fibres were observed in developing (P10–13) SCA684Q/84Q mice (Fig. 4) and have been associated with ataxia (e.g. Offermanns et al. 1997; Shuvaev et al. 2011), we wondered whether multiple climbing fibre innervation persisted in weanling (P21–24) SCA684Q/84Q mice. To address this, we made whole‐cell recordings from Purkinje cells from weanling WT and SCA684Q/84Q mice and elicited all‐or‐nothing climbing fibre responses with extracellular stimulation (Fig. 8 A; see Fig. 3 A and Methods for further information). We found that the majority of WT Purkinje cells were now mono‐innervated (18/25 or 72% of neurons), although with a significant fraction, 7/25 or 28% of cells, innervated by two climbing fibres (Fig. 8 B). Similarly, we found that weanling SCA684Q/84Q Purkinje cells showed the same pattern of climbing fibre innervation as WT (19/27 SCA684Q/84Q cells or 70% were mono‐innervated, and 8/27 or 30% were innervated by two climbing fibres, not significantly different from control, Mann–Whitney U test, P = 0.79; Fig. 8 B). There were furthermore no significant changes observed in SCA684Q/84Q synaptic properties including the maximal and minimal amplitudes of EPSCs, rise time and decay time constant (Table 2). These results are in contrast to multiple climbing fibre innervation observed in SCA1 that is present both during development and also persists into weanling ages, although ataxic symptoms are evident in these SCA1 mice as early as 3 weeks old (Ebner et al. 2013).
Figure 8. Climbing fibre innervation onto Purkinje cells has recovered in weanling SCA684Q/84Q mice.
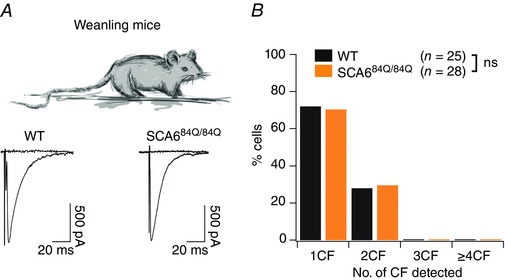
A, top, recordings were made from Purkinje cells from cerebellar slices from weanling (P21–24) mice. Bottom, sample recordings showing only 1 climbing fibre innervating both P22 WT (left) and SCA684Q/84Q (right) Purkinje cells. Properties of EPSCs were not significantly different (see Table 2). B, bar graph showing distribution of Purkinje cells from WT (black) and SCA684Q/84Q (orange) mice that are innervated by 1, 2, 3, or more climbing fibres (CF). Most WT and SCA684Q/84Q Purkinje cells are innervated by 1 climbing fibre; distributions not significantly different; Mann–Whitney U test, P = 0.79.
Table 2.
Synaptic properties for climbing fibres recorded from Purkinje cells in weanling (P21–24) mice
| Condition | |||
|---|---|---|---|
| Weanling (P21–24) Purkinje cell synaptic properties | WT | SCA684Q/84Q | P |
| CF‐EPSC rise time (ms) | 0.42 ± 0.004 | 0.42 ± 0.003 | 0.80 |
| CF‐EPSC τ decay (ms) | 10.32 ± 0.05 | 10.33 ± 0.04 | 0.92 |
| CF max. amplitude (nA) | 1.85 ± 0.08 | 1.92 ± 0.07 | 0.54 |
| CF disparity index | 1.35 ± 0.02 | 1.36 ± 0.01 | 0.68 |
| CF disparity ratio | 0.058 ± 0.020 | 0.044 ± 0.010 | 0.56 |
| n for CF data (subset of cells analysed for disparity)a | 25 (7) | 27 (8) | |
Mean ± SEM is shown for each condition, and the P value for a Student's t test comparison is shown. aOnly the subset of multiply‐innervated climbing fibres can be analysed for the Disparity index and ratio, and the n for this analysis is indicated in the parentheses.
These results demonstrate that the developmental alterations in firing rate and climbing fibre innervation in developing SCA684Q/84Q mice were recovered by weanling age (P21–24), suggesting that cerebellar‐related behaviour was likely to be unaffected as well. To test this, we assayed cerebellar‐related performance using a standard accelerating Rotarod assay over successive trial days, since we have found this is the most sensitive assay for motor coordination deficits in older SCA684Q/84Q mice (Fig. 9 A) (Jayabal et al. 2015). We found that, as expected, SCA684Q/84Q mice showed a day‐on‐day improvement of performance that was indistinguishable from WT mice (no significant differences between WT and SCA684Q/84Q mice with time to fall on the rod on any days, n = 10 for both WT and SCA684Q/84Q weanlings on day 4: P = 0.77; Fig. 9 B).
Figure 9. Cerebellar‐related motor coordination is normal in weanling (P21–24) SCA684Q/84Q mice.
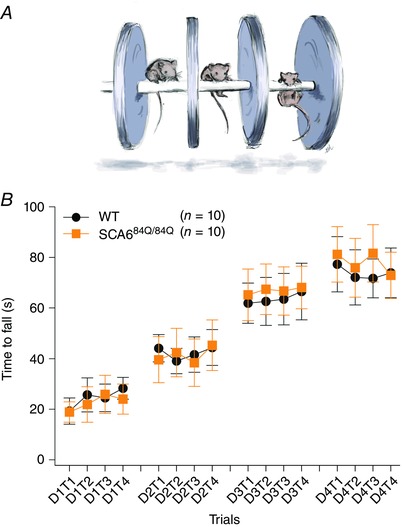
A, schematic diagram illustrating Rotarod assay. B, motor performance of weanlings in an accelerating Rotarod assay conducted in 4 trials (T1–T4) over 4 days of testing (D1–D4) is indistinguishable between WT (black circles) and SCA684Q/84Q mice (orange squares, day 4, WT: 73.75 ± 9.83 s, n = 10; SCA684Q/84Q: 77.90 ± 10.77 s, n = 10, Student's t test, P = 0.77).
Discussion
In this study, we report transient developmental abnormalities in the cerebellum in a mouse model of SCA6. We observe that firing rate and precision are increased in SCA684Q/84Q Purkinje cells during postnatal development, contrary to changes occurring later when disease symptoms are observed (Mark et al. 2015; Jayabal et al. 2016). Changes in the excitatory climbing fibre input were also observed in developing (P10–13) SCA684Q/84Q mice, while no changes were observed for inhibitory synaptic inputs. Interestingly, these alterations in intrinsic spiking properties and excitatory inputs during development were not accompanied by detectable changes in cerebellar‐related motor behaviour, in keeping with observations that children have normal motor behaviour prior to SCA6 onset. Finally, we found that these changes were transient, and had recovered in weanlings (P21–24) when the bulk of cerebellar development has already occurred (McKay & Turner, 2005; van Welie et al. 2011; Hashimoto & Kano, 2013; White & Sillitoe, 2013). These findings demonstrate that alterations in brain development in disease models do not necessarily give rise to behavioural abnormalities, and also may exist only transiently during development.
Purkinje cell firing is affected in several mouse models of ataxias, including EA2 (Walter et al. 2006), SCA1 (Hourez et al. 2011; Dell'Orco et al. 2015), SCA2 (Kasumu et al. 2012; Hansen et al. 2013), SCA3 (Shakkottai et al. 2011), and SCA6 (Mark et al. 2015; Jayabal et al. 2016). However, the changes associated with ataxia tend to manifest as reduced firing rates (Hourez et al. 2011; Hansen et al. 2013), reduced firing precision (Walter et al. 2006; Mark et al. 2015), or both (Jayabal et al. 2016). It is surprising then to observe enhancement of both firing rate and precision in developing Purkinje cells from pre‐motor symptom SCA684Q/84Q mice since these changes are in the opposite direction of firing changes at disease onset (Jayabal et al. 2016), and suggest that the mechanistic underpinnings of these changes is likely to be distinct from that seen at disease onset.
The changes we observe during development in SCA684Q/84Q mice were in fact somewhat counterintuitive: increases in firing rate and precision suggest an enhancement of Purkinje cell maturation, and perhaps heightened function, since such changes have not been shown to be associated with disease in the past, while persistent multi‐climbing fibre innervation suggests a late maturation of Purkinje cells, and suggests impaired function, although we detected no impairment of motor behaviour. Although the heterogeneous nature of these developmental alterations may simply reflect that the changes are unrelated to each other, it is possible that they are linked. For instance, perhaps Purkinje cells respond to persistent climbing fibre innervation, which is deleterious in adult mice as it is often associated with ataxic‐like symptoms (e.g. Offermanns et al. 1997), by enhancing the firing rate and regularity of Purkinje cells to restore normal climbing fibre properties and thus promote normal circuit function. Alternatively, altered climbing fibre elimination might arise due to changes in Purkinje cell firing properties; for example, changes in activity‐dependent synaptic plasticity at climbing fibre synapses (Bosman & Konnerth, 2009), which critically depend on the precise timing of postsynaptic action potentials, might result in surplus climbing fibres. A deeper understanding of the regulatory sequence of events during development in the SCA6 brain may help uncover key regulatory processes that will inform us about normal development as well.
What can we understand from our observation that the changes in the cerebellar circuit during the second postnatal week in SCA684Q/84Q mice do not produce detectable changes in motor coordination? One possible implication from this data is that the cerebellum may not shape motor behaviour at this age, perhaps due to immature wiring downstream of the cerebellar cortex, such as the deep cerebellar nuclei (DCN). Although immunocytochemistry suggests that Purkinje cell–DCN neuron synapses are present by the second postnatal week of development (Garin & Escher, 2001), to our knowledge it is unknown whether developing Purkinje cell–DCN neuron synapses are functional. A recent study by Alvina et al. (2016) found that DCN neurons have remarkably different intrinsic properties from developing mice compared to adult mice, showing that this brain region is likely to undergo dramatic functional changes over the course of development. Further studies of, for example, the development of DCN connectivity, may help to clarify the extent of cerebellar involvement in behaviour during postnatal development. However, alterations in development without an associated detectable behavioural change are frequently observed (e.g. Erickson et al. 2007; Posner et al. 2013; Peixoto et al. 2016) suggesting that the nervous system may have multiple compensatory mechanisms for developmental alterations.
Could developmental changes like the ones we observe contribute to the pathophysiology of SCA6? A recent study from Mark et al. (2015) shows that overexpressing the pathological α1ACT fragment in adult mice can lead to ataxic symptoms, arguing that developmental changes are not necessary for later ataxic symptoms. However, even if not strictly required for ataxia, SCA6 is a genetic disease where the mutated gene is present since conception, meaning that developmental changes may be relevant to understanding its pathophysiology. Indeed, characterizing developmental changes that occur prior to disease onset may help unravel the changes giving rise to disease symptoms from changes in the cerebellum that do not contribute to pathophysiology.
Often, when changes in brain circuits occur prior to the onset of disease‐related behavioural symptoms, they represent a mild initial stage of disease that will progressively worsen over time (e.g. Bibb et al. 2000; Tang et al. 2010; Dougherty et al. 2012; Hansen et al. 2013). The results reported here are distinct from such mechanisms, since we observe changes in firing precision and rate that are the opposite to those observed when ataxic symptoms are present (Mark et al. 2015; Jayabal et al. 2016). Interestingly, a recent study has identified developmental alterations in basal ganglia circuits in a mouse model of autism that are both transient and the opposite of changes observed in the adult: developing basal ganglia neurons exhibit enhanced excitatory synaptic drive, while adult neurons show reduced drive (Peixoto et al. 2016). This study, combined with results presented here, hints that transient developmental abnormalities in disease models that differ from changes seen in adult mice during a disease state might occur more commonly than hitherto appreciated.
Our data from weanling mice suggest that circuit function is rapidly restored during development, and the cerebellum functions normally. Whether this persists into adulthood remains to be seen, since there are likely to be developmental changes still taking place at the weanling age. Thus, the normal synaptic and firing properties we observe in weanlings may not persist into adulthood. Furthermore, even if normal circuit function persists, it is possible, nonetheless, that there are changes in weanling mice that we did not detect, such as changes in ion channel density or calcium dynamics in the cell. Recent computational modelling of a simple brain circuit suggests that homeostatic regulation of ion channels may contribute to pathological loss of function in temporally complex manners: a circuit may adapt to an initial insult, but later this homeostatic compensation can itself become pathological (O'Leary et al. 2014). It will be of interest to determine whether or not such homeostatic adaptation occurs in the SCA6 brain. Interestingly, homeostatic adaptations just prior to disease onset have been observed in a mouse model of SCA1 (Dell'Orco et al. 2015), suggesting that such mechanisms may function in ataxias. A deeper understanding of cerebellar circuits in the SCA6 brain both before and after motor coordination deficits are observed will help us unravel the complex pathophysiology underlying SCA6.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
S.J. performed all electrophysiological and behavioural experiments, imaged, reconstructed and analysed dendritic morphology, made the figures and helped to write the paper. L.L. performed neuronal reconstructions, climbing fibre immunocytochemistry experiments and did analyses. A.J.W. conceived of the project, analysed electrophysiological experiments, made figures and wrote the paper. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by Start‐up Funds from McGill University, a CFI Leaders Opportunity Fund (29127), and a CIHR Operating Grant (MOP 130570), a Royal Society (UK) Equipment Grant, and two Returning Student Awards from the Integrated Program in Neuroscience (IPN) to S.J.
Acknowledgements
We thank Anne McKinney, Keith Murai, Jesper Sjöström, Edward Ruthazer, Moushumi Nath, Sabrina Quilez, Brenda Toscano‐Márquez, Visou Ady, Autumn Metzger, Daneck Lang‐Ouellette, William Todd Farmer, Adele Tufford, Sara El Jaouhari, Oliver Russell, Ahmed Hamam and Gabriel Devlin for helpful discussions and input on the manuscript and/or figures. We thank David Dankort for the use of his fluorescence microscope, Jesper Sjöström for custom Igor software, Rüdiger Krahe for the use of his vibrating blade microtome, and Tiago Ferreira for help with Sholl analysis. We thank Eleanor Milman for artwork included in Figures 1, 4, 6, 7, 8 and 9.
Linked articles This article is highlighted by a Perspective by Miterko & Sillitoe. To read this Perspective, visit http://dx.doi.org/10.1113/JP273301.
References
- Alviña K & Khodakhah K (2010). The therapeutic mode of action of 4‐aminopyridine in cerebellar ataxia. J Neurosci 30, 7258–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvina K, Tara E & Khodakhah K (2016). Developmental change in the contribution of voltage‐gated Ca2+ channels to the pacemaking of deep cerebellar nuclei neurons. Neuroscience 322, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancillo M, White JJ, Lin T, Stay TL & Sillitoe RV (2015). In vivo analysis of Purkinje cell firing properties during postnatal mouse development. J Neurophysiol 113, 578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizawa T, Figueroa KP, Perlman SL, Gomez CM, Wilmot GR, Schmahmann JD, Ying SH, Zesiewicz TA, Paulson HL, Shakkottai VG, Bushara KO, Kuo SH, Geschwind MD, Xia G, Mazzoni P, Krischer JP, Cuthbertson D, Holbert AR, Ferguson JH, Pulst SM & Subramony SH (2013). Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis 8, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A & Greengard P (2000). Severe deficiencies in dopamine signalling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci USA 97, 6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman LW & Konnerth A (2009). Activity‐dependent plasticity of developing climbing fibre‐Purkinje cell synapses. Neuroscience 162, 612–623. [DOI] [PubMed] [Google Scholar]
- Dell'Orco JM, Wasserman AH, Chopra R, Ingram MA, Hu YS, Singh V, Wulff H, Opal P, Orr HT & Shakkottai VG (2015). Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J Neurosci 35, 11292–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty SE, Reeves JL, Lucas EK, Gamble KL, Lesort M & Cowell RM (2012). Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington disease. Exp Neurol 236, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, Hansel C & Gomez CM (2013). Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 154, 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner BA, Ingram MA, Barnes JA, Duvick LA, Frisch JL, Clark HB, Zoghbi HY, Ebner TJ & Orr HT (2013). Purkinje cell ataxin‐1 modulates climbing fibre synaptic input in developing and adult mouse cerebellum. J Neurosci 33, 5806–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MA, Haburcak M, Smukler L & Dunlap K (2007). Altered functional expression of Purkinje cell calcium channels precedes motor dysfunction in tottering mice. Neuroscience 150, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ & van Meyel DJ (2014). Neuronal morphometry directly from bitmap images. Nat Methods 11, 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin N & Escher G (2001). The development of inhibitory synaptic specializations in the mouse deep cerebellar nuclei. Neuroscience 105, 431–441. [DOI] [PubMed] [Google Scholar]
- Gruol DL, Deal CR & Yool AJ (1992). Developmental changes in calcium conductances contribute to the physiological maturation of cerebellar Purkinje neurons in culture. J Neurosci 12, 2838–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS & Pulst SM (2013). Changes in Purkinje cell firing and gene expression precede behavioural pathology in a mouse model of SCA2. Hum Mol Genet 22, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K & Kano M (1998). Presynaptic origin of paired‐pulse depression at climbing fibre‐Purkinje cell synapses in the rat cerebellum. J Physiol 506, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K & Kano M (2003). Functional differentiation of multiple climbing fibre inputs during synapse elimination in the developing cerebellum. Neuron 38, 785–796. [DOI] [PubMed] [Google Scholar]
- Hashimoto K & Kano M (2013). Synapse elimination in the developing cerebellum. Cell Mol Life Sci 70, 4667–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Tsujita M, Miyazaki T, Kitamura K, Yamazaki M, Shin HS, Watanabe M, Sakimura K & Kano M (2011). Postsynaptic P/Q‐type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc Natl Acad Sci USA 108, 9987–9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyser CJ ( 2004). Assessment of developmental milestones in rodents. Curr Protoc Neurosci Chapter 8, Unit 8 18. [DOI] [PubMed] [Google Scholar]
- Hillman D, Chen S, Aung TT, Cherksey B, Sugimori M & Llinas RR (1991). Localization of P‐type calcium channels in the central nervous system. Proc Natl Acad Sci USA 88, 7076–7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt GR, Softky WR, Koch C & Douglas RJ (1996). Comparison of discharge variability in vitro and in vivo in cat visual cortex neurons. J Neurophysiol 75, 1806–1814. [DOI] [PubMed] [Google Scholar]
- Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d'Exaerde A, Cheron G, Orr HT, Pandolfo M & Schiffmann SN (2011). Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 31, 11795–11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indriati DW, Kamasawa N, Matsui K, Meredith AL, Watanabe M & Shigemoto R (2013). Quantitative localization of Cav2.1 (P/Q‐type) voltage‐dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium‐activated potassium channels. J Neurosci 33, 3668–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Owada K, Ishida K, Fujigasaki H, Li Shun M, Tsunemi T, Ohkoshi N, Toru S, Mizutani T, Hayashi M, Arai N, Hasegawa K, Kawanami T, Kato T, Makifuchi T, Shoji S, Tanabe T & Mizusawa H (2001). Cytoplasmic and nuclear polyglutamine aggregates in SCA6 Purkinje cells. Neurology 56, 1753–1756. [DOI] [PubMed] [Google Scholar]
- Jayabal S, Chang HHV, Cullen KE & Watt AJ (2016). 4‐Aminopyridine alleviates ataxia and reverses cerebellar output deficiency in a mouse model of spinocerebellar ataxia type 6. Sci Rep 6, 29489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayabal S, Ljungberg L, Erwes T, Cormier A, Quilez S, El Jaouhari S & Watt AJ (2015). Rapid onset of motor deficits in a mouse model of spinocerebellar ataxia type 6 precedes late cerebellar degeneration. eNeuro 2, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, Eriksen BL, Strobaek D, Liang X, Egorova P, Vorontsova D, Christophersen P, Ronn LC & Bezprozvanny I (2012). Selective positive modulator of calcium‐activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol 19, 1340–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Oh SH, Lee JH, Chang SO, Kim J & Kim SJ (2012). Lobule‐specific membrane excitability of cerebellar Purkinje cells. J Physiol 590, 273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark MD, Krause M, Boele HJ, Kruse W, Pollok S, Kuner T, Dalkara D, Koekkoek S, De Zeeuw CI & Herlitze S (2015). Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J Neurosci 35, 8882–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markvartova V, Cendelin J & Vozeh F (2010). Changes of motor abilities during ontogenetic development in Lurcher mutant mice. Neuroscience 168, 646–651. [DOI] [PubMed] [Google Scholar]
- Matsumura R, Futamura N, Fujimoto Y, Yanagimoto S, Horikawa H, Suzumura A & Takayanagi T (1997). Spinocerebellar ataxia type 6. Molecular and clinical features of 35 Japanese patients including one homozygous for the CAG repeat expansion. Neurology 49, 1238–1243. [DOI] [PubMed] [Google Scholar]
- McKay BE & Turner RW (2005). Physiological and morphological development of the rat cerebellar Purkinje cell. J Physiol 567, 829–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Hashimoto K, Shin HS, Kano M & Watanabe M (2004). P/Q‐type Ca2+ channel α1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci 24, 1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocholi E, Ballester‐Lurbe B, Arque G, Poch E, Peris B, Guerri C, Dierssen M, Guasch RM, Terrado J & Perez‐Roger I (2011). RhoE deficiency produces postnatal lethality, profound motor deficits and neurodevelopmental delay in mice. PloS One 6, e19236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Miyazaki T, Kitamura K, Hashimoto K, Yanagawa Y, Obata K, Sakimura K, Watanabe M & Kano M (2012). GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron 74, 384–396. [DOI] [PubMed] [Google Scholar]
- O'Leary T, Williams AH, Franci A & Marder E (2014). Cell types, network homeostasis, and pathological compensation from a biologically plausible ion channel expression model. Neuron 82, 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M & Simon MI (1997). Impaired motor coordination and persistent multiple climbing fibre innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc Natl Acad Sci USA 94, 14089–14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orduz D & Llano I (2007). Recurrent axon collaterals underlie facilitating synapses between cerebellar Purkinje cells. Proc Natl Acad Sci USA 104, 17831–17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y & Sabatini BL (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B mice. Nat Neurosci 19, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL & Svoboda K (2003). ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner M, Skiba J, Brown M, Liang JO, Nussbaum J & Prior H (2013). Loss of the small heat shock protein αA‐crystallin does not lead to detectable defects in early zebrafish lens development. Exp Eye Res 116, 227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell'Orco JM, Sankaranarayanan A, Wulff H & Paulson HL (2011). Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 31, 13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SL, Hoebeek FE, Schonewille M, De Zeeuw CI, Aertsen A & De Schutter E (2007). Regular patterns in cerebellar Purkinje cell simple spike trains. PloS One 2, e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuvaev AN, Horiuchi H, Seki T, Goenawan H, Irie T, Iizuka A, Sakai N & Hirai H (2011). Mutant PKCγ in spinocerebellar ataxia type 14 disrupts synapse elimination and long‐term depression in Purkinje cells in vivo. J Neurosci 31, 14324–14334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara I ( 2005). Microzonal projection and climbing fibre remodelling in single olivocerebellar axons of newborn rats at postnatal days 4–7. J Comp Neurol 487, 93–106. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Niimi K & Itakura C (2009). Motor coordination impairment in aged heterozygous rolling Nagoya, Cav2.1 mutant mice. Brain Res 1279, 50–57. [DOI] [PubMed] [Google Scholar]
- Tang CC, Poston KL, Dhawan V & Eidelberg D (2010). Abnormalities in metabolic network activity precede the onset of motor symptoms in Parkinson's disease. J Neurosci 30, 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno T, Wakamori M, Koike M, Uchiyama Y, Ishikawa K, Kubota H, Yoshida T, Sasakawa H, Peters C, Mizusawa H & Watase K (2012). Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc Natl Acad Sci USA 109, 17693–17698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Warrenburg BP, Sinke RJ, Verschuuren‐Bemelmans CC, Scheffer H, Brunt ER, Ippel PF, Maat‐Kievit JA, Dooijes D, Notermans NC, Lindhout D, Knoers NV & Kremer HP (2002). Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology 58, 702–708. [DOI] [PubMed] [Google Scholar]
- Van Leuven W, Van Dam D, Moens L, De Deyn PP & Dewilde S (2013). A behavioural study of neuroglobin‐overexpressing mice under normoxic and hypoxic conditions. Biochim Biophys Acta 1834, 1764–1771. [DOI] [PubMed] [Google Scholar]
- van Welie I, Smith IT & Watt AJ (2011). The metamorphosis of the developing cerebellar microcircuit. Curr Opin Neurobiol 21, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C & Khodakhah K (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci 9, 389–397. [DOI] [PubMed] [Google Scholar]
- Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, Unno T, Sun Y, Kasai S, Watanabe M, Gomez CM, Mizusawa H, Tsien RW & Zoghbi HY (2008). Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age‐dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci USA 105, 11987–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Cuntz H, Mori M, Nusser Z, Sjöström PJ & Häusser M (2009). Traveling waves in developing cerebellar cortex mediated by asymmetrical Purkinje cell connectivity. Nat Neurosci 12, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP & Catterall WA (1995). Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci 15, 6403–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ & Sillitoe RV (2013). Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol 2, 149–164. [DOI] [PubMed] [Google Scholar]
- Womack MD, Chevez C & Khodakhah K (2004). Calcium‐activated potassium channels are selectively coupled to P/Q‐type calcium channels in cerebellar Purkinje neurons. J Neurosci 24, 8818–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe I, Sasaki H, Yamashita I, Takei A, Fukazawa T, Hamada T & Tashiro K (1998). [Initial symptoms and mode of neurological progression in spinocerebellar ataxia type 6 (SCA6)]. Rinsho Shinkeigaku 38, 489–494. [PubMed] [Google Scholar]
- Yang Q, Hashizume Y, Yoshida M, Wang Y, Goto Y, Mitsuma N, Ishikawa K & Mizusawa H (2000). Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol 100, 371–376. [DOI] [PubMed] [Google Scholar]
- Yu F, Liao Y, Jin Y, Zhao Y, Ren Y, Lu C, Li G, Li Y & Yang J (2008). Effects of in utero meso‐2,3‐dimercaptosuccinic acid with calcium and ascorbic acid on lead‐induced fetal development. Arch Toxicol 82, 453–459. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chung SK & Chow BK (2014). The knockout of secretin in cerebellar Purkinje cells impairs mouse motor coordination and motor learning. Neuropsychopharmacology 39, 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY & Lee CC (1997). Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A‐voltage‐dependent calcium channel. Nat Genet 15, 62–69. [DOI] [PubMed] [Google Scholar]