

Abstract
Background
Bardet-Biedl syndrome (BBS) is a rare genetic disorder that features retinal degeneration, obesity, polydactyly, learning disabilities and renal abnormalities. The diagnosis is often missed at birth, the median age at diagnosis being 9 years. In the attempt to shed light on BBS and improve its diagnosis and treatment, we evaluated the genotype-phenotype relationship in patients with a molecular diagnosis of BBS.
Methods
We analyzed three common BBS genes, BBS1, BBS10 and BBS2, in 25 Italian patients fulfilling the clinical criteria of BBS. In 12 patients, we identified gene-specific biallelic variants and thus correlated genotype to the ophthalmic, renal and audio-vestibular phenotypes.
Results
At least one sequence variant was found in 60% of patients. The most common mutated gene was BBS1 followed by BBS10. Of the 17 sequence variants we found, 11 have not previously been associated with BBS. In 12 patients, we identified biallelic pathogenic variants; they had retinitis pigmentosa with early onset of visual impairment. However, retinal dystrophy was less severe in patients with BBS1 than in those with BBS10 variants. Overall, we found a high prevalence of renal dysmorphism and dysfunction. Notably, patients with BBS10 variants had the most severe renal impairment, which resulted in a critical decline in renal function. All the patients who underwent audio-vestibular evaluation had dysfunction of the cochlear outer hair cells, thus confirming the presence of hearing defects.
Conclusion
BBS1, BBS2 and BBS10 are major causative genes in Italian BBS patients. BBS10 was associated with the worse outcome in terms of the renal, ocular and audiovestibular phenotypes. Cochlear dysfunction should be included among the hallmarks of BBS.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-017-0372-0) contains supplementary material, which is available to authorized users.
Keywords: Bardet-Biedl syndrome; BBS1, BBS2 and BBS10 gene variants; Ciliopathy; Renal, ocular and audiovestibular phenotype
Background
Bardet-Biedl syndrome (BBS) is a systemic hereditary disorder characterized by the coexistence of rod-cone dystrophy, polydactyly, obesity, cognitive impairment, and renal dysfunction. Its prevalence varies among geographic areas, ranging from 1:160,000 in North Europe to 1:13,500 in Kuwait and Newfoundland [1]. The phenotype is heterogeneous and the diagnosis is often missed at birth. Polydactyly or syndactyly are generally the first signs recognized at birth, while visual defects, obesity and cognitive impairment develop during the first year of age [2].
Retinal degeneration occurs in over 90% of BBS patients and visual prognosis is poor [3]. Retinal dystrophy in BBS is progressive and varies in severity. Patients experience progressive night blindness, followed by photophobia and loss of central and color vision. At clinical level, they show marked reduction of electroretinogram (ERG) amplitude, which depends on a primary loss of rod photoreceptors followed by cone death [4]. An in vivo micro-structural analysis of retinal layers in patients with BBS revealed macular involvement [5]. The prevalence of renal impairment varies among studies, which however ultimately indicated that few BBS patients have a reduced glomerular filtration rate (GFR), while most have an abnormal renal structure and a normal GFR [2, 6]. Whether an abnormal kidney structure predisposes to progressive renal failure remains to be elucidated. Interestingly, hearing loss is not considered a main feature of BBS, and has been reported in only two studies [7, 8].
BBS is considered an autosomal recessive disease characterized by genetic heterogeneity, which at least partly explains the clinical variability of this condition, although oligogenic inheritance has also been hypothesized [1, 9, 10]. Indeed, biallelic mutations have been reported in 19 genes, namely BBS1, BBS2, ARL6, BBS4, BBS5, BBS6 (MKKS), BBS7, BBS8 (TTC8), BBS9, BBS10, BBS11 (TRIM32), BBS12, MKS1, CEP290, C2ORF86, SDCCAG8, LZTFL1, BBIP1, IFT27, which account for 70–80% of BBS cases [11, 12]. The various BBS-associated genes encode proteins involved in the regulation of ciliary structure, biogenesis and function [1]. Therefore, cilia dysfunction is the main hypothesis of BBS pathogenesis [1]. In this context, it is notable that BBS shares several characteristic features with such other ciliopathies as Meckel, Joubert, and Senior-Locken syndromes.
Here, we report the genetic and clinical findings (ocular, renal and audio-vestibular phenotypes) in a cohort of Italian patients who fulfilled the diagnostic criteria of BBS. We focused the molecular analysis on the most common disease-associated genes in Caucasians, namely, BBS1, BBS10 and BBS2 [13]. Our study aims to expand the spectrum of pathogenic variants associated to BBS in Italy and, by analyzing genotype-phenotype correlations, to improve the diagnosis and treatment of this complex genetic disorder.
Methods
Patients
Twenty-five patients referring to the Eye Clinic of the Second University of Naples (Italy) met the clinical diagnostic criteria for BBS according to Beales et al. [7]. The female/male ratio was 2:3 and mean age was 25.6 years (range: 9–65 years). All procedures were conducted according to international guidelines and to the tenets of the Helsinki Declaration 2008 and 2013. Each patient (or parent or legal guardian) gave written consent to undergo DNA analysis, which was performed according to the guidelines for genetic testing approved by the Ministero della Salute, Rome, Italy (G.U. n. 224, 23th September 2004).
Molecular study
Genomic DNA was extracted from peripheral blood leukocytes with the automated MagNA Pure LC system (Roche Diagnostics, Milan, Italy). DNA samples were first analyzed with the BBS–ALMS1 mutation array (Asper Biotech, Tartu, Estonia) that detects 253 sequence variants in the BBS1-7, BBS9, BBS10, BBS12 and ALMS1 genes. All exons and flanking intronic sequences of the BBS1, BBS2 and BBS10 genes were amplified with M13-tailed primer pairs and fully sequenced with M13 primers by using the Big Dye™ Terminator v.3.1 Sequencing kit and the ABI Prism 3730 DNA Analyzer (Applied Biosystems-Life Technologies Italia, Monza, Italy). Mutation numbering is based on the genomic and transcript reference sequences of BBS1 (NG_009093.1, NM_024649.4), BBS2 (NG_009312.1, NM_031885.3) and BBS10 (NG_016357.1, NM_024685.3).
To predict the impact of the novel sequence variants on the expression of BBS1, BBS2 and BBS10, we used the online tools Variant Effect Predictor (VEP) [14] and MutationTaster [15] that predict the effect of known and new variants (single nucleotide polymorphisms [SNPs], insertions, deletions, copy number variations or structural variants) on genes, transcripts, and protein sequences, as well as on regulatory regions. In the case of missense changes, VEP also assigns scale-invariant feature transform (SIFT) and polymorphism phenotyping v2 (PolyPhen-2) probability scores of the pathogenetic effect on the putative protein variant. VEP and MutationTaster not only handle single amino acid substitutions, but also insertions and deletions; they also identify non-canonical splice sites.
Ophthalmological study
All 25 patients underwent a complete ophthalmological examination including best-corrected visual acuity (BCVA) measured using the Snellen chart, slit-lamp anterior segment examination, fundus examination, fundus photography, Goldmann visual field examination, standard ERG and optical coherence tomography (OCT). The ERG was recorded with a Ganzfield stimulator following the guidelines of the International Society of Clinical Electrophysiology of Vision [16]. OCT was performed with new generation tomography, which uses spectral domain-based techniques that allow the acquisition scans or 5 linear or a retinal area of 6 × 6 mm2 through 512 (horizontal) × 128 (vertical) scans (SD-OCT, Cirrus HD OCT, Carl Zeiss, Dublin, CA, USA).
Renal study
Renal function was evaluated in 21 patients. Glomerular function was evaluated by estimating the GFR and the urine albumin-to-creatinine ratio (ACR). Albumin was measured in the early morning urine sample with a standard immunochemical method and expressed as urine ACR (mg/g). GFR was estimated according to the Modification of Diet In Renal Disease study group and the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) [17], using standardized plasma creatinine measurement. In children, GFR was estimated with the Schwartz formula: GFR [(mL/min/1.73 m 2) = k × height (cm)/serum creatinine (mg/dL)]. The normal estimated GFR (eGFR) is 80–120 mL/min/1.73 m2; eGFR < 90 mL/min/1.73 m2 indicates impaired renal function and eGFR < 60 mL/min/1.73 m2 represents an increased risk factor for an adverse renal outcome. Moreover, to track longitudinal changes in GFR, we assessed eGFR 3 years after the first visit in patients with biallelic mutation of BBS1 and BBS10. Tubular function was analyzed in patients with eGFR > 60 ml/min/1.73 m2. Acid-base balance was evaluated on arterial blood; urinary and plasma electrolytes were also measured. Renal concentrating ability was assessed by measuring urine osmolality 12 h after water restriction (Osmometer model 3320, A. De Mari, Italy). Urine osmolality < 750 mOsm/kg indicates a defect in urine concentrating capability. Renal morphology was determined by ultrasound.
Audio-vestibular study
Six genotyped patients (5 men and 1 woman) aged between 13 and 38 years underwent audio-vestibular function testing. Information about drug history, pre- peri- and post-partum problems, previous audiological disorders, head trauma or neurological defects (e.g., history of migraine, epilepsy, vertigo) was obtained for all patients.
The hearing threshold was evaluated by liminal pure tone audiometry and assessment of perception by verbal speech audiometry. Subjects with a hearing threshold > 20 dB hearing level on the middle and/or high frequencies were considered to have hearing loss. The impedance analysis to evaluate the middle ear functioning was performed according to the guidelines of the American Speech-Language-Hearing Association. The presence of inner ear damage was verified by means of distortion product otoacustic emission (DPOAE). DPOAE was measured with a Madsen Cappella instrument, which generates two primary frequency tones, 2f1 and 2f2, with a stimulus frequency separation of f1/f2. Intensity of the custom stimulus was 40 dB sound pressure level (SPL) at both frequencies. The DPOAE was recorded by automatic scanning of the 250–8000 Hz frequency interval focused on the pure tone audiometric test frequencies. Auditory evoked potentials were evaluated with standard parameters. Three chloride silver electrodes were located in the vertex (active), mastoid (right or left) and forehead (ground) positions. Electrode impedances were maintained at ≤ 7 kΩ. Stimuli for auditory brainstem response recording were digitized at a rate of 20 kHz, and presented over headphones. Stimuli were 100 μs clicks presented monaurally at 110 dB SPL. Broadband noise at 70 dB SPL was presented to the opposite ear to mask any stimulation via acoustic cross talk. Clicks were presented at a rate of 21/s in 4-min runs. A conventional method of alternating click stimulus polarity was used to reduce stimulus artifacts in the average waveforms. Band-pass filtered the signals between 30 and 3000 Hz. The average waveform was focused on a period extending from 10 ms before the stimulus to 10 ms after the stimulus. Vestibular function was evaluated with statokinetic tests (Romberg, sensitized Romberg), detection of spontaneous nystagmus with video nystagmography, detection of evoked nystagmus with the head shaking test (HST) and the bithermal caloric test. The latter was performed with the Fitzgerald Hallpike method [18]. The ears were stimulated by irrigation with hot water (44 °C) and cold (30 °C) water. Patients were placed in a supine position with head flexed forward 30°, so that the straight line that joins the tragus to the outer canthus of eye was vertical. Irrigation was carried out with 250 ml of water at a flow rate about 5–8 ml/s for 40 s. The nystagmic reaction was induced in a few seconds and reached the maximum about 60 s after stimulation.
Results
We enrolled 25 patients from 24 unrelated families. In addition to retinitis pigmentosa (RP), the main features of our patients that were consistent with the clinical diagnosis of BBS were postaxial polydactyly (21/25 patients), obesity (15/25 patients), a history of obesity (2/25 patients), renal abnormalities (18/21 patients). We found that 8/15 males had a history of hypogonadism, four of whom had cryptorchidism; 4/10 females had congenital abnormalities of the urogenital tract. Moreover, 18/25 patients showed intellectual disabilities including delay in learning development during early childhood and the need for educational support teachers. Consanguinity or presumed consanguinity (geographic isolates) was reported in 24% of patients (6/25).
All 25 patients underwent molecular analysis. First, we applied the BBS–ALMS1 mutation array, which revealed known sequence variants in 6/25 patients. These mutations affected the BBS1, BBS2 and BBS10 genes; therefore, we sequenced these genes in patients with one or no array-detected mutation. Sequencing confirmed the array data and revealed 12 additional sequence changes. Overall, our analysis revealed at least one sequence variant in 15/25 patients (60%). We found that 12/25 patients (about 48%) had biallelic putative disease-causing variants, which supported the clinical diagnosis.
Table 1 illustrates the molecular data. Seven unrelated patients have sequence variants in BBS1; 5 of them have biallelic variants. Of the 8 independent BBS1 variant alleles, 4 have sequence changes not previously linked to BBS. In summary, among the 10 independent BBS1 alleles that we sequenced in the patients with BBS1 variants, 30% carry known sequence variants, 50% carry sequence changes not previously linked to BBS, and 20% are normal (Fig. 1). The patient who carries the monoallelic p.V568M missense change in BBS1 also has new biallelic pathogenic variants in BBS2. Overall, 5 patients carry BBS2 sequence variants, but only 2 have a BBS2 genotype (biallelic variants) that is consistent with the syndrome. In fact, two other patients have BBS2 monoalleic variants, i.e., the new c.2144G > A (p.R715Q) and the c.986 T > C (p.M329T) (rs201146063) SNP, respectively, and no mutation in BBS1 or BBS10. Another patient has a new monoallelic intronic sequence variant in BBS2 (c.535-79_90del) and also biallelic, likely pathogenic variants in BBS10. In summary, among the 10 independent BBS2 alleles that we sequenced, 30% carry known sequence variants, 40% carry sequence changes not previously linked to BBS, and 30% are normal (Fig. 1). Overall, 4 patients have biallelic pathogenic variants in BBS10 (about 16%). We found that 4/6 (67%, Fig. 1) independent BBS10 alleles carry new sequence variants, i.e., c.641 T > A (p.V214E), c.235dupA (p.T79Nfs*17), c.962A > G (p.Y321C) and c.2137_2140del (p.K713Ffs11*724Iext*1). Except for one case (Table 1, P.9), our homozygous BBS patients have consanguineous parents. In all cases, Mendelian segregation of the variant alleles was confirmed in parents and, when available, in unaffected siblings.
Table 1.
Sequence variants identified in the BBS patients who tested positive to the molecular analysis
| Patient ID | Genotype | ||
|---|---|---|---|
| BBS1 | BBS2 | BBS10 | |
| P.1a | c.664G>C/c.664G>C (p.G222R) | N/N | N/N |
| P.2a | c.664G>C/c.664G>C (p.G222R) | na | na |
| P.3a,b | c.1169T>G/c.1169T>G (p.M390R) | na | na |
| P.4a,b | c.1169T>G / c.1169T>G (p.M390R) | na | na |
| P.5a | c.1169T>G / c.1169T>G (p.M390R) | na | na |
| P.6 | c.1169T>G/c.1642delC (p.M390R/p.L548Wfs*31) | na | na |
| P.7 | c.592-59G>A/N | N/N | N/N |
| P.8 | c.1702G>A/N (p.V568M) | c.84delC/c.1059dupT (p.P29Rfs*50/p.N354X) | N/N |
| P.9 | N/N | c.225T>G/c.225T>G (p.V75G) | N/N |
| P.10 | N/N | c.2144G>A/N (p.R715Q) | N/N |
| P.11 | N/N | c.986T>C/N (p. M329T) | N/N |
| P.12 | N/N | c.535-79_90del/N | c.2137_2140del/c.962A>G (p.K713Ffs*16/p.Y321C) |
| P.13 | na | na | c.235dupA/c.271dupT (T79Nfs*17/p.C91LfsX5) |
| P.14a | na | na | c.509T>C/c.509T>C (p.L170S) |
| P.15a | N/N | N/N | c.641T>A/c.641T>A (p.V214E) |
In bold, variants not previously linked to the BBS phenotype. For cDNA numbering, +1 corresponds to the A of the ATG translation initiation codon, which is codon 1. Reference gene sequences were BBS1 (NG_009093.1, NM_024649.4), BBS2 (NG_009312.1, NM_031885.3), BBS10 (NG_016357.1, NM_024685.3)
N gene-specific normal allele, n.a. not analyzed
aWith consanguineous parents
bPatients 3 and 4 are siblings
Fig. 1.
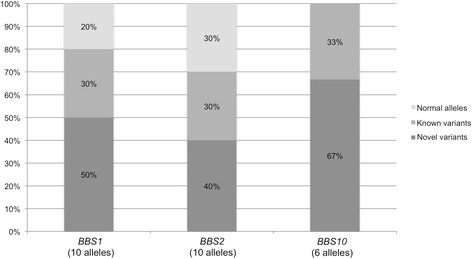
Prevalence of known vs the novel BBS1, BBS2, BBS10 alleles in our genotyped BBS patients. Bars indicate, for the three groups of patients (BBS1, BBS2, BBS10), the percentage of independent alleles with new (dark grey), known (intermediate grey), normal alleles (light grey)
Bioinformatic prediction indicated that all the new sequence variants in the coding regions of BBS1, BBS2 and BBS10 were very likely “disease-causing”, whereas the two intronic variants were possible polymorphisms (Additional file 1: Table S1). Notably, many of the variants we identified are listed in the SNP database (National Center for Biotechnology Information, NCBI), all with a minor allele frequency < 0.0001 (Additional file 1: Table S1); however, this is the first report that links them to the BBS phenotype.
Genotype to ocular phenotype correlation
All patients with biallelic mutations (12/12) had visual defects (Table 2). Nine of these patients were affected by legal blindness having a visual acuity less than or equal to 20/200 (0.1 decimals); 3/12 patients had a visual acuity between 20/100 (0.2 decimals) and 20/70 (about 0.3 decimals). All 12 patients were able to perceive light. Visual impairment was not congenital, however 66.6% of patients had horizontal nystagmus, 58.3% strabismus and 50.0% had cataracts. At fundus examination, nine patients had osteoblast-like pigment clusters, mainly located on the equator, narrowing of retinal blood vessels, and optic disc pallor, which indicate diffuse retinal pigment epithelial dystrophy (Fig. 2a); three of them had macular dystrophy. In the remaining three patients, we observed widespread tapetoretinal degeneration and the absence of retinal pigment epithelium (Fig. 2b). All the 12 genotyped patients underwent ERG; they showed an extinguished, not age-related scotopic and photopic electroretinogram. Despite the nystagmus and fixation instability and low visual acuity, OCT images were acquired in nine patients, and these showed reduced macular thickness and retinal pigment epithelium dystrophy. Four patients showed an epiretinal membrane and three patients had signs of vitreomacular traction syndrome (Fig. 2c). One patient had a macular lamellar hole (Fig. 2d).
Table 2.
Comparative genotype-phenotype correlation analysis in BBS patients
| Patient ID | Phenotype | Genotype | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ocular | Renal | Audio-vestibular | Gene | Mutation | |||||||||
| Eye vision R/L | Fundus finding | Macular alterations | eGFR <60 ml/min/1.73 m2 |
Max uOsm <750 mOsm/kg | Ultrasound abnormalities | Hearing impairment | Timpanogram type | DPOAE L/R | Vestibular test | ABR alteration | |||
| P.1 | 0.25/0.2 | RP sp | Yes | No | Yes | Yes | No | AA | +/− | spontaneous NY | No | BBS1 | c.664G>C/c.664G>C |
| P.2 | 0.3/0.2 | RP sp | No | No | No | Yes | mixed | BB | + /- | positional NY | only V wave | BBS1 | c.664G>C/c.664G>C |
| P.3 | 0.2/0.1 | RP | NP | NP | NP | NP | NP | NP | NP | NP | NP | BBS1 | c.1169T>G /c.1169T>G |
| P.4 | LP/LP | RP | NP | NP | NP | Yes | NP | NP | NP | NP | NP | BBS1 | c.1169T>G /c.1169T>G |
| P.5 | LP/LP | RP | Yes | No | No | Yes | No | AA | +/− | No | No | BBS1 | c.1169T>G /c.1169T>G |
| P.6 | 0.05/0.03 | RP | Yes | No | No | No | NP | NP | NP | NP | NP | BBS1 | c.1169T>G/c.1642delC |
| P.8a | 0.02/0.008 | RP sp | Yes | No | Yes | Yes | NP | NP | NP | NP | NP | BBS2 | c.84delC/c.1059dupT |
| BBS1 | c.1702G>A/N | ||||||||||||
| P.9 | 0.02/0.02 | RP | No | No | No | Yes | sensorineural | AA | −/− | positional NY | > latency I,III,V | BBS2 | c.225T>G/c.225T>G |
| P.12 | LP/LP | RP | Yes | No | Yes | Yes | NP | NP | NP | NP | NP | BBS10 | c.2137_2140del/c.962A>G |
| BBS2 | c.535-79_90del/N | ||||||||||||
| P.13 | 0.03/0.08 | RP | Yes | Yes | Yes | Yes | sensorineural | AA | +/+ | No | No | BBS10 | c.235dupA/c.271dupT |
| P.14 | LP/LP | RP | NP | NP | NP | NP | NP | NP | NP | NP | NP | BBS10 | c.509T>C/c.509T>C |
| P.15 | LP/LP | RP | Yes | No | Yes | Yes | sensorineural | AA | +/+ | positional NY | No | BBS10 | c.641T>A/c.641T>A |
F female, M male, R right, L left, RP retinitis pigmentosa, sp sine pigmento, + partially present, − absent, NY nystagmus, LP light perception, NP not performed, N normal sequence
aDied of heart failure
Fig. 2.
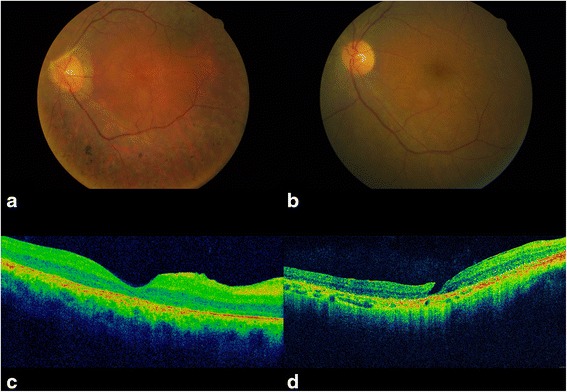
Representative images of the ocular findings in the BBS patients analyzed. Fundus photography showing a narrowing of retinal blood vessels, diffuse retinal pigment epithelial dystrophy with pigment clusters in mid-periphery; b narrowing of retinal blood vessels, widespread tapetoretinal degeneration and absence of pigment clusters. OCT scan showing c vitreomacular traction syndrome with retinal pigment epithelium dystrophy; (d) a macular lamellar hole with retinal pigment epithelium dystrophy
Genotype-phenotype correlation revealed that the six patients with biallelic mutations in BBS1 (aged 9 to 70 years) had a BCVA ranging from light perception to 0.3 decimals; four of them had a BCVA equal to or better than 1/10, three patients had nystagmus and five exotropia. Typical RP associated with subcapsular cataract was present in four patients, all over 25 years of age. The six BBS1-mutated patients presented extinguished scotopic and photopic ERG responses. OCT examination was performed only in four patients, three of whom showed vitreoretinal abnormalities (Table 2).
The two patients with biallelic mutations in BBS2 had a BCVA ≤ 0.02 decimal and nystagmus; one of them also developed exotropia in the second decade of life. Both patients had diffuse retinal pigment epithelium dystrophy without cataract, and the oldest developed vitreoretinal abnormalities at OCT at the age of 19 years. In both cases, the scotopic and photopic ERG responses were extinguished.
We found that three of the four patients with biallelic variants in BBS10 showed a BCVA of light perception; the remaining one had nystagmus associated with exotropia. All four showed typical RP; in the two eldest subjects (31 and 40 years old, respectively), subcapsular cataract also appeared at 30 years of age. Scotopic and photopic ERG responses were extinguished in all cases; OCT examination revealed vitreoretinal abnormalities in the three subjects we analyzed (Table 2). Despite the relatively small number of patients with a known genotype, genotype-phenotype correlation analysis revealed that, although BCVA reduction was age-related, BBS1-mutated patients had a significantly better visual acuity (p ≤ 0.006), with a slower progression of BCVA reduction (0.03 decimals/year; p < 0.01), compared with our BBS10- and BBS2-mutated patients.
Genotype to renal phenotype correlation
We evaluated the renal phenotype in 9/12 BBS patients with a positive molecular test (Table 2). We obtained by telephone interview information about the renal functionality of the three patients who did not undergo renal examination. Interestingly, a 70-year-old woman, who was homozygote for the common BBS1 mutation p.M390R (patient P.4, Table 2), underwent radical nephrectomy for a suspicious renal mass, 20 years earlier. Figure 3a correlates the patients’ eGFR with genotype. A young BBS10 patient with congenital multicystic renal dysplasia, who was diagnosed with end-stage renal disease at the age of 23 years, had the lowest eGFR, and the eGFR reduction was associated to albuminuria and hypertension. The eGFR exceeded 90 ml/min/1.73 m2 in the other two BBS10 patients, which however manifested tubular dysfunctions. In the patients with biallelic mutations in BBS1 or BBS2, eGFR exceeded 90 ml/min/1.73 m2.
Fig. 3.
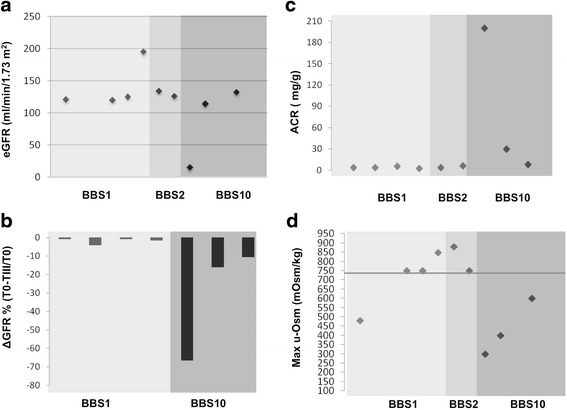
Analysis of renal function. a eGFR, calculated according to the CKD-EPI formula, in our BBS patients. The most severe renal dysfunction occurred in a patient carrying biallelic BBS10 mutations. b Relative eGFR decline in 3 years. eGFR was estimated at baseline (T0) and after 3 years (TIII). Relative eGFR change (ΔGFR) during 3 years is expressed as percentage (%) of the T0. c Urine albumin-to-creatinine ratio (ACR). Two of three mutated patients showed ACR above 30 mg/g; the other patient showed normal ACR. d Maximal urine concentrating ability. Urine osmolality was measured in the second morning urine void, after overnight fasting and water restriction
We also evaluated changes in eGFR in BBS1 and BBS10 patients (Fig. 3b) three years after baseline. The rate of decline (ΔGFR) correlated with GFR at baseline. In fact, renal dysfunction progressed faster in the patient with the lowest GFR. Interestingly, all patients with BBS10 mutations had ΔGFR higher than 10%, whereas it was lower than 10% in the BBS1-mutated patients. The ACR, a marker of glomerular damage, exceeded 30 mg/g only in two patients, both with biallelic mutations in BBS10 (Fig. 3c), whereas it was lower than 30 mg/g in our BBS1 and BBS2 patients.
Tubular function was evaluated only in subjects with an eGFR >60 ml/min/1.73 m2 (8/9 genotyped patients). The most common tubular dysfunction we found was a defect in urine concentrating ability, in agreement with a previous report [17]. All the three patients with biallelic mutations in BBS10 had hyposthenuria even 12 h after water restriction (Fig. 3d) and therefore they were affected by renal dysfunction. In contrast, urine concentrating ability was normal in 3/4 patients with mutated BBS1 and in both the BBS2-mutated patients (Fig. 3d).
Plasma electrolytes were within normal range in all patients (Additional file 1: Table S2). The mean concentrations of Na+ and K+ were normal, and Na+ and Cl− fractional excretions (FE%) were lower than 1% in all patients except one with the BBS10 mutation, who showed a slight increase in the fractional excretion of sodium, FENa+, and of chlorine, FECl−, (1.04 and 1.41%, respectively). Acid-base status was normal in all patients, except in one BBS1 patient who was affected by metabolic acidosis. The urine Ca+2/creatinine (UCa/Cr) ratio was <0.2 mg/mg in all patients except one.
Abnormal kidney ultrasound appearance is a common feature of BBS patients [17]. Accordingly, in our cohort, 8/9 genotyped patients showed a broad range of kidney anatomical variations, i.e., parenchymal and peri-pelvic cysts, renal hypoplasia, pelvic dilation and fetal lobulations (Additional file 1: Table S3). We noted that the severity of structural abnormalities correlated with the severity of renal dysfunction.
Genotype to audiovestibular phenotype correlation
Tonal audiometry analysis revealed cochlear sensorineural hearing loss (SNHL) in 3/6 patients (Additional file 1: Table S4); one had moderate bilateral SNHL, one mild bilateral SNHL, and one mild SNHL in the right ear. Another patient had a mixed moderate hearing loss more pronounced in the right ear. Hearing impairment in two patients required a hearing aid. Impedentiometry showed a type “A” normal tympanogram and normal stapedial reflexes in five patients (Table 2). However, at the Metz recruitment test, patient P.9 who had moderate SNHL had a considerably reduced dynamic range, i.e. the gap between the acoustic reflex threshold and the pure-tone audiometry hearing threshold level, which indicates cochlear pathology. The patient with mixed hearing loss showed a type “B” tympanogram and absent reflexes. All six patients had no otoacustic emission (Additional file 1: Table S5).
In the six patients we tested, speech audiometry headphones were normal; two patients had difficulty in verbal comprehension. No patient experienced true rotatory vertigo or dizziness during the vestibular test. Spontaneous nystagmus occurred in one patient. Positional nystagmus was observed in three patients (50%). In one patient, HST was positive and the caloric test showed a pathological caloric weakness in the right ear. The morphology and latency of the auditory brain response waves were normal in four patients. The latency of the I, III and V waves was increased in one patient, and only the V wave was observed at 80 dB hearing level in another patient.
Discussion
Bardet-Biedl syndrome, a rare clinically and genetically heterogeneous disorder, is often misdiagnosed due to the high phenotypic variability and mainly because it shares several characteristic features with other ciliopathies [1]. Here we describe genetic and some clinical features of 12 BBS Italian patients and report the genotype-phenotype correlation.
Twenty-five Italian patients fulfilling the clinical criteria of BBS underwent molecular analysis of the BBS1, BBS10 and BBS2 genes because pathogenic variants in these genes have been found in 23%, 20% and 8% of BBS patients, respectively worldwide [13, 19]. We identified 21 independent alleles. Twelve families had biallelic genotypes that were consistent with the disease phenotype. Overall, we identified 17 different sequence variants, 6 known and 11 not previously linked to the disease, in about 60% of our patients, which indicates BBS1, BBS2 and BBS10 are frequently mutated in Italian patients. We also found monoallelic variants in BBS1 and BBS2 in two patients with biallelic pathogenic variants in BBS2 and BBS10, respectively, which suggests the possibility of triallelism. We were unable to verify triallelic inheritance because these patients are the only affected members of their families. BBS1 is the most frequently mutated gene in our cohort (28% of patients). Notably, two unrelated patients are homozygous for the p.G222R substitution in BBS1, which therefore represents 20% of BBS1-variant alleles in our cohort. The minor allele frequency reported in the ExAC database (Additional file 1: Table S1) indicates this variant is very rare worldwide. Therefore, p.G222R might be a founder disease allele, in Italy. Also p.M390R was frequent in our cohort of patients (30% of our BBS1 variant alleles). Notably, the Exome Variant Server database records p.M390R in 0.25% (G = 23/T = 8567) of BBS1 alleles of individuals of European-African descent. It has also been reported in approximately 80% of Caucasian BBS1-positive patients [19, 20]. Therefore, we cannot consider p.M390R a founder allele in Italy.
Surprisingly, we identified seven independent variant alleles in BBS2 and only six in BBS10. However, whereas all the BBS10-mutated patients have genotypes fully consistent with the disease, only 2/5 BBS2-mutated patients have unambiguous pathogenic genotypes; the remaining three patients are heterozygous carriers of variants with a predicted low pathogenic significance (Additional file 1: Table S1).
Twelve of the variants we identified were not in the Human Gene Mutation Database (Version 2015.4) or in the Exome Variant Server. Five are point/subtle deletions or duplications that lead to a frameshift and/or premature stop codon, and therefore can be reasonably considered pathogenic. A further five are nucleotide substitutions that cause missense changes, and, in all cases, bioinformatic tools predicted that they exert potential pathogenic effects. Although these five substitutions are listed in the SNP database, they have not previously been found in BBS patients. The remaining two intronic sequence variants are predicted to be possible polymorphisms. In this context, segregation of the new variant alleles we found in the “biallelic” families strongly supports their pathogenic role.
In the two patients with potential triallelism [21], the clinical picture of the patient with biallelic mutations in BBS2 and one putative pathogenic missense change in BBS1 was particularly serious; in fact, this patient was born with a severe congenital aortic stenosis. This finding, which is unusual in BBS, negatively affected prognosis and caused the patient’s death at the age of 18 years. Since we limited our genetic analysis to three genes, we cannot exclude that other genotyped patients may have mutations in other genes that could exert an epistatic effect. In particular, two adult siblings (P.3 and P.4 in Tables 1 and 2) homozygous for the common BBS1 variant p.M390R have different BBS phenotypes. This finding could be consistent with triallelism, also based on reports that some homozygotes for the p.M390R variant may or may not manifest the disease [20, 21]. However, it is noteworthy that the elder of our two siblings was the most severely affected and that BBS has an age-dependent penetrance and variable expressivity [22].
Ophthalmologic analysis revealed that visual acuity was age-related in the 12 genotyped patients. In fact, the visual defect was more severe in patients over 19 years of age (visual acuity ≤ 20/200) than in children (visual acuity between 20/100 and 20/70). No patient lost the ability to perceive light. Genotype-phenotype correlation indicated a severe reduction in BCVA in all patients except the two BBS1-mutated children probably because of their young age. Also the appearance of fundus abnormalities correlated with age. In fact, pigment-type osteoblasts, narrowing of the retinal vessels and pallor of the optic disc occurred late in the disease, namely, at a mean age 35.8 years in 8/12 (75%) patients. Instead, a “salt and pepper” fundus, which is considered a harbinger of retinal disease, appeared at a mean age of 15 years in 4/12 (25%) patients. In most cases, macular changes started in the early teens, whereas bone spicule pigments occurred mainly in early adulthood.
The scotopic and photopic components of ERG were altered in all the 12 genotyped patients. Eight of these patients (75%) underwent OCT study, which confirmed that the most frequent findings were outer retina thinning in the macular region and dystrophy of the pigmented epithelium [5]. Unlike a previous report [23], loss of “lamination” of the retina was not gene-related in our cohort. The retinal structure abnormalities in our patients did not correlate with genotype, age or disease severity.
We previously reported that the prevalence of vitreoretinal abnormalities in BBS is twice that in RP patients [24]. The most frequent abnormalities in RP patients were cystoid macular edema (20%) followed by epiretinal membrane (16%); vitreo-macular traction was reported in only 5% of patients [24]. Forty-four per cent of our BBS patients show an epiretinal membrane and 33.3% vitreo-macular traction, while no patient had cystoid macular edema.
Renal dysfunction was frequent in our cohort. The four BBS1-mutated patients we analyzed had mild renal abnormalities, normal eGFR and a normal electrolyte balance. One patient was affected by chronic metabolic acidosis. The ultrasound appearance of the kidney was unremarkable in one patient, while the others showed fetal lobulation or isolated parenchymal cysts. Kidney size and cortical thickness were normal in all patients of this BBS1 subgroup. Only one BBS1 patient had hyposthenuria. Modifier genes may have contributed to the onset of this dysfunction, which however is the most frequent renal dysfunction in BBS.
The two BBS2-mutated patients had a normal eGFR and tubular function, and a mild renal phenotype, similar to the BBS1-mutated patients. However, the ultrasound renal appearance differed greatly between the two groups of patients. The former had typical pelvic dilation/peripelvic cysts, whereas the latter had only mild fetal lobulation.
The three BBS10-mutated patients we analyzed have renal dysfunction. The most severely affected patient has two frameshift mutations, both leading to a non-functional protein. He was born with a renal malformation and was diagnosed with end stage renal disease at the age of 23 years. The other two BBS10-mutated adults have normal eGFR. However, one has a high ACR, which is a marker of glomerular damage. Both these BBS10-mutated patients had defective urine concentrating ability and a normal eGFR. We recently reported that BBS10 knock-down affected forskolin-dependent AQP2 trafficking to the apical membrane of epithelial tubular cells, thus providing a potential explanation for hyposthenuria [25]. In addition, our finding that the decline of eGFR in three years was more severe in BBS10-mutated patients than in BBS1-mutated patients indicates that BBS10 deficiency is related to a poor renal prognosis [26]. Therefore, we conclude that, in our cohort of patients, BBS10 variant alleles are associated to severe kidney dysfunction.
The audiological study revealed that two BBS10-mutated patients and one BBS2-mutated patient had cochlear SNHL. In contrast, our BBS1-mutated patients have no or a mixed hearing impairment. These results differ from previous reports that hearing loss, mainly due to conductive loss, is a minor sign of BBS [8]. Two of our patients, affected by sensorineural and mixed hearing loss, respectively, were successfully treated with hearing aids. Therefore, patients with suspected BBS should undergo hearing evaluation. In addition, our DPOAE results shed light on the BBS phenotype. In fact, because all our BBS patients had abnormal DPOAEs, we concluded that they had alteration of outer hair cell function. This agrees with evidence that the BBS phenotype arises from a ciliary dysfunction and consequently it would affect tissues in which hair cells are present. Therefore, DPOAEs may be useful for the early detection of cochlear damage in BBS patients.
Vestibular function analysis suggested abnormalities in the nystagmographic framework in 4/6 patients (67%), which could be explained by the sharp decline in visual acuity. In one case, we hypothesized the presence of a unilateral peripheral vestibular lesion. Notably, two young patients have the same BBS1 genotype (homozygous p.G222R), but very different hearing phenotypes even though they are of about the same age. Also in this case, the variable expressivity of BBS could reflect an epistatic effect of a putative triallelism.
Conclusion
In our cohort of Italian BBS patients there is a high prevalence of RP with early onset of visual impairment, a high prevalence of renal dysmorphism and dysfunction, and of subclinical hearing defects that, although generally poorly substantiated, are a useful hallmark of BBS. BBS1, BBS2 and BBS10 are major causative genes also in Italian BBS patients and the identification of new mutations demonstrates a high allelic heterogeneity. Pathogenic variants of BBS10 correlated with a worse outcome, at least in terms of renal, ocular and audiovestibular phenotypes. As BBS10 variants severely affect renal structure and function, patients manifesting kidney malformation should be scanned for mutations of this gene. Overall, our study may help to improve the identification of this complex disorder.
Acknowledgments
We are grateful to Jean Ann Gilder (Scientific Communication srl, Naples, Italy) for revising and editing the text.
Funding
This work has been supported by Grant PON03PE_00146_1 2007/2013 (BIBIOFAR) from the Italian Ministry of University and Research (to FS); Grant POR Campania FSE 2007/2013 (CAMPUS-Bioframe and project DIAINTECH) from the Regione Campania, Italy (both to FS).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Authors’ contributions
GE: designed the genetic study, interpreted the genetic variants and drafted the manuscript. FT: participated in the recruitment of patients, summarized the ophthalmologic clinical data, performed the ocular phenotype-genotype correlation and helped to draft the manuscript. MZ: carried out renal function analysis, summarized the nephrologic clinical data, performed the renal phenotype-genotype correlation and helped to draft the manuscript. AAC, MDA and TF: performed the genetic analyses, and evaluated and summarized the APEX array and Sanger sequencing data. VDI and SR: participated in the recruitment of patients in the Eye Clinic and performed ocular tests. GC and LR: performed the renal imaging analysis. PI: performed the audiovestibular tests. AF: evaluated the audiovestibular data and participated in the phenotype-genotype correlation. EM: coordinated the audiovestibular analysis and participated in the critical review of the manuscript. GC: coordinated the nephrological analysis and participated in the critical review of the manuscript. SF: coordinated the recruitment the BBS patients at the eye clinic, supervised the ocular phenotype-genotype correlations and participated in the critical review of the manuscript. FS: coordinated and supervised the entire study and critically reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All participants (or their parents) gave consent to publish data in anonymous form.
Ethics approval and consent to participate
The study was approved by the institutional ethics committee of the Seconda Università di Napoli. Each participant provided written informed consent.
Abbreviations
- ACR
Albumin-to-creatinine ratio
- BBS
Bardet-Biedl syndrome
- BCVA
Best-corrected visual acuity
- CKD-EPI
Chronic Kidney Disease Epidemiology Collaboration
- DPOAE
Distortion product otoacustic emission
- eGFR
Estimated GFR
- ERG
Electroretinogram
- ExAC
Exome Aggregation Consortium
- FECl
Fractional excretion of chlorine
- FENa
Fractional excretion of sodium
- GFR
Glomerular filtration rate
- OCT
Optical coherence tomography
- PolyPhen
Polymorphism phenotyping
- RP
Retinitis pigmentosa
- SIFT
Scale-invariant feature transform
- SNHL
Sensorineural hearing loss
- SNP
Single nucleotide polymorphism
- UCa/Cr
Urine Ca+2/creatinine
- VEP
Variant Effect Predictor
Additional file
Bioinformatically-predicted putative effects of the variants linked to the BBS phenotype. Table S2 Electrolytes and acid base balance in BBS patients. Table S3 Correlation between renal structural alterations and genotype in BBS patients. Table S4 Results of pure tone audiometry. Table S5 DPOAE results. (DOCX 94 kb)
Contributor Information
Gabriella Esposito, Email: gabriella.esposito@unina.it.
Francesco Testa, Email: testa.francesco@gmail.com.
Miriam Zacchia, Email: miriamzacchia@virgilio.it.
Anna Alessia Crispo, Email: crispo@ceinge.unina.it.
Valentina Di Iorio, Email: valedior@inwind.it.
Giovanna Capolongo, Email: giovi.capolongo@gmail.com.
Luca Rinaldi, Email: lucarinaldi@hotmail.it.
Marcella D’Antonio, Email: dantonio@ceinge.unina.it.
Tiziana Fioretti, Email: tiziana.fioretti@gmail.com.
Pasquale Iadicicco, Email: pasquale_iadicicco@libero.it.
Settimio Rossi, Email: settimio.rossi@unina2.it.
Annamaria Franzè, Email: annamaria.franze@unina.it.
Elio Marciano, Email: marciano@unina.it.
Giovanbattista Capasso, Phone: +39 081 566 6652, Email: gb.capasso@unina2.it.
Francesca Simonelli, Phone: +39 081 566 6762, Email: francesca.simonelli@unina2.it.
Francesco Salvatore, Phone: +39 081 746 3133, Email: salvator@unina.it.
References
- 1.Zaghloul NA, Katsanis N. Mechanistic insights into Bardet–Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–37. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet–Biedl syndrome. Am J Kidney Dis. 1996;27:776–83. doi: 10.1016/S0272-6386(96)90513-2. [DOI] [PubMed] [Google Scholar]
- 3.Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12:238–49. doi: 10.2174/138920211795860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Héon E, Westall C, Carmi R, Elbedour K, Panton C, Mackeen L, Stone EM, Sheffield VC. Ocular phenotypes of three genetic variants of Bardet–Biedl syndrome. Am J Med Genet A. 2005;132:283–7. doi: 10.1002/ajmg.a.30466. [DOI] [PubMed] [Google Scholar]
- 5.Gerth C, Zawadzki RJ, Werner JS, Héon E. Retinal morphology in patients with BBS1 and BBS10 related Bardet–Biedl Syndrome evaluated by Fourier–domain optical coherence tomography. Vision Res. 2008;48:392–9. doi: 10.1016/j.visres.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zacchia M, Zacchia E, Zona E, Capolongo G, Raiola I, Rinaldi L, Trepiccione F, Ingrosso D, Perna A, Di Iorio V, Simonelli F, Moe OW, Capasso G. Renal phenotype in Bardet-Biedl syndrome: a combined defect of urinary concentration and dilution is associated with defective urinary AQP2 and UMOD excretion. Am J Physiol Renal Physiol. 2016;311(4):F686–F694. doi: 10.1152/ajprenal.00224.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–46. [PMC free article] [PubMed] [Google Scholar]
- 8.Deveault C, Billingsley G, Duncan JL, Bin J, Theal R, Vincent A, Fieggen KJ, Gerth C, Noordeh N, Traboulsi EI, Fishman GA, Chitayat D, Knueppel T, Millán JM, Munier FL, Kennedy D, Jacobson SG, Innes AM, Mitchell GA, Boycott K, Héon E. BBS genotype–phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum Mutat. 2011;32:610–9. doi: 10.1002/humu.21480. [DOI] [PubMed] [Google Scholar]
- 9.Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL, Lupski JR. Triallelic inheritance in Bardet–Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–9. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 10.Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet–Biedl syndrome. Nat Genet. 2008;40:443–8. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 11.Scheidecker S, Etard C, Pierce NW, Geoffroy V, Schaefer E, Muller J, Chennen K, Flori E, Pelletier V, Poch O, Marion V, Stoetzel C, Strähle U, Nachury MV, Dollfus H. Exome sequencing of Bardet–Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18) J Med Genet. 2014;51:132–6. doi: 10.1136/jmedgenet-2013-101785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aldahmesh MA, Li Y, Alhashem A, Anazi S, Alkuraya H, Hashem M, Awaji AA, Sogaty S, Alkharashi A, Alzahrani S, Al Hazzaa SA, Xiong Y, Kong S, Sun Z, Alkuraya FS. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet–Biedl syndrome. Hum Mol Genet. 2014;23:3307–15. doi: 10.1093/hmg/ddu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forsythe E, Beales PL. Bardet–Biedl syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet] Seattle: University of Washington, Seattle; 1993–2015; 2003. [Google Scholar]
- 14.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70. [DOI] [PMC free article] [PubMed]
- 15.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep–sequencing age. 2014;11(4):361–2. [DOI] [PubMed]
- 16.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full–field clinical electroretinography (2008 update) Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 17.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J. CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rundle FW, McCabe BF. A clinical evaluation of caloric testing by the Fitzgerald–Hallpike–Cawthorne method. Laryngoscope. 1961;71:1186–95. doi: 10.1288/00005537-196110000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Muller J, Stoetzel C, Vincent MC, Leitch CC, Laurier V, Danse JM, Hellé S, Marion V, Bennouna-Greene V, Vicaire S, Megarbane A, Kaplan J, Drouin-Garraud V, Hamdani M, Sigaudy S, Francannet C, Roume J, Bitoun P, Goldenberg A, Philip N, Odent S, Green J, Cossée M, Davis EE, Katsanis N, Bonneau D, Verloes A, Poch O, Mandel JL, Dollfus H. Identification of 28 novel mutations in the Bardet–Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet. 2010;127:583–93. doi: 10.1007/s00439-010-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox KF, Kerr NC, Kedrov M, Nishimura D, Jennings BJ, Stone EM, Sheffield VC, Iannaccone A. Phenotypic expression of Bardet–Biedl syndrome in patients homozygous for the common M390R mutation in the BBS1 gene. Vision Res. 2012;75:77–87. doi: 10.1016/j.visres.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Badano JL, Kim JC, Hoskins BE, Lewis RA, Ansley SJ, Cutler DJ, Castellan C, Beales PL, Leroux MR, Katsanis N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet–Biedl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12:1651–9. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 22.Beales PL, Badano JL, Ross AJ, Ansley SJ, Hoskins BE, Kirsten B, Mein CA, Froguel P, Scambler PJ, Lewis RA, Lupski JR, Katsanis N. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non–Mendelian Bardet–Biedl syndrome. Am J Hum Genet. 2003;72:1187–99. doi: 10.1086/375178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eichers ER, Abd-El-Barr MM, Paylor R, Lewis RA, Bi W, Lin X, Meehan TP, Stockton DW, Wu SM, Lindsay E, Justice MJ, Beales PL, Katsanis N, Lupski JR. Phenotypic characterization of Bbs4 null mice reveals age–dependent penetrance and variable expressivity. Hum Genet. 2006;120:211–26. doi: 10.1007/s00439-006-0197-y. [DOI] [PubMed] [Google Scholar]
- 24.Azari AA, Aleman TS, Cideciyan AV, Schwartz SB, Windsor EA, Sumaroka A, Cheung AY, Steinberg JD, Roman AJ, Stone EM, Sheffield VC, Jacobson SG. Retinal disease expression in Bardet–Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina–wide degeneration. Invest Ophthalmol Vis Sci. 2006;47:5004–10. doi: 10.1167/iovs.06-0517. [DOI] [PubMed] [Google Scholar]
- 25.Testa F, Rossi S, Colucci R, Gallo B, Di Iorio V, Corte della M, Azzolini C, Melillo P, Simonelli F. Macular abnormalities in Italian patients with retinitis pigmentosa. Br J Ophthalmol. 2014;98:946–50. doi: 10.1136/bjophthalmol-2013-304082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zacchia M, Esposito G, Carmosino M, Barbieri C, Zacchia E, Crispo AA, Fioretti T, Trepiccione F, Di Iorio V, Simonelli F, Salvatore F, Capasso G, Svelto M, Procino G. Knockdown of the BBS10 gene product affects apical targeting of AQP2 in renal cells: a possible explanation for the polyuria associated with Bardet–Biedl syndrome. J Genet Syndr Gene Ther. 2014;5:222. doi: 10.4172/2157-7412.1000222. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.