

ABSTRACT
Zika virus (ZIKV) has emerged as a cause of congenital brain anomalies and a range of placenta-related abnormalities, highlighting the need to unveil the modes of maternal-fetal transmission. The most likely route of vertical ZIKV transmission is via the placenta. The earliest events of ZIKV transmission in the maternal decidua, representing the maternal uterine aspect of the chimeric placenta, have remained unexplored. Here, we show that ZIKV replicates in first-trimester human maternal-decidual tissues grown ex vivo as three-dimensional (3D) organ cultures. An efficient viral spread in the decidual tissues was demonstrated by the rapid upsurge and continued increase of tissue-associated ZIKV load and titers of infectious cell-free virus progeny, released from the infected tissues. Notably, maternal decidual tissues obtained at midgestation remained similarly susceptible to ZIKV, whereas fetus-derived chorionic villi demonstrated reduced ZIKV replication with increasing gestational age. A genome-wide transcriptome analysis revealed that ZIKV substantially upregulated the decidual tissue innate immune responses. Further comparison of the innate tissue response patterns following parallel infections with ZIKV and human cytomegalovirus (HCMV) revealed that unlike HCMV, ZIKV did not induce immune cell activation or trafficking responses in the maternal-fetal interface but rather upregulated placental apoptosis and cell death molecular functions. The data identify the maternal uterine aspect of the human placenta as a likely site of ZIKV transmission to the fetus and further reveal distinct patterns of innate tissue responses to ZIKV. Our unique experimental model and findings could further serve to study the initial stages of congenital ZIKV transmission and pathogenesis and evaluate the effect of new therapeutic interventions.
IMPORTANCE In view of the rapid spread of the current ZIKV epidemic and the severe manifestations of congenital ZIKV infection, it is crucial to learn the fundamental mechanisms of viral transmission from the mother to the fetus. Our studies of ZIKV infection in the authentic tissues of the human maternal-fetal interface unveil a route of transmission whereby virus originating from the mother could reach the fetal compartment via efficient replication within the maternal decidual aspect of the placenta, coinhabited by maternal and fetal cells. The identified distinct placental tissue innate immune responses and damage pathways could provide a mechanistic basis for some of the placental developmental abnormalities associated with ZIKV infection. The findings in the unique model of the human decidua should pave the way to future studies examining the interaction of ZIKV with decidual immune cells and to evaluation of therapeutic interventions aimed at the earliest stages of transmission.
KEYWORDS: congenital HCMV, congenital Zika virus, decidua, decidual innate immune response, intrauterine transmission, organ culture, placenta
INTRODUCTION
Zika virus (ZIKV), a mosquito-borne flavivirus, has recently emerged as a cause of severe birth defects (1, 2). Congenital ZIKV infection is associated with microcephaly and a range of neurological anomalies (2–6), as well as with placenta-related abnormalities, including placental calcifications, intrauterine growth restriction (IUGR), and fetal loss (2, 3, 5–11). Since its introduction into Brazil in 2015, ZIKV has been rapidly spreading worldwide, highlighting the urgent need to unveil the mode(s) of maternal-fetal transmission and develop preventive measures.
The developing fetal brain has been identified as the end-organ target for ZIKV infection (10, 12–16), Yet, the mechanism of vertical transmission has remained largely unknown. Whereas no other flavivirus has been definitively associated with birth defects in humans, a similar range of congenital malformations is caused by human cytomegalovirus (HCMV) (17), a leading cause of congenital infection (18–20). HCMV is known to infect and injure the human placenta (19, 20), and it is likely that lessons learned from studies of HCMV transplacental transmission would facilitate the understanding of congenital ZIKV infection.
ZIKV RNA and antigens have been detected in amniotic fluid and placental biopsy specimens from cases of ZIKV-related fetal malformations and loss (2, 6, 21–23). These findings, coupled with recent studies in mouse vertical ZIKV transmission models (13, 15, 24, 25) and in nonhuman primate models (26), suggest that the placenta is the most likely route of ZIKV transmission to the fetus.
The human placenta is a chimeric organ, containing both maternal and fetal structures: the maternal decidua—the specialized endometrium of pregnancy, constituting the uterine implantation site—and the fetus-derived chorionic villi (Fig. 1A) (20). During normal placental development, invasive fetal cytotrophoblasts (CTBs), originating from anchoring chorionic villi, invade the maternal decidua, which constitutes a unique multi-cell-type tissue, coinhabited by the invasive CTBs and maternal epithelial, stromal, endothelial, and immune cells (Fig. 1). Importantly, the placenta is known to be armed with physical and innate immune barriers against invading pathogens (20, 27), and recent studies have revealed the importance of both systemic and local placental innate immune responses in the defense against ZIKV (27–31).
FIG 1.

Schematic presentation of the maternal-fetal interface, depicting potential ZIKV transmission routes. (A) Chimeric maternal-fetal interface: the fetus-derived placental villi, composed of floating villi and anchoring villi, invading the maternal decidua. The solid and dashed squares mark a placental villus and the maternal decidua, shown in detail in panels B and C, respectively. (B) Placental anchoring and floating villi, bathed in maternal blood within the intervillous space. The surface of the villous tree is composed of a multinucleated syncytiotrophoblast cell layer, covering a subjacent layer of cytotrophoblasts. The villous core contains stromal cells, Hofbauer cells, and fetal blood capillaries. Extravillous trophoblasts invade and anchor the placenta to the maternal decidua. (C) Overview of the maternal decidua. Invasive extravillous trophoblasts, originating from anchoring villi (see also panel B), partially replace the resident maternal endothelium and commingle with multiple types of maternal cells, including epithelial, decidual, endothelial, and immune cells: decidual NK cells, macrophages, dendritic cells, and T cells. The enlarged insets in panels B and C depict potential routes of maternal-fetal viral transmission: from the maternal blood via the placental villi (B) or through the maternal decidua (C) aspects.
Thus far, what has been experimentally learned about ZIKV infection of the human placenta comes from studies in isolated primary placental cells, demonstrating the susceptibility of placental macrophages (Hofbauer cells) and CTBs to ZIKV (1, 32, 33). Importantly, a recent study showed that ZIKV replicates in an explant model of first-trimester chorionic villi (32). Together, these studies have addressed crucial infection patterns at the fetal aspect of the placenta. Yet, it has remained unknown how ZIKV spreads from the mother to the fetal compartment to reach these cells. Here, we reasoned that the initial events of transmission could occur at the maternal decidua, where invasive fetal CTBs are buried within a multitude of maternal cells. To gain insight into these earliest events of viral tissue interplay within the authentic uterine environment, we have employed ZIKV infection in decidual tissues maintained as integral three-dimensional (3D) organ cultures. This ex vivo infection model has been successfully employed by us for studies of HCMV transmission (20, 34). In the present study, we show that ZIKV efficiently infects and spreads through both early- and midgestation human maternal decidual tissues. Employing parallel infections with ZIKV and HCMV, we further identify distinctive virus-specific innate tissue response patterns in the maternal-fetal interface.
RESULTS
ZIKV infects and spreads through early- and midgestation decidual tissues.
To evaluate the susceptibility of decidual tissues to ZIKV, first-trimester decidual tissues were infected with ZIKV strain PRVABC59 (derived from the current epidemic). We have already shown that these tissues remain viable and retain their natural histology for ∼2 weeks under optimized growth conditions and that they are susceptible to HCMV replication (34). Organ cultures of chorionic villus tissues obtained from the same donors were infected in parallel. We monitored the kinetics of viral infection and spread, first measuring the accumulation of viral RNA in the infected tissues by quantitative real-time PCR. As shown in Fig. 2A, there was a rapid and consistent increase in decidual tissue viral RNA over time, with an ∼2-log increase measured over 7 days postinfection (dpi)—suggesting viral replication in the tissues. We next quantified the viral RNA and determined the titers of the infectious virus in the respective cleared supernatants of the same infected tissues. As shown in Fig. 2B and C, there was a surge in extracellular viral RNA, and increasing titers of infectious cell-free viral progeny were consistently released from the infected decidual tissues. Together, these findings revealed active ZIKV replication in the decidual cultures.
FIG 2.

ZIKV infection kinetics in maternal-decidual and fetus-derived chorionic villus organ cultures. Decidual and chorionic villus cultures were infected in parallel with ZIKV or HCMV (5 × 104 PFU/well). (A, B, and E) Levels of ZIKV RNA, determined by quantitative RT-PCR, at the indicated times postinfection. The levels of tissue-associated ZIKV RNA in infected tissues, normalized to β-actin, are shown in panels A and E. The copy number of extracellular ZIKV RNA measured in the supernatants of the early-gestation-infected tissues is shown in panel B. (C) Infectious ZIKV progeny titers in the supernatants of the same infected tissues at the indicated times postinfection, determined by a standard plaque assay. (D) Levels of HCMV DNA, normalized to RNase P, in tissues infected in parallel to those in panel A. (E) Levels of tissue-associated ZIKV RNA following infection of tissues obtained at midgestation. (F) Comparison of normalized tissue-associated ZIKV and HCMV genome copies following infection of tissues obtained at early gestation versus midgestation. The data shown are representative of 3 independent experiments. Each point represents the mean ± SEM from 5 biological replicates. PR, ZIKV strain PRVABC59, isolated in Puerto Rico in 2015; AFR, ZIKV strain MP1751, isolated in Uganda, Africa, in 1962.
Similarly, and in agreement with a recent report (32), ZIKV replication was demonstrated in first-trimester chorionic villi infected in parallel (Fig. 2A to C).
There are separate African and Asian lineages of the virus (35), and the latter strain has caused the current epidemic, associated with congenital infection and birth defects. In comparative experiments examining parallel infections with strains PRVABC59 and MP1751 (previously isolated in Africa), we found that the two ZIKV strains similarly replicated in both the decidual and chorionic villus tissues (Fig. 2C).
In accordance with previously reported findings by us and by others, we showed that the same tissues, infected in parallel, were susceptible to HCMV infection and spread (Fig. 2D) (34, 36). There was a 1- to 1.6-log increase in viral DNA accumulation in the tissues (Fig. 2D). It should be noted that the pattern of HCMV infection and spread has been found to be highly consistent among hundreds of tissues which we have studied so far and has been described in detail before (34). The kinetics and pattern of infection and spread differed between ZIKV and HCMV, which may be due to the much-shorter replication cycle of ZIKV and to the different modes of spread of the two viruses in the same tissues: following HCMV infection, individual HCMV-infected cells were detected at 2 dpi, with gradual progression of infection between 3 and 7 dpi. In contrast with the cell-free pattern of ZIKV spread (as revealed by the large amounts of released infectious virus [Fig. 2B and C]), HCMV demonstrated a dominant cell-associated pattern of viral spread, and no infectious HCMV could be detected in the supernatants of infected cultures even at late times postinfection.
Temporal patterns of ZIKV susceptibility in decidual and chorionic villus tissues during gestation.
While a strong temporal link of severe congenital abnormalities with first-trimester infection was revealed (2, 37, 38), adverse pregnancy outcomes have been reported following infection later during gestation (3, 5, 8). Having demonstrated ZIKV infection in early-gestation tissues, we also examined the kinetics of ZIKV replication in decidual and chorionic villus tissues obtained at midgestation (weeks 19 to 21) (Fig. 2E). Interestingly, we found that midgestation decidual tissues remained susceptible to ZIKV replication, whereas midgestation chorionic villi (obtained from the same donors and infected in parallel) demonstrated significantly reduced viral replication (∼2-log reduction) compared to the decidual tissues (P = 0.001) (Fig. 2E and F). This finding was consistently observed in 3 tissues from independent donors and appeared to be specific to ZIKV, as the susceptibility of the same tissues to HCMV was not altered by the gestation stage (Fig. 2F). The combined results suggest the potential role of the maternal decidua as an ongoing site of ZIKV transmission beyond early gestation.
We further evaluated the in situ distribution of infected cells (Fig. 3). Immunohistochemical analysis of the infected tissue sections at 2 dpi revealed multiple newly appearing ZIKV antigen-positive cells distributed throughout the decidual tissue sections. In the chorionic villus tissue sections, fetal CTBs (positive for cytokeratin 7) were positive for ZIKV antigen (in agreement with reference 32), whereas the adjacent superficial syncytiotrophoblast layer covering the chorionic villi was consistently negative for ZIKV antigen. Control tissues infected for 1 h and then washed extensively were negative for ZIKV E antigen staining, indicating that the observed staining at 2 dpi is likely to reflect viral replication. Control infected tissue sections reacted with secondary antibodies only were negative by immunohistochemical staining.
FIG 3.

Immunohistochemistry analysis of ZIKV-infected maternal-decidual and chorionic villus tissues. Decidual and chorionic villus organ cultures were mock infected or infected with ZIKV (5 × 104 PFU/well). Sections were obtained at 2 days postinfection (dpi) and subjected to immunohistochemistry analysis for ZIKV glycoprotein E (gE) and cytokeratin 7 (CK7; an epithelial/cytotrophoblast cell marker). Red indicates positive antibody staining. The arrows point to the ZIKV E antigen- and CK7 antigen-positive cytotrophoblast cell layer in the corresponding panels of chorionic villi. Arrowheads point to the ZIKV E antigen-negative superficial syncytiotrophoblast layer in placental villi. Bar, 20 μm.
ZIKV induces distinct innate immune responses in the maternal-fetal interface.
Having shown that ZIKV effectively replicates in the maternal and fetal facets of the placenta, we proceeded to study the innate tissue responses to the virus. Analysis of the innate response within the authentic environment of multicellular integral tissues might closely recapitulate the tissue responses in vivo. We further compared the innate tissue responses between ZIKV and HCMV, demonstrated to replicate in the same tissues (see above) (Fig. 2) (34, 39). To this end, we performed a genome-wide transcriptome analysis of infected versus mock-infected tissues (Fig. 4). Decidual and chorionic villus tissues were infected in parallel by ZIKV and HCMV, using the same viral inoculum titer, and the transcriptome analysis was conducted at 2 dpi. This time point was chosen based on the observed rapid replication kinetics of ZIKV in these tissues, showing peak viral replication at 2 dpi (Fig. 2), and on our recent demonstration that while HCMV infection and spread are slower in the tissues (Fig. 2D and reference 34), the innate immune response in the decidual tissue is already induced at early times postinfection and reaches its peak at 24 to 48 h postinfection (39).
FIG 4.

Decidual and chorionic villus tissue innate responses to ZIKV and HCMV infection. Decidual and chorionic villus organ cultures were mock infected or infected in parallel with ZIKV or HCMV (5 × 104 PFU/well). At day 2 postinfection, RNA was extracted and subjected to transcriptome analysis. To account for the potential donors' tissue-to-tissue variability, decidual and chorionic villus tissues from the same donors were infected in parallel with ZIKV and HCMV. Two pools (each representing a mixture of 5 independent donor tissues) for each experimental condition were used, together representing 10 tissues from different individuals. (A) Number of overlapping and uniquely differentially expressed genes under the different indicated conditions. The numbers in parentheses represent the total number of significantly differentially expressed genes under each of the indicated experimental conditions. The number in each cell represents the number of significantly differentially expressed genes in the corresponding category. (B) Number of genes upregulated or downregulated under each indicated condition shown in relation to the magnitude of the fold change from mock-infected tissue. (C and D) Molecular functions enriched under each condition. Ingenuity pathway analysis was used to summarize the molecular functions most significantly changed in infected decidual (C) and placental villus (D) tissues.
Decidual tissue innate responses.
We found that infection with both ZIKV and HCMV substantially dysregulated the gene expression profile in the decidual tissue (Fig. 4A). Despite the more robust replication of ZIKV observed in the tissue, ZIKV exerted a relatively restricted decidual tissue response compared to HCMV (Fig. 4A and B). This was reflected by the lower number of differentially expressed genes following ZIKV infection (Fig. 4A and B), along with a lesser extent of gene upregulation, as measured by the fold changes from mock-infected tissues (Fig. 4B), compared to the effect of HCMV on the decidua. Interestingly, while the HCMV response (as revealed by gene ontology analysis) was dominated by upregulation of immune cell activation, proliferation, and cell trafficking pathways, these pathways were not significantly altered by ZIKV (Fig. 4C; see also Fig. S1 in the supplemental material). Nonetheless, ZIKV significantly upregulated some innate immunity genes in the decidual tissue, in particular genes related to the antiviral interferon signaling pathways (Fig. 4C and 5). Of note, gamma interferon (IFN-γ), which was upregulated by ∼78-fold following HCMV infection (increase from mock), was not affected by ZIKV (Fig. 5; also see Fig. S1 in the supplemental material). The HCMV-mediated upregulation of IFN-γ was similarly observed in tissues from HCMV-seronegative and -seropositive donors (39). The absence of IFN-γ induction following ZIKV infection was further confirmed by quantitative real-time reverse transcription-PCR (RT-PCR) (Fig. 6A) and implies that unlike HCMV, ZIKV did not activate the decidual tissue immune cell response. In contrast, ZIKV infection substantially induced the expression of type I and type III IFNs, IFN-α, IFN-β, and IFN-λ, in the decidual tissues (Fig. 6A). Heat maps representing cytokine and chemokine expression levels in infected and mock-infected decidual tissues showed the distinctive upregulation of cytokines, such as IFN-α, migration inhibition factor (MIF), CXCL6, AIMP1, and leukemia inhibitory factor (LIF), by ZIKV, whereas HCMV upregulated a wide array of different cytokines/chemokines, including (in addition to the above-mentioned IFN-γ), CXCL10 (IP10), CXCL9, tumor necrosis factor (TNF), interleukin-6 (IL-6), IL-1β, and IL-15 (for detailed representation of the cytokines and chemokines with the greatest expression variability, see Fig. S1A in the supplemental material).
FIG 5.

The interferon signaling pathway as differentially regulated by ZIKV and HCMV in maternal-decidual and fetus-derived chorionic villus tissues. Pathway enrichment of differentially expressed genes (FDR of <0.1) in decidual tissue (A) and placental villus (B) was carried out with ingenuity pathway analysis (IPA). Gene upregulation is depicted in shades of red, from white (not significantly changed, to dark red (highly upregulated).
FIG 6.
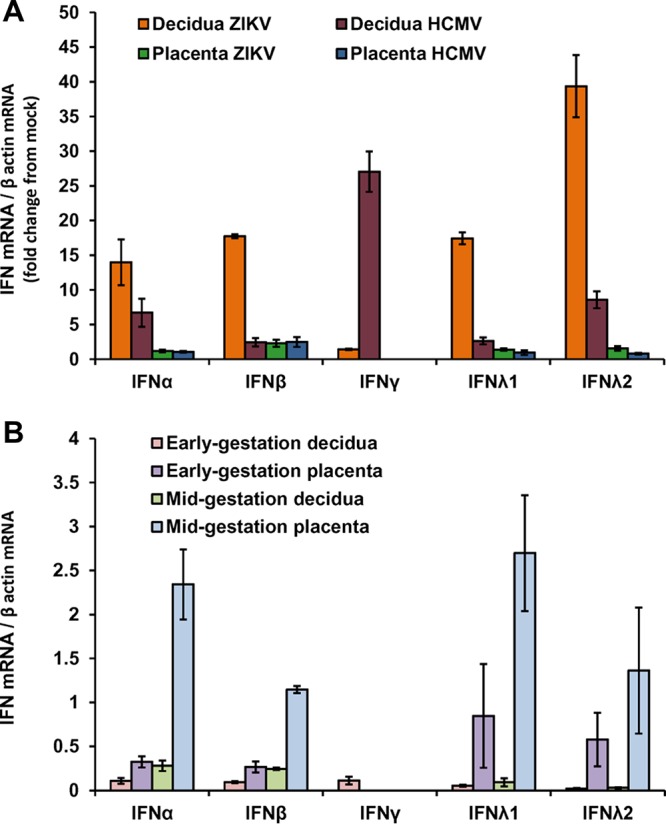
IFN expression in infected and mock-infected decidual and placental tissues. (A) Decidual and placental cultures were infected in parallel with ZIKV or HCMV. RNA was extracted at 7 dpi, and the indicated IFN mRNA levels were analyzed by quantitative RT-PCR and normalized by the housekeeping gene β-actin. Results are shown as fold change from mock-infected tissues. The data shown are representative of at least three independent experiments, each tested in 5 replicates. (B) RNA was extracted at 7 days postculture from mock-infected organ cultures of decidual and placental tissues obtained at early gestation or midgestation. The indicated IFN mRNA levels were analyzed by quantitative RT-PCR and normalized by the housekeeping gene β-actin. Results are shown as averages from 3 different tissues obtained from different individuals.
Chorionic villus tissue innate responses.
Compared with the broad effect on the decidual tissues, the impact of both viruses on the chorionic villus tissue transcriptome was relatively restricted, as revealed by the much smaller number of differentially regulated genes (Fig. 4A and B). Notably, ZIKV and HCMV induced divergent cytokines, i.e., type I and type III IFNs were induced by ZIKV, whereas multiple other cytokines/chemokines were specifically affected by HCMV (see Fig. S1B in the supplemental material) along with divergent regulatory pathways in placental villi (Fig. 4A and D). In fact, the differences between the innate responses to the two viruses were even more pronounced in chorionic villus tissues than in the decidual tissues (Fig. 4A, C, and D). Chorionic villus responses to ZIKV were distinctively enriched for apoptosis, cell death, and necrosis molecular functions (the enriched gene transcripts related to these functions included MUC1, SP110, PNPT1, EIF2AK2, IRF9, MX1, ISG15, S100A4, IFIH1, IRF7, GEM, GLS, PLSCR1, OAS1, DDX58, KRT17, USP18, IER3, HSH2D, DHX58, NT5C3A, and IFI27), whereas HCMV-triggered responses were dominated by upregulation of leukocyte migration, mobilization, and homing functions (Fig. 4D; also see Fig. S1B in the supplemental material).
Temporal patterns of IFN expression in decidual and chorionic villus tissues during gestation as related to ZIKV susceptibility.
Having shown the significantly reduced ZIKV replication in midgestation chorionic villus tissues compared with concurrently obtained decidual tissues and with early-gestation tissues (Fig. 2E and F), we further sought to examine whether differences in the tissue innate immune state could be related to the differences in ZIKV susceptibility. We examined the expression of type I, type II, and type III IFNs in mock-infected decidual and chorionic villus tissues obtained at early gestation and midgestation. As shown in Fig. 6B, while there was an expected tissue-to-tissue variation among different donors, higher expression levels of type I IFNs were found in midgestation chorionic villus tissues than in midgestation decidual tissues (P = 0.004 for IFN-β; P = 0.068 for IFN-α) and compared to early-gestation chorionic villus tissues (P = 0.003 for IFN-β; P = 0.018 for IFN-α). IFN-λ mRNA levels were also found to be higher in midgestation chorionic villi (Fig. 6B), and yet these differences did not reach statistical significance. This inverse correlation between the levels of IFN expression and the temporal pattern of ZIKV susceptibility suggests that the increased expression of innate tissue antiviral factors during gestation could be related to the reduced ZIKV susceptibility of midgestation chorionic villi.
DISCUSSION
In light of the rapid spread of the current ZIKV epidemic and the severe manifestations of congenital ZIKV infection, it is crucial to learn the mechanisms of viral transmission through the maternal-fetal interface.
While extensive experimental data supporting transplacental transmission have been recently obtained from mouse and nonhuman primate models (25, 26), from studies in primary human placental cells (27, 33), and from an explant model of first-trimester chorionic villi (32) (representing the fetal part of the human placenta), the earliest events of ZIKV transmission in the maternal uterine environment have remained unexplored. Our findings identify the human maternal decidua as a likely route of vertical ZIKV transmission to the fetus.
Active viral replication in the decidual tissues was demonstrated by the rapid upsurge and continued increase of tissue-associated ZIKV RNA load, along with the production and release (to the medium) of large amounts of infectious progeny virus from the infected decidual tissues. The rapid kinetics of ZIKV spread as demonstrated in both the decidual and chorionic villus tissues differed from the slower kinetics of HCMV infection in the same tissues (Fig. 2 and reference 34), a difference which may be partly due to the much shorter replication cycle of ZIKV than of HCMV. In particular, the efficient cell-free mode of spread of ZIKV contrasted with the predominant cell-to-cell mode of spread of HCMV in the same tissues, revealed by the complete absence of cell-free virus release (see also reference 34), and could have implications for the potential effectiveness of antibody-mediated preventive approaches.
We show that ZIKV targets the fetal CTBs within first-trimester chorionic villi (positive for ZIKV antigens and for cytokeratin 7 [Fig. 3]). This finding is in accordance with a recent report which showed that ZIKV infects CTBs in first-trimester chorionic villi (32). ZIKV replication has been also demonstrated in placental macrophages (Hofbauer cells) within first-trimester chorionic villi (32), as well as in human primary amniotic epithelial cells, trophoblast progenitor cells, placental fibroblasts, endothelial cells, Hofbauer cells, and CTBs isolated from mid- and late-gestation placenta (5, 32, 33). Likewise, ZIKV was found to infect trophoblasts of the maternal and fetal placenta along with placental endothelial cells in a mouse transplacental transmission model (24). Importantly, we also found that the superficial syncytiotrophoblast layer, covering the villous core and directly contacting the maternal blood in the intervillous space (Fig. 1B), was consistently spared from ZIKV infection (Fig. 3). This finding is in agreement with the lack of ZIKV NS3 protein production in these cells, as reported by Tabata et al. (32). The resistance of the syncytiotrophoblast cell layer to ZIKV infection could be related to the recent finding that syncytialized trophoblasts from term placenta are refractory to ZIKV infection due to their constitutive secretion of type III IFNs (27). It remains unclear how ZIKV could bypass this placental barrier layer to access the subjacent fetal cells. In this regard, the hereby-demonstrated viral replication in the maternal decidua, where fetal cells directly interact with maternal cells, could represent at least one mechanism whereby virus originating from the mother could circumvent innate placental defenses (Fig. 1C).
Routes of vertical transmission and their relative impact can vary with the gestational stage (5). While a compelling association of severe outcome with first-trimester infection was identified (2, 37, 38), it is now realized that ZIKV can also infect and damage the fetus later during gestation (3, 5, 8). Our finding that midgestation decidual tissues remain susceptible to ZIKV replication, in contrast to the reduced susceptibility of chorionic villi at this gestational stage (Fig. 2E and F), highlights the potential role of the decidua as a site of ongoing ZIKV transmission beyond early gestation.
Given the central role of placental CTBs in anchoring and perfusion placental functions (20), it is plausible that direct targeting of these cells by ZIKV would significantly impair placental development and function (19). In addition to the deleterious effects of direct infection per se, it is increasingly recognized that the innate tissue responses to the virus can play a crucial role in the pathogenesis and outcome of congenital viral infections (29, 39–41). We therefore examined the global impact of ZIKV on decidual and chorionic villus tissues, in comparison with HCMV, employing genome-wide transcriptome analysis following parallel infection of the same tissues. In evaluating the decidual tissue response, one should bear in mind that the maternal decidua constitutes a specialized immunological site, geared to allow immune tolerance of the semiallogeneic fetus (20), and that the immune-privileged environment in the decidua is tightly controlled by the composition of its immune cell population, dominated by decidual NK (dNK) cells (20, 42–44) (Fig. 1). While both ZIKV and HCMV generally upregulated the decidual tissue antiviral innate immune responses and interferon signaling pathway (Fig. 4 to 6; see also Fig. S1 in the supplemental material), HCMV innate responses and the cytokine alteration profile were dominated by upregulation of immune cell activation, proliferation, and trafficking pathways, pathways which were not modified by ZIKV (Fig. 4C). Importantly, IFN-γ, known to be exclusively expressed by immune cells, was among the most upregulated genes in HCMV-infected decidual tissues and was not affected by ZIKV (Fig. 5 and 6; also Fig. S1 in the supplemental material), further implying that unlike HCMV, ZIKV does not appear to activate decidual tissue immune cell response and trafficking. Conversely, ZIKV stimulated the expression of type I and type III IFNs, not remarkably affected by HCMV (Fig. 6A; also Fig. S1 in the supplemental material). These differences further argue for the important role of IFNs and interferon-stimulated genes (ISGs) rather than immune cell activation, in the early responses to the rapidly replicating ZIKV within the maternal-fetal interface tissue.
In line with the divergent decidual tissue responses to the two viruses, chorionic villus tissue response to ZIKV was distinctively enriched for apoptosis, cell death, and necrosis molecular functions, whereas the HCMV-triggered responses were dominated by upregulation of leukocyte migration, mobilization, and homing functions (Fig. 4D; also Fig. S1 in the supplemental material). The direct relation of these findings to clinical pathogenesis should be further studied. In view of these findings, it is noteworthy that the histopathology of naturally infected placenta in congenital HCMV cases is indeed characterized by immune cell infiltration (40, 41), whereas limited pathological investigations of congenital ZIKV infection did not show increased inflammatory cell infiltration (23). Based on our findings, it is tempting to speculate that following HCMV infection, innate immune responses may dysregulate the immunotolerance of the maternal-fetal interface, whereas at least some of the adverse pregnancy outcomes associated with ZIKV could be mediated by the virus-induced direct tissue damage pathways.
In summary, we have shown that ZIKV efficiently replicates in decidual tissues, identifying the maternal uterine aspect of the human placenta as a likely route of ZIKV transmission to the fetus. The data further reveal distinct patterns of placental-tissue innate immune responses to ZIKV and HCMV. Our unique ex vivo experimental model and findings could further serve to study the initial stages of transmission and pathogenesis of congenital ZIKV infection and evaluate the effect of new therapeutic interventions in the otherwise inaccessible human maternal-fetal interface.
MATERIALS AND METHODS
Cells and viruses.
Vero cells (obtained from the American Type Culture Collection) were used for ZIKV propagation. Cells were infected with ZIKV strain PRVABC59, a low-passage-number, sequence-verified strain isolated from an infected patient in Puerto Rico in December 2015 (5) (generously provided by S. Lanciotti, U.S. Centers for Disease Control and Prevention), or with ZIKV strain MP1751, isolated from Aedes africanus mosquitoes captured in the Zika Forest, Uganda, in November 1962 (45) (purchased from Public Health England), at a multiplicity of infection (MOI) of 0.01. At 2 days postinfection (dpi), supernatants were collected, centrifuged to remove cellular debris, and frozen at −80°C. Virus titers were determined by a standard plaque assay. Primary human foreskin fibroblasts (HFF; provided by A. Lifshitz, Hadassah Medical Center Clinical Virology Laboratory) were used for HCMV propagation (34). The HCMV strain used was TB40/E expressing IE2-fused enhanced yellow fluorescent protein (EYFP) (strain RV1164; generously provided by M. Winkler, Germany [34]). All cell lines have been tested for mycoplasma contamination.
Preparation and infection of decidual and chorionic villus organ cultures.
Decidual and chorionic villus organ cultures were prepared as previously described (34). In brief, decidual and chorionic villus tissues, obtained from women undergoing first-trimester elective pregnancy terminations, were kept on ice until sectioning, washed with phosphate-buffered saline (PBS), sectioned by a microtome into thin slices, and incubated in decidual medium: Dulbecco's modified Eagle's medium (DMEM) with 25% Ham's F-12 medium, 10% fetal bovine serum, 5 mM HEPES, 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B (Biological Industries), at 37°C with 5% CO2. Tissue viability was monitored as described previously (34). For infection of organ cultures, the tissues were placed in 48-well plates and inoculated with the indicated virus (5 × 104 PFU/well) for 12 h, for optimized viral adsorption. Following viral adsorption, the cultures were washed extensively and further incubated for the duration of the experiment, with medium replacement every 2 to 3 days.
Immunohistochemistry analysis.
Mock- and ZIKV-infected decidual and chorionic villus tissues were formaldehyde fixed, paraffin embedded, and sectioned (5-μm thickness). Sections were placed in 0.01 M citrate buffer, warmed in a pressure cooker to 110°C for 5 min, and cooled to room temperature, followed by incubation with primary mouse monoclonal antibodies (MAbs) to flavivirus glycoprotein E (E; clone D1-4G2-4-15; Millipore) and rabbit monoclonal antibody to the epithelial/cytotrophoblast cell marker cytokeratin 7 (CK7; clone EPR1619Y; Abcam) diluted in CAS-Block (Zymed Laboratories) or with CAS-Block containing no primary antibody to serve as a negative control. Sections were then washed and incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit secondary antibodies (Biocare Medical). The sections were washed again, and the antigens were detected by the HRP substrate 3-amino-9-ethylcarbazole (AEC; Sigma), followed by counterstaining with hematoxylin.
RNA and DNA purification and quantification.
Infected and mock-infected decidual and chorionic villus tissues were washed and stored at −80°C until assayed. DNA-free RNA was extracted (NucleoSpin RNA isolation kit; Macherey-Nagel) and subjected to reverse transcription (GoScript; Promega), followed by quantitative real-time PCR (7900HT; Applied Biosystems). ZIKV oligonucleotide sequences were as follows: forward, 5′ CCACTAACGTTCTTTTGCAGACAT 3′; reverse, 5′ CCGCTGCCCAACACAAG 3′; probe, 5′ 6-FAM (carboxyfluorescein)/AG CCT ACC T/ZEN/T GAC AAG CAG TCA GAC ACT CAA/3′ IABkFQ (35). β-Actin oligonucleotide sequences were as follows: forward, 5′ CCTGGCACCCAGCACAAT 3′; reverse, 5′ GCCGATCCACACGGAGTACT 3′; probe, 5′ 6-FAM/AT CAA GAT C/ZEN/A TTG CTC CTC CTG AGC GC/3′ IABkFQ. Interferon (IFN) oligonucleotide sequences were as follows: IFN-α, forward, 5′ GGCTGTGAGGAAATACTTCCAAAGAA 3′; reverse, 5′ GATCTCATGATTTCTGCTCTGACAAC 3′; IFN-β, forward, 5′ TGGGAGGCTTGAATACTGCCTCAA 3′; reverse, 5′ TGCGGCGTCCTCCTTCTGGA 3′; IFN-γ, forward, 5′ GCAACAAAAAGAAACGAGATGACTTCG 3′; reverse, 5′ TGAGTTCATGTATTGCTTTGCGTTG 3′; IFN-λ1, forward, 5′ CGCCTTGGAAGAGTCACTCA 3′; reverse, 5′ GAAGCCTCAGGTCCCAATTC 3′; IFN-λ2, forward, 5′ ACATAGCCCAGTTCAAGTC 3′; reverse, 5′ GACTCTTCTAAGGCATCTTTG 3′ (27, 39).
DNA was extracted using the NucleoSpin tissue kit (Macherey-Nagel) and quantified as previously described (34).
cDNA library preparation, deep sequencing, and bioinformatics analysis.
Transcriptome libraries were prepared using the Illumina TruSeq RNA library preparation kit (Illumina RS-122-2001), according to the manufacturer's recommended protocol, starting with around 450 ng of total RNA. The amplified indexed libraries were quantified using an Invitrogen Qubit fluorometer and equally pooled according to pool design. Pooled libraries were run on a 4% agarose gel, and DNA around 270 bp (the length of RNA inserts plus the 3′ and 5′ adapters) was size selected and recovered in 15 μl elution buffer (Qiagen). Size-selected libraries were then quantified again using the Qubit fluorometer. Size was verified using the High Sensitive DNA gels on an Agilent 2200 TapeStation instrument. Libraries were sequenced on a NextSeq 500 instrument using the NextSeq 500 High-Output V2 sequencing kit (FC-404-2005), in a single-end configuration, reading 75 bases.
Raw reads were quality trimmed at both ends, using in-house Perl scripts, with a quality threshold of 32. Adapter sequences were removed using Cutadapt (46) (version 1.7.1; https://cutadapt.readthedocs.org/en/stable/). The remaining reads were further filtered to remove low-quality reads, using the fastq_quality_filter program of the FASTX package (version 0.0.14; http://hannonlab.cshl.edu/fastx_toolkit/), with a quality threshold of 20 in at least 90% of the read's positions. The processed fastq files were mapped to the human genome GRCh38 using TopHat (47) (v2.0.14), with Ensembl release 78 annotations. Quantification of raw counts per gene was done with the Cufflinks package (48, 49) (v2.2.1), using the cuffquant and cuffnorm programs. Normalization and differential expression were done with the DESeq2 package (50) (version 1.6.3) of the Bioconductor project separately for each tissue. Quality control assays were performed to ensure that the data meet the assumptions of the statistical tests and to assess the inter- and intraconditional group variations. Genes with a total of less than 2 counts in all samples were filtered out prior to normalization. Differential expression was run with default parameters, where the significance threshold for a gene was set to a Padj of <0.1.
Differential expression results were visualized in R (version 3.1.1, using the packages VennDiagram_1.6.16′ and pheatmap_1.0.8′) (51). Pathway and molecular function and disease enrichment analysis of the significantly differentially expressed genes (false discovery rate [FDR] of <0.1) was carried out using Qiagen's ingenuity pathway analysis (IPA; Qiagen, Redwood City, CA).
Statistical analysis.
All data (means ± standard errors of the means [SEM]) were analyzed using unpaired, two-tailed t tests for comparisons between two groups; P values of <0.05 were considered significant. Statistical analysis of the transcriptome data was done as described in the Fig. 4 legend.
Ethics statement.
The study was approved by the Hadassah Medical Center Institutional Review Board (0138-08-HMO) and performed according to the Declaration of Helsinki, Good Clinical Practice guidelines, and the Human-Experimentation Guidelines of the Israeli Ministry of Health. All participants signed written informed consent.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Israel Science Foundation, the European Union Seventh Framework Programme 562 FP7/2012-2016 (grant agreement number 316655), and the Israeli Ministry of Health.
We thank Eyal Mishani for his help in moving this project forward and Kelly Shacham, Ori Goldstein, Anita Rotenberg-Steiner, Yulia Yachnin, and Marina Dorozko for technical assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01905-16.
REFERENCES
- 1.Lazear HM, Diamond MS. 2016. Zika virus: new clinical syndromes and its emergence in the Western Hemisphere. J Virol 90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. 2016. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med 374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 3.Franca GV, Schuler-Faccini L, Oliveira WK, Henriques CM, Carmo EH, Pedi VD, Nunes ML, Castro MC, Serruya S, Silveira MF, Barros FC, Victora CG. 2016. Congenital Zika virus syndrome in Brazil: a case series of the first 1501 livebirths with complete investigation. Lancet 388:891–897. doi: 10.1016/S0140-6736(16)30902-3. [DOI] [PubMed] [Google Scholar]
- 4.Russell K, Oliver SE, Lewis L, Barfield WD, Cragan J, Meaney-Delman D, Staples JE, Fischer M, Peacock G, Oduyebo T, Petersen EE, Zaki S, Moore CA, Rasmussen SA. 2016. Update: interim guidance for the evaluation and management of infants with possible congenital Zika virus infection—United States, August 2016. MMWR Morb Mortal Wkly Rep 65:870–878. doi: 10.15585/mmwr.mm6533e2. [DOI] [PubMed] [Google Scholar]
- 5.Coyne CB, Lazear HM. 2016. Zika virus—reigniting the TORCH. Nat Rev Microbiol 14:707–715. doi: 10.1038/nrmicro.2016.125. [DOI] [PubMed] [Google Scholar]
- 6.Martines RB, Bhatnagar J, de Oliveira Ramos AM, Davi HP, Iglezias SD, Kanamura CT, Keating MK, Hale G, Silva-Flannery L, Muehlenbachs A, Ritter J, Gary J, Rollin D, Goldsmith CS, Reagan-Steiner S, Ermias Y, Suzuki T, Luz KG, de Oliveira WK, Lanciotti R, Lambert A, Shieh WJ, Zaki SR. 2016. Pathology of congenital Zika syndrome in Brazil: a case series. Lancet 388:898–904. doi: 10.1016/S0140-6736(16)30883-2. [DOI] [PubMed] [Google Scholar]
- 7.Adibi JJ, Marques ET Jr, Cartus A, Beigi RH. 2016. Teratogenic effects of the Zika virus and the role of the placenta. Lancet 387:1587–1590. doi: 10.1016/S0140-6736(16)00650-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brasil P, Pereira JP Jr, Raja Gabaglia C, Damasceno L, Wakimoto M, Ribeiro Nogueira RM, Carvalho de Sequeira P, Machado Siqueira A, Abreu de Carvalho LM, Cotrim da Cunha D, Calvet GA, Neves ES, Moreira ME, Rodrigues Baiao AE, Nassar de Carvalho PR, Janzen C, Valderramos SG, Cherry JD, Bispo de Filippis AM, Nielsen-Saines K. 2016. Zika virus infection in pregnant women in Rio de Janeiro—preliminary report. N Engl J Med doi: 10.1056/NEJMoa1602412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martines RB, Bhatnagar J, Keating MK, Silva-Flannery L, Muehlenbachs A, Gary J, Goldsmith C, Hale G, Ritter J, Rollin D, Shieh WJ, Luz KG, Ramos AM, Davi HP, Kleber de Oliveria W, Lanciotti R, Lambert A, Zaki S. 2016. Notes from the field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses—Brazil, 2015. MMWR Morb Mortal Wkly Rep 65:159–160. doi: 10.15585/mmwr.mm6506e1. [DOI] [PubMed] [Google Scholar]
- 10.Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, Vizjak A, Pizem J, Petrovec M, Avsic Zupanc T. 2016. Zika virus associated with microcephaly. N Engl J Med 374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 11.Meaney-Delman D, Rasmussen SA, Staples JE, Oduyebo T, Ellington SR, Petersen EE, Fischer M, Jamieson DJ. 2016. Zika virus and pregnancy: what obstetric health care providers need to know. Obstet Gynecol 127:642–648. doi: 10.1097/AOG.0000000000001378. [DOI] [PubMed] [Google Scholar]
- 12.Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK. 2016. Zika virus impairs growth in human neurospheres and brain organoids. Science 352:816–818. doi: 10.1126/science.aaf6116. [DOI] [PubMed] [Google Scholar]
- 13.Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, Polonio CM, Cunha I, Freitas CL, Brandao WN, Rossato C, Andrade DG, Faria DDP, Garcez AT, Buchpigel CA, Braconi CT, Mendes E, Sall AA, Zanotto PM, Peron JP, Muotri AR, Beltrao-Braga PC. 2016. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin CF, Xu Z. 2016. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19:120–126. doi: 10.1016/j.stem.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 15.Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY, Cao WC, Qin CF, Luo ZG. 2016. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 26:645–654. doi: 10.1038/cr.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, Christian KM, Didier RA, Jin P, Song H, Ming GL. 2016. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, Soroceanu L, Cobbs CS, Hcmv Gliomas S. 2012. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol 14:246–255. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boppana SB, Ross SA, Fowler KB. 2013. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis 57(Suppl 4):S178–S181. doi: 10.1093/cid/cit629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira L, Maidji E, McDonagh S, Tabata T. 2005. Insights into viral transmission at the uterine-placental interface. Trends Microbiol 13:164–174. doi: 10.1016/j.tim.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Weisblum Y, Panet A, Haimov-Kochman R, Wolf DG. 2014. Models of vertical cytomegalovirus (CMV) transmission and pathogenesis. Semin Immunopathol 36:615–625. doi: 10.1007/s00281-014-0449-1. [DOI] [PubMed] [Google Scholar]
- 21.Calvet G, Aguiar RS, Melo AS, Sampaio SA, de Filippis I, Fabri A, Araujo ES, de Sequeira PC, de Mendonca MC, de Oliveira L, Tschoeke DA, Schrago CG, Thompson FL, Brasil P, Dos Santos FB, Nogueira RM, Tanuri A, de Filippis AM. 2016. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 16:653–660. doi: 10.1016/S1473-3099(16)00095-5. [DOI] [PubMed] [Google Scholar]
- 22.Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jaaskelainen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G, Timofeev J, Rodriguez FJ, Levanov L, Razak J, Iyengar P, Hennenfent A, Kennedy R, Lanciotti R, du Plessis A, Vapalahti O. 2016. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 374:2142–2151. doi: 10.1056/NEJMoa1601824. [DOI] [PubMed] [Google Scholar]
- 23.van der Eijk AA, van Genderen PJ, Verdijk RM, Reusken CB, Mogling R, van Kampen JJ, Widagdo W, Aron GI, GeurtsvanKessel CH, Pas SD, Raj VS, Haagmans BL, Koopmans MP. 2016. Miscarriage associated with Zika virus infection. N Engl J Med 375:1002–1004. doi: 10.1056/NEJMc1605898. [DOI] [PubMed] [Google Scholar]
- 24.Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, Diamond MS. 2016. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165:1081–1091. doi: 10.1016/j.cell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mysorekar IU, Diamond MS. 2016. Modeling Zika virus infection in pregnancy. N Engl J Med 375:481–484. doi: 10.1056/NEJMcibr1605445. [DOI] [PubMed] [Google Scholar]
- 26.Dudley DM, Aliota MT, Mohr EL, Weiler AM, Lehrer-Brey G, Weisgrau KL, Mohns MS, Breitbach ME, Rasheed MN, Newman CM, Gellerup DD, Moncla LH, Post J, Schultz-Darken N, Schotzko ML, Hayes JM, Eudailey JA, Moody MA, Permar SR, O'Connor SL, Rakasz EG, Simmons HA, Capuano S, Golos TG, Osorio JE, Friedrich TC, O'Connor DH. 2016. A rhesus macaque model of Asian-lineage Zika virus infection. Nat Commun 7:12204. doi: 10.1038/ncomms12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB. 2016. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19:705–712. doi: 10.1016/j.chom.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie X, Shan C, Shi PY. 2016. Restriction of Zika virus by host innate immunity. Cell Host Microbe 19:566–567. doi: 10.1016/j.chom.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 30.Pierson TC, Graham BS. 2016. Zika virus: immunity and vaccine development. Cell 167:625–631. doi: 10.1016/j.cell.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL. 2016. The IFITMs inhibit Zika virus replication. Cell Rep 15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- 32.Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, Harris E, Pereira L. 2016. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 20:155–166. doi: 10.1016/j.chom.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, Schinazi RF, Chakraborty R, Suthar MS. 2016. Zika virus infects human placental macrophages. Cell Host Microbe 20:83–90. doi: 10.1016/j.chom.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weisblum Y, Panet A, Zakay-Rones Z, Haimov-Kochman R, Goldman-Wohl D, Ariel I, Falk H, Natanson-Yaron S, Goldberg MD, Gilad R, Lurain NS, Greenfield C, Yagel S, Wolf DG. 2011. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J Virol 85:13204–13213. doi: 10.1128/JVI.05749-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, Stanfield SM, Duffy MR. 2008. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis 14:1232–1239. doi: 10.3201/eid1408.080287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher S, Genbacev O, Maidji E, Pereira L. 2000. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol 74:6808–6820. doi: 10.1128/JVI.74.15.6808-6820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansson MA, Mier-y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL. 2016. Zika and the risk of microcephaly. N Engl J Med 375:1–4. doi: 10.1056/NEJMp1605367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleber de Oliveira W, Cortez-Escalante J, De Oliveira WT, do Carmo GM, Henriques CM, Coelho GE, Araujo de Franca GV. 2016. Increase in reported prevalence of microcephaly in infants born to women living in areas with confirmed Zika virus transmission during the first trimester of pregnancy—Brazil, 2015. MMWR Morb Mortal Wkly Rep 65:242–247. doi: 10.15585/mmwr.mm6509e2. [DOI] [PubMed] [Google Scholar]
- 39.Weisblum Y, Panet A, Zakay-Rones Z, Vitenshtein A, Haimov-Kochman R, Goldman-Wohl D, Oiknine-Djian E, Yamin R, Meir K, Amsalem H, Imbar T, Mandelboim O, Yagel S, Wolf DG. 2015. Human cytomegalovirus induces a distinct innate immune response in the maternal-fetal interface. Virology 485:289–296. doi: 10.1016/j.virol.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 40.Gabrielli L, Bonasoni MP, Santini D, Piccirilli G, Chiereghin A, Petrisli E, Dolcetti R, Guerra B, Piccioli M, Lanari M, Landini MP, Lazzarotto T. 2012. Congenital cytomegalovirus infection: patterns of fetal brain damage. Clin Microbiol Infect 18:E419–E427. doi: 10.1111/j.1469-0691.2012.03983.x. [DOI] [PubMed] [Google Scholar]
- 41.Pereira L, Petitt M, Fong A, Tsuge M, Tabata T, Fang-Hoover J, Maidji E, Zydek M, Zhou Y, Inoue N, Loghavi S, Pepkowitz S, Kauvar LM, Ogunyemi D. 2014. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J Infect Dis 209:1573–1584. doi: 10.1093/infdis/jiu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Bouteiller P. 2013. Human decidual NK cells: unique and tightly regulated effector functions in healthy and pathogen-infected pregnancies. Front Immunol 4:404. doi: 10.3389/fimmu.2013.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nancy P, Tagliani E, Tay CS, Asp P, Levy DE, Erlebacher A. 2012. Chemokine gene silencing in decidual stromal cells limits T cell access to the maternal-fetal interface. Science 336:1317–1321. doi: 10.1126/science.1220030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Red-Horse K, Drake PM, Fisher SJ. 2004. Human pregnancy: the role of chemokine networks at the fetal-maternal interface. Expert Rev Mol Med 6:1–14. [DOI] [PubMed] [Google Scholar]
- 45.Haddow AJ, Williams MC, Woodall JP, Simpson DI, Goma LK. 1964. Twelve isolations of Zika virus from Aedes (Stegomyia) Africanus (Theobald) taken in and above a Uganda forest. Bull World Health Organ 31:57–69. [PMC free article] [PubMed] [Google Scholar]
- 46.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17:10–12. doi:http://dx.doi.org/10.14806/ej.17.1.200. [Google Scholar]
- 47.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Boutros PC. 2011. VennDiagram: a package for the generation of highly customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.