

ABSTRACT
Simian arteriviruses are a diverse clade of viruses infecting captive and wild nonhuman primates. We recently reported that Kibale red colobus virus 1 (KRCV-1) causes a mild and self-limiting disease in experimentally infected crab-eating macaques, while simian hemorrhagic fever virus (SHFV) causes lethal viral hemorrhagic fever. Here we characterize how these viruses evolved during replication in cell culture and in experimentally infected macaques. During passage in cell culture, 68 substitutions that were localized in open reading frames (ORFs) likely associated with host cell entry and exit became fixed in the KRCV-1 genome. However, we did not detect any strong signatures of selection during replication in macaques. We uncovered patterns of evolution that were distinct from those observed in surveys of wild red colobus monkeys, suggesting that these species may exert different adaptive challenges for KRCV-1. During SHFV infection, we detected signatures of selection on ORF 5a and on a small subset of sites in the genome. Overall, our data suggest that patterns of evolution differ markedly among simian arteriviruses and among host species.
IMPORTANCE Certain RNA viruses can cross species barriers and cause disease in new hosts. Simian arteriviruses are a diverse group of related viruses that infect captive and wild nonhuman primates, with associated disease severity ranging from apparently asymptomatic infections to severe, viral hemorrhagic fevers. We infected nonhuman primate cell cultures and then crab-eating macaques with either simian hemorrhagic fever virus (SHFV) or Kibale red colobus virus 1 (KRCV-1) and assessed within-host viral evolution. We found that KRCV-1 quickly acquired a large number of substitutions in its genome during replication in cell culture but that evolution in macaques was limited. In contrast, we detected selection focused on SHFV ORFs 5a and 5, which encode putative membrane proteins. These patterns suggest that in addition to diverse pathogenic phenotypes, these viruses may also exhibit distinct patterns of within-host evolution both in vitro and in vivo.
KEYWORDS: evolution, simian arterivirus, simian hemorrhagic fever virus, virology, within-host diversity
INTRODUCTION
In 1964, acute outbreaks of a highly lethal viral hemorrhagic fever caused by a previously unknown agent swept through captive nonhuman primate research facilities in the former Soviet Union and the United States (1–4). Although the origin and natural reservoir of this agent were not unambiguously determined, serological and epizootiological studies implicated primates of African origin as possible hosts (2). Patas monkeys (African origin) housed in the same facilities as infected rhesus monkeys (Asian origin) were shown to harbor this infectious agent asymptomatically during the outbreak. Experiments performed after the outbreak showed that infected blood from asymptomatic patas monkeys caused hemorrhagic fever when used to infect naive macaques via needle inoculation (2). These data spawned the hypotheses that this outbreak was facilitated by shared tattoo needles used on multiple animal species in the facility and that wild African primates may be the natural reservoirs (2). Sporadic outbreaks of this “simian hemorrhagic fever” (SHF) continued to occur among captive macaques from the 1960s to the 1990s (5–8). Until recently, it was believed that all SHF outbreaks were caused by a single virus, simian hemorrhagic fever virus (SHFV), of the nidoviral family Arteriviridae.
Using deep sequencing to screen for the presence of RNA viruses in plasma collected from apparently healthy wild monkeys in Kibale National Park, Uganda (9), we previously discovered coinfection of a red colobus monkey (Piliocolobus rufomitratus tephrosceles) with two novel arteriviruses highly divergent from SHFV. These two viruses were subsequently named Kibale red colobus viruses 1 and 2 (KRCV-1 and -2) (10, 11). KRCV-1 has not been detected in any other primate species of African or Asian origin besides red colobus monkeys, either in captivity or in the wild, despite screens of captive macaques and wild nonhuman primates from Kibale National Park. However, additional highly divergent simian arteriviruses have recently been discovered in apparently healthy wild red-tailed monkeys (Cercopithecus ascanius) (12), yellow baboons (Papio cynocephalus) (13), hybrid kinda × grayfooted-chacma baboons (Papio kindae × Papio ursinus griseipes) (14), malbroucks (Chlorocebus cynosuros), African green monkeys (Chlorocebus aethiops) (15), vervet monkeys (Chlorocebus pygerythrus) (15), and De Brazza's monkeys (Cercopithecus neglectus) (10), indicating that these viruses are widespread among African monkeys. We recently analyzed samples collected during three separate SHF outbreaks that occurred in captive macaque colonies in the 1960s and late 1980s. Sequencing analysis revealed that only one of these outbreaks was in fact caused by SHFV, whereas the other two outbreaks were caused by phylogenetically distinct, and previously undiscovered, viruses now referred to as simian hemorrhagic encephalitis virus (SHEV) and Pebjah virus (PBJV) (16). These findings indicate that diverse members of this expanding viral group can cause severe disease in macaques and that some simian arteriviruses do naturally reside in primate species of African origin.
Understanding the evolution of simian arteriviruses in their natural host species may inform the mechanisms by which they are able to cause asymptomatic infection. An analysis of KRCV-1 isolates from 60 wild red colobus monkeys from Kibale National Park, Uganda, uncovered signatures of diversifying selection in two regions of the open reading frame (ORF) 5-encoded major arterivirus surface glycoprotein GP5, possibly due to immune pressure or “mutational hot spots” (11, 13). A recent comparative analysis of those same KRCV-1 isolates suggested that regions under diversifying selection are present in ORFs TF, 3′, 4′, 3, 4, and 5 (unpublished data). Whether these or other regions also experience positive selection during cross-species transmission of simian arteriviruses remains unknown. Notably, the ways in which natural selection impacts within-host evolution of simian arterivirus infections have not been characterized for any virus that has caused SHF.
We recently infected two groups of four crab-eating macaques (Macaca fascicularis) with KRCV-1 or SHFV and examined clinical signs, viral replication kinetics, and pathology. Crab-eating macaques are unlikely to naturally harbor KRCV-1 because they are of Asian origin and would not encounter African primate species in the wild. Furthermore, simian arteriviruses have been detected only in African primates, and KRCV-1 has never been detected in either captive or wild macaques. Therefore, replication in crab-eating macaques likely poses a novel host environment for KRCV-1 and SHFV. In these animals, SHFV caused the classically described SHF, whereas KRCV-1 caused a relatively mild and self-limiting disease (17). Here we characterize the evolution of KRCV-1 and SHFV during replication in cell culture and during infection in crab-eating macaques. Our data reveal evolutionary processes by which these viruses adapt to replication in crab-eating macaques, a species not known to naturally harbor simian arteriviruses. Infection of macaques with simian arteriviruses may also provide a model for studying cross-species transmission that can be applied to understanding human zoonotic viral emergence.
RESULTS
KRCV-1 infection. (i) KRCV-1 replicates in cell culture.
Prior to our study of KRCV-1 infection in crab-eating macaques (17), SHFV was the only simian arterivirus that had been isolated in cell culture. Culturing SHFV has been successful only on primary nonhuman primate cell lines and the immortalized grivet kidney cell line MA-104 and its subclones (e.g., MARC-145) (18). Other, nonsimian, arteriviruses have been successfully isolated using primary macrophages (19). Therefore, we grew KRCV-1 from a wild Ugandan red colobus monkey (KRCV-1-RC01 isolate) using rhesus monkey (Macaca mulatta) leukocytes collected by bronchoalveolar lavage (BAL) and then expanded this isolate on a variety of primary and immortalized cells (Fig. 1). A schematic of the full passage history is shown in Fig. 1 and is detailed in Materials and Methods of reference 17. After passage in rhesus monkey BAL fluid leukocytes, MARC-145 cells, and rhesus monkey peripheral blood mononuclear cells (PBMCs), we obtained an expanded, high-titer stock (7.37 × 107 genome copies/ml) that caused visible cytopathic effects in cell culture (Fig. 2a).
FIG 1.

Passage history of KRCV-1 in cell culture. KRCV-1 was isolated from the blood of a wild Ugandan red colobus monkey (RC01), and this isolate was subsequently referred to as KRCV-1-RC01. KRCV-1-RC01 went through a series of passages on primary rhesus monkey broncheoalveolar lavage (BAL) fluid leukocytes (pink-shaded boxes), grivet kidney cells (MARC-145 cells; blue-shaded boxes), and primary rhesus monkey peripheral blood mononuclear cells (PBMCs; yellow-shaded boxes). Each passage is designated by a passage number (e.g., P1, P2) and a day of passage (e.g., D1, D2). A microcentrifuge tube icon indicates attempts to sequence an isolate; a MiSeq instrument icon indicates that sequencing yielded data. Viral RNA content analysis was performed on each day of the P6 passage (data not shown). CPE, cytopathic effect.
FIG 2.

KRCV-1 cell culture passage results in the fixation of SNPs in ORFs associated with host cell entry. (a) Images taken during KRCV-1 passage in rhesus monkey leukocytes from bronchoalveolar lavage (BAL) fluid on passage 1, day 3 (P1-D3), and in MARC-145 cells from passage 2, day 18 (P2-D18). The top panel shows two photographs of cytopathic effect (CPE) in BAL fluid leukocytes. The bottom panel shows mock-infected MARC-145 cells on the left and CPE in KRCV-1-infected MARC-145 cells as observed on day 18 in passage 2 (P2-D18) on the right. (b) The numbers of synonymous (blue) and nonsynonymous (red) SNPs detected in each ORF (with reference to KRCV-1-RC01) are shown as a histogram, with the ORF shown on the x axis and the number of SNPs detected in that ORF shown on the y axis. (c) The number of SNPs divided by the number of synonymous or nonsynonymous sites present in each ORF is shown.
During cell culture passage, KRCV-1 acquired single-nucleotide polymorphisms (SNPs) in ORFs likely involved in host cell entry and exit. To assess whether cell culture propagation resulted in substitutions in the KRCV-1 genome, we isolated viral RNA from the final cell culture KRCV-1 stock (KRCV-1 from rhesus monkey peripheral blood mononuclear cells [rh-KRCV-1]) and sequenced it on an Illumina MiSeq instrument. For a direct comparison with the diversity present in the original KRCV-1-RC01 virus isolate, we used the same unbiased, random-hexamer priming approach as previously described (9). We attempted to isolate and sequence viral RNA in cell culture supernatants from an additional four time points, but we were unable to obtain even genome coverage, likely due to low sample titers and volumes (Fig. 1). We deposited the sequencing reads from these cell culture samples in the Short Read Archive database with the rest of the study samples (see Materials and Methods), but we excluded them from further analyses.
We mapped the reads for rh-KRCV-1 to the KRCV-1-RC01 consensus sequence (i.e., the viral genome obtained and sequenced directly from the infected host red colobus monkey, RC01) and enumerated SNPs present at a frequency of ≥1% of sequencing reads. We also estimated nucleotide diversity by computing the π statistic (20), which is the average number of pairwise differences in a set of sequences. π provides a different metric than counting SNPs. In assessing π, SNPs are weighted such that a population with more high-frequency polymorphisms will be estimated to be more diverse than a population with the same number of low-frequency variants. Therefore, π is less sensitive to errors in low-frequency SNP calling.
After cell culture passage, nucleotide diversity across all rh-KRCV-1 ORFs was reduced by an average of 94% (range, 88 to 98%) in comparison to that observed in KRCV-1-RC01. This observation is consistent with decreases in genetic diversity documented during the passage of other viruses, such as HIV-1 (21) and influenza A virus (22). By the end of cell culture passage, rh-KRCV-1 harbored 68 SNPs fixed across the genome (Fig. 2b), with a high proportion of nonsynonymous (amino acid-changing) SNPs per site accumulating in ORFs 2a, 3, 4, 5, 6, and 7 (Fig. 2c). In other, nonsimian, arteriviruses, these ORFs are thought to encode envelope proteins that are likely involved in host cell receptor binding, host cell entry, and virion budding (19, 23–25).
(ii) Nonsynonymous diversity is highest in KRCV-1 ORFs 2a′, 2a, and 7 during replication in crab-eating macaques.
We previously infected four crab-eating macaques intramuscularly with 109 genome copies of rh-KRCV-1 to determine the disease progression (17). As the minimum infectious dose of KRCV-1 in macaques is not known, we chose to inoculate with this high dose to maximize the likelihood of infection. We collected blood from each animal on days 0, 3, 5, 8, 10, 14, 21, and 28 postinoculation and quantified viral loads by quantitative reverse transcription-PCR (qRT-PCR). KRCV-1 replicated to high titers in all four animals, with an average viral load of 1.26 × 107 copies/ml plasma (Fig. 3a; replotted from data reported previously [17]). KRCV-1 infection caused clinical signs (e.g., mild depression of activity and dehydration, moderate reduction in appetite, recumbent posture) in all four animals by day 3 postinoculation (17). None developed signs of classic viral hemorrhagic fever, including cyanosis, fever, diarrhea, dyspnea, facial edema, lymphadenopathy, weight loss, or splenomegaly, and viral RNA was undetectable in plasma by day 21 or 28 postinoculation (Fig. 3a).
FIG 3.

KRCV-1 selection in infected macaques is weak. (a) Log10 viral load in viral RNA copies/ml plasma from each crab-eating macaque infected with KRCV-1. Viral loads were quantified using qRT-PCR. The numbers 1 to 4 indicate the crab-eating macaque animal number. Data taken from reference 17 were replotted here. (b) π was calculated using either the original assemblies or assemblies subsampled to produce even coverage of 1,000× across the entire genome. For each ORF, animal, and time point, the corresponding π value calculated from the original assemblies is plotted on the x axis versus the π value for the same sample calculated from the subsampled assembly on the y axis. The results obtained were very similar, suggesting that uneven genome coverage likely did not impact diversity estimates. (c) KRCV-1 genome nucleotide diversity (π) was estimated for each ORF, day of infection, and infected crab-eating macaque (numbered 1 to 4). (d) The log-transformed π values for every ORF are plotted against the log-transformed viral load values. Regression analysis was performed in R.
For this study, we characterized KRCV-1 genetic diversity using deep sequencing. To assess whether selection shaped the viral population in crab-eating macaques, we estimated nucleotide diversity (π) across all 14 ORFs in the KRCV-1 genome on each day of macaque infection. To ensure that uneven coverage depth did not impact our diversity estimates, we estimated π using our original reference-based assemblies and assemblies that were subsampled to produce even coverage across the genomes (see Materials and Methods for details).
We found that diversity estimates obtained using both methods produced highly concordant results (Fig. 3b), and the data from our nonsubsampled assemblies are presented below (Fig. 3c and d). By day 3 postinoculation, nucleotide diversity increased across all ORFs in comparison to that of the rh-KRCV-1 stock (Fig. 3c). However, nucleotide diversity fluctuated only slightly throughout the remainder of infection, and no single ORF exhibited consistently higher diversity in all four animals (Table 1; see Tables S1 to S4 in the supplemental material) (in crab-eating macaque 4, no single ORF exhibited significantly higher diversity, so these values are not reported). Diversity was slightly, but significantly, correlated with viral load (R2 = 0.078, P = 1.267 × 10−6) (Fig. 3d).
TABLE 1.
Comparison of nucleotide diversities among ORFs in SHFV- or KRCV-1-infected crab-eating macaquesa
| Comparison group | F value | P valueb | Significant ORFs |
|---|---|---|---|
| All SHFV-infected macaques | 46.20 (45.74) | <0.0001 (<0.0001) | 5a (5a) |
| All KRCV-1-infected macaques | 10.18 (10.65) | <0.0001 (<0.0001) | 2a′ and 4′ (2a′ and 4′) |
| KRCV-1-infected macaque 1 | 12.82 (11.35) | <0.0001 (<0.0001) | 2a′ and 4′ (2a′ and 4′) |
| KRCV-1-infected macaque 2 | 3.609 (2.873) | 0.0004 (0.0038) | 4′ (4′) |
| KRCV-1-infected macaque 3 | 13.42 (12.12) | <0.0001 (<0.0001) | 2a′, 3′ and 4′ (2a′, 3′ and 4′) |
| KRCV-1-infected macaque 4 | 2.012 (2.068) | 0.0454 (0.0394) | None (none) |
Data for all time points were combined for the individual crab-eating macaque analyses. For the “All KRCV-1” and “All SHFV” analyses, data for all time points for all macaques infected with either KRCV-1 or SHFV were combined for each respective analysis. The far-right column summarizes the results of pairwise comparisons using Tukey's multiple-comparison test as detailed in Tables S1 to S4 and S6. ORFs for which comparisons against all or most other ORFs were significantly different by Tukey's multiple-comparison test are listed. Values in parentheses indicate those derived from performing the same analysis on assemblies that were subsampled to 1,000× coverage.
Significance values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. ns, not significant.
Analyses of nucleotide diversity can also take into account the relative rates of nonsynonymous and synonymous polymorphisms within an ORF, yielding nonsynonymous (πN) and synonymous (πS) diversity, respectively. Comparing the relative values of πN and πS within a gene can be used to infer the type and degree of selection pressure. πN > πS typically indicates that diversifying (positive) selection is favoring new substitutions; πN < πS suggests that purifying selection is acting to remove new variation. πN ≈ πS indicates that selection is not playing a major role in determining changes in allele frequencies at that locus over time or that positive and purifying selection are equally prevalent within the analyzed region.
We calculated πN and πS for each KRCV-1 ORF in all four infected crab-eating macaques at every time point of infection. Because mutation is inherently random, at a single time point, any given gene could stochastically exhibit unusually high or low diversity due to new mutations, recent loss of deleterious variants, or inefficient purging of deleterious variants. To reduce spurious detection of elevated diversity and recover any major trends conserved across all animals in this study, we calculated πN and πS values for each animal and time point separately and then averaged these values across all animals and time points. These combined, average πN and πS values are shown in Table 2 and represent overall gene-wise diversity estimates. Averaging across the entire gene provides a relatively stringent metric for detecting diversifying positive selection, because only a small subset of sites are expected to be under positive selection, in the background of purifying selection across the majority of a gene. Furthermore, combining data for all infected monkeys for this analysis specifically tests for selection that is common to crab-eating macaques as a species, rather than selection imposed by factors (e.g., innate immune alleles) unique to each individual monkey.
TABLE 2.
Comparison of nonsynonymous to synonymous nucleotide diversities by open reading frame from KRCV-1-infected crab-eating macaquesa
| ORF | πN/πS | Mean πN ± SD | Mean πS ± SD | Significanceb |
|---|---|---|---|---|
| 1a | 0.59 | 0.00114 ± 0.000272 | 0.00194 ± 0.000688 | **** |
| 1b | 0.56 | 0.00116 ± 0.000341 | 0.00207 ± 0.000646 | **** |
| 2a′ | 1.47 | 0.00263 ± 0.00141 | 0.00178 ± 0.00119 | * |
| 3′ | 0.58 | 0.00145 ± 0.000653 | 0.00249 ± 0.00188 | * |
| 4′ | 0.85 | 0.00223 ± 0.00108 | 0.00262 ± 0.00170 | ns |
| 2a | 1.86 | 0.00258 ± 0.00243 | 0.00139 ± 0.000778 | ns |
| 2b | 0.52 | 0.00102 ± 0.000444 | 0.00199 ± 0.00139 | ** |
| 3 | 0.65 | 0.00100 ± 0.000347 | 0.00153 ± 0.000668 | **** |
| 4 | 0.61 | 0.00123 ± 0.000414 | 0.00203 ± 0.00139 | * |
| 5a | 0.56 | 0.00126 ± 0.000176 | 0.00224 ± 0.00136 | ** |
| 5 | 0.70 | 0.00122 ± 0.000299 | 0.00176 ± 0.000614 | *** |
| 6 | 0.53 | 0.00103 ± 0.000283 | 0.00196 ± 0.000538 | **** |
| 7 | 1.03 | 0.00174 ± 0.000724 | 0.00169 ± 0.000492 | ns |
KRCV-1, Kibale red colobus virus 1; π, nucleotide diversity; ORF, open reading frame. Data for all time points and animals are combined for this analysis. ORFs with an πN/πS ratio of >1 are shown in bold.
Significance values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. ns, not significant.
We found that the majority of the KRCV-1 genome was under purifying selection when data for all animals and time points were combined (πN/πS ratios = 0.39 to 0.80, P < 0.05, paired t test) (Table 2). In ORF 4′, πN was lower than πS but not significantly so (πN/πS = 0.85, P = 0.437). In ORFs 2a′, 2a, and 7, we observed πN/πS ratios of >1 that were either not statistically significant or only marginally so (πN/πS = 1.47 for ORF 2a′, P = 0.042; 1.86 for ORF 2a, P = 0.086; and 1.03 for ORF 7, P = 0.653) (Table 2). ORF 2a′ is absent in all nonsimian arterivirus genomes, and the function of the protein it encodes is unknown. ORF 2a encodes the E protein, which likely functions as an ion channel incorporated into the virion that may be important for host cell entry (26). Interactions among the “minor” (GP2, -3, and -4) arterivirus surface glycoproteins and E protein were reported as being important for arterivirion assembly (23, 24). Interestingly, ORF 2a was also affected by high rates of nonsynonymous substitutions in rh-KRCV-1 (Fig. 2c), suggesting that changes in this ORF may be selected during virus adaptation to new host environments. Importantly, πN and πS results obtained using assembly files that were subsampled to produce even genome coverage matched those obtained using our original data (Table S5).
Contrary to what was observed in wild red colobus monkeys (11), we did not detect signatures of diversifying selection acting in ORF 5. Moreover, results of a recent study of KRCV-1 infection in red colobus monkeys did not detect any peaks of nonsynonymous diversity in any of the ORFs we analyzed here in which πN was greater than πS. These results further suggest that these two hosts (red colobus monkeys and crab-eating macaques) may present different adaptive challenges for the virus (C. W. Nelson, A. L. Bailey, J. M. Dinis, L. H. Moncla, S. D. Sibley, M. Lauck, T. L. Goldberg, D. H. O'Connor, and A. L. Hughes, submitted for publication), as would be expected for a natural versus nonnatural host.
To ensure that signals of diversifying selection were not drowned out by calculating averages across the entire ORFs, we repeated our analyses using a sliding window approach (Fig. 4). We calculated πN and πS in windows of nine codons with a step size of one codon, similar to previous analysis of ORFs 3 and 5 of KRCV-1 in wild red colobus monkeys (11). These analyses confirmed that the vast majority of KRCV-1 genome nucleotide sites in crab-eating macaques are under purifying selection, with the exception of a few small regions scattered across the genome (Fig. 4a). A number of peaks in both synonymous and nonsynonymous diversity were apparent in the regions encompassing ORFs 2a′, 2b′, 3′, 4′, and 2a (Fig. 4b), suggesting that these regions may be relatively unconstrained, experience an elevated mutation rate, or are subject to diversifying selection.
FIG 4.

Sliding window analysis reveals peaks in KRCV-1 diversity To quantify KRCV-1 diversity on a finer scale, we calculated πN and πS measurements in sliding windows of nine codons, with a step size of one codon. Because several ORFs overlap, we performed these analyses separately for each ORF and then plotted results from every ORF as shown here. Regions with overlapping ORFs will therefore have multiple πN and πS values plotted over top each other. (a) Data for all crab-eating macaques and time points were pooled for this analysis. The πN and πS values represent the mean values at that codon across all macaques and time points. πN is shown as a red line, and πS is shown as a blue line. Below the plot is a cartoon depiction of the organization of the KRCV-1 genome. Replicase genes are shown in gray, ORFs absent in the nonsimian arteriviruses are shown in blue, and ORFs encoding structural proteins are shown in orange. (b) An enlarged rendering of the graph from panel a spanning KRCV-1 genome nucleotide 10,000 to nucleotide 13,000, between which numerous peaks in both synonymous and nonsynonymous diversity are apparent.
(iii) No SNPs are consistently selected during KRCV-1 replication in macaques.
We next examined whether any particular SNPs increased in frequency over time. We enumerated SNPs present in ≥1% of sequencing reads in all four infected macaques throughout infection and plotted the frequencies of all SNPs as a heat map for each animal (Fig. S1). Because every SNP that became fixed in the rh-KRCV-1 stock virus during cell culture passage remained fixed for the duration of replication in vivo, we mapped all reads to the rh-KRCV-1 reference sequence so that only newly arising mutations are shown.
These analyses revealed that a number of SNPs increased in frequency during replication in each macaque (shown by an increase in dark red rectangles on the heat map in Fig. S1). To assess whether any conserved SNPs rose to a high frequency in multiple animals, we compared all SNPs present at the last time point when virus was detectable (either day 14 or 21 postinoculation) for all four infected macaques (Fig. 5). Although a number of SNPs became fixed in each macaque, no single constellation of SNPs increased to a high frequency or was shared at a high frequency across all macaques (Fig. 5). In fact, no single mutation was shared at a frequency of >50% by all four macaques at the last time point of infection (Fig. 5). Overall, these data do not suggest that any single mutation or combination of mutations was selected during replication in vivo. Despite prolonged infection with high viral loads, we did not find evidence that KRCV-1 is under strong selection to adapt in crab-eating macaques.
FIG 5.
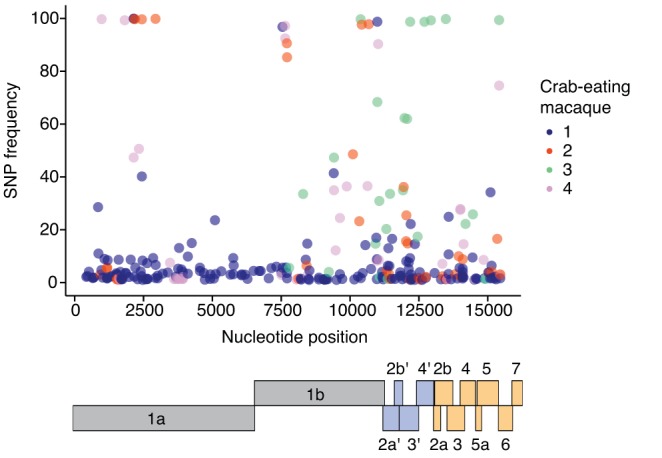
No set of SNPs is consistently selected in KRCV-1-infected macaques. For each of the four crab-eating macaques, every SNP detected at the last time point at which virus was detectable is plotted. Each dot represents one SNP; different colors represent the different macaques. No SNPs were found at a high frequency at the last time point of infection in all four macaques. Below the plot is a cartoon depiction of the organization of the KRCV-1 genome. Replicase genes are shown in gray, ORFs absent in the nonsimian arteriviruses are shown in blue, and ORFs encoding structural proteins are shown in orange.
SHFV infection. (i) ORF 5a exhibited elevated nucleotide diversity in SHFV replicating in crab-eating macaques.
Unlike KRCV-1, SHFV is known to cause severe viral hemorrhagic fever in macaques (4). In our previous study, the four crab-eating macaques initially inoculated with KRCV-1 were infected with SHFV after KRCV-1 clearance to assess whether infection with KRCV-1 would elicit immune protection from SHFV (17). These macaques were not protected from SHFV infection. To compare within-host evolution of a hemorrhagic fever-causing virus to that of KRCV-1, an additional four macaques were infected with SHFV alone (without prior infection with KRCV-1). Plasma viral loads remained high for the duration of infection (mean = 8.33 × 109 copies/ml) (Fig. 6a; data replotted from reference 17). All four macaques reached endpoint criteria (specified in reference 17) between days 4 and 8 postinoculation and were euthanized.
FIG 6.

SHFV infection of crab-eating macaques leads to high diversity in ORFs 5a and 5. (a) The plasma viral RNA load is shown for each crab-eating macaque infected with SHFV. Viral loads were quantified using qRT-PCR, with the dotted line indicating the lower limit of quantification for our assay. Animals infected with KRCV-1 prior to SHFV infection are represented by the same colors and shapes (filled circles) as depicted in Fig. 2 but are shown with dashed lines. Animals infected with SHFV only are represented by solid lines with triangles. Data taken from reference 17 were replotted here. (b) π was calculated using either the original assemblies or assemblies subsampled to produce even coverage of 1,000× across the entire genome. For each ORF, animal, and time point, the corresponding π value calculated from the original assemblies is plotted on the x axis versus the π value for the same sample calculated from the subsampled assembly on the y axis. The results obtained were very similar, suggesting that uneven genome coverage likely did not impact diversity estimates. (c) Nucleotide diversity (π) was estimated for each macaque, time point, and ORF and for both SHFV stocks. (d) πN and πS were estimated using a sliding window for ORFs 5 and 5a, and the results for the region of ORF 5-ORF 5a overlap are shown in panel d. Solid lines depict πN values, while dotted lines represent πS values; ORF 5 results are shown in gray, while ORF 5a results are shown in blue.
For this study, we investigated SHFV evolution during infection in crab-eating macaques and the impact of preexisting immune responses to KRCV-1 on SHFV within-host evolution. The SHFV stock virus used in the macaque study (17) was passaged twice on immortalized grivet kidney cells (MA-104 cells) before inoculation. We sequenced the two stock virus passages (designated P1 and P2) and analyzed them together with SHFV sequences from infected animals. Diversity estimates of the original and subsampled assemblies were equivalent (adjusted R2 = 1) (Fig. 6b), so we chose to plot the results of the original assemblies (Fig. 6c).
π calculations revealed that after a slight increase after inoculation, diversity levels of most ORFs remained stable throughout infection (Fig. 6c). The exception was SHFV ORF 5a, which exhibited higher diversity in all eight macaques studied (Fig. 6c, light blue traces). Comparison of π calculations for each ORF by one-way analysis of variance (ANOVA) revealed that diversity levels were significantly different from each other (P < 0.0001) (Table 1). We then made pairwise comparisons between every combination of ORFs using Tukey's multiple-comparison test, pooling the data for all animals and time points for each ORF. Diversity was higher in ORF 5a than in any other ORF (P < 0.0001) (Table S6).
To determine whether this elevated diversity resulted in amino acid-changing (nonsynonymous) or silent (synonymous) polymorphisms, we calculated measures of πN and πS for each ORF as we did for KRCV-1-infected animals (Table 3). Almost all of the SHFV genome was under purifying selection (πN/πS = 0.40 to 0.63, P < 0.001). In ORF 7, the πN/πS ratio was 0.72, but this ratio was not significantly different from 1, suggesting that selection on ORF 7 might have been weak (P = 0.088) (Table 2). Again, repeating these analyses using subsampled assemblies produced highly similar results (Table S5).
TABLE 3.
Comparison of nonsynonymous to synonymous nucleotide diversities by open reading frame from SHFV-infected crab-eating macaquesa
| ORF | πN/πSa | Mean πN ± SD | Mean πS ± SD | Significanceb |
|---|---|---|---|---|
| 1a | 0.59 | 0.00143 ± 0.000138 | 0.00242 ± 0.000326 | **** |
| 1b | 0.42 | 0.00115 ± 0.000118 | 0.00276 ± 0.000562 | **** |
| 2a′ | 0.58 | 0.00153 ± 0.000591 | 0.00263 ± 0.00114 | *** |
| 2b′ | 0.63 | 0.00142 ± 0.000243 | 0.00223 ± 0.000638 | **** |
| 3′ | 0.53 | 0.00116 ± 0.000556 | 0.00221 ± 0.00101 | **** |
| 4′ | 0.54 | 0.00140 ± 0.000463 | 0.00259 ± 0.00127 | *** |
| 2a | 0.40 | 0.00108 ± 0.000163 | 0.00271 ± 0.00116 | **** |
| 2b | 0.54 | 0.00124 ± 0.000349 | 0.00229 ± 0.000938 | **** |
| 3 | 0.67 | 0.00128 ± 0.000255 | 0.00190 ± 0.000419 | **** |
| 4 | 0.66 | 0.00122 ± 0.000286 | 0.00185 ± 0.000447 | **** |
| 5a | 1.05 | 0.00497 ± 0.00224 | 0.00474 ± 0.00376 | ns |
| 5 | 0.46 | 0.00136 ± 0.000308 | 0.00298 ± 0.00126 | **** |
| 6 | 0.40 | 0.00127 ± 0.000197 | 0.00317 ± 0.00115 | **** |
| 7 | 0.72 | 0.00211 ± 0.00137 | 0.00293 ± 0.001763 | ns |
ORFs with an πN/πS ratio of >1 are shown in bold.
Significance values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. ns, not significant.
In contrast, ORF 5a had a slightly elevated πN/πS ratio across the entire ORF (πN/πS = 1.05, P = 0.09) (Table 3), although again, this trend was not statistically significant. ORF 5a is 65 codons in length and resides entirely within ORF 5, so the elevated diversity we observed may impact both ORFs. Because ORF 5 and ORF 5a are not in the same reading frame, we next wanted to determine whether the elevated nonsynonymous diversity we observed in ORF 5a also resulted in nonsynonymous changes in ORF 5. We calculated πN and πS measurements using a sliding window as we did for KRCV-1 and plotted the region of overlap between ORFs 5 and 5a (Fig. 6d). These analyses revealed two distinct peaks in nonsynonymous diversity in ORF 5a, corresponding to codons 2 to 16 and 33 to 41 (Fig. 6d, blue traces). In ORF 5, nonsynonymous peaks were apparent in codons 6 to 15 and 37 to 63, significantly overlapping the peaks observed in ORF 5a (Fig. 6d, gray traces). Interestingly, these analyses also revealed a large peak in synonymous diversity in ORF 5 in codons 11 to 19, overlapping the region of ORF 5a with the most pronounced peak in nonsynonymous diversity. Conversely, a large peak in ORF 5a synonymous diversity was apparent in codons 44 to 58, aligning with a peak in nonsynonymous diversity in ORF 5 codons 48 to 63. Overall, these data suggest that nonsynonymous changes in ORF 5a frequently cause nonsynonymous changes in ORF 5 and that selection may be shaping diversity differently within these two ORFs.
(ii) Three SNPs increase in frequency during SHFV replication in crab-eating macaques.
Heat map analysis of SHFV from infected macaques revealed that three SNPs increased in frequency in multiple macaques over the course of infection (Fig. S2, indicated by arrows and boxes). The first SNP, C14032T, is situated in both ORF 5 and ORF 5a, encoding Pro11Ser and Ser8Phe, respectively. This SNP was detectable in 20% of viruses in the P2 stock and rapidly increased in frequency in all SHFV-infected macaques (Fig. 7a). C14032T remained at a high frequency in all but one macaque (animal 6) and was present at 54 to 96% in the remaining seven macaques at the end of infection (Fig. 7a). For unknown reasons, C14032T declined in frequency in animal 6 on day 5 postinoculation and rebounded to 45% frequency by day 8 postinoculation. This drop in frequency correlates with a drop in viremia in animal 6 at the same time points. Interestingly, standard amino acid distance measures based on volume and polarity (27) suggest that the Pro11Ser change encoded in ORF 5 is relatively conservative (distance = 0.56), while the Ser8Phe change encoded in ORF 5a is relatively radical (distance = 3.45).
FIG 7.

Three SNPs increase in frequency during SHFV replication. (a, b, and c) Three SNPs that increased in frequency in multiple macaques during replication were identified. (d) A single site was simulated for 5 × 106 virions, for which 76% harbored an A allele and 24% harbored a G allele. Samples of 1,000 alleles were then taken without replacement from this “parental population” to represent the dilution of the stock SHFV to 1,000 PFU. The frequency of the A allele was calculated in each of the 1,000 allele samples and is displayed as a histogram. The y axis represents the number of simulated replicates, and the x axis is the change in frequency of the A allele between the parental and sample populations. A total of 100,000 simulations were performed. The maximum change in frequency observed among all 100,000 replicates was 6%.
The second SNP we identified, G1828A, encodes a glycine-to-glutamate change at amino acid position 540 in ORF 1a. While G1828A was not detectable in the SHFV stock virus P1 or P2, this SNP was detectable in all macaques at day 3 postinoculation and continued to increase in frequency in all animals except for animals 6 and 8 (Fig. 7b). G1828A remained at a very high frequency in 5 animals (71 to 92% frequency). In contrast, this SNP was maintained at a frequency of ∼17% in animal 6 and declined in animal 8 at the end of infection; animal 5 did not survive past day 4. Importantly, viral loads remained high throughout infection in all eight macaques infected with SHFV (Fig. 6a), suggesting that the fixation of both G1828A and C14032T SNPs is unlikely to be due to random bottlenecks caused by a shrinking population size at the end of infection.
Intriguingly, the final SNP we identified exhibited different trajectories in the animals infected with KRCV-1 prior to SHFV infection (KRCV-1 + SHFV group) and those that were not exposed to KRCV-1 (SHFV-only group). In ORF 2a′, G11594A encodes an alanine-to-threonine change at position 37. This SNP was present in 25% of viruses in the P1 stock and increased to 76% frequency in the P2 stock (Fig. 7c). Although present at a high frequency in the P2 inoculum, G11594A rapidly decreased in frequency in the KRCV-1 + SHFV macaques and was detected in only 5 to 38% of viruses at the time of euthanasia (Fig. 7c, dashed lines). Conversely, G11594A remained at a high frequency in SHFV-only macaques and was present in 70 to 99% of viruses at the time of euthanasia (Fig. 7c, solid lines).
To determine whether these different trajectories were a result of random changes in the frequency of G11594A during SHFV stock virus preparation, we wrote a custom script to simulate the effect of randomly sampling the original SHFV stock (titer = 5 × 106 PFU/ml) and dilutions down to the 1,000 PFU used for the inoculum. We performed 100,000 simulations of a population of 5 × 106 virions harboring G11954A at 76% frequency, from which 1,000 virions were randomly sampled. The maximum change in frequency observed for G11594A due to random sampling alone was 6% (Fig. 7d), suggesting that the differing trajectories we observed between the KRCV-1 + SHFV and SHFV-only groups are not due to random sampling during SHFV stock dilution (P < 1 × 10−5) (Fig. 7d). This analysis does not take into account selection for particular viral variants at the start of infection (a selective bottleneck), which could have skewed the frequency of G11594A in vivo.
DISCUSSION
At least three simian arteriviruses (PBJV, SHEV, and SHFV) caused severe historical outbreaks of viral hemorrhagic fever (simian hemorrhagic fever [SHF]) in macaques of different species, and many more arteriviruses have recently been discovered in wild African nonhuman primates (2, 3, 8, 9, 11–14, 16, 24, 28). Despite the prevalence and veterinary significance of simian arteriviruses, their within-host evolution has not been widely studied. In our study, we found that KRCV-1 fixed 68 substitutions during cell culture propagation but exhibited surprisingly limited signatures of selection during replication in vivo. In contrast, SHFV evolution was characterized by selection acting at a few specific sites in the genome and signatures of diversifying (positive) selection in ORF 5a.
During cell culture passage of KRCV-1, 68 SNPs became fixed across the genome. A number of these substitutions may have been fixed due to random bottlenecks introduced by passaging (22, 29) or due to cell culture-specific selective pressure. None of the SNPs that became fixed in the rh-KRCV-1 isolate reverted during replication in vivo, suggesting that whether they were fixed due to selection, bottlenecking, or hitchhiking, these SNPs were not strongly detrimental to KRCV-1 replicative fitness in vivo. We did not find any sites in KRCV-1 that were consistently under positive selection in all infected macaques and instead found that a unique constellation of substitutions became fixed in each macaque. The fixation of these individually distinct sets of SNPs may be driven by animal-specific immune responses or differential expression of intrinsic antiviral factors. As the macaques used in this study were outbred, each one might have harbored unique major histocompatibility complex types and intrinsic antiviral factor alleles. Alternatively, a bottleneck during infection, as viral population size decreased, might have promoted random fixation of different SNPs in each macaque (30). The correlation between diversity and viral population size (Fig. 3c), the drop in viral load at the last time point of infection (Fig. 3a), and the loss of a large number of SNPs at the last time point of infection (Fig. S1b, c, and d) are consistent with this explanation (30). It is also possible that some necessary adaptation occurred during cell culture passage, resulting in the limited signatures of positive selection observed in vivo.
Previous analyses of KRCV-1 sequences from wild Ugandan red colobus monkeys identified high levels of nonsynonymous nucleotide diversity in ORF 5 (11). Here we found that ORF 5 in KRCV-1 was consistently under purifying selection. KRCV-1 RNA was present in our infected macaques for 14 to 21 days at high concentrations, suggesting that the lack of diversifying selection we detected here is not likely to be due simply to a smaller population size or inefficient within-host selection in our study. ORF 5 is thought to encode the major surface glycoprotein, GP5, which has been documented as a target for neutralizing antibodies and is likely involved in host cell entry (19). An anti-GP5 adaptive immune response would explain the signatures of diversifying selection detected in this ORF previously (11), as would diversification for broad cell tropism within naturally infected hosts. We also found that eight nonsynonymous SNPs accumulated in ORF 5 of KRCV-1 after cell culture passage, all of which were maintained during in vivo replication, perhaps indicating adaptation of KRCV-1 to the macaque receptor.
In contrast to what we found for ORF 5, we detected elevated nonsynonymous diversity in ORFs 2a′, 2a, and 7 during both in vitro and in vivo KRCV-1 replication. ORF 2a′ is unique to the simian arteriviruses, and although it is necessary for infectious virion production (31), its precise function is not known. ORF 2a encodes the E protein, a putative ion channel that interacts with the minor arterivirus surface glycoproteins to facilitate virion assembly and host cell entry (23, 24). Interactions with host-specific proteins during virion assembly and budding or differential pH conditions during host cell entry might select for changes in E protein.
ORF 7 encodes the nucleoprotein that encapsidates viral RNA. Host species-specific selection on other viral nucleoproteins has been reported, such as that on influenza A virus nucleoprotein, which is restricted by the myxovirus resistance protein A (MxA) differentially in birds, humans, and nonhuman primates (32, 33). Polymorphisms in host-specific restriction factors may be one possible mechanism for the diversity we observed in ORF 7 in our study. Taken together, our data suggest that the selective forces shaping KRCV-1 within-host diversity likely differ between colobus monkeys (11) and crab-eating macaques. It is unclear whether this reflects evolution within a natural versus a nonnatural host or inherent differences between these two species.
Within-host evolution of SHFV was markedly different than that of KRCV-1, with this difference most evident in ORFs 5a and 5. ORF 5a encodes a short protein that is incorporated at low levels into the virion envelope, and deletion of ORF 5a in nonsimian arteriviruses reduces the viral titer or abolishes replication completely (25). Although little is known about the function of ORF 5a in simian arteriviruses, our data suggest that the encoded protein may be a target of selection during replication in crab-eating macaques. Intriguingly, we observed a peak in nonsynonymous diversity in codons 37 to 63 in ORF 5, spanning putative GP5 epitope sites (codons 27 to 31 and 37 to 52) described in other arteriviruses (34–38). If the location of these epitope sites is conserved for SHFV, then immune selection might have driven diversification in this region. As C14032T causes a relatively conservative amino acid change in the ORF 5-encoded protein but a more radical one in the ORF 5a-encoded protein, differential selection on the two overlapping ORFs is also possible. Purifying selection in that region of ORF 5 may be relaxed, while positive selection may be acting more strongly on ORF 5a.
In porcine reproductive and respiratory syndrome virus (PRRSV) infection in pigs, PRRSV-specific antibodies can be detected as early as 14 days postexposure, and specific T-cell responses may appear by 2 to 8 weeks postexposure (19). Equine arteritis virus (EAV) infection in horses shows similar patterns, with neutralizing antibodies appearing as early as 7 to 14 days after exposure (19). Given the short time between infections with KRCV-1 and SHFV, cross-reactive immune responses may have been generated in the macaques previously infected with KRCV-1. Interestingly, we identified an SNP, G11594A, which resides in ORFs 2a′ and 3, and exhibited distinct trajectories in SHFV-infected animals that were initially infected with KRCV-1 versus those that were KRCV-1-naive. We speculate that G11594A might have been differentially selected due to immune pressure because of immune priming by KRCV-1 infection, though we cannot determine from our data whether putative immune selection might have been acting on ORF 2a′, ORF 3, or both.
Every simian arterivirus isolated from wild nonhuman primates thus far has been found only in primates of a single species (9, 11–13), despite the fact that host species occupy overlapping habitats and frequently interact (39). Although KRCV-1 may naturally infect crab-eating macaques, we suspect that natural KRCV-1 infection in these primates is unlikely. Natural simian arterivirus infections have been documented only in African primate species, not in Asian ones; Asian primates are geographically separated from red colobus monkeys. KRCV-1 has not been detected in red-tailed guenons, despite extensive interactions between red colobus monkeys and red-tailed guenons, suggesting that KRCV-1 may not readily infect other host species. However, our data show that KRCV-1 is capable of replicating in macaques of Asian origin.
An outstanding question is why such species specificity is observed in nature given the frequent interaction of nonhuman primate species in Kibale National Park, where these viruses were originally detected. One explanation is that simian arteriviruses and their respective hosts may have coevolved long enough to establish competitive exclusion and preclude natural cross-species transmission. The competitive exclusion principle proposes that organisms of two species competing for the same resources cannot coexist at equal population sizes and has been used to explain the establishment of viruses in specific hosts to the exclusion of other viruses (40–42). Our data show that different genes experienced higher nonsynonymous diversity during replication in cell culture and in crab-eating macaques than during surveys of wild red colobus monkeys. These data suggest that each primate species may exert a unique constellation of selective forces on infecting simian arteriviruses, which could favor host-specific coevolution and explain the high divergence observed among contemporaneous simian arteriviruses. Alternatively, it is possible that some of these viruses can and do infect primates of other species but simply have not yet been detected. Future work resolving phylogenetic signals of coevolution combined with enhanced surveillance may begin to answer these questions. Finally, it remains unclear which host or viral factors are responsible for the wide range of phenotypes associated with simian arterivirus infections. Understanding these factors remains a ripe area of research that is important for improving captive and wild nonhuman primate health.
MATERIALS AND METHODS
Virus isolation and cell culture passage.
Isolation of Kibale red colobus virus 1 (KRCV-1) was described previously (17). The complete passage history of the original RC01 isolate, designated KRCV-1-RC01, is shown in Fig. 1.
Simian hemorrhagic fever virus (SHFV) variant NIH LVR42-0/M6941 was obtained in 2011 from the American Type Culture Collection, Manassas, VA (ATCC VR-533). The SHFV animal challenge stock was grown on grivet (Chlorocebus aethiops) fetal kidney MA-104 C-1 cells (ATCC CRL-2378.1) as previously described (43) in Eagle's minimal essential medium (EMEM; Lonza, Walkersville, MD) supplemented with 10% heat-inactivated fetal bovine serum (FBS; SAFC Biosciences, Lenexa, KS) at 37°C in a humidified atmosphere of 5% CO2. The genomic sequence of the prepared SHFV P2 stock was determined experimentally by deep sequencing (28) and was validated against the prototype sequence (GenBank accession no. AF180391.2). SHFV and KRCV-1 animal challenge stocks were judged sterile after negative blood agar streaks, the absence of mycoplasma with Mycosensor (Agilent Technologies, Santa Clara, CA), detection of endotoxins in the normal range using the Limulus test (Endosafe-PTS; Charles River, Wilmington, MA). In addition, inoculation of grivet Vero E6 (ATCC CRL-1586) cells under MA-104 growth conditions (SHFV and KRCV-1 grow only in MA-104 or MA-104-derived cells) with such challenge stocks did not result in cytopathic effects. Titers of SHFV stocks were determined by plaque assay on MA-104 cells as previously described (43, 44).
Animal infections.
Experimental infection of crab-eating macaques (Macaca fascicularis) was described previously (17). Briefly, eight male crab-eating macaques, 7 to 9 years old and weighing 5.95 to 8.7 kg, were injected intramuscularly with 1-ml inocula containing 1,000 PFU of SHFV (equivalent to 106 genome copies) or 109 genome copies of KRCV-1 in sterile phosphate-buffered saline (pH 7.4). After KRCV-1 clearance, these animals were rechallenged with 1,000 PFU of SHFV. The experiments were performed in a biosafety level 4 (BSL-4) containment facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Experimental procedures were approved by the Animal Care and Use Committee (ACUC) of the Division of Clinical Research (DCR), National Institute of Allergy and Infectious Diseases (NIAID), and were in compliance with the Animal Welfare Act regulations, Public Health Service policy, and the Guide for the Care and Use of Laboratory Animals recommendations.
Viral load measurement.
Viral titers were measured using a qRT-PCR assay specific for each virus. The assay to measure KRCV-1 was performed as described previously (11). Briefly, viral RNA was reverse transcribed and quantified using a SuperScript III One-Step qRT-PCR system (Life Technologies, Carlsbad, CA) with virus-specific primers and probe on a LightCycler 480 instrument (Roche, Indianapolis, IN). SHFV was measured with a similar assay using forward primer 5′-AGTCCAGAGGAGGGAATAGGCT-3′, reverse primer 5′-CAGGAGCAGCTCTACGTTGTTG-3′, and probe 5′-6fam-AACGACCTCGCCGCAGCACT-BHQ1-3′. Reverse transcription was performed at 37°C for 15 min and then 50°C for 30 min, followed by 2 min at 95°C and 50 amplification cycles consisting of 95°C for 15 s and 63°C for 1 min. Primers were used at a final concentration of 600 nM; probe was used at a final concentration of 100 nM. The reaction mixture also contained 150 ng of random primers (Promega, Madison, WI) and MgSO4 at a final concentration of 3.0 mM.
Deep-sequencing sample preparation.
Viral RNA was extracted from cell culture supernatant using a MinElute viral RNA minikit (Qiagen, Germantown, MD). Viral RNA from the rh-KRCV-1 cell culture stock was prepared for sequencing using an unbiased sample preparation method with random hexamer primers as previously described (9). Plasma from infected macaques was stored in TRIzol. Viral RNA was extracted from plasma samples using standard TRIzol extraction with chloroform and 10 μg of glycogen as a carrier, followed by purification with an RNeasy MinElute cleanup kit (Qiagen). Clean RNA was reverse transcribed using a SuperScript III first-strand synthesis kit (Invitrogen, Grand Island, NY) for qRT-PCR. Due to the length of simian arterivirus genomes (∼16 kb), reverse transcription was improved by using two primers: oligo(dT) and a virus-specific custom primer that binds approximately halfway through the genome (KRCV-1-RT, CGGAGACGGGAGATAGATTG; SHFV-RT, TGTGCTGGACATAGAGAACCA).
Genomes were amplified using 10 overlapping amplicons (see primers listed in Table S7) with Phusion HotStart 2× Mastermix (New England BioLabs, Ipswich, MA) in accordance with the manufacturer's recommended cycling conditions. Primers were annealed at 56°C for KRCV-1 and 58°C for SHFV, and genes were amplified in 40 (KRCV-1) or 35 (SHFV) PCR cycles. To ensure that we included the maximum number of cDNA input copies into our PCRs, we optimized our viral RNA extraction volume, reverse transcription RNA input volume, and cDNA input volume for each sample. The estimated number of cDNA viral input copies used for each sample for sequencing is listed in Table S8. PCR products were purified from a 1% agarose gel using a QIAquick gel extraction kit (Qiagen) and eluted in nuclease-free water. All reverse transcription and PCR experiments were performed with negative water controls to exclude contamination, and each virus genome (run on an agarose gel as 10 separate amplicons) was run in its own gel tray, which was washed with bleach, ethanol, and fresh Tris-acetate-EDTA (TAE) buffer between each use.
Cleaned PCR product was quantified with a Qubit dsDNA HS assay kit (Invitrogen) and diluted in diethylpyrocarbonate-treated water to a concentration of 0.2 ng/μl. Library preparation was performed with a Nextera XT sample preparation kit (Illumina, San Diego, CA) in accordance with the manufacturer's protocol. The average fragment length was determined with a high-sensitivity DNA kit and a 2100 Bioanalyzer (Agilent, Santa Clara, CA). Sample libraries were pooled in equimolar concentrations to a total final concentration of 2 nM and prepared for Illumina sequencing according to the Nextera XT DNA library prep reference guide (Illumina). To prevent carryover between runs on the MiSeq instrument (Illumina), we changed barcode tags such that no two samples from this study ever had the same tag. Also, we alternated simian arterivirus runs with other users such that simian arteriviruses were never sequenced in two sequential runs. All samples were run on the Illumina MiSeq instrument with a 600-cycle kit as a 12 pM library with 1% PhiX control. Output files were generated in fastq format.
Sequence data analysis.
Fastq files were imported into CLC Genomics Workbench version 7 (CLC Bio, Aarhus, Denmark) for analysis. Reads were trimmed using a quality score threshold of Q30 and a minimum length of 100 bases. All reads were mapped to a reference sequence requiring that at least a 50% of the contiguous read was 80% identical to the reference. To ensure that our assemblies did not contain any extraneous contaminant reads (e.g., due to instrument carryover), all mapped reads were also de novo assembled. Contigs generated by de novo assembly were used as the input to BLAST, such that any contaminant reads could be identified and removed. We did not detect any contaminant reads in our data set. The SHFV that was amplified in cell culture to generate passage 1 (P1) and P2 stocks was sequenced, and fastq files were mapped to the reverse genetics sequence used to generate the stocks. A consensus sequence was derived from the P1 stock virus and used as the reference sequence for all subsequent SHFV mappings. ORF coordinates were obtained from sequence KM655766 from GenBank and manually inspected to ensure that they resulted in the correct reading frame, start and stop sites, and gene length. The average sequence depth acquired for this data set was 5,330 ± 2,694 for SHFV genomes.
KRCV-1 from rhesus monkey peripheral blood mononuclear cells (rh-KRCV-1) was mapped to the KRCV-1-RC01 consensus sequence, which was generated previously using unbiased sequencing (9). Therefore, SNPs reported for this cell culture isolate reflects nucleotide changes with regard to the KRCV-1-RC01 consensus sequence. Deep-sequencing reads from the rh-KRCV-1 isolate were used to generate a consensus sequence for this isolate by using the majority base call at every position. Conflicts were resolved by choosing the base with the greatest number of reads with the highest quality scores (highest calculated Q-score). Regions with no coverage were filled in from the reference sequence. Because the rh-KRCV-1 virus stock was used to inoculate macaques, sequences from infected macaques were mapped to this rh-KRCV-1 consensus sequence. The average sequence depth acquired for this data set was 7,585 ± 4,921 (mean ± standard deviation) for KRCV-1 genomes.
Variant calling on all mappings required a variant to be present at a frequency of ≥1%, covered by a minimum of 100 reads, and have a central base quality score of ≥Q30. This cutoff has previously been determined as a conservative cutoff given the intrinsic error of our pipeline and instrument (45). We noted an increase in the number of low-frequency (<10%) variants called when we removed duplicate reads from our assemblies. Thus, to ensure conservative SNP calls, duplicate reads were not removed for the analyses presented in this article.
Diversity calculation.
The π statistic for measuring nucleotide diversity, which quantifies the average number of pairwise differences between a set of DNA sequences without regard to a reference sequence, was calculated using open-source PoPoolation version 1.2.2 (Institute of Population Genetics, University of Veterinary Medicine, Vienna, Austria) (20). Sequence Alignment/Map (SAM) files of the reference-based mappings were exported from CLC Genomics Workbench and converted to sorted pileup files using SAMtools (46). π was calculated using the Variance-at-position.pl script, and πN and πS estimates were calculated for each ORF using the Syn-nonsyn-at-position.pl script. Sliding window analysis was performed using the Syn-nonsyn-sliding.pl script, with a window size of 27 nucleotides (9 codons) and a step size of 3 nucleotides (1 codon). Because several KRCV-1 and SHFV ORFs overlap and some ORFs are in different reading frames, sliding window analysis was performed for each ORF separately. Sliding window plots show the πN or πS value corresponding to the nucleotide site in the middle codon of the window. To ensure that uneven genome coverage did not impact our estimates of diversity, we calculated π, πN, and πS for each sample using both the original and subsampled assemblies. For the subsampled assemblies, we used the subsample-pileup.pl script implemented in PoPoolation version 1.2.2 (20) to subsample our assemblies to 1,000× coverage. These subsampled assemblies were analyzed in parallel with our original assemblies and are shown in parentheses in Tables S1 to S4 and S6. The subsampled assemblies were used to calculate πN and πS in Table S5. Corrections were disabled for all π analyses.
Statistical analysis.
Paired t tests comparing πN and πS values for all ORFs were performed in R by combining data for all animals and time points and testing the null hypothesis that πN equals πS. One-way analysis of variance (ANOVA) was used to test the null hypothesis that mean diversity estimates in all ORFs were equal. Following a significant one-way ANOVA result, we used Tukey's multiple-comparison test to perform pairwise comparisons between each ORF to correct for multiple comparisons. These analyses were performed in Prism version 7.0a for Mac OS X (GraphPad Software, La Jolla, CA). Regression analyses comparing viral load with nucleotide diversity (π) was performed by first log10-transforming all viral load and diversity estimates, which improved the normality of the data (data not shown). These analyses, as well as regression analyses comparing π from original assemblies with π from subsampled assemblies, were performed in R.
Simulation of G11594A in SHFV P2 stock virus.
The original SHFV P2 stock virus had a titer of 5 × 106 PFU/ml. For animal inoculations, this stock was diluted such that each animal received 1,000 PFU. To simulate the effect of this dilution on the frequency of G11594A, we used a custom Python script to simulate a population of 5 × 106 virions, each with a single site, for which 76% of the population harbored an A allele and 24% of the population harbored a G allele. We then took random samples of 1,000 sites, without replacement, from this parental population and quantified the frequency of the A allele in each sampled population. We performed 100,000 simulated replicates and plotted the overall change in the frequency of the A allele observed in our simulations in Fig. 6f by using R.
Accession number(s).
All consensus sequences and fastq files described in the article are publicly available in GenBank. Consensus sequences were deposited in BankIt as fasta files under GenBank accession numbers KX656700 to KX656748. Fastq files have been archived with BioProject PRJNA336562 and are available in the Short Read Archive (SRA) under project accession number SRP080965.
Supplementary Material
ACKNOWLEDGMENTS
We thank Laura Bollinger at IRF-Frederick for critically editing the manuscript.
L.H.M. performed experiments and analyzed data. A.M.W., G.B., J.T.W., J.M.D., O.C., M.L., V.W.-J., J.C.J., Y.C., and J.H.K. performed experiments to generate data. C.W.N., A.M.W., D.H.O., J.H.K., L.H.M., T.L.G., and T.C.F. analyzed data. V.W.-J., J.C.J., Y.C., A.L.B., M.L., T.L.G., D.H.O., P.B.J., J.H.K., and T.C.F. conceived the experimental design. L.H.M., J.M.D., A.L.B., C.W.N., T.L.G., D.H.O., J.H.K., and T.C.F. wrote the manuscript.
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services or the institutions and companies affiliated with the authors.
L.H.M. was supported in part by NIH National Research Service Award T32 GM07215 and through a Wisconsin Distinguished Graduate Fellowship provided by John and Tashia Morgridge. This work was funded in part through Battelle Memorial Institute's prime contract with the U.S. National Institute of Allergy and Infectious Diseases (NIAID) under contract no. HHSN272200700016I. J.C.J. performed this work as an employee of Battelle Memorial Institute. Subcontractors to Battelle Memorial Institute who performed this work are V.W.-J., Y.C., and J.H.K., employees of Tunnell Government Services, Inc. This work was also funded by National Institutes of Health (NIH) grant TW009237 as part of the joint NIH-NSF Ecology of Infectious Disease program, grant R01 AI077376, and Office of Research Infrastructure Programs (ORIP) grant P51OD011106. C.W.N. was funded by the Gerstner Scholars Fellowship from the Gerstner Family Foundation at the American Museum of Natural History.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02231-16.
REFERENCES
- 1.Allen AM, Palmer AE, Tauraso NM, Shelokov A. 1968. Simian hemorrhagic fever. II. Studies in pathology. Am J Trop Med Hyg 17:413–421. [DOI] [PubMed] [Google Scholar]
- 2.London WT. 1977. Epizootiology, transmission and approach to prevention of fatal simian haemorrhagic fever in rhesus monkeys. Nature 268:344–345. doi: 10.1038/268344a0. [DOI] [PubMed] [Google Scholar]
- 3.Palmer AE, Allen AM, Tauraso NM, Shelokov A. 1968. Simian hemorrhagic fever. I. Clinical and epizootiologic aspects of an outbreak among quarantined monkeys. Am J Trop Med Hyg 17:404–412. [PubMed] [Google Scholar]
- 4.Tauraso NM, Shelokov A, Palmer AE, Allen AM. 1968. Simian hemorrhagic fever. 3. Isolation and characterization of a viral agent. Am J Trop Med Hyg 17:422–431. [PubMed] [Google Scholar]
- 5.Abildgaard C, Harrison J, Espana C, Spangler W, Gribble D. 1975. Simian hemorrhagic fever: studies of coagulation and pathology. Am J Trop Med Hyg 24:537–544. [DOI] [PubMed] [Google Scholar]
- 6.Dalgard DW, Hardy RJ, Pearson SL, Pucak GJ, Quander RV, Zack PM, Peters CJ, Jahrling PB. 1992. Combined simian hemorrhagic fever and Ebola virus infection in cynomolgus monkeys. Lab Anim Sci 42:152–157. [PubMed] [Google Scholar]
- 7.Espana C. 1971. Review of some outbreaks of viral disease in captive nonhuman primates. Lab Anim Sci 21:1023–1031. [PubMed] [Google Scholar]
- 8.Renquist D. 1990. Outbreak of simian hemorrhagic fever. J Med Primatol 19:77–79. [PubMed] [Google Scholar]
- 9.Lauck M, Hyeroba D, Tumukunde A, Weny G, Lank SM, Chapman CA, O'Connor DH, Friedrich TC, Goldberg TL. 2011. Novel, divergent simian hemorrhagic fever viruses in a wild Ugandan red colobus monkey discovered using direct pyrosequencing. PLoS One 6:e19056. doi: 10.1371/journal.pone.0019056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bao Y, Ng TF, LeBreton M, Schneider BS, Gillis A, Tamoufe U, Diffo Jle D, Takuo JM, Kondov NO, Coffey LL, Wolfe ND, Delwart E, Clawson AN, Postnikova E, Bollinger L, Lackemeyer MG, Radoshitzky SR, Palacios G, Wada J, Shevtsova ZV, Jahrling PB, Lapin BA, Deriabin PG, Dunowska M, Alkhovsky SV, Rogers J, Friedrich TC, O'Connor DH, Goldberg TL. 2016. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol 161:755–768. doi: 10.1007/s00705-015-2672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey AL, Lauck M, Weiler A, Sibley SD, Dinis JM, Bergman Z, Nelson CW, Correll M, Gleicher M, Hyeroba D, Tumukunde A, Weny G, Chapman C, Kuhn JH, Hughes AL, Friedrich TC, Goldberg TL, O'Connor DH. 2014. High genetic diversity and adaptive potential of two simian hemorrhagic fever viruses in a wild primate population. PLoS One 9:e90714. doi: 10.1371/journal.pone.0090714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauck M, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Chapman CA, Ting N, Switzer WM, Kuhn JH, Friedrich TC, O'Connor DH, Goldberg TL. 2013. Exceptional simian hemorrhagic fever virus diversity in a wild African primate community. J Virol 87:688–691. doi: 10.1128/JVI.02433-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailey AL, Lauck M, Sibley SD, Pecotte J, Rice K, Weny G, Tumukunde A, Hyeroba D, Greene J, Correll M, Gleicher M, Friedrich TC, Jahrling PB, Kuhn JH, Goldberg TL, Rogers J, O'Connor DH. 2014. Two novel simian arteriviruses in captive and wild baboons (Papio spp.). J Virol 88:13231–13239. doi: 10.1128/JVI.02203-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey AL, Lauck M, Sibley SD, Friedrich TC, Kuhn JH, Freimer NB, Jasinska AJ, Phillips-Conroy JE, Jolly CJ, Marx PA, Apetrei C, Rogers J, Goldberg TL, O'Connor DH. 2015. Zoonotic potential of simian arteriviruses. J Virol 90:630–635. doi: 10.1128/JVI.01433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey AL, Lauck M, Ghai RR, Nelson CW, Heimbruch K, Hughes AL, Goldberg TL, Kuhn JH, Jasinska AJ, Freimer NB, Apetrei C, O'Connor DH. 2016. Arteriviruses, pegiviruses, and lentiviruses are common among wild African monkeys. J Virol 90:6724–6737. doi: 10.1128/JVI.00573-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauck M, Alkhovsky SV, Bao Y, Bailey AL, Shevtsova ZV, Shchetinin AM, Vishnevskaya TV, Lackemeyer MG, Postnikova E, Mazur S, Wada J, Radoshitzky SR, Friedrich TC, Lapin BA, Deriabin PG, Jahrling PB, Goldberg TL, O'Connor DH, Kuhn JH. 2015. Historical outbreaks of simian hemorrhagic fever in captive macaques were caused by distinct arteriviruses. J Virol 89:8082–8087. doi: 10.1128/JVI.01046-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wahl-Jensen V, Johnson JC, Lauck M, Weinfurter JT, Moncla LH, Weiler AM, Charlier O, Rojas O, Byrum R, Ragland DR, Huzella L, Zommer E, Cohen M, Bernbaum JG, Cai Y, Sanford HB, Mazur S, Johnson RF, Qin J, Palacios GF, Bailey AL, Jahrling PB, Goldberg TL, O'Connor DH, Friedrich TC, Kuhn JH. 2016. Divergent simian arteriviruses cause simian hemorrhagic fever of differing severities in macaques. mBio 7:e02009-15. doi: 10.1128/mBio.02009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HS, Kwang J, Yoon IJ, Joo HS, Frey ML. 1993. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol 133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- 19.Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 20.Kofler R, Orozco-terWengel P, De Maio N, Pandey RV, Nolte V, Futschik A, Kosiol C, Schlotterer C. 2011. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One 6:e15925. doi: 10.1371/journal.pone.0015925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voronin Y, Holte S, Overbaugh J, Emerman M. 2009. Genetic drift of HIV populations in culture. PLoS Genet 5:e1000431. doi: 10.1371/journal.pgen.1000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foll M, Poh YP, Renzette N, Ferrer-Admetlla A, Bank C, Shim H, Malaspinas AS, Ewing G, Liu P, Wegmann D, Caffrey DR, Zeldovich KB, Bolon DN, Wang JP, Kowalik TF, Schiffer CA, Finberg RW, Jensen JD. 2014. Influenza virus drug resistance: a time-sampled population genetics perspective. PLoS Genet 10:e1004185. doi: 10.1371/journal.pgen.1004185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brinton MA, Di H, Vatter HA. 2015. Simian hemorrhagic fever virus: recent advances. Virus Res 202:112–119. doi: 10.1016/j.virusres.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vatter HA, Donaldson EF, Huynh J, Rawlings S, Manoharan M, Legasse A, Planer S, Dickerson MF, Lewis AD, Colgin LM, Axthelm MK, Pecotte JK, Baric RS, Wong SW, Brinton MA. 2015. A simian hemorrhagic fever virus isolate from persistently infected baboons efficiently induces hemorrhagic fever disease in Japanese macaques. Virology 474:186–198. doi: 10.1016/j.virol.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veit M, Matczuk AK, Sinhadri BC, Krause E, Thaa B. 2014. Membrane proteins of arterivirus particles: structure, topology, processing and function. Virus Res 194:16–36. doi: 10.1016/j.virusres.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee C, Yoo D. 2006. The small envelope protein of porcine reproductive and respiratory syndrome virus possesses ion channel protein-like properties. Virology 355:30–43. doi: 10.1016/j.virol.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyata T, Miyazawa S, Yasunaga T. 1979. Two types of amino acid substitutions in protein evolution. J Mol Evol 12:219–236. doi: 10.1007/BF01732340. [DOI] [PubMed] [Google Scholar]
- 28.Lauck M, Palacios G, Wiley MR, L Y, Fang Y, Lackemeyer MG, Cai Y, Bailey AL, Postnikova E, Radoshitzky SR, Johnson RF, Alkhovsky SV, Deriabin PG, Friedrich TC, Goldberg TL, Jahrling PB, O'Connor DH, Kuhn JH. 2014. Genome sequences of simian hemorrhagic fever virus variant NIH LVR42-0/M6941 isolates (Arteriviridae: Arterivirus). Genome Announc 2:e00978-14. doi: 10.1128/genomeA.00978-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195–205. doi: 10.1038/nrg2526. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton MB, Matrosovich TY, Gray T, Roberts NA, Klenk HD. 2012. Population genetics. Wiley-Blackwell, Oxford, United Kingdom. [Google Scholar]
- 31.Vatter HA, Di H, Donaldson EF, Baric RS, Brinton MA. 2014. Each of the eight simian hemorrhagic fever virus minor structural proteins is functionally important. Virology 462-463:351–362. doi: 10.1016/j.virol.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manz B, Dornfeld D, Gotz V, Zell R, Zimmermann P, Haller O, Kochs G, Schwemmle M. 2013. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog 9:e1003279. doi: 10.1371/journal.ppat.1003279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moncla LH, Ross TM, Dinis JM, Weinfurter JT, Mortimer TD, Schultz-Darken N, Brunner K, Capuano SV, Boettcher C, Post J, Johnson M, Bloom CE, Weiler AM, Friedrich TC. 2013. A novel nonhuman primate model for influenza transmission. PLoS One 8:e78750. doi: 10.1371/journal.pone.0078750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balasuriya UB, Patton JF, Rossitto PV, Timoney PJ, McCollum WH, MacLachlan NJ. 1997. Neutralization determinants of laboratory strains and field isolates of equine arteritis virus: identification of four neutralization sites in the amino-terminal ectodomain of the G(L) envelope glycoprotein. Virology 232:114–128. doi: 10.1006/viro.1997.8551. [DOI] [PubMed] [Google Scholar]
- 35.Ostrowski M, Galeota JA, Jar AM, Platt KB, Osorio FA, Lopez OJ. 2002. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J Virol 76:4241–4250. doi: 10.1128/JVI.76.9.4241-4250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plagemann PGW. 2004. The primary GP5 neutralization epitope of North American isolates of porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol 102:263–275. doi: 10.1016/j.vetimm.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 37.Plagemann PG. 2001. Complexity of the single linear neutralization epitope of the mouse arterivirus lactate dehydrogenase-elevating virus. Virology 290:11–20. doi: 10.1006/viro.2001.1139. [DOI] [PubMed] [Google Scholar]
- 38.Plagemann PGW, Rowland RRR, Faaberg KS. 2002. The primary neutralization epitope of porcine respiratory and reproductive syndrome virus strain VR-2332 is located in the middle of the GP5 ectodomain. Arch Virol 147:2327–2347. doi: 10.1007/s00705-002-0887-2. [DOI] [PubMed] [Google Scholar]
- 39.Chapman CA, Chapman LJ. 1996. Mixed-species primate groups in the Kibale Forest: ecological constraints on association. Int J Primatol 17:31–50. doi: 10.1007/BF02696157. [DOI] [Google Scholar]
- 40.Amaku M, Coutinho FA, Chaib E, Massad E. 2013. The impact of hepatitis A virus infection on hepatitis C virus infection: a competitive exclusion hypothesis. Bull Math Biol 75:82–93. doi: 10.1007/s11538-012-9795-0. [DOI] [PubMed] [Google Scholar]
- 41.Bahl J, Vijaykrishna D, Holmes EC, Smith GJ, Guan Y. 2009. Gene flow and competitive exclusion of avian influenza A virus in natural reservoir hosts. Virology 390:289–297. doi: 10.1016/j.virol.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Domingo E, Escarmis C, Sevilla N, Moya A, Elena SF, Quer J, Novella IS, Holland JJ. 1996. Basic concepts in RNA virus evolution. FASEB J 10:859–864. [DOI] [PubMed] [Google Scholar]
- 43.Caì Y, Postnikova EN, Bernbaum JG, Yu SQ, Mazur S, Deiuliis NM, Radoshitzky SR, Lackemeyer MG, McCluskey A, Robinson PJ, Haucke V, Wahl-Jensen V, Bailey AL, Lauck M, Friedrich TC, O'Connor DH, Goldberg TL, Jahrling PB, Kuhn JH. 2015. Simian hemorrhagic fever virus cell entry is dependent on CD163 and uses a clathrin-mediated endocytosis-like pathway. J Virol 89:844–856. doi: 10.1128/JVI.02697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson RF, Dodd LE, Yellayi S, Gu W, Cann JA, Jett C, Bernbaum JG, Ragland DR, St Claire M, Byrum R, Paragas J, Blaney JE, Jahrling PB. 2011. Simian hemorrhagic fever virus infection of rhesus macaques as a model of viral hemorrhagic fever: clinical characterization and risk factors for severe disease. Virology 421:129–140. doi: 10.1016/j.virol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilker PR, Dinis JM, Starrett G, Imai M, Hatta M, Nelson CW, O'Connor DH, Hughes AL, Neumann G, Kawaoka Y, Friedrich TC. 2013. Selection on haemagglutinin imposes a bottleneck during mammalian transmission of reassortant H5N1 influenza viruses. Nat Commun 4:2636. doi: 10.1038/ncomms3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.