

Abstract
Previous studies identified the Ser/Thr protein kinase, AKT, as a therapeutic target in thrombo-inflammatory diseases. Here we report that specific inhibition of AKT with ARQ 092, an orally-available AKT inhibitor currently in phase Ib clinical trials as an anti-cancer drug, attenuates the adhesive function of neutrophils and platelets from sickle cell disease patients in vitro and cell-cell interactions in a mouse model of sickle cell disease. Studies using neutrophils and platelets isolated from sickle cell disease patients revealed that treatment with 50–500 nM ARQ 092 significantly blocks αMβ2 integrin function in neutrophils and reduces P-selectin exposure and glycoprotein Ib/IX/V-mediated agglutination in platelets. Treatment of isolated platelets and neutrophils with ARQ 092 inhibited heterotypic cell-cell aggregation under shear conditions. Intravital microscopic studies demonstrated that short-term oral administration of ARQ 092 or hydroxyurea, a major therapy for sickle cell disease, diminishes heterotypic cell-cell interactions in venules of sickle cell disease mice challenged with tumor necrosis factor-α. Co-administration of hydroxyurea and ARQ 092 further reduced the adhesive function of neutrophils in venules and neutrophil transmigration into alveoli, inhibited expression of E-selectin and intercellular adhesion molecule-1 in cremaster vessels, and improved survival in these mice. Ex vivo studies in sickle cell disease mice suggested that co-administration of hydroxyurea and ARQ 092 efficiently blocks neutrophil and platelet activation and that the beneficial effect of hydroxyurea results from nitric oxide production. Our results provide important evidence that ARQ 092 could be a novel drug for the prevention and treatment of acute vaso-occlusive complications in patients with sickle cell disease.
Introduction
Sickle cell disease (SCD) is an inherited blood disorder caused by a homozygous Glu6Val mutation at position 6 of β-globin, resulting in hemoglobin S (HbS). HbS is polymerized upon deoxygenation, resulting in sickling and hemolysis of red blood cells, endothelial cell activation, and chronic inflammation.1 In addition, there are several heterozygous forms of SCD,2 such as HbS/β0-thalassemia, which is often clinically similar to sickle cell anemia. The many clinical manifestations in SCD patients include recurrent vaso-occlusive episodes mediated by heterotypic cell-cell adhesion/aggregation, which cause pain crises and increase mortality due to organ damage and acute chest syndrome.3,4 Hydroxyurea, an important therapy for SCD, induces production of fetal hemoglobin and also has other beneficial effects, including increasing nitric oxide (NO) species and decreasing the level of soluble vascular cell adhesion molecule 1.5–7 Consistently, in vivo studies showed that intravenous infusion of hydroxyurea increases the level of plasma NO metabolites (NOx) and has beneficial effects on vaso-occlusive events in Berkeley mice, a model of SCD.8,9 However, SCD patients on hydroxyurea therapy often suffer from vaso-occlusive crises, suggesting that a novel or supplemental therapy is required.
Intravital microscopy provided strong evidence that neutrophil-platelet interactions on activated endothelial cells can cause microvascular occlusion under thrombo-inflammatory conditions, including SCD and ischemia/reperfusion injury.9–12 Among several receptors and counter-receptors, the neutrophil-platelet association is primarily mediated by the interaction of neutrophil P-selectin glycoprotein ligand-1 (PSGL-1) and αMβ2 integrin with platelet P-selectin and glycoprotein Ibα (GPIbα), respectively.13 We have shown that AKT2 positively regulates the function of αMβ2 integrin and P-selectin during vascular inflammation12 and that combining hydroxyurea with AKT2 inhibition has immediate benefits in acute vaso-occlusive events and improves survival in SCD mice.9 Although these results suggest that AKT2 inhibition may be a supplemental therapy for SCD patients with vaso-occlusive crises, no AKT2-specific inhibitor is currently available in the clinic.
As a Ser/Thr protein kinase, AKT regulates numerous cellular processes, such as cell growth, survival, and metabolism.14 Its activity is controlled by phosphorylation of the Thr308 and Ser473 residues by 3-phosphoinositide-dependent kinase 1 and mammalian target of rapamycin complex 2, respectively.15 Activated AKT then phosphorylates Ser/Thr residues on a variety of substrates.16 Despite 80% sequence homology of the three isoforms, each AKT isoform plays a partially overlapping but distinct role in platelet activation and aggregation.17–19 In neutrophils, which express AKT1 and AKT2, only AKT2 regulates cell migration, NADPH oxidase 2 activation, β2 integrin function, and neutrophil-platelet interactions under inflammatory conditions.12,20 As a major isoform in endothelial cells, AKT1 modulates the activity of endothelial NO synthase and is involved in angiogenesis, acute inflammation, and atherosclerosis.21–23 Human AKT isoforms share around 98% sequence homology with mouse proteins. These studies suggest the importance of each AKT isoform in the pathophysiology of vascular diseases and identify AKT as an attractive therapeutic target.
Several AKT inhibitors are being developed as anti-cancer drugs.24,25 ARQ 092 has been reported to be an orally-available, highly-selective AKT inhibitor.26,27 Recent studies show that ARQ 092 blocks the activity of AKT1, AKT2, and AKT3 with IC50 values of 5.0, 4.5, and 16 nM, respectively, and has excellent selectivity (>1,000-fold) over other kinases.26 As an allosteric inhibitor, this compound blocks membrane translocation of inactive AKT and even dephosphorylates the membrane-associated active form, thereby perturbing AKT activity.26 Using cells and tissues isolated from patients with Proteus syndrome harboring AKT1-E17K mutations, a previous study demonstrated effective inhibition of the mutant AKT1 by ARQ 092.27 This compound is currently in phase Ib clinical studies for the treatment of lymphoma, breast and endometrial cancers, and tumors with AKT or phosphoinositide 3-kinase (PI3K) mutations, and is well tolerated at a continuous daily dose of 60 mg or a dose of 600 mg when administered once a week, for several months.28
In the present study, we demonstrate that ARQ 092 decreases the activation state of neutrophils and platelets isolated from SCD patients, thereby reducing platelet-neutrophil interactions in vitro. Furthermore, in vivo studies revealed that oral administration of hydroxyurea and ARQ 092 efficiently blocks neutrophil-endothelial cell and neutrophil-platelet interactions in venules and impairs neutrophil infiltration into the alveoli, thereby improving survival in SCD mice challenged with tumor necrosis factor (TNF)-α. Our results warrant further study of ARQ 092 in a clinical trial to treat acute vaso-occlusive crises in SCD patients.
Methods
Mice
Wild-type (C57BL/6, 6-week old, male and female), hemizygous [Tg(Hu-miniLCRα1 GγAγδβS) Hba−/− Hbb+/−] and Berkeley sickle [Tg(Hu-miniLCRα1 GγAγδβS) Hba−/− Hbb−/−] mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). SCD mice (20–24 weeks old) were generated by transplantation of bone marrow cells isolated from Berkeley mice into lethally irradiated wild-type mice as described previously.9,29 Three to 4 months after transplantation, polymerase chain reaction and electrophoresis analyses showed that all chimeric mice expressed the transgene (human HbS) (Online Supplementary Figure S1). In contrast, mouse hemoglobin was not detected. These chimeric Berkeley mice are hereafter referred to as SCD mice. Both male and female SCD mice (20–24 weeks old) were used in this study. The University of Illinois Institutional Animal Care and Use Committee approved all animal care and experimental procedures.
Sickle cell disease patients
Sixteen homozygous (HbSS) and six HbS/β0-thalassemia patients (20–52 years, 9 men and 13 women) who had not taken aspirin or ibuprofen within 5 days were included in our studies. None of the patients had been treated with hydroxyurea prior to blood donation. No significant differences were observed in the levels of surface markers of resting and stimulated platelets and neutrophils in patients with HbSS or HbS/β0-thalassemia. Blood from all patients was drawn at routine clinic visits without a pain crisis. Multiple experiments were performed using one patient’s blood sample and each experiment was repeated with blood from three or four different patients. All patients enrolled in this study provided informed consent. The collection and use of blood samples for laboratory analysis were approved by the Institutional Review Board of the University of Illinois at Chicago.
ARQ 092 preparation
The process used to synthesize ARQ 092 and the compound’s molecular properties are described elsewhere.30
Isolation of neutrophils and platelets
Platelets were isolated from mice and SCD patients as we have previously described.11 Platelets were suspended in HEPES-Tyrode buffer (20 mM HEPES, pH 7.3, 136 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 1 mM MgCl2, and 5.5 mM glucose without CaCl2 and bovine serum albumin) at a concentration of 3 × 108 platelets/mL. Neutrophils were isolated from SCD patients’ blood and mouse bone marrow as described previously.12 The concentration of neutrophils was adjusted to 1×107 cells/mL in RPMI1640 medium. Human and mouse neutrophils were stimulated for 10 min at 37°C with 0.5 and 10 μM N-formylmethionylleucyl-phenylalanine (fMLP), respectively, and platelets were activated with 0.025 U/mL thrombin for 5 min at 37°C, unless otherwise stated.
Intravital microscopy
Male SCD mice were fasted overnight and treated with saline or 250 mg/kg of hydroxyurea (50 mg/mL) by oral gavage and subsequently with intraperitoneal injection of TNF-α (500 ng), 3 h prior to imaging. Phosphoric acid (0.01 M) or ARQ 092 (100 mg/10 mL/kg) was administered orally 30 min before imaging. Platelets and neutrophils were monitored via infusion of DyLight 488-conjugated anti-CD42c (0.1 μg/g body weight) and Alexa Fluor 647-conjugated anti-Ly-6G antibodies (0.05 μg/g body weight), respectively. Fluorescence and bright-field images were recorded using an Olympus BX61W microscope with a 60 × 1.0 NA water immersion objective and a Hamamatsu C9300 high-speed camera through an intensifier (Video Scope International, Sterling, VA, USA), and data were analyzed using Slidebook v6.0 (Intelligent Imaging Innovations, Denver, CO, USA). Real-time images were captured in the inflamed cremaster venules, which had a diameter of 25–40 μm. The rolling influx of neutrophils (rolling cells/min) and number of adherent neutrophils were determined over a 5-min period (number/field/5 min). Five to six different venules were monitored in each mouse. Since most platelets adhered to the top of adherent neutrophils,12 the kinetics of platelet accumulation was determined by the integrated median fluorescence intensities of the anti-CD42c antibody which were normalized to the number of adherent neutrophils and plotted over time.
Other methods
Genotyping and chimerism analysis of the SCD mice, neutrophil-platelet aggregation assays, survival times, plasma NOx (nitrites/nitrates) level, flow cytometry, platelet aggregation/agglutination assays, immunohistochemistry, reactive oxygen species (ROS) generation, and Ca2+ mobilization are described in detail in the Online Supplementary Methods.
Statistics
Data were analyzed using GraphPad Prism 6 software by ANOVA with the Tukey test, Student t-test, and Mantle-Cox log-rank test (survival curve). P values less than 0.05 were considered statistically significant.
Results
ARQ 092 inhibits activation of neutrophils and platelets isolated from patients with sickle cell disease
We previously reported that the basal levels of AKT phosphorylation are significantly higher in neutrophils and platelets isolated from SCD patients under stable conditions than in those from healthy donors.12 We found that AKT in human neutrophils and platelets is maximally phosphorylated 2 min after stimulation with fMLP or thrombin, respectively (Online Supplementary Figure S2A,B). Similar results were obtained with mouse neutrophils and platelets (Online Supplementary Figure S2C,D). To determine the inhibitory effect of ARQ 092 on AKT phosphorylation, neutrophils isolated from SCD patients were pretreated with ARQ 092 and further incubated with or without fMLP for 2 min, followed by immunoblotting. We observed that ARQ 092, at doses of 50 and 500 nM, markedly reduced phosphorylation of AKT but not of PI3K p85α/β or Src, following fMLP treatment (Figure 1A). Treatment of patients’ neutrophils with 50 and 500 nM ARQ 092 significantly inhibited the surface amount of αMβ2 integrin following fMLP stimulation (Figure 1B). Furthermore, binding of anti-activated αMβ2 antibodies (CBRM1/5) was decreased by treatment with ARQ 092 (Figure 1C). Although 5 nM ARQ 092 caused a moderate and significant inhibition of AKT phosphorylation, no inhibitory effect on αMβ2 integrin function was observed (data not shown). Consistent with impaired αMβ2 integrin function, 50 or 500 nM ARQ 092 significantly inhibited binding of soluble fibrinogen, a ligand for αMβ2 integrin, to fMLP-stimulated neutrophils (Figure 1D). The surface expression of PSGL-1 was not affected by 500 nM ARQ 092 (data not shown). These results suggest that specific AKT inhibition by ARQ 092 attenuates the membrane translocation and ligand-binding function of αMβ2 integrin in stimulated neutrophils isolated from SCD patients.
Figure 1.

ARQ 092 inhibits activation of neutrophils and platelets isolated from SCD patients in vitro. (A–D) Neutrophils or (E–J) platelets isolated from SCD patients were pretreated with vehicle (0.1% DMSO), or 50 or 500 nM ARQ 092 and then incubated with or without 0.5 μM fMLP or 0.025 U/mL thrombin for 2 min, respectively. (A) Immunoblotting was performed using equal amounts (50 μg) of neutrophil lysate protein, followed by densitometry (n = 3). (B–D) Flow cytometry was performed using phycoerythin-conjugated control IgG or antibodies against total (ICRF44) or activated αMβ2 (CBRM1/5), or DyLight 488-conjugated fibrinogen. The geometric mean fluorescence intensity of antibodies was normalized to that of control IgG, and data are presented as fold increase compared with vehicle-treated, unstimulated cells. (E) Immunoblotting was performed using equal amounts (50 μg) of platelet lysate protein, followed by densitometry (n = 3). (F) Flow cytometry was performed to measure P-selectin exposure. (G–H) Platelets were stimulated with (G) 0.025 or (H) 0.05 U/mL thrombin. The surface amount of GPIbα was measured by flow cytometry. (I) Platelet agglutination was induced by 0.5 μg/mL vWF and 0.1 mg/mL ristocetin. (J) Platelet aggregation was induced by 0.025 U/mL thrombin. The representative agglutination or aggregation trace was obtained from three independent experiments. Data represent the mean ± SD (n = 3–4). *P<0.05, **P< 0.01, ***P<0.001, and ****P<0.0001 versus unstimulated vehicle control (or between two groups), ANOVA and the Tukey test.
We also tested whether ARQ 092 inhibits the function of platelet surface molecules. We found that treatment with 50 and 500 nM ARQ 092 inhibited AKT phosphorylation in thrombin-stimulated platelets from SCD patients, without affecting PI3K or Src phosphorylation (Figure 1E). Treatment of patients’ platelets with 50 and 500 nM ARQ 092 significantly reduced P-selectin exposure, an indicator of α granule secretion, after stimulation with 0.025 U/mL thrombin (Figure 1F). As seen in neutrophils, while 5 nM ARQ 092 caused a significant but not complete inhibition of AKT phosphorylation, minimal inhibitory effects on P-selectin exposure were observed (data not shown). We found that treatment with ARQ 092 did not affect the surface expression of GPIbα but impaired platelet agglutination induced by binding of von Willebrand factor to GPIbα of the GPIb/IX/V complex (Figure 1G,I). This suggests that AKT inhibition impairs the ligand-binding function of GPIbα. In addition, thrombin-induced aggregation of patients’ platelets was also inhibited by 50 and 500 nM ARQ 092 (Figure 1J). However, a higher concentration of thrombin attenuated the inhibitory effect of ARQ 092 (Online Supplementary Figure S3A), implying that the increased concentration of thrombin induces other signaling pathways which are independent of AKT. Similar results were obtained with cross-linked collagen-related peptide, a GPVI-selective ligand (Online Supplementary Figure S3B,C). Our results indicate that specific AKT inhibition by ARQ 092 effectively blocks α-granule secretion and the adhesive function of activated platelets isolated from SCD patients.
ARQ 092 attenuates heterotypic aggregation of sickle cell patients’ neutrophils and platelets under stirring conditions
Heterotypic cell-cell interactions can cause vaso-occlusion in SCD patients.13 Since ARQ 092 inhibited the function of surface molecules on neutrophils and platelets isolated from SCD patients, we investigated whether ARQ 092 affects heterotypic cell-cell aggregation in vitro. Neutrophils and platelets isolated from SCD patients aggregated under stirring conditions mimicking venous shear, creating a new cell population (R1 gate) in which most cells were positive for both L-selectin (a leukocyte marker) and CD41a (αIIb, a platelet marker) (Figure 2A). As quantified by the number of cell-cell aggregates (R3) in the R1 gate, pre-treatment of neutrophils or platelets with 500 nM ARQ 092 moderately but significantly decreased neutrophil-platelet aggregation (Figure 2A–B). When both cell types were treated with ARQ 092, the number of aggregates was further reduced. Since aggregated platelets without associated neutrophils are not detected in the R1 gate, we further measured the fluorescence intensities of anti-CD41a antibodies in the gate. Pretreatment of neutrophils or platelets with ARQ 092 significantly inhibited the antibody signal, and the inhibitory effect was further increased when both cell types were treated with the inhibitor (Figure 2C). As a control, neutrophil-platelet aggregation was not affected by either anti-L-selectin or anti-CD41a antibodies at a concentration used for cell labeling or even at a 10-fold higher concentration (Online Supplementary Figure S4). Thus, these results show that ARQ 092 effectively blocks heterotypic aggregation of patients’ neutrophils and platelets in vitro.
Figure 2.
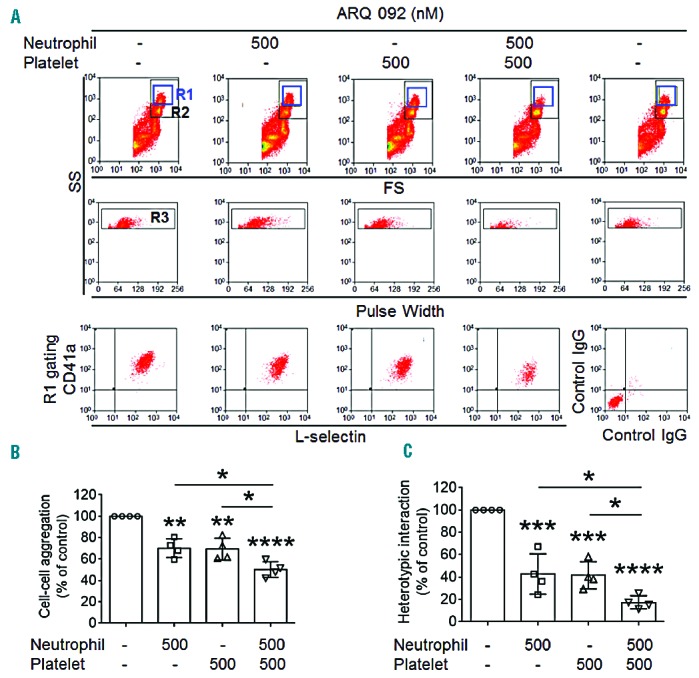
ARQ 092 perturbs heterotypic aggregation of neutrophils and platelets from SCD patients under stirring conditions. Neutrophils and platelets isolated from SCD patients were pretreated with vehicle [0.1% DMSO (−)] or 500 nM ARQ 092 and then incubated with FITC-conjugated anti-L-selectin and APC-conjugated anti-CD41a antibodies, respectively. Thrombin-activated platelets were mixed with neutrophils under stirring conditions, followed by flow cytometric analysis. R1, leukocyte-platelet aggregates; R2, neutrophils; and R3, the number of cell aggregates in the R1 gate. (B) Cell-cell aggregation was measured by the number of cell-cell aggregates (R3). (C) Heterotypic (neutrophil-platelet) interaction was measured by the fluorescence intensities of anti-CD41a antibodies in the R1 gate. Data represent the mean ± SD (n = 4). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 versus vehicle control (or between two groups), ANOVA and the Tukey test.
ARQ 092 specifically inhibits AKT phosphorylation in neutrophils and platelets and reduces cell activation in sickle cell disease mice ex vivo
Recent studies using mouse xenograft tumor models demonstrated that oral administration of 75–100 mg/kg of ARQ 092 significantly delays tumor growth.26 Importantly, the authors showed that after oral administration of 100 mg/kg of ARQ 092 to mice, the maximum plasma concentration reached around 2 μM at 30 min and the plasma level remained around 300 nM at 8 h.26 Thus, we sought to determine the ex vivo effect of ARQ 092 on AKT phosphorylation in neutrophils and platelets isolated from SCD mice. Vehicle or 100 mg/kg of ARQ 092 was administered orally to the mice. Blood and bone marrow were collected, 30 min after treatment, to isolate platelets and neutrophils, respectively. No spontaneous bleeding was observed in the abdominal cavity of ARQ 092-treated SCD mice. As a control, ARQ 092 at 100 mg/kg did not alter the number of circulating blood cells in wild-type mice (Table 1) and had no inhibitory effect on splenomegaly in SCD mice (data not shown). We found that phosphorylation of AKT, but not of Src and PI3K, was disrupted in fMLP-stimulated neutrophils isolated from SCD mice treated with ARQ 092, compared to those treated with vehicle (Figure 3A). The membrane translocation of αMβ2 integrin and soluble fibrinogen binding were significantly impaired in neutrophils from ARQ 092-treated mice (Figure 3B, C). Consistently, oral administration of ARQ 092 abrogated AKT phosphorylation in thrombin-activated platelets without affecting Src or PI3K phosphorylation (Figure 3D). P-selectin exposure and platelet aggregation were significantly reduced in platelets, activated with either thrombin or cross-linked collagen-related peptide, from ARQ 092-treated SCD mice, (Figure 3E,F). As a control, treatment of isolated neutrophils and platelets with 50 and 500 nM ARQ 092 in vitro specifically inhibited AKT phosphorylation following agonist stimulation (Online Supplementary Figure S5). In addition, treatment with ARQ 092 also inhibited AKT phosphorylation in TNF-α-stimulated neutrophils in vitro (Online Supplementary Figure S6). Together, these results indicate that oral administration of 100 mg/kg of ARQ 092 significantly attenuates the activation state of neutrophils and platelets in SCD mice by specific inhibition of AKT.
Table 1.
The number of circulating blood cells in wild-type mice after oral administration of ARQ 092.
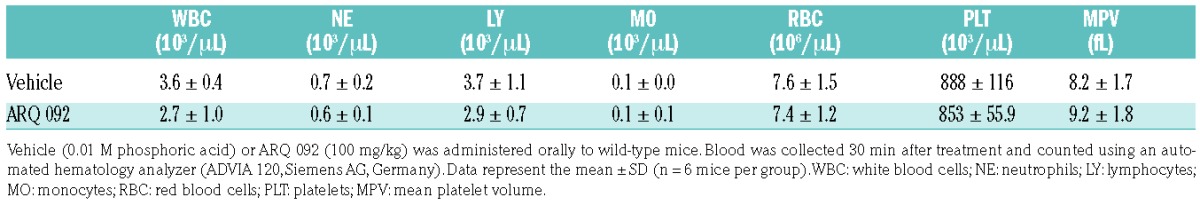
Figure 3.
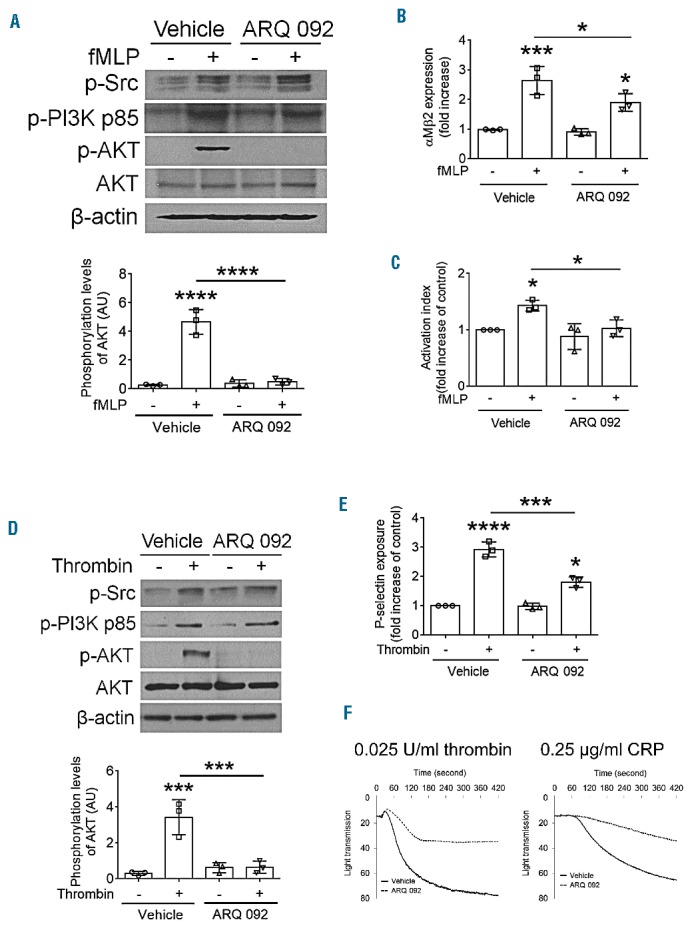
Oral administration of ARQ 092 blocks AKT phosphorylation and activation of neutrophils and platelets isolated from SCD mice ex vivo. Vehicle (0.01 M phosphoric acid) or ARQ 092, 100 mg/kg, was given orally to SCD mice. Blood and bone marrow were collected 30 min after treatment. Isolated (A–C) neutrophils or (D–F) platelets were incubated with or without 10 μM fMLP or 0.025 U/mL thrombin for 2 min, respectively. (A and D) To determine the phosphorylation levels of Src, PI3K, and AKT, equal amounts (50 μg) of cell lysate protein were immunoblotted, followed by densitometry (n = 3). (B and C) The surface amount of αMβ2 and soluble fibrinogen binding were measured by flow cytometry. (E) P-selectin exposure was measured by flow cytometry. (F) Platelet aggregation was induced by 0.025 U/mL thrombin or 0.25 μg/mL cross-liked collagen-felated peptide (CRP). The representative aggregation traces were obtained from two independent experiments. All other data represent the mean ± SD (n = 3). *P<0.05, ***P<0.001, and ****P<0.0001 versus unstimulated vehicle control (or between two groups), ANOVA and Tukey test.
Oral administration of hydroxyurea and ARQ 092 reduces cell-cell interactions in sickle cell disease mice challenged with tumor necrosis factor-α
A previous study showed that oral administration of 250 mg/kg of hydroxyurea partially inhibits leukocyte adhesion to the venules of SCD mice.8 Thus, we investigated whether oral administration of a single dose of ARQ 092 (100 mg/kg) or co-administration with hydroxyurea (250 mg/kg) affects neutrophil-endothelial cell and neutrophil-platelet interactions in the cremaster venules of SCD mice challenged with TNF-α which induces acute vaso-occlusive events in the mice.8,29 Due to the different pharmacokinetics and mechanisms of action,8,26,31 SCD mice were treated with saline or hydroxyurea by oral gavage prior to intraperitoneal injection of TNF-α. For ARQ 092, vehicle or the compound was given orally 2.5 h after the TNF-α injection, followed by surgical procedures (Figure 4A). No spontaneous bleeding was observed at the surgical site in any of the mice. We found that compared to the vehicle control, treatment with hydroxyurea or ARQ 092 alone significantly decreased the number of adherent neutrophils with a minimal increase in the rolling influx (Figure 4B–D, Online Supplementary Videos 1–4). No differences were observed between the effects of the two vehicle controls. Compared to hydroxyurea or ARQ 092 treatment, co-administration of both hydroxyurea and ARQ 092 significantly enhanced the rolling influx and decreased the number of adherent neutrophils in the venules (Online Supplementary Video 5). To investigate platelet-neutrophil interactions, we measured the fluorescence intensities of anti-CD42c antibodies. Treatment with hydroxyurea, ARQ 092 or both abrogated platelet-neutrophil interactions, compared to vehicle controls (Figure 4E). These results suggest that oral administration of both hydroxyurea and ARQ 092 efficiently inhibits acute vaso-occlusive events in TNF-α-challenged SCD mice.
Figure 4.
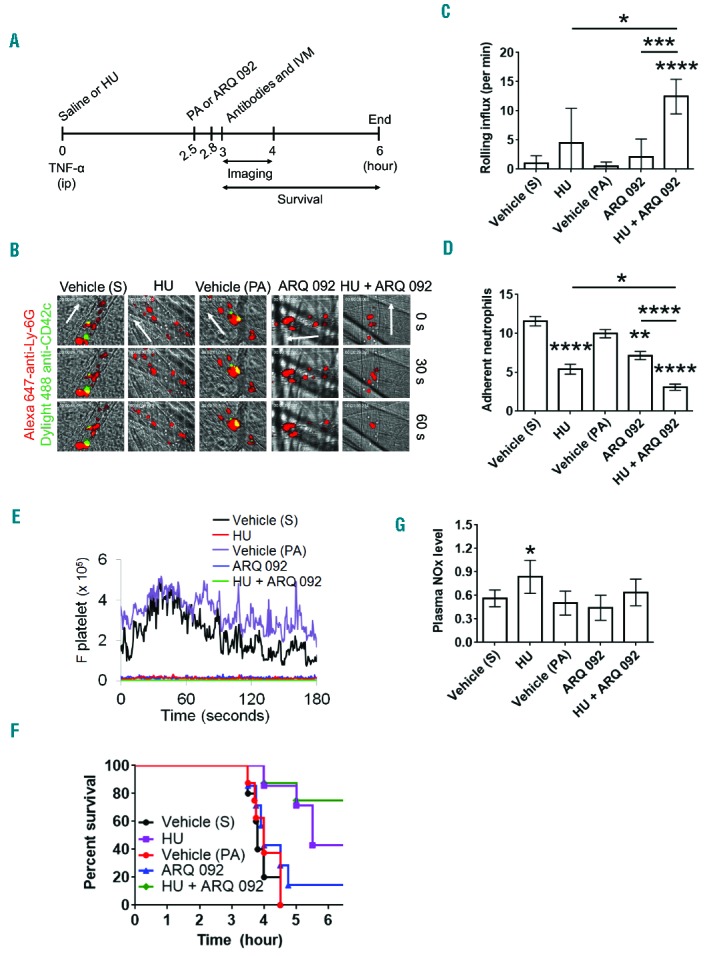
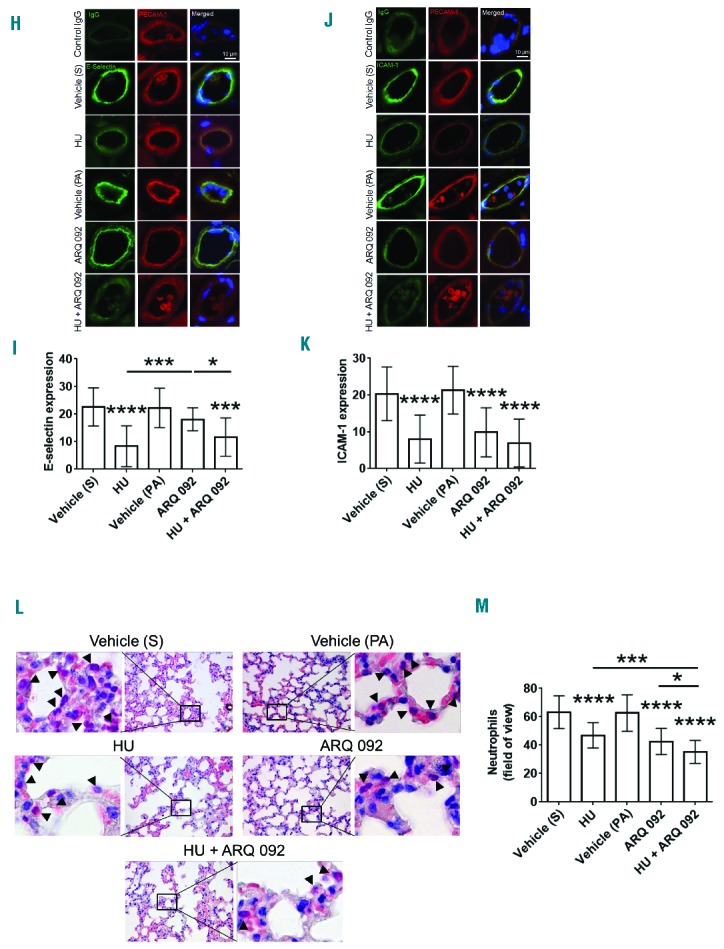
(A–G). Oral administration of hydroxyurea and ARQ 092 has numerous beneficial effects in TNF-α-challenged SCD mice. Intravital microscopy was performed as described in the Methods. Neutrophils and platelets were labeled by infusion of Alexa Fluor 647-conjugated anti-Ly-6G and DyLight 488-conjugated anti-CD42c antibodies, respectively. (A) Timeline for each treatment and surgery for intravital microscopy (IVM) in SCD mice. S: saline; HU: hydroxyurea; PA: 0.01 M phosphoric acid. (B) Representative images at various time-points. The time “0” was set as the image capture was initiated for each vessel. (C, D) Number of rolling (for 1 min) and adherent neutrophils (for 5 min). Data represent the mean ± SEM (n = 7–8 mice). (E) The integrated median fluorescence intensities of anti-CD42c antibodies (F platelets) were plotted over time. (F) Survival curves. Survival was significantly improved in the groups treated with hydroxyurea alone (P = 0.0028) or both hydroxyurea and ARQ 092 (P=0.0017), compared to each vehicle control. Mantel-Cox log-rank test. (G) Plasma NOx levels were measured as described in the Methods. Data represent the mean ± SD (n = 7–8 mice per group). (H–M). Oral administration of hydroxyurea and ARQ 092 has numerous beneficial effects in TNF-α-challenged SCD mice. (H–K) After recording survival times, the cremaster muscle was taken out for immunohistochemistry. Muscle sections were labeled with control IgG or rat monoclonal antibodies against E-selectin or ICAM-1 and then with DyLight 488-conjugated anti-rat IgG antibodies, followed by incubation with APC-conjugated anti-PECAM-1 antibodies and a mounting reagent containing DAPI. (H and J) Representative images. Bar = 10 μm. (I and K) The fluorescence intensities of antibodies. Data represent the mean ± SD (n = 18–22 vessels in 5 mice per group). (L–M) After recording survival times, lungs were taken out for histochemistry. Neutrophils were stained with naphthol AS-D chloroacetate. (L) Representative images. (M) The number of transmigrated neutrophils (arrow heads) was quantified in the field of view (110 mm2). Data represent the mean ± SD (n = 36–42 sections in 5–6 mice per group). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 versus each vehicle control (or between two groups), ANOVA and the Tukey test. S: saline; HU: hydroxyurea; PA: phosphoric acid.
Hydroxyurea alone or with ARQ 092, but not ARQ 092 alone, prolongs survival times of sickle cell disease mice challenged with tumor necrosis factor-α
We and others reported that a surgical procedure to expose the cremaster muscle after intraperitoneal injection of TNF-α in SCD mice leads to death within several hours as a result of acute vaso-occlusive events.8,9,29 Compared to the vehicle controls (saline, S; 0.01 M phosphoric acid, PA), treatment with hydroxyurea or both hydroxyurea and ARQ 092 significantly prolonged survival times in TNF-α–challenged SCD mice, whereas treatment with ARQ 092 alone did not improve survival (S versus hydroxyurea, P=0.0028; PA versus hydroxyurea + ARQ 092, P=0.0017; ARQ 092 versus hydroxyurea + ARQ 092, P=0.0087; and hydroxyurea versus hydroxyurea + ARQ 092, P=0.31) (Figure 4F). Fifty percent of SCD mice treated with vehicle (S), hydroxyurea, vehicle (PA), ARQ 092, and both hydroxy urea and ARQ 092 died at 4.0, 5.5, 4.0, 4.1, and >6 hours, respectively, after TNF-α injection.
Hydroxyurea alone, but not with ARQ 092, enhances plasma levels of nitric oxide metabolites in sickle cell disease mice challenged with tumor necrosis factor-α
We recently reported that a single intravenous infusion of hydroxyurea significantly enhances plasma NOx levels in hemizygous control (Hbb+/−) and Berkeley (Hbb−/−) mice.9 We further measured the plasma NOx levels in TNF-α-challenged SCD mice after oral administration of hydroxy urea, ARQ 092, or both. The plasma NOx levels were significantly elevated in the mice after oral administration of hydroxyurea, but not ARQ 092, compared to the vehicle control (Figure 4G). Co-administration of hydroxyurea and ARQ 092 increased the plasma NOx level by 1.3-fold relative to the vehicle (PA) control, but this increase was not significantly different from that following administration of vehicle controls or hydroxyurea alone.
Co-administration of hydroxyurea and ARQ 092 downregulates the expression of E-selectin and intercellular adhesion molecule-1 in sickle cell disease mice challenged with tumor necrosis factor-α
E-selectin and intercellular adhesion molecule-1 (ICAM-1) expressed on activated endothelial cells are required for neutrophil rolling and adhesion, respectively, during vascular inflammation.32 Immunohistochemistry of cremaster muscle sections showed that, compared to the vehicle control, treatment with hydroxyurea or both hydroxyurea and ARQ 092, but not ARQ 092 alone, significantly reduced the expression of E-selectin (Figure 4H,I). ICAM-1 expression was significantly decreased in mice treated with hydroxyurea, ARQ 092, or both, compared to each vehicle control (Figure 4J,K). These results imply that, unlike AKT2-specific inhibition which perturbs expression of both E-selectin and ICAM-1,9 inhibition of all AKT isoforms may have additional effects on vascular endothelial cells of TNF-α-challenged SCD mice.
Co-administration of hydroxyurea and ARQ 092 efficiently blocks neutrophil transmigration in sickle cell disease mice
Neutrophils rapidly transmigrate from the pulmonary microvasculature and cause lung injury during inflammation. We found that oral administration of hydroxyurea or ARQ 092 significantly blocked neutrophil transmigration into the alveoli of TNF-α-challenged SCD mice (Figure 4L,M). Co-administration of hydroxyurea and ARQ 092, compared to hydroxyurea or ARQ 092 alone, further decreased the number of transmigrated neutrophils. These results suggest that hydroxyurea with AKT inhibition efficiently reduces lung inflammation in TNF-α-challenged SCD mice.
Co-administration of hydroxyurea and ARQ 092 efficiently blocks numerous functions of neutrophils and platelets isolated from sickle cell disease mice challenged with tumor necrosis factor-α ex vivo
We further examined which neutrophil and platelet functions are efficiently inhibited by both drugs. Hydroxyurea, ARQ 092, or both were given orally to TNF-α-challenged SCD mice as described in Figure 4A without surgery. Blood and bone marrow were collected 3 h after TNF-α treatment to isolate platelets and neutrophils, respectively. We found that following stimulation of neutrophils with fMLP, the surface amount of αMβ2 integrin and soluble fibrinogen binding were significantly inhibited by ARQ 092 or both hydroxyurea and ARQ 092, but not hydroxyurea alone, compared with vehicle control (Figure 5A,B). Although studies showed that an elevation in cytosolic Ca2+ levels is critical for neutrophil activation and that deletion or inhibition of neutrophil AKT2 impairs Ca2+ mobilization following fMLP stimulation,33 Ca2+ release and influx in fMLP-stimulated neutrophils from SCD mice were not affected by any treatment (Figure 5C). Since hydroxyurea produces NO species protecting against oxidative stress5,6 and neutrophil AKT2 is important for ROS generation by activating the NADPH oxidase 2 complex,20 we also measured ROS generation. As measured by the DCF signal, intracellular ROS generation was not reduced in fMLP-stimulated neutrophils isolated from TNF-α-challenged SCD mice after oral administration of hydroxyurea or ARQ 092 alone (Figure 5D). When the mice were treated with both drugs, however, the DCF signal was significantly decreased in stimulated neutrophils. Similar results were obtained using Amplex red which detects extracellular H2O2 (Figure 5E,F).
Figure 5.

Co-administration of hydroxyurea and ARQ 092 efficiently inhibits αMβ2 integrin function and ROS generation in stimulated neutrophils isolated from SCD mice ex vivo. Vehicle (0.01 M phosphoric acid, −), hydroxyurea (HU), ARQ 092, or both were given orally to TNF-α-challenged SCD mice as described in Figure 4A. Bone marrow was collected at 3 h after TNF-α injection. Isolated neutrophils were incubated with or without fMLP. (A, B) The surface level of αMβ2 integrin and soluble fibrinogen binding were measured by flow cytometry. (C) Neutrophils were pretreated with Ca2+ dye and incubated with fMLP for 200 s and 2 mM CaCl2 was then added. A representative graph was obtained from three independent experiments. (D) Neutrophils were incubated with DCFH-DA prior to fMLP stimulation. Intracellular ROS generation was measured by the DCF signal in flow cytometry. (E, F) Neutrophils were mixed with Amplex red reaction solution and treated with or without fMLP. Extracellular H2O2 was detected by absorbance (560 nm) as described in the Methods. The quantification graph was obtained at the 15 min mark of the reaction. Data represent the mean ± SD (n = 4–5). *P<0.05, **P<0.01, and ***P<0.001 versus unstimulated vehicle control (or between two groups), ANOVA and the Tukey test.
We further investigated the combined effect of hydroxyurea and ARQ 092 on platelet function ex vivo. P-selectin exposure was significantly reduced in thrombin-activated platelets isolated from TNF-α-challenged SCD mice treated with hydroxyurea or ARQ 092 (Figure 6A). The inhibitory effect was further enhanced in platelets from the mice treated with both drugs. In platelets, unlike neutrophils, we observed that treatment with hydroxyurea, ARQ 092, or both equally inhibited Ca2+ influx with little effect on Ca2+ release during thrombin activation (Figure 6B–D). We also found that oral administration of hydroxyurea or ARQ 092 did not affect the surface expression of GPIbα but impaired von Willebrand factor-mediated agglutination of platelets isolated from TNF-α-challenged SCD mice (Online Supplementary Figure S7 and Figure 6E). Co-administration of both drugs slightly increased the inhibitory effect on agglutination. Similar results were obtained from platelet aggregation assays (Figure 6F). Moreover, oral administration of hydroxyurea, ARQ 092, or both markedly reduced the generation of intracellular ROS in thrombin-activated platelets isolated from TNF-α-challenged SCD mice (Figure 6G). Extracellular H2O2 generation was weakly decreased by hydroxyurea or ARQ 092 but significantly impaired by both drugs (Figure 6H,I). Although the precise mechanism(s) by which hydroxyurea inhibits platelet functions remains to be determined, our results suggest that the combination therapy of hydroxyurea and ARQ 092 efficiently inhibits numerous platelet functions in TNF-α-challenged SCD mice.
Figure 6.

Co-administration of hydroxyurea and ARQ 092 efficiently inhibits numerous functions of activated platelets isolated from SCD mice ex vivo. Vehicle (0.01 M phosphoric acid, −) or drugs were given orally to TNF-α-challenged SCD mice as described in Figure 5. Blood was collected 3 h after TNF-α injection. Isolated platelets were treated with or without thrombin. (A) Flow cytometry was performed to measure P-selectin exposure. (B–D) Platelets were pretreated with Ca2+ dye and incubated with thrombin for 5 min and 2 mM CaCl2 was then added. Ca2+ release (C) and influx (D) were measured and quantified by the AUC (area under the curve, arbitrary units). (E) Platelet agglutination was induced by 10 μg/mL vWF and 10 μg/mL botrocetin. (F) Platelet aggregation was induced by thrombin. The representative agglutination or aggregation trace was obtained from three independent experiments. (G) Platelets were incubated with DCFH-DA prior to thrombin stimulation. Intracellular ROS generation was measured by the DCF signal in flow cytometry. (H–I) Platelets were mixed with Amplex red reaction solution and treated with or without thrombin. Extracellular H2O2 was detected by absorbance (560 nm) as described in the Methods. The quantification graph was obtained at the 30 min mark of the reaction. Data represent the mean ± SD (n = 3–4). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 versus unstimulated vehicle control (or between two groups), ANOVA and the Tukey test.
The short-term beneficial effect of hydroxyurea results from nitric oxide production
Previous studies suggested that the inhibitory effect of hydroxyurea on cell-cell interactions results from NO production in vivo.8 Thus, we sought to determine whether hydroxyurea-generated NO is important for the inhibitory mechanism of the drug. Using water-soluble carboxy-PTIO (a NO scavenger that has no effect on NO synthase activity),34 we repeated our ex vivo studies shown in Figures 5 and 6. We found that PTIO treatment itself did not affect the membrane translocation and ligand binding of αMβ2 integrin in fMLP-stimulated neutrophils (Figure 7A, B) or P-selectin exposure in thrombin-activated platelets (Figure 7C). In neutrophils, hydroxyurea alone had a small effect on the function of αMβ2 integrin, and PTIO treatment nullified the effect of hydroxyurea, but not ARQ 092, on αMβ2 membrane translocation and fibrinogen binding (Figure 7A,B). The potentiated inhibitory effect of both drugs was restored with PTIO treatment to a level similar to the inhibitory effect of ARQ 092 alone. We also found that administration of PTIO reversed the inhibitory effect of hydroxyurea, but not ARQ 092, on P-selectin exposure in activated platelets (Figure 7C). The synergistic effect of both hydroxyurea and ARQ 092 was abolished by PTIO treatment, and the inhibitory effect of both drugs with PTIO treatment was similar to that of ARQ 092 alone. Thus, these results imply that the synergistic effects of both hydroxyurea and ARQ 092 on neutrophil-platelet interactions result from two different signaling pathways: direct NO production by hydroxyurea and AKT inhibition by ARQ 092.
Figure 7.

Hydroxyurea (HU) reduces neutrophil and platelet activation via NO production. SCD mice were pretreated by intravenous injection of PTIO, a NO scavenger (1 mg/kg) 30 min prior to TNF-α challenge. Vehicle (0.01 M phosphoric acid, −) or drugs were given orally to TNF-α-challenged SCD mice as described in Figure 5. Blood and bone marrow were collected 3 h after TNF-α injection. Isolated (A, B) neutrophils and (C) platelets were treated with or without fMLP (for 10 min) or thrombin (for 5 min), respectively. Flow cytometry was performed to measure (A, B) the surface level of αMβ2 integrin and soluble fibrinogen binding in stimulated neutrophils and (C) P-selectin exposure in stimulated platelets. Data represent the mean ± SD (n = 3). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 versus unstimulated vehicle control (or between two groups), ANOVA and the Tukey test.
Discussion
ARQ 092 is an orally-available, selective AKT inhibitor which is currently in phase Ib clinical trials for the treatment of certain cancers.28 In the present study, we showed that ARQ 092 reduces the adhesive function of neutrophils and platelets isolated from SCD patients in vitro. Importantly, oral administration of a single dose of ARQ 092 abrogated AKT phosphorylation in isolated neutrophils and platelets following agonist stimulation, significantly reduced cell-cell interactions in cremaster venules, and decreased neutrophil transmigration into the alveoli of TNF-α-challenged SCD mice. The inhibitory effects and survival were increased when the mice were pretreated with hydroxyurea. Thus, our studies provide important evidence that a specific inhibitor of AKT may be beneficial to treat acute vaso-occlusive events in SCD patients. Since AKT signaling is crucial for the function of intravascular cells during numerous vascular diseases,12,17–21,23 ARQ 092 is likely to inhibit the activity of all AKT isoforms in intravascular cells and thereby attenuate the process of thrombosis and inflammation in SCD patients.
In addition to sickled red blood cells and activated endothelial cells, activation and adhesion of neutrophils and platelets contribute to the vaso-occlusive complications of SCD.35 Our study shows that ARQ 092 reduces the ligand-binding function of αMβ2 integrin in stimulated neutrophils isolated from SCD patients. Importantly, we found that ARQ 092 inhibits αMβ2 integrin function in neutrophils isolated from SCD mice after oral administration of the inhibitor. Although oral administration of hydroxyurea or ARQ 092 alone did not affect intracellular ROS generation in fMLP-stimulated neutrophils isolated from the mice, co-administration of hydroxyurea and ARQ 092 significantly decreased ROS generation, suggesting that combined therapy may efficiently attenuate oxidative stress conditions in SCD patients. Consistent with previous reports showing that the activation and adhesive function of platelets is regulated by AKT,17–19,36 we found that ARQ 092 significantly impairs P-selectin exposure and GPIbα-mediated agglutination in platelets from SCD patients in vitro and in SCD mouse platelets ex vivo. In support of the importance of neutrophil αMβ2 integrin and platelet P-selectin and GPIbα for neutrophil-platelet interactions,13 we observed that specific AKT inhibition in both neutrophils and platelets isolated from SCD patients effectively decreases heterotypic cell-cell aggregation. Thus, our results suggest that co-administration of hydroxyurea and ARQ 092 is beneficial to inhibit cell-cell interactions during vaso-occlusion in SCD. Most AKT inhibitors, including ARQ 092, are being developed as anti-cancer drugs.25 Given the fact that some cancer patients are at high risk of thrombogenesis,37 our results also provide indirect evidence that ARQ 092 may be an effective drug in cancer patients with thrombotic complications.
It is believed that leukocyte adhesion to endothelial cells initially mediates cell-cell interactions and aggregation in the vessels of SCD patients and induces the inflammatory process.35 Our studies demonstrate that oral administration of a single dose of both hydroxyurea and ARQ 092 efficiently inhibits neutrophil-endothelial cell and neutrophil-platelet interactions in cremaster venules and reduces neutrophil recruitment into the alveoli of TNF-α-challenged SCD mice. Interestingly, survival was improved by hydroxyurea alone or both hydroxyurea and ARQ 092, but not ARQ 092 alone. We previously reported that intravenous infusion of an AKT2 inhibitor decreases E-selectin and ICAM-1 expression on cremaster vessels of TNF-α–challenged SCD mice.9 However, oral administration of ARQ 092 resulted in a significant reduction in the expression of ICAM-1, but not E-selectin. Although we cannot eliminate the possibility of off-target effects of these AKT inhibitors, these results imply that inhibition of endothelial cell AKT1 and/or AKT3 may regulate E-selectin expression during inflammation. Furthermore, our findings that ARQ 092 alone significantly decreases cell-cell interactions but has no benefit on survival in TNF-α-challenged SCD mice may limit usage of this inhibitor as a supplement to hydroxyurea therapy in SCD patients.
Oxidative stress results in reduced NO bioavailability in SCD patients and mice, which in turn aggravates inflammatory conditions.38,39 Hydroxyurea has many beneficial effects in SCD patients. Importantly, preclinical and clinical studies demonstrated that hydroxyurea increases the plasma level of NO species and stimulates a cGMP-signaling pathway.5,6,8,9 As seen in SCD mice after intravenous infusion of hydroxyurea,9 plasma NOx levels were also elevated after oral administration of hydroxyurea to SCD mice. Although combination therapy with hydroxyurea and ARQ 092 showed beneficial effects on acute vaso-occlusive events and survival in TNF-α-challenged SCD mice, the plasma NOx levels increased by hydroxyurea treatment seemed to be slightly decreased by the dual therapy (hydroxyurea versus hydroxyurea + ARQ 092¸ P=0.067, Student t-test.). Studies showed that endothelial cell AKT1 plays an important role in acute inflammation and angiogenesis, which is associated with phosphorylation of endothelial NO synthase.21,23,40,41 Although ARQ 092 may inhibit endothelial cell AKT1-NO synthase signaling, the plasma NOx level was not changed by ARQ 092 treatment alone in TNF-α-challenged SCD mice, implying that endothelial NO synthase-derived NO production may not be important for maintaining the basal level of plasma NOx in SCD mice. Alternatively, our finding that a NO scavenger, PTIO, does not alter the inhibitory effect of ARQ 092 on platelet and neutrophil function (Figure 7) suggests that ARQ 092 is unlikely to affect cellular NO generation. Since hydroxyurea serves as a NO donor, further studies are required to determine how ARQ 092 influences the production of hydroxyurea-induced NOx in TNF-α–challenged SCD mice.
In addition to hydroxyurea, many drugs targeting cell adhesion, inflammation, induction of fetal hemoglobin, coagulation, or platelet activation/aggregation are currently in clinical trials for the treatment of vaso-occlusive crises in SCD patients.35,42 In particular, preclinical and clinical studies with rivipansel (GMI-1070, a pan-selectin inhibitor) revealed that inhibition of leukocyte-endothelial cell interactions attenuates vaso-occlusion and improves survival in SCD mice,29 and reduces time to resolution of vaso-occlusive events and requirement for opioid analgesia in SCD patients.43 Nevertheless, complete inhibition of leukocyte-endothelial cell interactions could cause side effects by disrupting immune responses. We found that oral administration of ARQ 092 significantly, but not completely, inhibited cell-cell interactions in microvessels of TNF-α-challenged SCD mice. Importantly, when combined with hydroxyurea, ARQ 092 showed synergistic effects on acute vaso-occlusive events and improved survival of the mice. In addition, our studies demonstrated that 50–500 nM ARQ 092 is efficacious in inhibiting platelet and neutrophil functions. In cancer patients, the plasma concentration of ARQ 092 reached 2.6, 6.4, and 8.1 μM after oral administration of 20, 40, and 60 mg per day, respectively, for 15 days (unpublished data). Thus, a minimal oral dose of ARQ 092 may be sufficient to attenuate vaso-occlusive events in SCD patients. We reported that neutrophils and platelets isolated from SCD patients, compared to healthy donor cells, exhibit a significant increase in the basal level of AKT phosphorylation without altering its expression.12 Since the AKT signaling pathway is aberrantly activated in many types of cancers,44 our studies provide strong evidence that in addition to anti-cancer therapy, ARQ 092 has the potential to be developed for the treatment of acute vaso-occlusive episodes in SCD patients, possibly in combination with hydroxyurea.
Supplementary Material
Acknowledgements
The authors thank Dr. Lewis Hsu for his helpful comments. This work was in part supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL109439 and HL130028, JC), American Society of Hematology Bridge Fund (JC) and Scholar Award (JL), and American Heart Association postdoctoral (KK) and predoctoral fellowship (AT).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/2/246
References
- 1.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364(9442):1343–1360. [DOI] [PubMed] [Google Scholar]
- 2.Habara A, Steinberg MH. Minireview: genetic basis of heterogeneity and severity in sickle cell disease. Exp Biol Med (Maywood). 2016;241(7):689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 2013;122(24):3892–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potoka KP, Gladwin MT. Vasculopathy and pulmonary hypertension in sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cokic VP, Smith RD, Beleslin-Cokic BB, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest. 2003;111(2):231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nahavandi M, Wyche MQ, Perlin E, Tavakkoli F, Castro O. Nitric oxide metabolites in sickle cell anemia patients after oral administration of hydroxyurea; hemoglobinopathy. Hematology. 2000;5(4):335–339. [DOI] [PubMed] [Google Scholar]
- 7.Saleh AW, Hillen HF, Duits AJ. Levels of endothelial, neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1999;102(1):31–37. [DOI] [PubMed] [Google Scholar]
- 8.Almeida CB, Scheiermann C, Jang JE, et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood. 2012;120(14):2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barazia A, Li J, Kim K, Shabrani N, Cho J. Hydroxyurea with AKT2 inhibition decreases vaso-occlusive events in sickle cell disease mice. Blood. 2015;126(22):2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hidalgo A, Chang J, Jang JE, et al. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15(4):384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim K, Li J, Tseng A, Andrews RK, Cho J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood. 2015;126(16):1952–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Kim K, Hahm E, et al. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. J Clin Invest. 2014;124(4):1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Kim K, Barazia A, Tseng A, Cho J. Platelet-neutrophil interactions under thromboinflammatory conditions. Cell Mol Life Sci. 2015;72(14):2627–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23(10):1515–1527. [DOI] [PubMed] [Google Scholar]
- 15.Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010;9(3):487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. 2010;3(1):81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, De S, Damron DS, et al. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104(6):1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woulfe D, Jiang H, Morgans A, et al. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest. 2004;113(3):441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien KA, Stojanovic-Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118(15):4215–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood. 2010;115(21):4237–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Lorenzo A, Fernandez-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci USA. 2009;106(34):14552–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez-Hernando C, Ackah E, Yu J, et al. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6(6):446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MY, Luciano AK, Ackah E, et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci USA. 2014;111(35):12865–12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;19(11):1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nitulescu GM, Margina D, Juzenas P, et al. Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (Review). Int J Oncol. 2016;48(3):869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Y, Savage RE, Eathiraj S, et al. Targeting AKT1-E17K and the PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS One. 2015;10(10):e0140479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindhurst MJ, Yourick MR, Yu Y, et al. Repression of AKT signaling by ARQ 092 in cells and tissues from patients with Proteus syndrome. Sci Rep. 2015;5:17162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tolcher A, Harb W, Sachdev JC, et al. Results from a phase 1 study of ARQ 092, a novel pan AKT-inhibitor, in subjects with advanced solid tumors or recurrent malignant lymphoma. Eur J Cancer. 2015;51:S66. [Google Scholar]
- 29.Chang J, Patton JT, Sarkar A, et al. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood. 2010;116(10):1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lapierre JM, Eathiraj S, Vensel D, et al. Discovery of 3-(3-(4-(1-aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): an orally bioavailable, selective, and potent allosteric AKT inhibitor. J Med Chem. 2016;59(13):6455–6469. [DOI] [PubMed] [Google Scholar]
- 31.Gwilt PR, Tracewell WG. Pharmacokinetics and pharmacodynamics of hydroxyurea. Clin Pharmacokinet. 1998;34(5):347–358. [DOI] [PubMed] [Google Scholar]
- 32.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17(11): 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Kim K, Hahm E, et al. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. J Clin Invest. 2014;124(4):1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akaike T, Yoshida M, Miyamoto Y, et al. Antagonistic action of imidazolineoxyl N-oxides against endothelium-derived relaxing factor/.NO through a radical reaction. Biochemistry. 1993;32(3):827–832. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin H, Stojanovic A, Hay N, Du X. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. 2008;111(2):658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falanga A, Russo L, Verzeroli C. Mechanisms of thrombosis in cancer. Thromb Res. 2013;131(Suppl 1):S59–62. [DOI] [PubMed] [Google Scholar]
- 38.Dasgupta T, Fabry ME, Kaul DK. Antisickling property of fetal hemoglobin enhances nitric oxide bioavailability and ameliorates organ oxidative stress in transgenic-knockout sickle mice. Am J Physiol Regul Integr Comp Physiol. 2010;298(2): R394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–1389. [DOI] [PubMed] [Google Scholar]
- 40.Ackah E, Yu J, Zoellner S, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115(8):2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauer PM, Fulton D, Boo YC, et al. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278(17):14841–14849. [DOI] [PubMed] [Google Scholar]
- 42.Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood. 2016;127(7):810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125(17):2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.