

Abstract
Autoimmune lymphoproliferative syndrome is a rare genetic disorder characterized by defective FAS-mediated apoptosis, autoimmune disease, accumulation of mature T-cell receptor alpha/beta positive, CD4 and CD8 double-negative T cells and increased risk of lymphoma. Despite frequent hematologic abnormalities, literature is scarce regarding the bone marrow pathology in autoimmune lymphoproliferative syndrome. We retrospectively reviewed 3l bone marrow biopsies from a cohort of 240 patients with germline FAS mutations. All biopsies were performed for the evaluation of cytopenias or to rule out lymphoma. Clinical information was collected and morphological, immunohistochemical, flow cytometric and molecular studies were performed. Bone marrow lymphocytosis was the predominant feature, present in 74% (23/31) of biopsies. The lymphoid cells showed several different patterns of infiltration, most often forming aggregates comprising T cells in 15 cases, B cells in one and a mixture of T and B cells in the other seven cases. Double-negative T cells were detected by immunohistochemistry in the minority of cases (10/31; 32%); significantly, all but one of these cases had prominent double-negative T-lymphoid aggregates, which in four cases diffusely replaced the marrow space. One case showed features of Rosai-Dorfman disease, containing scattered S-100+ cells with emperipolesis and double-negative T cells. No clonal B or T cells were detected by polymerase chain reaction in any evaluated cases. Classical Hodgkin lymphoma was identified in three cases. Our results demonstrate that infiltrates of T cells, or rarely B cells, can be extensive in patients with autoimmune lymphoproliferative syndrome, mimicking lymphoma. A multi-modality approach, integrating clinical, histological, immunohistochemical as well as other ancillary tests, can help avoid this diagnostic pitfall. This study is registered at Clinicaltrials.gov ID # NCT00001350
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is a rare genetic disorder which typically presents in childhood with chronic non-malignant lymphadenopathy, splenomegaly, hypergammaglobulinemia, autoimmune multilineage cytopenias and increased risk of lymphoma.1–3 A pathognomonic feature of ALPS, which is also considered an important mediator of the disease, is the accumulation of T-cell receptor-alpha/beta positive, CD4 and CD8 double-negative T lymphocytes (DNT cells).4,5 ALPS is caused by defective FAS-mediated lymphocyte apoptosis, which in turn leads to disruption of lymphocyte homeostasis and immune dysregulation.6,7 The majority of ALPS patients have germline mutations of the FAS gene (ALPS-FAS).7,8 Others may acquire a somatic FAS mutation in lymphocyte subsets9,10 and a small number of patients carry mutations involving other apoptotic pathway genes, including FASL and CASPASE 10.11–15 Patients with ALPS, particularly those with FAS mutations, generally have a good prognosis for survival to adulthood. However, recent studies suggest that with longer follow-up, the relative risk of lymphoma is significantly higher than previously reported.16
The clinical manifestations of ALPS, such as lymphadenopathy, splenomegaly and cytopenias, are not specific and can also be found in leukemia, lymphoma and other non-malignant lymphoproliferative disorders. Thus, the successful diagnosis of ALPS requires a high index of suspicion and a multifaceted approach, integrating clinical information, laboratory parameters and tissue morphology.17–19 Immunophenotyping is essential and genetic testing is necessary for diagnostic confirmation. In previous studies, we described the histopathological and immunophenotypic features seen in lymph nodes from ALPS patients.20 Histologically, lymph nodes show variable paracortical expansion with increased DNT cells.20 In addition, features of Rosai-Dorfman disease have been identified in a subset of the lymph nodes.21 While in some cases the expansion of DNT cells can be extensive, mimicking lymphoma, in other cases, the DNT cells constitute a relatively minor proportion and are difficult to identify. It is not uncommon for the diagnosis to be delayed in patients with unusual clinical presentations and/or atypical pathology.16 In addition to lymph node biopsies, bone marrow biopsies are sometimes performed in ALPS patients for the evaluation of worsening cytopenias or to rule out lymphoma. While lymph node manifestations of ALPS are well recognized, bone marrow findings have not been described.
Here, we present findings from bone marrow aspirate and biopsy specimens from a large series of ALPS patients with germline FAS mutations (ALPS-FAS). We highlight the clinicopathological features of these cases and emphasize the pitfalls and importance of using a multi-modal approach to establish the correct diagnosis.
Methods
Patients
The National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), ALPS program has studied the natural history of ALPS and identified 240 patients with FAS mutations who had clinical symptoms of ALPS.16 Out of these 240 patients, we identified 31 patients who had bone marrow biopsies obtained between 2000 and 2013. All marrow biopsies were taken prior to starting immunosuppressive treatments for cytopenias. Of the 31 bone marrow samples, 13 cases were from procedures undertaken at the NIH Clinical Center and 18 cases were samples sent from outside hospitals to the NIH for evaluation. Corresponding peripheral blood smears, flow cytometric and/or molecular genetic data were obtained when available. Medical records were retrospectively reviewed. Clinical and laboratory data including age, gender, symptoms, date of onset, laboratory parameters including complete blood counts, DNT cell counts, vitamin B12, interleukin-10 and sFasL levels, treatment regimens and follow-up information were collected. All patients and parents of minors signed consent documents for participation in the ALPS Natural History Study under the NIH/NIAID Institutional Review Board-approved research protocol.
Histological and immunohistochemical studies
All peripheral blood smears, bone marrow aspirates and biopsies were processed and stained according to standard protocols. Immunohistochemical and special stains for CD3 (Ventana, clone 2GV6), CD4 (Ventana, clone SP35), CD8 (Ventana, clone SP57), CD20 (Ventana, clone L26), CD45RO (Ventana, clone UCHL-1), S-100 (Ventana, polyclonal), CD15 (Ventana, clone MMA), CD21 (Dako, clone 1F8), CD30 (Ventana, clone Ber-H2), CD138 (Cell Marque, clone B-A38), kappa (Ventana, polyclonal), and lambda (Ventana, polyclonal) were performed on paraffin sections using a Ventana Benchmark Ultra automated platform (Ventana Medical Systems, AZ, USA) following the manufacturer’s instruction. Evaluation of DNT cell infiltration was performed semiquantitatively in serial sections, with examination of both nodular and diffuse lymphoid infiltrates as well as areas showing well preserved hematopoiesis. The lymphoid infiltrates were considered diffuse if there was an effacement of bone marrow fat cells and interstitial if there was a preservation of fat cells. Very extensive nodular cases were considered as diffuse. Reticulin fibrosis was graded using a previously published scheme.22 Cellularity was evaluated with regard to the age of the patients and included trilineage hematopoiesis as well as lymphoid infiltrates.
Molecular studies
DNA was extracted from bone marrow aspirates or formalin-fixed paraffin-embedded tissue using the QIAcube extraction system. Polymerase chain reaction (PCR) was performed to detect immunoglobulin (IGH and IGK loci) or T-cell receptor (TRG locus) gene rearrangements in cases in which B- or T-cell lymphoma was suspected. For the IGH locus, two separate reactions were performed targeting framework II and III sequences within the joining region, as described by Ramasamy et al.23 Two additional reactions were performed for the IGK locus using the Biomed II primer set described by van Dongen et al.24 For the TRG locus, a single multiplexed PCR reaction was performed, as described by Lawnicki et al.25 Following the PCR amplification, fluorescence-labeled PCR products were analyzed by capillary electrophoresis on an ABI 3130xl Genetic Analyzer, and electropherograms were analyzed using GeneMapper software version 4.0 (ABI).
Results
Clinical characteristics
The clinical features of the study cohort are summarized in Table 1. The cohort consisted of 31 patients (23 males and 8 females). The median age at onset of disease was 2 years (range, 0.1 to 28 years), while the median age at diagnosis was 10 years (range, 1 to 40 years). The median age at bone marrow biopsy was 15.5 years (range, 0.5 to 50 years). Three patients had a history of Rosai-Dorfman disease; none of the patients had a known past history of lymphoma. Of the 31 patients, many (n=17) had undergone splenectomy. All patients harbored heterozygous germline FAS mutations.
Table 1.
Clinical features of ALPS patients.
All patients had a history of single or multilineage cytopenias. Lymphadenopathy and splenomegaly were notable in all patients. Upon presentation, nearly all patients (30 out of 31) had elevated circulating DNT cell counts, ranging from 2.0% to 30.5% with a median percentage of 7.9%. In addition, nearly all patients had significantly elevated ALPS associated biomarkers, including serum vitamin B12, interleukin-10 and sFasL (Table 1). At the time of their bone marrow biopsy, 79% of the patients (19/23 evaluable patients) had cytopenias, most commonly anemia (52%) and thrombocytopenia (52%), while 22% had peripheral lymphocytosis and 22% had monocytosis.
Histology, immunophenotype and molecular studies
The bone marrow biopsies were hypercellular in eight cases, normocellular in 21 and hypocellular in four cases (Table 2). Seven cases showed erythroid hyperplasia and five demonstrated increased megakaryocytes. All cases were routinely examined for myelodysplasia. Occasional hypolobated megakaryocytes were noted in six cases, four of which were associated with megakaryocytic hyperplasia. Focal mild megakaryocytic clustering was noted in one case. There was no overt evidence of dysplasia. There was no evidence of hemophagocytosis in any of the bone marrow specimens.
Table 2.
Pathological features of bone marrows from ALPS patients.
In nearly half of the biopsies (16/33), mild eosinophilic hyperplasia was seen, while concurrent peripheral eosinophilia was present in only one case.
Reticulin staining demonstrated mild fibrosis (grade 1–2/4) in most cases assessed (8/l2). Moderate to marked fibrosis (grade 3–4/4) was identified in four cases (4/12), all with prominent lymphoid aggregates.
Bone marrow lymphocytosis was a common finding and was seen in more than two-thirds of cases (23, 70%). A summary of the morphological patterns and immunophenotypes of the lymphoid infiltrates is presented in Table 2. Of the 23 cases, 21 displayed multiple nodular lymphoid (15 cases) or lymphohistiocytic (6 cases) aggregates, and the remaining two cases exhibited interstitial lymphoid infiltrates. Immunostains showed that the lymphoid infiltrates were T-cell predominant in 15 cases, B-cell predominant in one case and seven cases were formed of mixed populations of T and B cells. A population of CD4 and CD8 double-negative and/or CD45RO-negative T cells (DNT cells) was identified in a total of ten cases (10 out of 23) by immunohistochemistry. Significantly, all but one case showing prominent DNT cells contained prominent lymphoid aggregates.
In most cases, the lymphoid aggregates were small to medium-sized, well-circumscribed, distributed in non-paratrabecular locations and lacked cytological atypia. However, in four cases (4/21, cases 9, 18, 19, 23), there were extensive lymphoid infiltrates coalescing to diffusely replace normal marrow elements, mimicking a lymphoproliferative disorder. Aspirate smears from these four patients showed abundant lymphocytes with slightly enlarged nuclei, smooth to mildly irregular nuclear contours and dispersed chromatin. Of note, all four patients presented with marked peripheral lymphocytosis in addition to cytopenias; three of the four also demonstrated concurrent monocytosis.
Immunohistochemistry showed that the lymphoid infiltrates in three of these four cases (cases 9, 19, 23) consisted mainly of T cells with markedly increased DNT cells, as shown in Figure 1 (case 9). In one case (case 18, Figure 2), there were tight clusters of medium to large B cells accompanied by CD21-positive follicular dendritic cell mesh-works reminiscent of B-cell follicles, also admixed with numerous small T cells. In all four cases, T-cell gene rearrangement studies were performed and showed no evidence of a clonal T-cell population. Additionally, a B-cell clonality study was performed in case 18 and it, too, was negative for clonal rearrangements. Interestingly, in case 23 (Figure 3), in addition to increased DNT cells, there was a histiocytic infiltrate, most prominent around lymphoid aggregates. The infiltrate contained scattered S-l00-positive histiocytes that showed evidence of emperipolesis in the aspirate smear, hematoxylin & eosin and immunostained sections, features seen in Rosai-Dorfman disease. Staining for CD138, kappa and lambda light chains demonstrated mild polyclonal plasmacytosis. Of note, this patient had a prior history of Rosai-Dorfman disease involving multiple lymph nodes. There are two additional patients in this cohort who also had ALPS-related Rosai-Dorfman disease identified by lymph node biopsies; however, no features of Rosai-Dorfman disease were detected in their bone marrow specimens.
Figure 1.
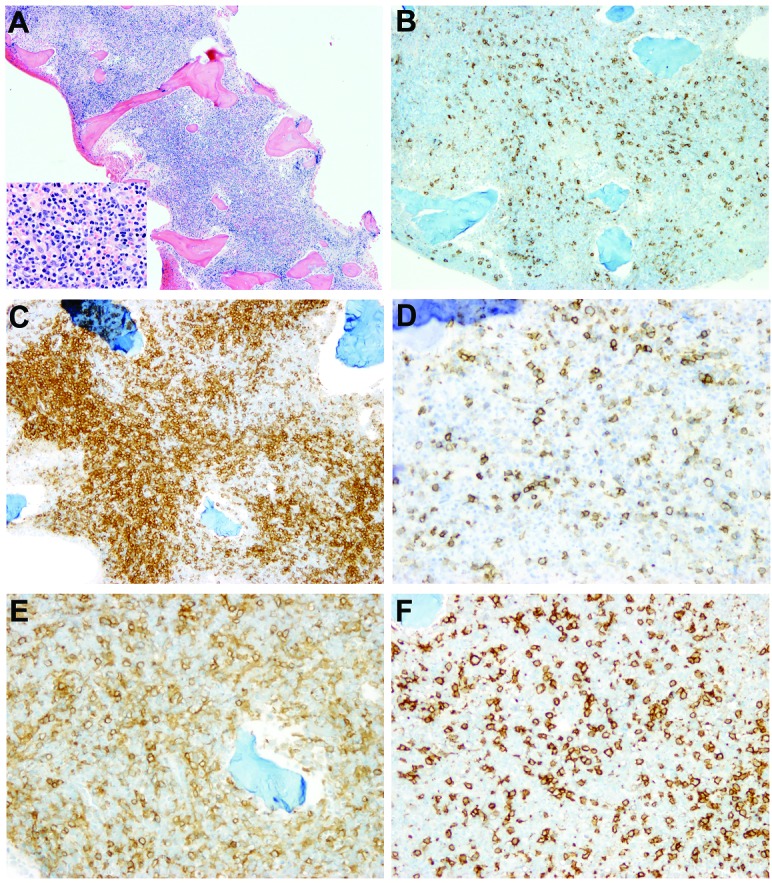
Extensive lymphoid infiltrates composed predominantly of T cells in a bone marrow with ALPS (case 9). (A) H&E, X 40 and X 500 (insert), (B) CD20, X 40, (C) CD3, X 40, (D) CD45RO, X 40, (E) CD4, X 40, (F) CD8, X 40. Note the markedly decreased expression of CD45RO in the T-cell infiltrates, consistent with the increase in DNT cells. The DNT cell population is less morphologically apparent using CD4 and CD8 staining.
Figure 2.
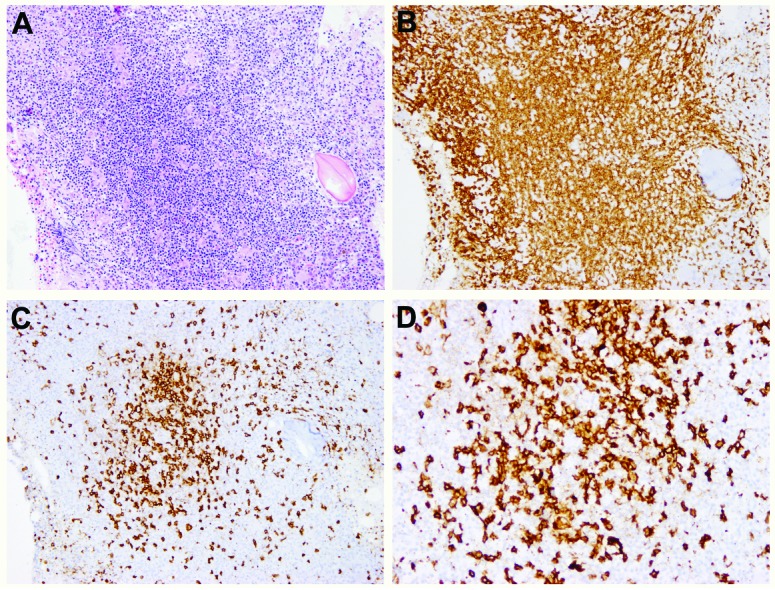
Nodular lymphoid infiltrates containing mixed T and B cells in a bone marrow with ALPS (case l8). (A) H&E, X 40, (B) CD3, X 40, (C) CD45RO, X 40, (D) CD20, X 200. Note the clusters of medium-sized to large B cells surrounded by numerous small T cells. There is loss of expression of CD45RO.
Figure 3.
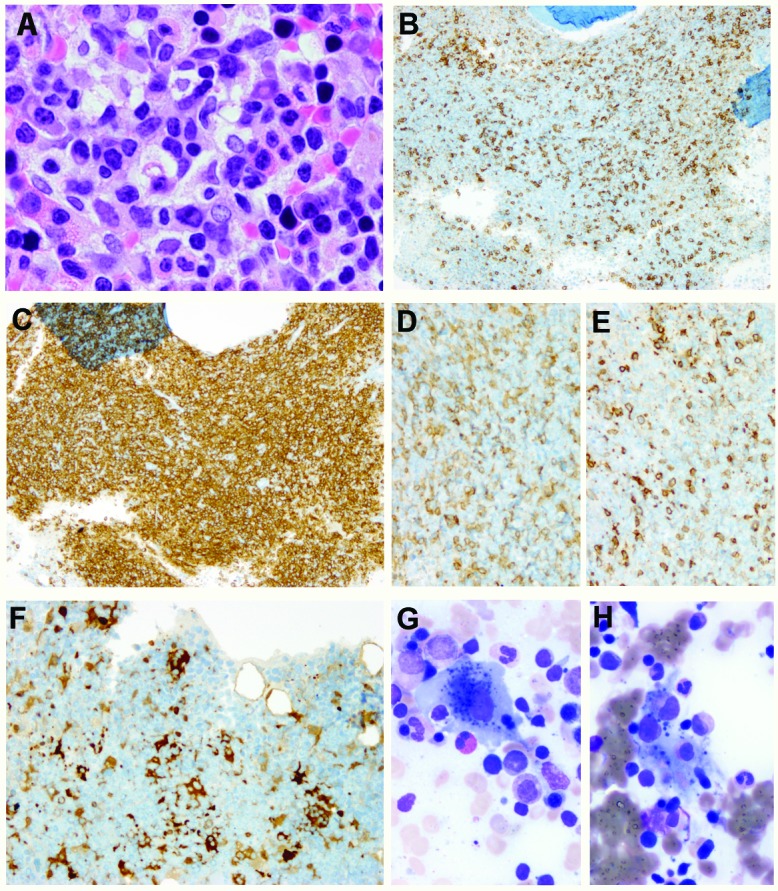
Lymphohistiocytic infiltrate with features of Rosai-Dorfman disease in a bone marrow with ALPS (case 23). (A) H&E, X 500, (B) CD20, X 40, (C) CD3, X 40, (D) CD4, X 100, (E) CD8, X 100, (F) Sl00, X 200, (G) and (H) Giemsa, X 500. Note the increased CD4 and CD8 double-negative T cells and histiocytes with emperipolesis.
The extent of CD3-positive lymphoid infiltrate in the bone marrow showed a statistically significant correlation with the number of DNT cells detected in the peripheral blood by flow cytometric analysis (P=0.04; Table 3). A correlation between DNT cells in the blood and bone marrow DNT cells detected by immunohistochemical analysis was also observed, but did not reach statistical significance.
Table 3.
Detection of DNT cells by immunohistochemisty in bone marrow biopsies correlates with the extent of T-lymphoid infiltrate and percent of peripheral blood DTN cells.

Of the two cases with interstitial lymphoid infiltrates, one case (case 11, Figure 4) exhibited a particularly pronounced proliferation of B cells throughout the marrow with very few intermixed T cells, leading to the suspicion of a B-cell lymphoma. However, the B cells did not show significant cytological atypia and lacked monoclonality by flow cytometry. Concurrent flow cytometric analysis demonstrated an expansion of maturing hematogones that co-expressed varying amounts of CD20, CD10, CD34 and TdT. Furthermore, no clonal B-cell gene rearrangements were detected by molecular studies and no lymphoma developed during a follow-up period of 9 years.
Figure 4.

Diffuse interstitial lymphoid infiltrates composed predominantly of B cells in a bone marrow with ALPS (case ll). (A) H&E, X l00, (B) CD3, X l00, (C) CD20, X l00.
Remarkably, among six cases with lymphohistiocytic infiltrates, three (cases 29, 30, 31) appeared unusually fibrotic and rare large atypical cells resembling Hodgkin/Reed Sternberg cells were identified amidst a variable number of inflammatory cells (Figure 5). The atypical cells were positive for CD30 and CD15, weakly positive for PAX5, but negative for CD20 and CD45, consistent with classical Hodgkin lymphoma. In situ hybridization for EBER was performed in two cases; in both cases, the Reed-Sternberg cells were EBER-positive, as well as LMP1-positive and EBNA2-negative. Of note, none of the three patients had a history of lymphoma; the bone marrow biopsy represented the first diagnostic tissue sample in these patients. Immunostains for DNT cells were also performed and no distinct DNT cell population was identified.
Figure 5.
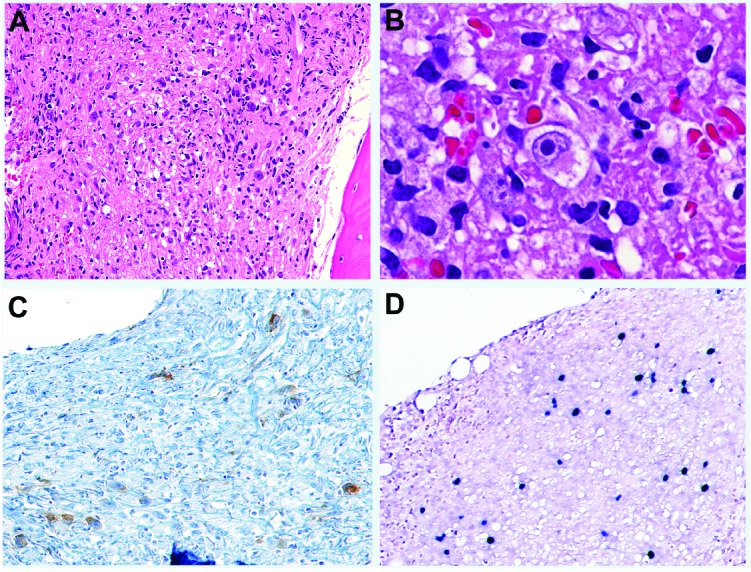
Classical Hodgkin lymphoma, EBV-positive, involving a bone marrow with ALPS. (A) H&E, X 100, (B) H&E, X 500, (C) CD30, X 100, (D) EBER ISH, X 100. Note the Reed-Sternberg cells positive for CD30 and EBER.
Clinical course and outcome
Clinical follow-up was available for all patients. The follow-up time ranged from 4 to 54 years, with a median of 15 years. Twenty-seven patients received multiple immunosuppressive treatment regimens consisting of corticosteroids, mycophenolate mofetil and rapamycin. Twenty-seven of the 31 patients were alive at the time of writing this manuscript. All three patients with classical Hodgkin lymphoma (CHL) died as a result of lymphoma. Additionally, one patient progressed to non-clonal, tumor-like growth of DNT cells with extensive involvement of bone marrow, lymph node and thymus, and died of bacterial sepsis and intracerebral hemorrhage.
Discussion
In this study, we report the spectrum of bone marrow pathology in ALPS patients with germline FAS mutations. To the best of our knowledge, this is the first report describing features of bone marrow pathology in ALPS. We demonstrate that lymphoid infiltrates are frequent and can be extensive, often containing a significant population of DNT cells, which may prompt consideration of a diagnosis of lymphoma.
Lymph nodes and bone marrow are the tissues most frequently examined pathologically in ALPS. The changes observed in bone marrow biopsies were most often secondary to lymphoid infiltration: 74% of the bone marrow biopsies contained lymphoid infiltrates that were nodular or diffuse and interstitial. The extent was variable from case to case. Although the majority of the lymphoid aggregates in these cases were small and well-circumscribed, large ill-defined lymphoid aggregates were occasionally found. Well-formed follicles with germinal centers were rare. In nearly half of the cases with lymphoid aggregates, DNT cells were identified using a panel of immunohistochemical markers including CD3, CD20, CD4, CD8 and CD45RO. The extent of lymphoid infiltrate in the bone marrow generally correlated with the number of DNT cells in peripheral blood. In rare cases with prominent B-cell germinal centers, distinct DNT cells were not identified by immunohistochemistry despite a large number of T cells in the marrow and a relatively high percentage of DNT cells in the peripheral blood.
Lymphoid aggregates are common findings in elderly patients.26,27 In young individuals, they are less common and are often associated with immune or inflammatory disorders, drug therapy or infections.28,29 In such cases the lymphoid aggregates are usually few in number and small.26 In our cohort, although the patients were predominantly children and adolescents, the frequency of lymphoid aggregates in the bone marrow was high. In four cases, the lymphoid infiltrates were particularly extensive, involving almost the entire marrow. Despite the morphological features suggesting lymphoma, immunohistochemistry and flow cytometry studies demonstrated the presence of a DNT cell population, consistent with ALPS. Furthermore, molecular studies in all four patients with extensive T-lymphoid infiltrates were negative for clonal rearrangements. Our results suggest that immunostaining for DNT cells in the bone marrow biopsy is a useful initial test to differentiate ALPS from other lymphoproliferative disorders. A multifaceted approach with judicious use of a variety of ancillary tests in combination with clinical correlation can help to establish the correct diagnosis and avoid pitfalls of lymphoma diagnosis and treatment.
Previous studies have shown that ALPS is associated with an increased risk of B-cell non-Hodgkin lymphoma and Hodgkin lymphoma.16,30 However, the reason for such propensity is not well understood. Several mechanisms have been proposed.30 One hypothesis is that lymphomagenesis in some of these patients may result from Epstein-Barr virus (EBV) infection exploiting defective T-cell surveillance resulting from impaired FAS-mediated apoptosis.31,32 For example, a rare case of EBV-positive lymphoma was reported in a patient with ALPS that was clinically heralded by progressively increasing EBV viremia.32 Previously, Price et al. documented ten cases of CHL among their cohort of 150 ALPS-FAS patients; six cases with known EBV status were EBV-positive. In our cohort, we identified three patients with CHL involving the marrow, two of whom were tested for EBV and both were positive. Overall it appears that CHL in patients with ALPS is often associated with EBV infection.
Case 23 is of particular interest: this patient displayed morphological and immunohistochemical features of Rosai-Dorfman disease in the bone marrow. Rosai-Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy (SHML), is a rare non-neoplastic histiocytic proliferation that may occur in lymph nodes and in a variety of extranodal sites, such as skin, nasal cavity, bone, salivary gland, central nervous system, etc.33,34 However, bone marrow involvement by SHML is rare.35–37 In our previous report, we identified histological features of SHML in lymph nodes from 40% of ALPS patients, suggesting an association between SHML and ALPS.21 While none of those patients in the previous study had clinical evidence of SHML-like proliferations in extranodal sites,21 our current case represents the first evidence of extranodal SHML-like features in the bone marrow of a patient with ALPS.
As regards other components in the bone marrow, trilineage hematopoiesis was mostly unaffected in our patients, except in those with space-occupying lymphoid infiltrates. Mild erythroid hyperplasia and granulocytic hypoplasia were found occasionally. Slight atypia in megakaryocytic maturation was noted in rare cases, although no cases fulfilled the criteria for myelodysplasia. Consistent with previous reports that eosinophilia is a common finding in ALPS,36 bone marrow evidence of eosinophilic hyperplasia was present in almost 50% of cases. Unlike the lymph nodes, which frequently demonstrated polyclonal plasmacytosis in ALPS patients,20 only a small subset of the bone marrows (19%) showed a slight increase of plasma cells, none over 5% of the nucleated cells.
In summary, we have described the pathological features of bone marrow in ALPS, which have not been reported previously. Generally, the bone marrow changes appear to recapitulate the morphological findings seen in the lymph nodes, with variable DNT lymphoid infiltrates, occasional histiocytic proliferation, and frequent eosinophilia and plasmacytosis. Our study demonstrates the need for use of immunohistochemistry as well as other ancillary studies to differentiate ALPS DNT lymphoid infiltrates from neoplastic lymphoid infiltrates. Recognizing these morphological features in the appropriate clinical setting can guide further laboratory evaluation and appropriate clinical management.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/2/364
References
- 1.Sneller MC, Straus SE, Jaffe ES, et al. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90(2):334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sneller MC, Wang J, Dale JK, et al. Clinical, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89(4):1341–1348. [PubMed] [Google Scholar]
- 3.Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999;130(7):591–601. [DOI] [PubMed] [Google Scholar]
- 4.Rieux-Laucat F. Inherited and acquired death receptor defects in human autoimmune lymphoproliferative syndrome. Curr Dir Autoimmun. 2006;9:18–36. [DOI] [PubMed] [Google Scholar]
- 5.Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr. 2012;24(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995; 81(6):935–946. [DOI] [PubMed] [Google Scholar]
- 7.Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–1349. [DOI] [PubMed] [Google Scholar]
- 8.Martin DA, Zheng L, Siegel RM, et al. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci USA. 1999;96(8):4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzelova E, Vonarbourg C, Stolzenberg MC, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351(14):1409–1418. [DOI] [PubMed] [Google Scholar]
- 10.Dowdell KC, Niemela JE, Price S, et al. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood. 2010;115(25):5164–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bi LL, Pan G, Atkinson TP, et al. Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome (ALPS) type Ib. BMC Med Genet. 2007;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clini Invest. 1996;98(5):1107–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del-Rey M, Ruiz-Contreras J, Bosque A, et al. A homozygous Fas ligand gene mutation in a patient causes a new type of autoimmune lymphoproliferative syndrome. Blood. 2006;108(4):1306–1312. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Zheng L, Lobito A, et al. Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98(1):47–58. [DOI] [PubMed] [Google Scholar]
- 15.Zhu S, Hsu AP, Vacek MM, et al. Genetic alterations in caspase-10 may be causative or protective in autoimmune lymphoproliferative syndrome. Hum Genet. 2006;119(3): 284–294. [DOI] [PubMed] [Google Scholar]
- 16.Price S, Shaw PA, Seitz A, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123(13):1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudman Spergel A, Walkovich K, Price S, et al. Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis. Pediatrics. 2013; 132(5):1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iyengar SR, Ebb DH, Yuan Q, Shailam R, Bhan AK. Case records of the Massachusetts General Hospital. Case 27-2013. A 6.5-month-old boy with fever, rash, and cytopenias. N Engl J Med. 2013; 369(9):853–863. [DOI] [PubMed] [Google Scholar]
- 19.Nomura K, Kanegane H, Otsubo K, et al. Autoimmune lymphoproliferative syndrome mimicking chronic active Epstein-Barr virus infection. Int J Hematol. 2011; 93(6):760–764. [DOI] [PubMed] [Google Scholar]
- 20.Lim MS, Straus SE, Dale JK, et al. Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol. 1998;153(5):1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maric I, Pittaluga S, Dale JK, et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol. 2005;29(7):903–911. [DOI] [PubMed] [Google Scholar]
- 22.Manoharan A, Horsley R, Pitney WR. The reticulin content of bone marrow in acute leukaemia in adults. Br J Haematol. 1979;43(2):185–190. [DOI] [PubMed] [Google Scholar]
- 23.Ramasamy I, Brisco M, Morley A. Improved PCR method for detecting monoclonal immunoglobulin heavy chain rearrangement in B cell neoplasms. Am J Clin Pathol. 1992;45(9):770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. [DOI] [PubMed] [Google Scholar]
- 25.Lawnicki LC, Rubocki RJ, Chan WC, Lytle DM, Greiner TC. The distribution of gene segments in T-cell receptor gamma gene rearrangements demonstrates the need for multiple primer sets. J Mol Diagn. 2003;5(2): 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thiele J, Zirbes TK, Kvasnicka HM, Fischer R. Focal lymphoid aggregates (nodules) in bone marrow biopsies: differentiation between benign hyperplasia and malignant lymphoma–a practical guideline. J Clin Pathol. 1999;52(4):294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girodon F, Favre B, Carli PM, et al. Minor dysplastic changes are frequently observed in the bone marrow aspirate in elderly patients without haematological disease. Clin Lab Haematol. 2001;23(5):297–300. [DOI] [PubMed] [Google Scholar]
- 28.Kreft A, Weber A, Springer E, Hess G, Kirkpatrick CJ. Bone marrow findings in multicentric Castleman disease in HIV-negative patients. Am J Surg Pathol. 2007;31(3): 398–402. [DOI] [PubMed] [Google Scholar]
- 29.Rosenthal NS, Farhi DC. Bone marrow findings in connective tissue disease. Am J Clin Pathol. 1989;92(5):650–4. [DOI] [PubMed] [Google Scholar]
- 30.Straus SE, Jaffe ES, Puck JM, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98(1): 194–200. [DOI] [PubMed] [Google Scholar]
- 31.Wakiguchi H. Overview of Epstein-Barr virus-associated diseases in Japan. Crit Rev Oncol Hematol. 2002;44(3):193–202. [DOI] [PubMed] [Google Scholar]
- 32.Pace R, Vinh DC. Autoimmune lymphoproliferative syndrome and Epstein-Barr virus-associated lymphoma: an adjunctive diagnostic role for monitoring EBV viremia? Case Reports Immunol. 2013; 2013:245893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Venkataraman G, McClain KL, Pittaluga S, Rao VK, Jaffe ES. Development of disseminated histiocytic sarcoma in a patient with autoimmune lymphoproliferative syndrome and associated Rosai-Dorfman disease. Am J Surg Pathol. 2010;34(4):589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neven B, Magerus-Chatinet A, Florkin B, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011;118(18): 4798–4807. [DOI] [PubMed] [Google Scholar]
- 35.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7(1):19–73. [PubMed] [Google Scholar]
- 36.Kim YJ, Dale JK, Noel P, et al. Eosinophilia is associated with a higher mortality rate among patients with autoimmune lymphoproliferative syndrome. Am J Hematol. 2007;82(7):615–624. [DOI] [PubMed] [Google Scholar]
- 37.Huang Q, Chang KL, Weiss LM. Extranodal Rosai-Dorfman disease involving the bone marrow: a case report. Am J Surg Pathol. 2006;30(9):1189–1192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.