

Abstract
Histone methylation and demethylation regulate B-cell development, and their deregulation correlates with tumor chemoresistance in diffuse large B-cell lymphoma, limiting cure rates. Since histone methylation status correlates with disease aggressiveness and relapse, we investigated the therapeutic potential of inhibiting histone 3 Lys27 demethylase KDM6B, in vitro, using the small molecule inhibitor GSK-J4. KDM6B is overexpressed in the germinal center B-cell subtype of diffuse large B-cell lymphoma, and higher KDM6B levels are associated with worse survival in patients with diffuse large B-cell lymphoma treated with R-CHOP. GSK-J4-induced apoptosis was observed in five (SU-DHL-6, OCI-Ly1, Toledo, OCI-Ly8, SU-DHL-8) out of nine germinal center B-cell diffuse large B-cell lymphoma cell lines. Treatment with GSK-J4 predominantly resulted in downregulation of B-cell receptor signaling and BCL6. Cell lines expressing high BCL6 levels or CREBBP/EP300 mutations were sensitive to GSK-J4. Our results suggest that B-cell receptor-dependent downregulation of BCL6 is responsible for GSK-J4-induced cytotoxicity. Furthermore, GSK-J4-mediated inhibition of KDM6B sensitizes germinal center B-cell diffuse large B-cell lymphoma cells to chemotherapy agents that are currently utilized in treatment regimens for diffuse large B-cell lymphoma.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is an aggressive form of non-Hodgkin lymphoma with an initial tumor response rate of 90% but a relapse rate of 40% at 5 years.1,2 DLBCL can be classified into germinal center B-cell-like (GCB) and activated B-cell-like (ABC) DLBCL based on distinct gene expression patterns.3 Although the 5-year survival rate of GCB-DLBCL patients given conventional R-CHOP treatment is superior, more than 30% of patients are not cured with initial chemotherapy.2 Histone methylation has been shown to correlate with disease aggressiveness, chemoresistance, and relapse in DLBCL.4–6 Furthermore, expression of several important proteins, such as BCL6, is also regulated by methylation status.7,8 BCL6 is essential for GCB-cell development, as it is critical to cell proliferation, suppression of the DNA damage response during antibody class switching, and acquisition of somatic hypermutations.9,10 A high level of BCL6 expression is consistently observed in GCB-DLBCL.3,11 Therefore, several approaches to inhibition of BCL6 have been exploited, such as targeting the BTB domain of BCL6, or using histone deacetylase inhibitors.12,13 In addition, tonic B-cell receptor (BCR) signaling, essential for the survival of B cells, also regulates BCL6 levels and approaches that target BCR signaling may, therefore, also have the potential to target BCL6.14
Epigenetic reprogramming during B-cell development is important for proper functioning of B cells.15 Histone methylation governs the outcomes of epigenetic reprogramming during B-cell activation and memory B-cell generation.16,17 Aberrant histone methylation or demethylation can result in inappropriate transcriptional silencing, which can drive chemoresistance and malignant transformation.18 An analysis of histone methylation in DLBCL samples suggests that aberrant methylation increases with disease aggressiveness.4 Importantly, abnormal methylation patterns in DLBCL are not randomly distributed, but rather are associated with specific genome regions that depend on the activity of target-specific transcriptional regulators.4
Lysine demethylase 6B (KDM6B) is induced by various stimuli such as the Epstein-Barr virus (EBV), while another lysine demethylase, KDM6A, is constitutively expressed, but both influence the methylation status of GCB cells.19 KDM6B expression increases in B-cell subsets with progressive stages of differentiation, and gene expression profiling shows that KDM6B transcriptional targets in GCB cells are significantly enriched for those differentially expressed during memory and plasma-cell differentiation, clearly implicating KDM6B in antigen-activated B-cell differentiation.19 KDM6B knockout mice die in utero, because of incomplete development of breathing control.20,21 Conditional knockout of KDM6B in the germinal center compartment reduces the number and size of B cells clearly indicating the role of this protein in germinal center-cell proliferation.19 Given the key role demethylation plays in the regulation of normal and malignant B cells, we explored the therapeutic potential of selective KDM6B inhibition in DLBCL. Our results suggest that targeting KDM6B with the small molecule inhibitor GSK-J4 has the potential to render DLBCL sensitive to numerous chemotherapeutic agents.
Methods
Cell culture and agents
DLBCL cell lines Karpas422, OCI-Ly1, and OCI-Ly8 (gifts from Dr. Riccardo Dalla-Favera) were grown in Iscove’s modified Dulbecco’s medium supplemented with 10% fetal bovine serum. The OCI-Ly4 (also a gift from Dr. Riccardo Dalla-Favera) was grown in Iscove’s modified Dulbecco’s medium supplemented with 10% human serum. DB, Pfeiffer, Toledo, SU-DHL-6, and SU-DHL-8, (purchased from American Type Cell Culture, Manassas, VA, USA) were grown in RPMI supplemented with 10% fetal bovine serum. GSK-J4 and its inactive analogue GSK-J5 (R&D Systems, Minneapolis, MN, USA) were used as KDM6B inhibitors.
Bioinformatic analysis
The Compagno lymphoma microarray dataset (GSE12195) was used to analyze KDM6B expression using Oncomine.22,23 Values of KDM6B expression were extracted for centroblast cells and DLBCL samples. Expression data values were plotted with prism (GraphPad Prism, GraphPad Software, Inc., La Jolla, CA, USA).
The correlation of survival outcome with KDM6B levels in DLBCL patients treated with R-CHOP was analyzed from a previously published gene expression dataset (GSE10846).24 Bioinformatics analysis was performed using PPISURV, a novel online tool that correlates gene expression with cancer survival rates using publicly available data (http://www.bioprofiling.de/PPISURV).25 PPISURV exploits only rank information from expression data sets. Gene expression rank reflects relative mRNA expression levels and is more consistent as it requires no normalization, and thus introduces no normalization bias. Samples were dichotomized by expression rank into “low expression” and “high expression” groups, corresponding to patients with an expression rank below or above the average expression rank across the data set, respectively. These groups along with survival information were used to find any statistical differences in survival outcome. The R statistical package was used to perform survival analyses26 and to draw Kaplan-Meier plots.
Cell proliferation
Cell proliferation was analyzed using WST-1 reagent (Roche, Indianapolis, IN, USA) as described previously.27 Briefly, cells were seeded in 96-well plates (10,000 cells per 100 μL) and treated with the indicated concentrations of drugs, such as GSK-J4, for 24 h. The metabolic viability of cells was evaluated with addition of 20 μL of WST-1 reagent in each well. Plates were incubated for 2 h and absorbance at 596 nm was measured by a VICTOR3 V plate reader (PerkinElmer, Waltham, MA, USA). Data were analyzed and reported as percentages of buffer-treated controls.
Apoptosis
Apoptosis was determined by the sub-G1 cell population (<2N) as described previously.28,29 Briefly, cell pellets were stained with a hypotonic solution containing 40 μg/mL of propidium iodide, 0.1% sodium citrate, and 0.1% Triton X-100 for 2 h. The DNA content in cell nuclei was subsequently analyzed with a flow cytometer (FACS Calibur; BD Biosciences, San Diego, CA, USA) and FlowJo software (Tree Star, Ashland, OR, USA).
Demethylase activity assay
KDM6B activity was measured using the Epigenase KDM6B/UTX demethylase activity assay calorimetric kit (Epigentek, Farmingdale, NY, USA). Briefly, GSK-J4-treated and buffer-treated control cells were harvested at the indicated times. Cytoplasmic and nuclear fractions were extracted and equal amounts of protein lysates were incubated with H3K27 substrate and assay buffer for 90 min in assay plates. KDM6B-mediated demethylated substrate products were detected following incubation with a specific capture antibody. Plates were washed and incubated with detection antibody for 30 min at room temperature. Plates were washed twice before 100 μL developer solution was added, and the reaction was stopped when positive control wells turned medium blue. Absorbance (450 nm) was measured using a VICTOR3 V plate reader (PerkinElmer). Data were analyzed and presented as GSK-J4 treatment-induced change in KDM6B activity [optical density(OD)/min/mg] using the following formula:
B-cell receptor analysis
CRISPR/Cas9 technology30 was used for knockout of the BCR. We designed three different target sites within the second exon of the immunoglobulin heavy chain μ gene (IGHM) coding for the constant region of the BCR heavy chain: IGHM C4 – CCCCCGCAAGTCCAAGCTCATCT, IGHM C5 – CAGGTGTCCTGGCTGCGCGAGGG, and IGHM C6 – ACCTGCCGCGTGGATCACAGGGG (sequences from 5’ to 3’ at the coding strand). Oligos with these sequences were cloned31 into the bicistronic expression vector px33030 coding for hSpCas9 endonuclease as well as subgenomic RNA. We used the Neon Transfection System (Life Technologies, Carlsbad, CA, USA) to electroporate px330 plasmids (12 μg per 1×106 cells) and subsequently immunostained with goat anti-human IgM pAb (Life Technologies, H15101) to detect BCR-positive and -negative fractions using a flow cytometer (LSR Fortessa; BD Biosciences, San Diego, CA, USA). Data were analyzed with FlowJo software (Tree Star). To determine the treatment response, transfected pools of cells were treated with different concentrations of GSK-J4 and immunostained to analyze the fraction of BCR-positive and -negative cells. Sphero Polystyrene Particles (6.8 μm; Spherotech, Lake Forest, IL, USA) were used for absolute cell number quantification of BCR wildtype and BCR knockout cells in mixed cultures.
Western blots
Western blots were performed using previously described protocols.32 Briefly, cells were harvested by centrifugation, washed with ice-cold phosphate-buffered saline and lysed with RIPA cell lysis buffer (Cell Signaling, Danvers, MA, USA). Protein concentration was determined by the Bradford assay (BioRad, Hercules, CA, USA) according to the manufacturer’s protocol. The primary antibodies were anti-human BCL6, BCOR, IRF4, pBTKTyr223, pCD79ATyr182, and pPLCϒTyr759 (1:1000; Cell Signaling). Anti-human H3K27(me)3, H3K27(me)1, H3 (1:500, Active Motif, Carlsbad, CA, USA), and horseradish peroxidase-labeled anti-human β-actin (1:40,000; Sigma-Aldrich, St. Louis, MO, USA) were used to compare loading of individual lanes. Horseradish peroxidase-labeled goat anti-rabbit and goat anti-mouse secondary antibodies were obtained from Jackson Laboratory (1:5,000; Bar Harbor, ME, USA). The intensity of protein bands was quantified through densitometry using Image J software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Experimental data are reported as means or medians with standard deviation, unless otherwise indicated. Differences between groups were calculated using the two-tailed Student t-test (GraphPad Prism). P values <0.05 were considered statistically significant.
Results
Demethylase KDM6B is overexpressed in diffuse large B-cell lymphoma
Bioinformatic analysis of microarray-based gene expression data (GSE12195),22,23 extracted from the Oncomine database, indicated that expression levels of KDM6B in samples from patients with GCB-DLBCL (n=9) were significantly higher (P<0.01) than those in samples from patients with ABC-DLBCL (n=17) or in normal germinal center centroblast B cells (n=7) (Figure 1A). Further analysis using the bioinformatic tool PPISURV considered whether KDM6B expression correlated with survival outcome after frontline therapy.25 In a cohort of 414 patients consisting of 181 patients on first-time therapy with CHOP chemotherapy and 233 treated with R-CHOP with a median follow-up of 2.8 years (GSE10846),24 patients with high expression levels (top 50%) of KDM6B had lower survival rates compared to patients expressing low levels of KDM6B (P<0.000489) (Figure 1B). The survival rate at 50 months was 48% for high KDM6B expressers and 71% for low KDM6B expressers. Furthermore, high KDM6B expression correlated with poor survival outcome in R-CHOP-treated (P<0.001) but not in CHOP-treated GCB-DLBCL (Figure 1C). CD20 and KDM6B are known to target NF-κB survival signaling, which may be the reason for differences in survival outcome between the two groups.19,33–35 These results indicate that KDM6B expression is associated with poor survival, suggesting a potential therapeutic benefit of its inhibition in DLBCL.
Figure 1.
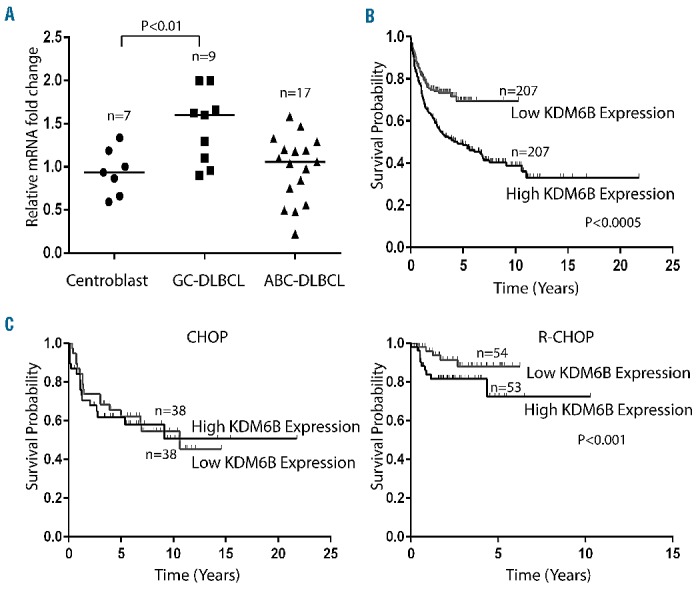
Demethylase KDM6B is overexpressed in DLBCL (A) Gene expression analysis of KDM6B in primary DLBCL samples (GSE 12195). Straight bars represent the median. The difference in KDM6B expression between GCB-DLBCL and normal centroblasts was statistically significant (P<0.01) (B) Kaplan-Meier plot of DLBCL patients (GSE10846) with low and high expression of KDM6B and survival outcome. Patients with higher KDM6B expression (top 50%) had a significantly lower survival rate (P<0.0005). (C) Kaplan-Meier plot of CHOP-treated (left panel) and R-CHOP-treated (right panel) GCB-DLBCL patients (GSE10846) with low and high expression of KDM6B and survival outcome. R-CHOP-treated GCB-DLBCL patients with higher KDM6B expression (top 50%) had a significantly lower survival rate (P<0.001), while no significant differences were found in CHOP-treated DLBCL patients.
Sensitivity of diffuse large B-cell lymphoma cell lines to GSK-J4
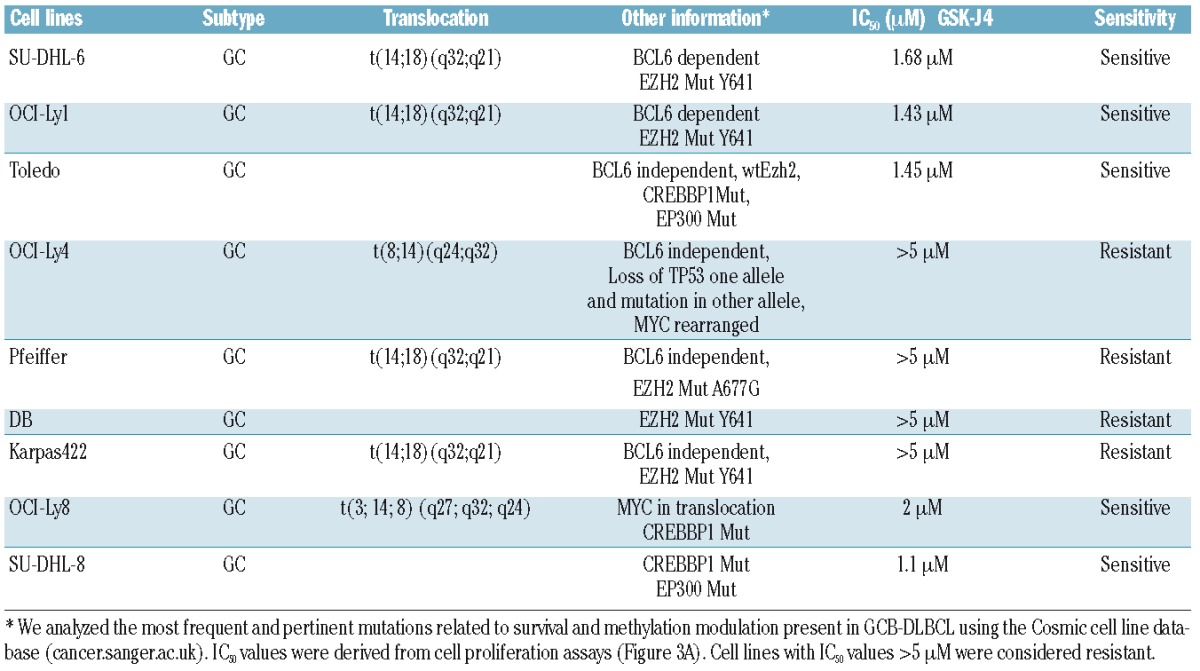
We first confirmed that low micromolar concentrations of GSK-J4 (1.5 μM) inhibit demethylase activity (Figure 2A) and produce a concurrent accumulation of H3K27(me)3 marker (Figure 2B). The dose-dependent correlated increase in H3K27(me)3 suggested that GSK-J4 can inhibit KDM6B and alter histone modification patterns. To test the potential of KDM6B inhibition, we treated a panel of GCB-DLBCL cell lines with different concentrations of GSK-J4 (0.5 μM – 5 μM) and analyzed viable cell numbers by using WST-1 reagent after 24 h. Cell lines fell clearly into resistant (Karpas-422, DB, Pfeiffer, OCI-Ly4) and sensitive (OCI-Ly1, OCI-Ly8, Toledo, SU-DHL-6, and SU-DHL-8) groups (Figure 3A). SU-DHL-8 was the most sensitive GCB-DLBCL cell line with an IC50 value of 1.1 μM. To elucidate whether GSK-J4 affected proliferation or induced apoptosis, we analyzed cells for DNA content after 24 h of incubation with 1.5 μM GSK-J4. Treatment with GSK-J4 significantly induced apoptosis in these cell lines and patients’ samples (Figure 3B). We then considered whether known genetic aberrations in cell lines were related to IC50 values of GSK-J4 (Table 1), and noted that GCB-DLBCL cell lines with the CREBBP1/EP300 mutation were sensitive to GSK-J4. In addition, previously known BCL6-dependent GCB-DLBCL cell lines, SU-DHL-6 and OCI-Ly1,13,36 were also sensitive to GSK-J4. BCL6 activity is known to be regulated by acetylation status37 or by BCR signaling.38 Thus one possibility is that GSK-J4 may target BCR-driven survival pathways, such as BCL6, in GCB-DLBCL. We, therefore, explored the effect of KDM6B inhibition on BCR-centered signaling, a common driver of DLBCL proliferation.
Figure 2.
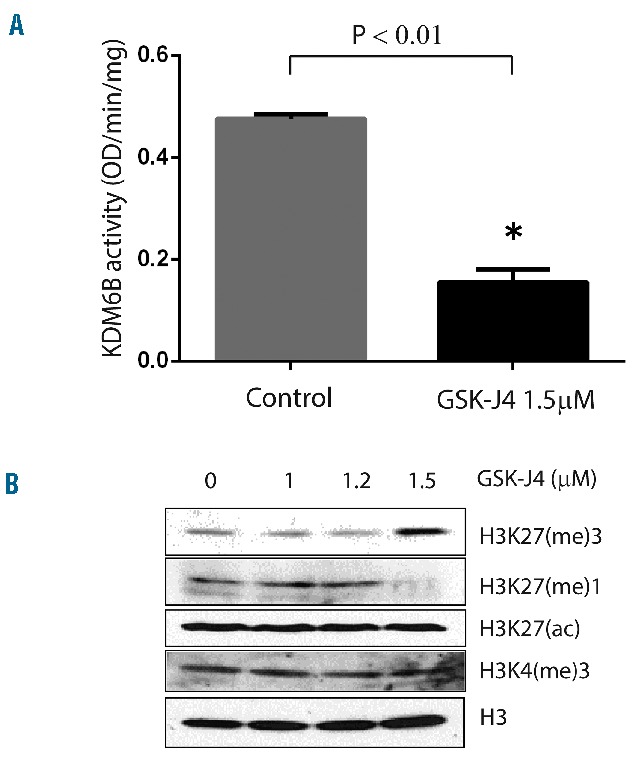
GSK-J4 targets enzymatic activity of KDM6B. (A) GSK-J4 induced alteration in KDM6B enzyme activity (OD/min/mg). SU-DHL-6 cells were incubated with GSK-J4 (1.5 μM) for 24 h and enzyme activity was calculated as described in the Methods section. (B) GSK-J4 treatment-induced changes in histone modifications as analyzed by western blot 24 h after GSK-J4 treatment in SU-DHL-6 cells.
Figure 3.
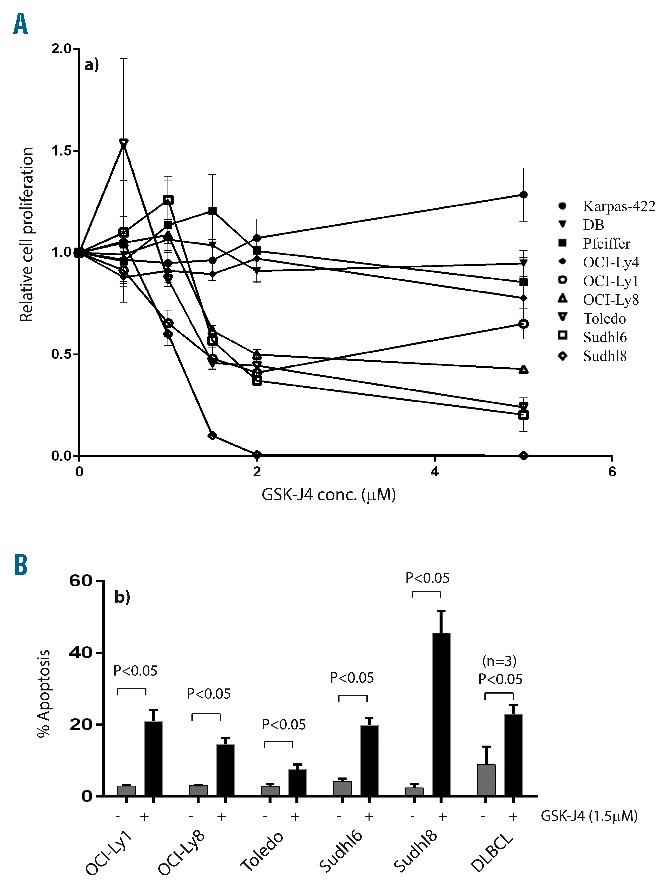
Sensitivity of DLBCL cell lines to GSK-J4. (A) WST-1 assay showing drug concentration-dependent effects of GSK-J4 (0.5–5 μM) on proliferation of DLBCL cell lines, Karpas-422, DB, Pfeiffer, OCI-Ly4, OCI-Ly1, OCI-Ly8, Toledo, SU-DHL-6, SU-DHL-8. (B) Sub-G1 apoptosis analysis of GSK-J4 (1.5 μM) induced cell death in sensitive DLBCL cell lines and fresh DLBCL patients’ samples (n=3). Apoptosis rates were compared to those of buffer-treated controls.
Table 1.
DLBCL cell lines characteristics.
KDM6B inhibition by GSK-J4 affects B-cell receptor-driven survival pathways
Treatment with GSK-J4 resulted in reduction of phosphorylation of proteins mediating BCR signaling, such as pBTKTYR223, pCD79ATYR182, and pPLCγTYR759, 24 h after treatment in SU-DHL-6 cells (Figure 4A). To show whether BCR signaling is affected by GSK-J4 treatment, we compared the sensitivity of BCR knockout and wildtype cells to GSK-J4 treatment. The BCR knockout cells were less sensitive to GSK-J4 than were the BCR wildtype SU-DHL-6 cells, indicating that a part of GSK-J4 cytotoxicity involves BCR signaling and that GSK-J4 requires wildtype BCR for its cytotoxic effect (Figure 4B). GSK-J4 treatment at concentrations that induce cell death also reduced protein levels of BCL6 in GCB-DLBCL (Figure 4C). To confirm that the BCL6 downregulation observed after GSK-J4 treatment was specifically due to KDM6B inhibition, we performed western blot analysis for BCL6 in KDM6B knockdown SU-DHL-6 cells. Inhibition of KDM6B by pooled short interfering RNA also resulted in downregulation of BCL6 levels (Figure 4D). A reduction in BCL6 levels was also observed in BCR knockout cells (Figure 4E). In addition, reduction in BCL6 levels was not observed in DB, a GSK-J4-insensitive cell line. It appears that downregulation of BCR signaling is responsible for reducing levels of survival pathway proteins, such as BCL6, in GCB-DLBCL.
Figure 4.

KDM6B inhibition by GSK-J4 affects BCR-driven survival pathways. (A) Analysis of B-cell/BCR signaling using western blot for phosphorylated/active forms of pBTKTYR223, pPLCγTYR759, and pCD79ATYR182. (B) Sensitivity of BCR wild type (WT) and BCR-knockout (KO) (generated with Crisper-Cas9 KO constructs) SU-DHL-6 cells to treatment of GSK-J4 was analyzed as change in absolute number of BCR WT and BCR-KO cells (normalized with equal numbers of beads) 24 h after treatment. (C) Effect of GSK-J4 on survival pathway proteins such as BCL6 in SU-DHL-6 cells analyzed by western blots. (D) KDM6B knockdown SU-DHL-6 cells generated using siRNA against KDM6B were used to analyze specific inhibition of BCL6 by western blot. (E) Effect of BCR knockdown on BCL6 levels in cell lines. BCR-KO was achieved using the Crisper-cas9 method.
Inhibition of KDM6B sensitizes diffuse large B-cell lymphoma cells to chemotherapy
In addition to studying the activity of GSK-J4 as a single agent, we also explored the molecule’s cytotoxic potential in combination with standard drugs. Treatment of SU-DHL-6 cells with GSK-J4 at a concentration of 1 μM induced negligible levels of apoptosis, whereas the apoptosis rates increased when GSK-J4 was given in combination with vincristine (4.47% to 34.8%), doxorubicin (2.71% to 24.63%), bortezomib (11.25% to 49.13%), carfilzomib (7.23% to 27.93%), vorinostat (1.89% to 23.33%) or panobinostat (3.6% to 30.3%) (Figure 5). Likewise, in another sensitive cell line, SU-DHL-8, treatment with GSK-J4 (1 μM) induced 11.66% apoptosis, whereas apoptosis rates increased when the GSK-J4 was given in combination with vincristine (7.16% to 78%), doxorubicin (21.7% to 69.43%), bortezomib (23.65% to 83.39%), carfilzomib (12.7% to 84.26%), vorinostat (18.17% to 42.56%) or panobinostat (14.14% to 57.7%). These results suggest that GSK-J4 treatment has the potential to sensitize DLBCL cells to components of CHOP therapy, proteasome inhibitors or histone deacetylase inhibitors. Our results clearly indicate that GSK-J4 sensitizes the killing effects of clinically approved drugs.
Figure 5.
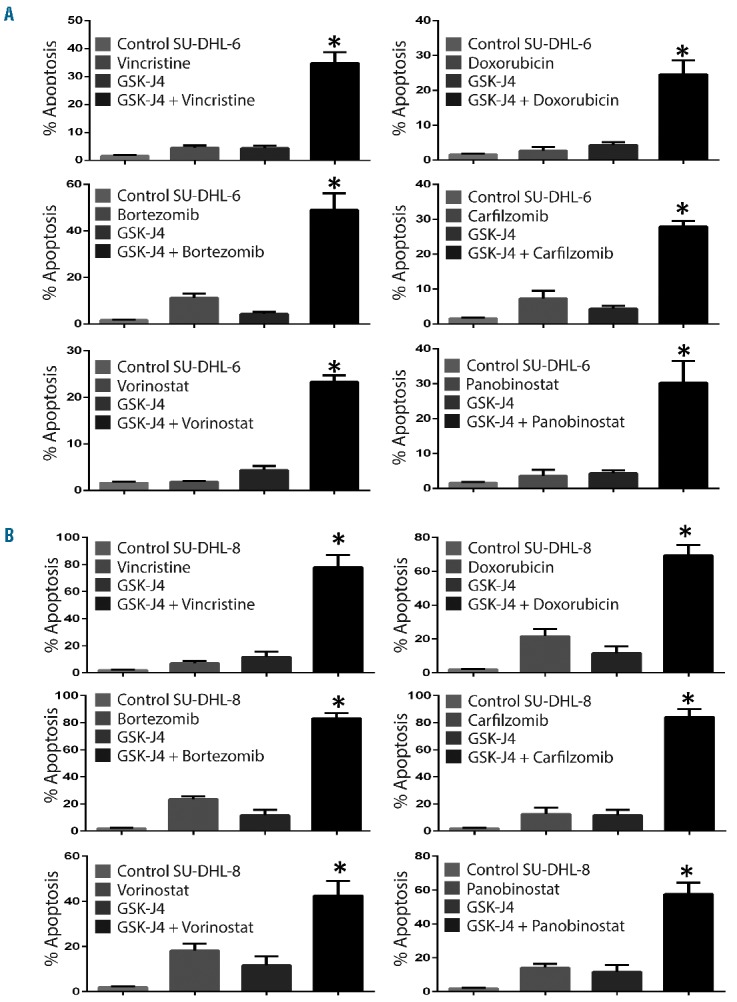
Inhibition of KDM6B sensitizes DLBCL cells to chemotherapy. (A) Analysis of apoptosis (sub-G1) induced by GSK-J4 (1 μM) in combination with various drugs i.e. vincristine (1 nM), doxorubicin (35nM), bortezomib (5 nM), carfilzomib (2 nM), vorinostat (10 nM) and panobinostat (1 nM) in SU-DHL-6 cells. (B) Analysis of apoptosis (sub-G1) induced by GSK-J4 (1 μM) in combination with various drugs i.e. vincristine (0.5 nM), doxorubicin (10 nM), bortezomib (5 nM), carfilzomib (2 nM), vorinostat (5 nM) and panobinostat (0.5 nM) in SU-DHL-8 cells. The percentage of sub-G1 was analyzed using propidium iodide staining. * Represents differences between the combination treatment and single drug treated groups that were statistically significant (P<0.05).
Discussion
Coexistence of epigenetic activation [H3K4 (me)3] and repression [H3K27(me)3] marks provides plasticity to a gene which can, therefore, be either activated or remain repressed under different situations.5,39,40 Several studies have suggested that both methylases (e.g., EZH2) and demethylases (e.g., KDM6B) coordinate to counterbalance changes in the epigenetic program during development, infection and malignancies.41–45 This would indicate the importance of KDM6B-derived demethylation in driving proliferation of DLBCL, even in the presence of an activated EZH2-dependent [H3K27(me)3] repressive environment. In addition, demethylases such as KDM2B are an essential component of the polycomb group complex required for EZH2-dependent methylation.46 Although the role of KDM6B in the polycomb complex needs further investigation, its high expression in GCB-DLBCL suggests that it has an important function. Since activating Y641 mutations of EZH2 are frequent in GCB-DLBCL cell lines and primary tumors,47 the demethylation by overexpressed KDM6B and consequent expression of genes related to cell proliferation are likely more important in GCB-DLBCL. Therefore, the demethylase KDM6B, which is overexpressed in GCB-DLBCL and associated with poor survival, is a rationale target in DLBCL.
We found that effects of GSK-J4 were specific, since the non-active analog GSK-J5 did not produce any significant toxicity. A recent study of GSK-J4 activity indicated that KDM5B, which removes H3K4(me)3 marks is also a target of GSK-J4, although with a lower affinity.48 However, our western blots showed that treatment of GCB-DLBCL cell lines with GSK-J4 did not influence the expression of H3K4(me)3 marks, thereby suggesting that KDM6B is the specific target of GSK-J4 in GCB-DLBCL.
Several studies have indicated the importance of demethylation in normal B-cell development and in malignant B cells, both of which are influenced by BCR signaling and BCL6.17 We showed that treatment with GSK-J4 leads to a reduction in BCL6 protein levels, perhaps through inhibition of BCR signaling. Indeed, a recent study suggested that KDM6B promotes the survival of DLBCL cells.49 BCR-mediated regulation of BCL6 activity in GCB-DLBCL has been recently reported.38 Our result corroborates the reported findings that knockdown of BCR leads to downregulation of BCL6. The clinical potential of targeting BCR in GCB-DLBCL has been recently elucidated.14,50–52 Our results suggest that GSK-J4 leads to a reduction in BCR activity and thereby downregulates survival pathways such as BCL6 in GCB-DLBCL.
In addition to the effect of KDM6B on BCR signaling observed in our study, several other mechanisms have been suggested in other tumor models, with implications for treatment strategies.53–57 Since epigenetic enzymes are associated with several genes, it is not surprising that inhibition of KDM6B may directly affect the expression of many genes. Despite the widespread effects at the genomic level, our results suggest a predominant downregulation of BCR signaling and provide the first evidence that demethylation can directly regulate BCR in DLBCL. Detailed investigations into the mechanism of KDM6B’s effect on BCR signaling are required to exploit this functional relationship.
Acetylation is known to inactivate BCL6;37 therefore, the high frequency of recurrent mutations in CREBBP/EP300, along with those in EZH2, further underscores the need for KDM6B activity to maintain BCL6 activity in GCB-DLBCL and explains the sensitivity of GCB-DLBCL cells with these mutations. Consistent with our observations, inhibition of BCL6 by retro-inverted BCL6 inhibitor (RI-BPI) is further sensitized by histone deacetylase inhibitors due to inhibition of EP300.36 Inhibition of KDM6B by GSK-J4, as a single agent, is directly toxic in GCB-DLBCL cells, but this small molecule inhibitor also sensitizes the cells to various chemotherapies. To conclude, our studies suggest that demethylase inhibitors can be useful in improving therapy for GCB-DLBCL.
Supplementary Material
Acknowledgments
This work was supported by grants from NCI/NIH (CA153170, and CA158692), NIDDK (DK091490), the Richard Spencer Lewis Memorial foundation, and the patients’ families. The University of Texas MD Anderson Cancer Center Flow Cytometry and Cellular Imaging Facility are supported by the NIH/NCI under award number P30CAQ16672.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/2/373
References
- 1.Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362(15):1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunleavy K, Grant C, Wilson WH. Using biologic predictive factors to direct therapy of diffuse large B-cell lymphoma. Ther Adv Hematol. 2013;4(1):43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. [DOI] [PubMed] [Google Scholar]
- 4.De S, Shaknovich R, Riester M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet. 2013;9(1):e1003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan H, Jiang Y, Boi M, et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun. 2015;6:6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinhardt JJ, Gartenhaus RB. Epigenetic approaches for chemosensitization of refractory diffuse large B-cell lymphomas. Cancer Discov. 2013;3(9):968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramachandrareddy H, Bouska A, Shen Y, et al. BCL6 promoter interacts with far upstream sequences with greatly enhanced activating histone modifications in germinal center B cells. Proc Natl Acad Sci USA. 2010;107(26):11930–11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green MR, Vicente-Dueñas C, Romero-Camarero I, et al. Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat Commun. 2014;5:3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol. 2010;105:193–210. [DOI] [PubMed] [Google Scholar]
- 10.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. 2012;247(1):172–183. [DOI] [PubMed] [Google Scholar]
- 11.Chen YW, Hu XT, Liang AC, et al. High BCL6 expression predicts better prognosis, independent of BCL6 translocation status, translocation partner, or BCL6-deregulating mutations, in gastric lymphoma. Blood. 2006;108(7):2373–2383. [DOI] [PubMed] [Google Scholar]
- 12.Parekh S, Prive G, Melnick A. Therapeutic targeting of the BCL6 oncogene for diffuse large B-cell lymphomas. Leuk Lymphoma. 2008;49(5):874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerchietti LC, Ghetu AF, Zhu X, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell. 2010;17(4):400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juszczynski P, Chen L, O’Donnell E, et al. BCL6 modulates tonic BCR signaling in diffuse large B-cell lymphomas by repressing the SYK phosphatase, PTPROt. Blood. 2009;114(26):5315–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barneda-Zahonero B, Roman-Gonzalez L, Collazo O, Mahmoudi T, Parra M. Epigenetic regulation of B lymphocyte differentiation, transdifferentiation, and reprogramming. Comp Funct Genomics. 2012; 2012:564381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10(5): 295–304. [DOI] [PubMed] [Google Scholar]
- 17.Lai AY, Mav D, Shah R, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. 2013;23(12):2030–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. [DOI] [PubMed] [Google Scholar]
- 19.Anderton JA, Bose S, Vockerodt M, et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and overexpressed in Hodgkin’s lymphoma. Oncogene. 2011;30(17):2037–2043. [DOI] [PubMed] [Google Scholar]
- 20.Burgold T, Voituron N, Caganova M, et al. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep. 2012;2(5):1244–1258. [DOI] [PubMed] [Google Scholar]
- 21.Jiang W, Wang J, Zhang Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res. 2013;23(1):122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brune V, Tiacci E, Pfeil I, et al. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J Exp Med. 2008;205(10):2251–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359(22):2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonov AV, Krestyaninova M, Knight RA, et al. PPISURV: a novel bioinformatics tool for uncovering the hidden role of specific genes in cancer survival outcome. Oncogene. 2014;33(13):1621–1628. [DOI] [PubMed] [Google Scholar]
- 26.Harrington DP, Fleming TR. A class of rank test procedures for censored survival data. Biometrika. 1982;69(3):553–566. [Google Scholar]
- 27.Wan G, Mathur R, Hu X, et al. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013;25(5):1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathur R, Chandna S, P NK, B SD. Peptidyl prolyl isomerase, Pin1 is a potential target for enhancing the therapeutic efficacy of etoposide. Curr Cancer Drug Targets. 2011;11(3):380–392. [DOI] [PubMed] [Google Scholar]
- 29.Mathur R, Sehgal L, Braun FK, et al. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J Hematol Oncol. 2015;8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sehgal L, Mathur R, Braun FK, et al. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia. 2014;28(12):2376–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jazirehi AR, Huerta-Yepez S, Cheng G, Bonavida B. Rituximab (chimeric anti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-{kappa}B signaling pathway in non-Hodgkin’s lymphoma B-cell lines: role in sensitization to chemotherapeutic drug-induced apoptosis. Cancer Res. 2005;65(1):264–276. [PubMed] [Google Scholar]
- 34.Yamamoto K, Tateishi K, Kudo Y, et al. Loss of histone demethylase KDM6B enhances aggressiveness of pancreatic cancer through downregulation of C/EBPalpha. Carcinogenesis. 2014;35(11):2404–2414. [DOI] [PubMed] [Google Scholar]
- 35.Das ND, Jung KH, Chai YG. The role of NF-kappaB and H3K27me3 demethylase, Jmjd3, on the anthrax lethal toxin tolerance of RAW 264.7 cells. PloS One. 2010;5(3): e9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerchietti LC, Hatzi K, Caldas-Lopes E, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J Clin Invest. 2010:4569–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bereshchenko OR, Gu W, Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002;32(4):606–613. [DOI] [PubMed] [Google Scholar]
- 38.Havranek O, Koehrer S, Comer JM, et al. The B-cell receptor is required for optimal viability, growth, and chemotherapy resistance of diffuse large B-cell lymphoma cell lines of the germinal center B-cell subtype. Blood. 2014;124(21):493.24904119 [Google Scholar]
- 39.Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27(12):1318–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. [DOI] [PubMed] [Google Scholar]
- 41.Siouda M, Frecha C, Accardi R, et al. Epstein-Barr virus down-regulates tumor suppressor DOK1 expression. PLoS Pathog. 2014;10(5):e1004125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol. 2013;3(3):227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci USA. 1993;90(19):9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alberghini F, Petrocelli V, Rahmat M, Casola S. An epigenetic view of B-cell disorders. Immunol Cell Biol. 2015;93(3):253–260. [DOI] [PubMed] [Google Scholar]
- 45.Pasini D, Hansen KH, Christensen J, et al. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and polycomb-repressive complex 2. Genes Dev. 2008;22(10):1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang GG, Konze KD, Tao J. Polycomb genes, miRNA, and their deregulation in B-cell malignancies. Blood. 2015;125(8):1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer cell. 2013;23(5):677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinemann B, Nielsen JM, Hudlebusch HR, et al. Inhibition of demethylases by GSK-J1/J4. Nature. 2014;514(7520):E1–2. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y. JMJD3 promotes survival of diffuse large B-cell lyphoma subtypes via distinct mechanisms. In: Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; 2014 Apr 5–9; San Diego, CA Philadelphia (PA): AACR; Cancer Res. 2014;74(19)348–348. [Google Scholar]
- 50.Cheng S, Coffey G, Zhang XH, et al. SYK inhibition and response prediction in diffuse large B-cell lymphoma. Blood. 2011;118(24): 6342–6352. [DOI] [PubMed] [Google Scholar]
- 51.L JY, Kenney T, Butterworth L, et al. Idelalisib has activity at clinically achievable drug concentrations in a subset of ABC and GCB diffuse large B-cell lymphoma and transformed follicular lymphoma cell lines. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22; Philadelphia, PA Philadelphia (PA): AACR; Cancer Res. 2015;75(15)2673–2673. [Google Scholar]
- 52.Zoellner AK, Bayerl S, Hutter G, et al. Temsirolimus inhibits cell growth in combination with inhibitors of the B-cell receptor pathway. Leuk Lymphoma. 2015;56(12): 3393–3400. [DOI] [PubMed] [Google Scholar]
- 53.Li Q, Zou J, Wang M, et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat Commun. 2014;5:5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hashizume R, Andor N, Ihara Y, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20(12): 1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams K, Christensen J, Rappsilber J, et al. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PloS One. 2014;9(5): e96545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Histone demethylase Jumonji D3 (JMJD3/KDM6B) at the nexus of epigenetic regulation of inflammation and the aging process. J Mol Med (Berl). 2014;92(10): 1035–1043. [DOI] [PubMed] [Google Scholar]
- 57.Barradas M, Anderton E, Acosta JC, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23(10):1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.