

Cardiac remodeling is frequently observed in patients with sickle cell disease (SCD), and contributes to cardiac dysfunction and premature death.1,2 Cardiac dysfunction in SCD has been related to chronic anemia and low oxygen saturation. However, the mechanisms whereby they induce cardiac dysfunction are not completely understood and additional factors still have to be identified; a potential candidate is the fibroblast growth factor 23 (FGF23). FGF23 is secreted by bone cells and controls calcitriol and serum phosphate concentrations by acting on the kidney, which is its main physiological target.3 The intact form of FGF23 (iFGF23) is the only form that circulates under physiological conditions and mediates its physiological effects. iFGF23 can be cleaved in an N- and a C-terminal fragment and it is not certain whether these fragments have any biological effects. Two different immunoassays can be used to measure iFGF23 or C-terminal FGF23 fragment (cFGF23) concentrations. In patients with chronic kidney disease (CKD), plasma FGF23 concentration progressively increases when glomerular filtration rate (GFR) declines. Elevated FGF23 concentration has been associated with cardiac hypertrophy and mortality in CKD patients4 and in the general population.5,6 Injection of iFGF23 in the heart of rats induces cardiac hypertrophy.7 In 5 of 6 nephrectomized rats (which have elevated iFGF23 plasma concentrations), treatment with an FGF receptor (FGFR) antagonist attenuated cardiac hypertrophy. The mechanism whereby iFGF23 can induce heart hypertrophy is still not completely understood but does not require the expression of the FGF23 co-receptor αKlotho. To determine whether FGF23 could contribute to cardiac remodeling in SCD patients, we measured plasma iFGF23 and cFGF23 concentrations in 77 young adult SCD patients and 172 healthy control subjects of the same ethnic background. A local ethic committee approved the study protocol (Comité de Protection des Personnes Ile de France II number: 2011531-RCEB). The main characteristics of the SCD patients and the control subjects are presented in Online Supplementary Table S1. Among the SCD patients, 53 had hemoglobin SS genotype (HbSS), 15 had hemoglobin SC (HbSC), 8 had sickle cell hemoglobin S-β thalassemia (HbS β), and one had hemoglobin SD (HbSD). Patients with SCD were younger, had lower body mass index (BMI), lower blood pressure, higher estimated GFR and lower hemoglobin concentration than controls. SCD patients had significantly higher cFGF23 plasma concentration than controls whatever their phenotypes (SS or non-SS) (Figure 1A). Fifty-eight (75.3%) SCD patients versus 18 (10.5%) controls had values above normal (150 RU/mL) (P<10−4). Patients with SS genotype had significantly higher cFGF23 concentration than non-SS patients (Figure 1A). Forty-five of the SS patients (86.5%) and 13 of the non-SS patients (52%) had cFGF23 concentration above the upper normal value (P=0.0013). We also measured FGF23 concentration with an assay that measures only iFGF23 in 50 SCD patients (33 with SS genotype and 17 with non-SS genotype). In this subgroup, cFGF23 concentration was above normal values in 39 patients (78%). By contrast, iFGF23 concentration was normal (<50 pg/mL) in all but 5 of these 50 SCD patients. Furthermore, in SCD patients, at variance with our observations in non-SCD subjects, iFGF23 and cFGF23 levels were not correlated (Online Supplementary Figure S1) suggesting that mainly cleaved FGF23 was present in the plasma of SCD patients. cFGF23 concentration correlated negatively with hemoglobin levels in SCD patients (r2=0.187, P<0.0001). We observed a negative correlation between hemoglobin levels and GFR (r2=0.062, P=0.0293) and a positive correlation between cFGF23 and GFR (r2=0.058, P=0.0343). There was no difference in serum phosphate concentration between SCD patients and controls (1.12±0.18 vs. 1.13±0.25 mmol/L; P=0.7267). Calcitriol concentration did not correlate with cFGF23 concentration in SCD patients (r2=0.038, P=0.0925). All these findings reinforce the hypothesis that elevated FGF23 in SCD patients could reflect the presence of cleaved FGF23 rather than the intact form.
Figure 1.

(A) Fibroblast growth factor 23 (FGF23) concentration in patients in sickle cell disease (SCD) and controls. C-terminal FGF23 (cFGF23) plasma concentration is higher in SCD patients with SS (circle) or non-SS (diamond) genotype than in control subjects (square). cFGF23 concentration was natural Log-transformed to obtain normal distribution. Anova P<10−4. Differences between groups were analyzed with Tukey-Kramer’s test: **P=0.0002 SS versus non-SS; ***P<10−4 versus control. (B) C-terminal FGF23 (cFGF23) induces adult rat ventricular cardiomyocyte (ARVMs) hypertrophy. cFGF23 induces cardiomyocyte hypertrophy in a dose-dependent manner. cFGF23-induced cardiomyocyte hypertrophy is abolished by the FGFR inhibitor (PD166866). FGF2 was used as a control. ARVMs in culture were treated for 24 hours with cFGF23, or FGF2, or with control medium. N=5–9 preparations. Anova P<0.0001. Comparison to control *P=0.005 Dunnet test.
We performed echocardiography in SCD patients and found that left ventricle mass index (LVMI) and left ventricle end diastolic diameter (LVEDD) were increased in 68.8% and 35.1% of SCD patients, respectively. Eight patients had an E/E’ ratio of more than 8 which is considered to be a good predictor of diastolic dysfunction (Online Supplementary Table S2).
Univariate analysis showed that LVMI, LVEDD and LVESD correlated significantly and positively with cFGF23 concentration and GFR, and negatively with hemoglobin levels (Table 1); this was seen in both SS and non-SS patients. Left ventricular ejection fraction (FE) and fraction of shortening (FS) did not correlate with FGF23, GFR, or hemoglobin values.
Table 1.
Univariate analysis of echocardiographic parameters.
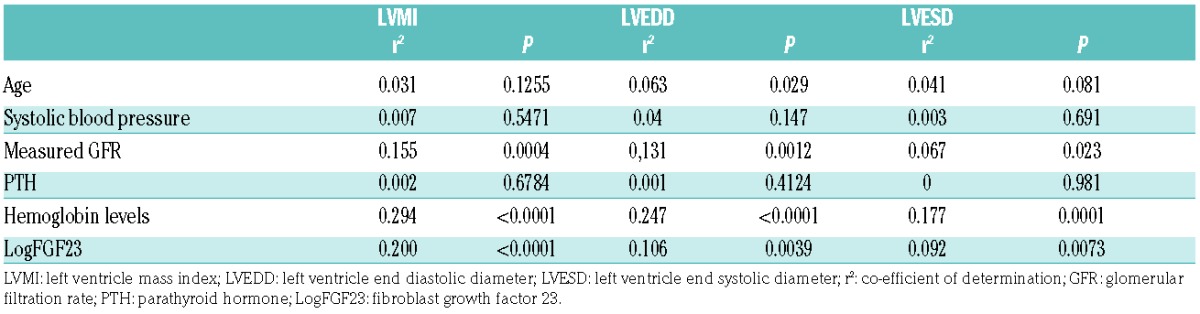
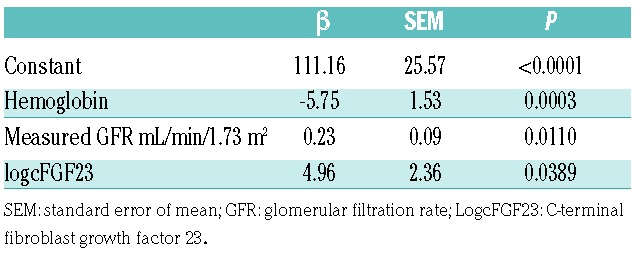
In multivariate regression analysis, the relationship between LVMI and cFGF23 remained significant after stepwise adjustment for hemoglobin and GFR (Table 2). After adjustment for GFR, LVEDD and LVESD no longer correlated with FGF23.
Table 2.
Multivariate linear regression analysis of left ventricular mass index.
To determine whether cFGF23 could induce cardiac hypertrophy, we expressed the cDNA sequence encoding amino acids 178 to 251 of FGF23 in Chinese hamster ovary cells and produced a purified protein using the Myc-tag of the protein. We cultivated adult rat ventricular myocytes (ARVMs) for 24 hours in the presence of the purified cFGF23. cFGF23 significantly increased the cell surface area of ARVMs in a concentration-dependent manner (Figure 1B). This effect was similar to that observed with FGF2 that is known to induce cardiac hypertrophy. ARVMs did not express αKlotho mRNA but did express various types of FGFR in western blot experiments (data not shown). To determine whether FGFR could mediate cFGF23-induced cardiomyocyte hypertrophy, ARVMs were incubated with cFGF23 or FGF2 alone or in the presence of an FGFR inhibitor (PD166866 from Sigma-Aldrich). The FGFR inhibitor fully prevented both cFGF23 and FGF2 induced hypertrophy (Figure 1B). Treatment of ARVMs with cFGF23 also resulted in a significant increase in the beta myosin heavy chain mRNA expression, a marker of cardiac hypertrophy (Online Supplementary Figure S2).
Our results indicate that a cleaved form of FGF23 is present in the blood of SCD patients and demonstrate for the first time that cFGF23 can induce cardiac hypertrophy via activation of a FGF receptor and may contribute to the cardiac remodeling in SCD patients. The mechanisms leading to the release of cFGF23 in the plasma of SCD patients require further investigation; however, we can make some hypotheses. cFGF23 correlated negatively with hemoglobin and positively with GFR, suggesting that cFGF23 concentration correlates with the consequences of the SCD, anemia and glomerular hyperfiltration that is an early marker of SCD renal dysfunction.8 In the present study, FGF23 elevation was not due to a decrease in GFR. Release of phosphate by hemolysis might have stimulated FGF23 release. However, several data do not support this hypothesis: there was no difference in serum phosphate concentration between control and SCD patients or between SS and non-SS patients. Iron deficit or overload can modify FGF23 concentration.9,10 A role for iron status in SCD patients is unlikely since we found no association between FGF23 and iron, transferrin or ferritin concentrations. Anemia induces tissue hypoxia that can stimulate cFGF23 production in mice.11 Bone cells UMR-106 cultured under hypoxic conditions increased FGF23 mRNA expression.11 Hypoxia-inducible factor-1α can bind to FGF23 promoter and stimulate its expression.12 In rats housed under hypobaric atmosphere, cFGF23 plasma concentration increased while iFGF23 concentration was unchanged,11 which is similar to our observations in SCD patients. In addition, cFGF23, but not iFGF23 plasma concentration, was increased in women with iron deficiency anemia.10 All these findings suggest that tissue hypoxia secondary to anemia may be the cause of the increase in cFGF23 in SCD patients. It is not known whether normalization of hemoglobin concentration would decrease cFGF23 concentration, since blood transfusion does not aim to normalize hemoglobin level in SCD patients.
Elevated FGF23 values were not associated with modifications of phosphate or calcitriol levels and this led us to suspect that FGF23 was not intact. This was confirmed by the comparison of two FGF23 assays that measure either intact FGF23 or the carboxy-terminal fragment. This lack of correspondence between these two assays was not observed in non-SCD-patients investigated in our department at the same time and in the same conditions as the SCD patients, making it very unlikely that the difference might be secondary to problems related to analytical methods.
Our data are thus consistent with the release of cFGF23 in SCD patients. One study reported an increase in iFGF23 plasma concentration in SCD children with elevated serum phosphate concentration and normal renal phosphate excretion, suggesting a renal resistance to iFGF23.13 In rat, cFGF23 can inhibit the iFGF23-induced phosphaturic effect,14 suggesting that a parallel increase in cFGF23 could explain the lack of effect of iFGF23 on phosphate excretion in these children. Unfortunately, cFGF23 concentration was not assessed in this study.
Our results in ARVMs confirm that cFGF23 could be responsible for the cardiac hypertrophy in SCD patients. While it has been demonstrated in mice and in neonate rat cardiomyocytes that iFGF23 can induce heart hypertrophy, the effect of cFGF23 per se has never been addressed. It is not known whether patients with familial calcinosis, who have elevated cFGF23 and low iFGF23 plasma concentrations, have cardiac hypertrophy. The present study is the first to provide evidence of a role of cFGF23 on heart. The C-terminal sequence is specific to FGF23, and differs from other FGFs, including FGF19 and FGF21.15 Our experiments indicate that the cFGF23 fragment by itself can increase ventricular cardiomyocyte size by stimulating an FGFR in the absence of αKlotho.
Supplementary Material
Acknowledgments
We thank Florence Lefebvre (Inserm UMR-S 1180 - LabEx LERMIT, Faculté de Pharmacie Université Paris-Sud) for her skillful help in preparing primary cultures of adult rat cardiomyocytes.
Footnotes
Funding: this work was supported by a grant from the Agence Nationale de la Recherche (ANR-13-BSV1-0002-01 CERF Labex Gr-Ex), from INSERM. This study was supported by grants from Laboratory of Excellence GR-Ex, reference ANR-11-LABX-0051. French National Research Agency, reference ANR-11-IDEX-0005-02.
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49(4):472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voskaridou E, Christoulas D, Terpos E. Sickle-cell disease and the heart: review of the current literature. Br J Haematol. 2012;157(6):664–673. [DOI] [PubMed] [Google Scholar]
- 3.Prié D, Torres PU, Friedlander G. Latest findings in phosphate homeostasis. Kidney International. 2009;75(9):882–889. [DOI] [PubMed] [Google Scholar]
- 4.Faul C. Fibroblast growth factor 23 and the heart. Curr Opin Nephrol Hypertens. 2012;21(4):369–375. [DOI] [PubMed] [Google Scholar]
- 5.Souma N, Isakova T, Lipiszko D, et al. Fibroblast Growth Factor 23 and Cause-Specific Mortality in the General Population: The Northern Manhattan Study. J Clin Endocrinol Metab. 2016;jc2016–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lutsey PL, Alonso A, Selvin E, et al. Fibroblast growth factor-23 and incident coronary heart disease, heart failure, and cardiovascular mortality: the Atherosclerosis Risk in Communities study. J Am Heart Assoc. 2014;3(3):e000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haymann JP, Stankovic K, Levy P, et al. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010;5(5):756–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braithwaite V, Prentice AM, Doherty C, Prentice A. FGF23 is correlated with iron status but not with inflammation and decreases after iron supplementation: a supplementation study. Int J Pediatr Endocrinol. 2012;2012(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res. 2013;28(8):1793–1803. [DOI] [PubMed] [Google Scholar]
- 11.Clinkenbeard EL, Farrow EG, Summers LJ, et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res. 2014;29(2):361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Doucet M, Tomlinson RE, et al. The hypoxia-inducible factor-1alpha activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res. 2016;4:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raj VM, Freundlich M, Hamideh D, et al. Abnormalities in renal tubular phosphate handling in children with sickle cell disease. Pediatr Blood Cancer. 2014;61(12):2267–2270. [DOI] [PubMed] [Google Scholar]
- 14.Goetz R, Nakada Y, Hu MC, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA. 2010;107(1):407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goetz R, Ohnishi M, Kir S, et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J Biol Chem. 2012;287(34):29134–29146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.